Abstract
Mitochondrial Cardiomyopathy (MCM) is a common manifestation of multi-organ Mitochondrial Diseases (MDs), occasionally present in non-syndromic cases. Diagnosis of MCM is complex because of wide clinical and genetic heterogeneity and requires medical, laboratory, and neuroimaging investigations. Currently, the molecular screening for MCM is fundamental part of MDs management and allows achieving the definitive diagnosis. In this article, we review the current genetic knowledge associated with MDs, focusing on diagnosis of MCM and MDs showing cardiac involvement. We searched for publications on mitochondrial and nuclear genes involved in MCM, mainly focusing on genetic screening based on targeted gene panels for the molecular diagnosis of the MCM, by using Next Generation Sequencing. Here we report twelve case reports, four case-control studies, eleven retrospective studies, and two prospective studies, for a total of twenty-nine papers concerning the evaluation of cardiac manifestations in mitochondrial diseases. From the analysis of published causal mutations, we identified 130 genes to be associated with mitochondrial heart diseases. A large proportion of these genes (34.3%) encode for key proteins involved in the oxidative phosphorylation system (OXPHOS), either as directly OXPHOS subunits (22.8%), and as OXPHOS assembly factors (11.5%). Mutations in several mitochondrial tRNA genes have been also reported in multi-organ or isolated MCM (15.3%). This review highlights the main disease-genes, identified by extensive genetic analysis, which could be included as target genes in next generation panels for the molecular diagnosis of patients with clinical suspect of mitochondrial cardiomyopathies.
1. Introduction
Cardiomyopathies (CM) represent a wide group of human disorders, extending from isolated cardiac diseases to cardiac involvement in the context of complex diseases, such as neuromuscular disorders, mitochondrial pathology, and metabolic/infiltrative/storage disease [1,2]. An extensive clinical examination, comprising a systematic screening of several organs, should be, initially, conducted to define whether the cardiomyopathy is isolated or it is part of a multisystem disorder. Furthermore, the family history, specific symptoms, and the presence of typical clinical and instrumental findings may help the physicians in the differential diagnosis between mitochondrial cardiomyopathy (MCM) and sarcomere disease. Indeed, mitochondrial diseases (MDs) may manifest with structural and clinical features of hypertrophic (HCM), dilated (DCM), or restrictive (RCM) cardiomyopathy, in absence of concomitant coronary artery disease, valvular disease, or hypertension [3,4,5,6,7]. MDs are a group of rare disorders (prevalence 5–12/100,000), due to both mitochondrial DNA (mtDNA) and nuclear genome (nDNA) alterations, characterized by defect of oxidative phosphorylation (OXPHOS), impairing the ATP production [8,9,10,11,12] (Supplementary data: mitochondrial disease).
MDs show an extremely clinical, biochemical, and genetical heterogeneous phenotype, depending on the involved tissue, the specific nuclear or mitochondrial DNA mutation, and mtDNA heteroplasmic level. Several degenerative, neuromuscular, ophthalmological and gastroenterological disorders, deafness, and endocrine manifestations [13,14,15,16], such as diabetes mellitus, are frequently observed in the affected individuals [11,13,17,18,19,20]. In MDs the heart, being a high-energy demand tissue, is one of the main affected organs, [21]. In fact, cardiac involvement in patients with MDs has been estimated to be approximately 20–25%, and up to 40% in children [19,22] and 30% in adults [23].
In mitochondrial cardiomyopathies, the biogenesis of mitochondria represents an adaptive response to the mitochondrial defects. In these pathologies, the energy deficiency, mechanical interference due to sarcomere misalignment and uneven contraction, increased oxidative stress, and uncoupled respiration are possible harmful factors to myocyte function [24].
In particular, HCM due to inter-myofibrillary proliferation of mitochondria and resulting in mechanical damage and deficit of sarcomere function, is the most common cardiac manifestation of MDs, occurring in approximately 40–50% of MCM cases [7,16,19]. Dilated or restrictive cardiomyopathy and cardiac conduction defects are less commonly identified in MDs [25].
Although some MDs may affect a single organ, most of them interest multiple organs, displaying a clinical syndrome (Supplementary data: mitochondrial disease).
In many individuals, the diagnosis is complex and requires a multidisciplinary approach including clinical, biochemical, histochemical, immunocytochemistry, and neuropathological investigations. Finally, the molecular analysis, by mtDNA and nDNA sequencing, may reveal the genetic basis of the pathology. To date, besides the 13 OXPHOS system proteins, 22 tRNA, and 2 rRNA, overall encoded by the mitochondrial genome, about 1500 nuclear-genome encoded proteins, encompass the mammalian mitochondrial proteome. These proteins constitute the structural subunits of the OXPHOS system, are responsible of the mtDNA maintenance, replication, and function, as well as are involved in mitochondrial assembly and stability of the five OXPHOS complexes. [1,16,26,27]. Because of this great genetic heterogeneity, the MDs genetic testing, through the candidate gene sequencing approach, may not identify the molecular alteration, leaving many patients without diagnosis. In recent years, high-throughput sequencing strategies, such as next-generation sequencing (NGS), are proving to be fundamental for the diagnosis of MD patients and for mechanistic MDs studies [28,29,30].
In this article, we reviewed the current genetic knowledge on MDs, focusing on diagnosis and management of MCM and MDs showing cardiac involvement. We searched for publications on mitochondrial and nuclear genes involved in mitochondrial cardiomyopathies, and identified a number of targeted genes, which should be included in genetic-panel for the molecular diagnosis of the mitochondrial cardiomyopathies, by using Next Generation Sequencing.
2. Mutations in Nuclear and Mitochondrial Genes
Several variations in mitochondrial and OXPHOS-related nuclear protein coding genes cause a reduction in enzyme function in one or more of the RC complexes, while mt-tRNA mutations may impair mitochondrial translation, by decreasing the availability of functional mt-tRNAs [31]. These dysfunctions, overall, cause defects of the oxidative phosphorylation system and subsequently impair the ATP production, leading to different phenotypes, depending on the tissue involved, type of pathogenic mutations and heteroplasmy level [10,11]. Estimates suggest that about 15–20% of RC defects are due to mtDNA mutations, while the remaining are caused by defects in nuclear genes [19,32].
Mitochondrial mutations may be inherited (~75% cases) or may occur de novo (~25% cases) [33]. MtDNA point mutations represent the most frequent causes of mtDNA alterations [8,11]. MtDNA point mutations (including small indel mutations) are pretty frequent as they are found in 1:200 newborns; however many individuals carry low heteroplasmic level of the mutant gene, thus they remain asymptomatic [34]. On the contrary, large-scale mtDNA deletions are generally rare (1.5/100,000) and typically arise as de novo mutations during embryonic development. Conversely to nDNA rearrangements, large-scale mtDNA deletions have a low recurrence risk (<10% risk of transmission), although the identification of the same deletion in mother and son suggests a matrilineal transmission [35,36,37]. In a more recent classification, the primary mitochondrial diseases (PMD) are caused by germline mutations, in mtDNA and/or nDNA genes encoding RC proteins or in nDNA genes involved in the complex protein-organization, needed for mitochondrial RC [38]. Conversely, secondary mitochondrial dysfunction (SMD) refers to abnormal mitochondrial function caused by mutations in non-OXPHOS genes affecting the mitochondrial dynamics and its capacity to produce ATP. SMD can be inherited or acquired (due to non-genetic causes such as environmental factors) which is an important distinction from PMD, which can only be inherited.
Furthermore, in addition to ATP deficiency, mitochondrial dysfunction, contributing to pathogenesis of MDs, may also affect calcium handling, ROS production, apoptosis dysregulation and nitric oxide (NO) deficiency [39].
Although the production of ROS and accumulation of their active metabolites is reported as the major known cause of mtDNA mutations [40], recent studies have reported mitochondria fission and fusion (mitochondrial dynamics) imbalance in cardiomyopathies [41]. In particular, the processes of mitochondrial fusion, fission, biogenesis and mitophagy, involved in mitochondrial morphology, quality, and abundance, have recently been implicated in cardiovascular disease. Studies suggest that changes in mitochondrial morphology may be an adaptive mechanism during cardiac pathological remodeling [42].
Genetic deletions of mitochondrial fusion-promoting genes, Mitofusin 1 and 2 (MFN1, MFN2), have been shown to induce mitochondrial fragmentation, whereas deletions of mitochondrial fission-promoting gene (Dynamin-1-like DRP1) cause mitochondrial enlargement. Likewise, because the inner mitochondrial membrane fusion protein OPA1 (optic atrophy factor 1 OPA1 gene) is essential for maintaining the shape of the mitochondria and normal cristae structure, cardiomyopathies, caused by OPA 1 gene deletions may determine defective mitochondrial inner membrane architecture [43]. Chen et al. [44] described small and fragmented mitochondria in both human and rat models of heart failure, which were associated with decreased OPA1 levels.
Furthermore, nuclear gene mutations, regulating mtDNA replication and maintenance, such as mitochondrial transcription factor A (TFAM), mtDNA polymerase γ (POLG), and PEO1 (Twinkle) genes have been associated with cardiac diseases [45,46].
3. Mitochondrial Cardiomyopathy (MCM)
The most frequent cardiac disorders refereed to mitochondrial dysfunction are cardiomyopathies [3,16,25]. Although the true prevalence of mtDNA-related CM is unknown, based on the prevalence of mtDNA disease and frequency of cardiac manifestations, about 1/10,000–15,000 of the general population is affected [7]. Furthermore, several studies report different prevalence between pediatric and adult population, with cardiomyopathy estimated to occur in 20–40% of children with MDs [16,22,47,48].
Mitochondrial cardiomyopathy is a myocardial disorder, without concomitant coronary artery disease, valvular disease, congenital heart disease, and hypertension, characterized by abnormal myocardial structure and/or function, subsequent to genetic alterations and resulting in the impairment of the mitochondrial respiratory chain [4]. Hypertrophic cardiomyopathy, dilated cardiomyopathy, and left ventricular non compaction (LVNC) are the main manifestations of MCM. Histiocytoid cardiomyopathy (HICMP) (Purkinje fiber dysplasia) and restrictive cardiomyopathy, as well as Takotsubo syndrome (TTS) have been also observed [16,19].
Moreover, MCM, in addition to the myocardium, may affect other cardiac tissue, such as the cardiac conduction system, cardiac valves, aortic root, coronary arteries or the pericardium, and connective tissue, thus leading impulse generation/conduction insufficiency, valvular diseases, systolic dysfunction, or heart failure and pulmonary arterial hypertension (PAH). Conduction system disorders arise in 5–10% of patients with MD, especially in patients with associated neuromuscular disease [49].
The abnormal heart-muscle structure and/or function is due to genetic defects, impairing the functionality of the mitochondrial RC complexes, their assembly and stability factors, tRNAs, rRNAs, mtDNA maintenance, and coenzyme Q10 (CoQ10) synthesis [19,50].
The cardiac involvement in MDs may be already evident at onset, but in some cases, it may develop over time. More rarely, in patients without or with mild multisystem involvement, the heart may be the only or the main clinically affected organ.
The imbalance of mitochondrial function in MCM is supported by a large amount of scientific publications, investigating the cardiological aspect both as one of the manifestations of a syndromic mitochondrial disorder and, in a smaller number of cases, as an isolated cardiac disease. In particular, Whabi et al. showed that myocardial involvement in mtDNA diseases is progressive and, in association with other cardiovascular manifestations and risk factors (i.e., intraventricular conduction block, diabetes, premature ventricular contractions, and left ventricular hypertrophy-LVH), is an independent predictor of morbidity and early mortality [23].
3.1. Hypertrophic Cardiomyopathy
Typical cardiac manifestation of MDs is HCM, occurring in about 50% of patients with MCM [22,51,52]. Generally, these subjects do not show left ventricular outflow tract obstruction and evolution toward left ventricular systolic dysfunction is more common than in sarcomere HCM [7,25]. Uncommonly, HCM appears as the only MDs manifestation, while frequently it’s part of a syndromic disorder, like mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS) (OMIM 540000), myoclonic epilepsy with ragged-red fibers (MERRF) (OMIM 545000), Chronic progressive external ophthalmoplegia (CPEO) (OMIM 157640), Leber hereditary optic neuropathy (LHON) (OMIM 535000), or neurogenic weakness with ataxia and retinitis pigmentosa (NARP) (OMIM 551500), diseases [53].
Mutations in nuclear and mitochondrial genes encoding mitochondrial RC complex subunits and mitochondrial RC complex assembly factors have been associated with HCM [54,55,56,57,58]. Furthermore, mutations in nuclear TSFM, encoding a mitochondrial translation elongation factor, have been identified in HCM or DCM with multi-organ disease [59].
Point mutations in mtDNA, particularly in mt-tRNA genes, seem to be more frequently associated with hypertrophic phenotype.
Left ventricular hypertrophy has been identified in 38–56% of patients harboring the m.3243A>G mutation, in the tRNA for Leucine gene (MTTL), which is the most common mutation involved in several MDs, also reported in DCM patient [25].
Furthermore, heart failure with impaired systolic function and ventricular dilatation, leading sudden death has been reported in patients with LVH bearing the mutations m.3243A>G in MTTL gene or m.8344A>G in MTTK gene [4,60]. Sudden death may occur in patients bearing the m.3243A>G mutation also in absence of myocardial involvement [61].
Table 1 shows the main nuclear and mitochondrial gene mutations associated with different cardiological phenotype [62,63,64,65].
3.2. More Rare Cardiomyopathies
Dilated cardiomyopathy occurs more rarely in MD patients, in which DCM may represent the initial manifestation of cardiac involvement [66]. Furthermore, some studies found DCM more frequently associated with mitochondrial diabetes (MIDD, Maternally Inherited Diabetes and Deafness) (OMIM 520000) [67]. More commonly, the cardiac chamber dilation and systolic dysfunction represent the progression of pre-existing hypertrophy, reported in about 10% of HCM patients [68]. Mutations in tRNA or in genes encoding subunits of the mitochondrial RC complexes as well as single or large-scale mtDNA deletion and/or depletion have been observed (Table 1) [7,60,69]. Mitochondrial cardiomyopathy may also appear as RCM, LVNC, or HICMP [70,71].
Mutations in mtDNA are common in LVNC, whereas sarcomere or ion channel genes variants account for few LVNC cases [72]. LVNC is caused by defects during cardiac development with abnormal myofibrils compaction and may evolve towards progressive ventricular dilatation and systolic dysfunction. This disease is commonly reported as a cardiac manifestation in multisystem diseases, especially in pediatric populations [4,73,74]. Histiocytoid cardiomyopathy is another rare cardiomyopathy, showing severe cardiac arrhythmias or dilated cardiomyopathy and arising in infancy or childhood [75]. Autosomal recessive, X-linked, and maternal transmission have been described; nevertheless, several reports indicate that HICMP is a cardiac manifestation of MDs and should be considered as MCM [76,77,78]. Histiocytoid cardiomyopathy is characterized by infiltration of the myocardium by pathognomonic histiocyte-like cells with foamy or granular cytoplasm full of morphologically abnormal mitochondria, showing significantly decreased succinate-cytochrome C reductase (SCR) or NADH-cytochrome C reductase activity [78]. Known molecular defects associated with HICMP are in Table 1.
3.3. Mitochondrial DNA in Conduction System and Coronary Heart Disease
MDs are responsible for defects of the contractile cardiac system, leading, indirectly, to dysfunction in cardiac electrical function. Arrhythmogenic cardiac diseases, including atrioventricular (AV) blocks, ventricular and supraventricular arrhythmias, associated with pre-excitation syndrome, represent the most common abnormalities of the cardiac electric system in MDs. Reduced ATP synthesis and increased ROS production, caused by the mitochondrial dysfunction, can directly damage the cardiomyocyte excitability.
In patients with mitochondrial dysfunction, the reduced ATP synthesis leads to decreased ATP/ADP which results in constitutive opening of the ATP-sensitive potassium (KATP) channels on the sarcoplasmic membrane; this reduces electrical transmission, by creating current sinks in the myocardium and decreases refractory periods, both increasing electrical instability and consequently, the arrhythmic risk [79].
Conduction system diseases, occurring in 5–10% of MDs patients are reported in association with MTTL and MTTK gene mutations, (Table 1) [49,60,80].
Intraventricular conduction anomalies may be responsible for sudden cardiac death (SCD) in these subjects [53,81].
In MDs patients, progression to high-grade AV block is often unexpected; hence, a fast identification of any conduction system disease is required for a timely intervention. In these patients, pacemaker is usually proposed on early PR-interval prolongation or bundle branch block [82].
Furthermore, coronary heart disease (CHD) may be a manifestation of MDs. CHD characterized by atherosclerosis of epicardial coronary arteries leading to vessel stenosis. Severe ischemia may lead to myocardial infarction and angina pectoris [83]. Genetic alterations and environmental factors increasing ROS production as well as insufficient antioxidant mechanisms, concur to the pathogenesis of atherosclerosis [84]. Numerous studies show that mtDNA mutations, altered mitochondrial DNA copy number (mtDNA-CN) and specific mtDNA haplogroups are involved in the CHD and myocardial infarction [85,86,87,88,89]. In particular, Kofler et al., found that mitochondrial haplogroup T was associated with CHD in Middle European Caucasian populations [86]. Mitochondrial mutations involved in atherosclerosis development and its clinical manifestations are showed in Table 1. These mutations cause defects in some mitochondrial respiratory chain proteins and tRNAs, leading their reduced concentration with deficit of mitochondrial function, and contributing to the development of atherosclerosis [90,91].
3.4. Other Mitochondrial Diseases with Cardiomyopathy
Other MDs with cardiological manifestations include Barth syndrome (OMIM 302060), Sengers syndrome (OMIM 212350), TMEM70-related mitochondrial complex V deficiency (OMIM 614052), and Friedreich ataxia (OMIM 229300) [92].
Barth syndrome (OMIM 302060) is an X-linked disorder, whose most common cardiac manifestations are LVNC and DCM, while HCM is uncommon. Barth syndrome is caused by mutations in the gene coding for tafazzin (TAZ), which is an inner mitochondrial membrane phospholipid transacylase with key role in remodeling of cardiolipin [93].
Sengers syndrome (acylglycerol kinase deficiency) is characterized by 3-methylglutaconic aciduria. Patients show HCM, myopathy, congenital cataracts, lactic acidosis, and exercise intolerance. Heart failure causes death in half of patients within the first year of life. The syndrome is caused by mutations in AGK gene, coding an acylglycerol kinase involved in the assembly of the mitochondrial adenine nucleotide transporter ANT1 [94]. The transmembrane protein 70, encoded by TMEM70 gene, is a protein located in the mitochondrial inner membrane involved in assembling and stabilizing of the mitochondrial respiratory chain Complex I and V proteins.
Alterations in TMEM70 cause Complex V deficiency but are reported also in the less severe Complex I deficiency and in combined OXPHOS deficiency [95].
Mutations in TMEM70 gene (Table 1) are the most common cause of ATP synthase deficiency, resulting in a multi-organ mitochondrial disease, known as neonatal mitochondrial encephalo-cardiomyopathy, which is characterized by a large variety of symptoms, including 3-methylglutaconic aciduria, lactic acidosis, mitochondrial myopathy, and HCM [96]. The main known mutations in this gene are c.317-2 A>G, located in the splice site of TMEM70 intron 2, which leads to aberrant splicing and loss of TMEM70 transcript, with most patients not surviving the neonatal period and the c.366A>T, (p.Tyr112Ter) resulting in Nuclear Type 2 Mitochondrial Complex V deficiency, showing HCM. The study of Cameron J.M. et colleagues on mitochondrial morphology in patients with mutations in this gene, revealed abnormal mitochondria, with whorled cristae and disrupted nucleoid clusters of mtDNA [97].
Finally, HCM is the cardiac manifestation of Friedreich ataxia, an autosomal recessive neurodegenerative disease caused by mutations in frataxin (FXN) gene (Table 1) encoding a mitochondrial iron-binding protein involved in the synthesis of the iron-sulphur (Fe–S) clusters (ISCs). ISCs are inorganic redox-active protein cofactors required for the activity of the OXPHOS complexes [98,99]. The absence of FXN protein debars the ISC-assembly complex and increases the iron deposition and its oxidation.
Cardiac dysfunction, mainly congestive heart failure or arrhythmia but also stroke, ischaemic heart disease, and pneumonia, are the most common causes of death in these patients [100].

Table 1.
List of known pathogenic mutations reported in association with cardiological phenotype, by MITOMAP database, literature data and using ACMG classification.
Table 1.
List of known pathogenic mutations reported in association with cardiological phenotype, by MITOMAP database, literature data and using ACMG classification.
| Cardiological Phenotype | Disorder | Mutations | Amino Acid Change | Ref |
|---|---|---|---|---|
| HCM | MELAS, MERRF, CPEO, LHON, NARP, MIDD, Sengers syndrome, Friedreich ataxia | ACAD9: c.797G>A | p.Arg266Gln | [101] |
| AGK: c.306T>G | p.Tyr102Ter | [102] | ||
| COX6B1: c.58C>T | p.Arg20Cys | [56] | ||
| FXN: GAA repeat expansion | - | [98] | ||
| MRPL3: c.950C>G | p.Pro317Arg | [63] | ||
| MRPL44: c.467T>G | p.Leu156Arg | [103] | ||
| MTCOX2: m.7896G>A | p.Trp104Ter | [58] | ||
| MTCYB: m.14849T>C | p.Ser35Pro | [104] | ||
| MTND1: m.3481G>A | p.Glu59Lys | [105] | ||
| MTND5: m.12338T>C | p.Met1Thr | [106] | ||
| MTRNR2: m.2336T>C | - | [107] | ||
| MTTK: m.8344A>G | - | [60] | ||
| MTTI: m.4300A>G | - | [65] | ||
| MTTL: m.3243A>G | - | [4] | ||
| NDUFS2: c.686C>A | p.Pro229Gln | [108] | ||
| NDUFV2: c.669_670insG | p.Ser224fs | [109] | ||
| NDUFA2: c.208+5G>A | - | [110] | ||
| NDUFAF1: c.631C>T | p.Arg211Cys | [111] | ||
| SCOX2: c.418G>A | p.Asp140Asn | [112] | ||
| SURF1: 845_846delCT | p.Ser282Cysfs | [113] | ||
| SDHD: c.275A>G | p.Asp92Gly | [114] | ||
| TMEM70: c.317-2A>G | - | [115] | ||
| TMEM70: c.366A>T | p.Tyr112Ter | [116] | ||
| TSFM: c.997C>T | p.Arg312Trp | [117] | ||
| DCM | MELAS, MIDD, LHON, Barth syndrome | MTTL: m.3243A>G | - | MITOMAP |
| MTTI: m.4300A>G | - | MITOMAP | ||
| MTTK: m.8344A>G | - | MITOMAP | ||
| MTND4: m.11778G>A | p.Arg340His | MITOMAP | ||
| TAZ: c.527A>G TSFM: c.355G>C | p.His176Arg | [118] | ||
| p.Val119Leu | [119] | |||
| RCM | Hearing loss and multi organ mitochondrial disorder, MELAS, MIDD | MTRNR1: m.1555A>G | - | MITOMAP |
| MTTL: m.3243A>G | - | MITOMAP | ||
| LVNC | MIDD | MTND1: m.3398T>C | p.Met31Thr | MITOMAP |
| HCM/LVNC | Leigh syndrome | MTND1: m.3697G>A | p.Gly131Ser | MITOMAP |
| HICMP | MERF | MTTK: m.8344A>G | - | MITOMAP |
| MTCYB: m.15498G>A | p.Gly251Asp | MITOMAP | ||
| Conduction system disease | KKS, CPEO | MTTL: m.3243A>G | - | MITOMAP |
| MTTK: m.8344A>G | - | MITOMAP | ||
| CHD | carotid atherosclerosis risk, HCM, Leigh syndrome, MELAS | MTCYB: m.15059G>A | p.Gly105Ter | MITOMAP |
| MTCYB: m.15243G>A MTND5: m.13513G>A | p.Gly166Glu | MITOMAP MITOMAP | ||
| p.Asp393Asn | ||||
| MTTL1: m.3256C>T | - | MITOMAP |
CPEO: Chronic progressive external ophthalmoplegia; CHD: Coronary heart disease; DCM: dilated cardiomyopathy; HCM: Hypertrophic Cardiomyopathy; HICMP: Histiocytoid cardiomyopathy; KKS: Kearns–Sayre syndrome; LHON: Leber hereditary optic neuropathy; LVNC: left ventricular non compaction; MELAS: mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes; MERRF: myoclonic epilepsy with ragged-red fibers; MIDD: Maternally Inherited Diabetes and Deafness; NARP: neurogenic weakness with ataxia and retinitis pigmentosa; RCM: restrictive cardiomyopathy. All the variants are reported as pathogenetic by using the American College of Medical Genetics and Genomics (ACMG) interpretations [120] and/or reported in MITOMAP (A Human Mitochondrial Genome Database. Available online: https://www.mitomap.org/; accessed on 2 October 2020), Clin Var (Clinical Variants Database. Available online: https://www.ncbi.nlm.nih.gov/clinvar/; accessed on 2 October 2020), HGMD (The Human Gene Mutation Database Professional. Available online: http://www.hgmd.cf.ac.uk/; accessed on 2 October 2020) databases and literature data.
3.5. Multiorgan Clinical Expression of Mitochondrial Cardiomyopathy
The clinical expression of MCM is often accompanied by multisystem manifestations presenting neuromuscular, endocrine, and neuro-sensorial features [121,122]. Most patients with neuromuscular signs show creatine kinase enzyme in reference values or slightly elevated levels, while higher liver enzyme levels have been found in up to 10% of patients. Renal features may include nephritic syndrome, tubulopathy, tubulointerstitial nephritis, and nonspecific renal failure. Endocrinopathies include hypothyroidism, hypoparathyroidism, diabetes mellitus, adrenocorticotropic hormone deficiency, and hypogonadism. Gastrointestinal symptoms (diarrhea, constipation, abdominal pain, nausea, and chronic intestinal pseudo-obstruction) may also be present. The main ophthalmologic manifestation is retinitis pigmentosa. Sensorineural hearing loss occurs in 7% to 26% of patients, and its prevalence increases with age [4].
4. Diagnosis
Diagnosis of MCM is particularly complex because of wide clinical and genetic heterogeneity and requires a multidisciplinary approach, including extensive medical, laboratory and neuroimaging investigations. Physicians should suspect a possible MCM in patients with signs and symptoms of cardiomyopathy and multisystem involvement without a clear cause and refer the patients to a geneticist or other specialists with MDs expertise.
Genetic counselling is a fundamental part of the diagnostic workup and should be performed by specialized personnel. A detailed pedigree of the patient’s family should be obtained, and all first-degree relatives should be referred to a specific cardiogenetic clinic. An integrated diagnostic approach, consisting of biochemical screening, histopathological studies, cardiological investigations, including magnetic resonance imaging, functional assays, and molecular genetic testing, should be conducted to reach a correct diagnosis. The identification of a causative mutation allows the presymptomatic identification and the follow up of family members. The advent of NGS techniques has revolutionized the diagnosis of MDs in terms of expanding the number of involved genes as well as the discovery of new genes potentially associated with the diseases.
4.1. Medical Examination
An initial careful clinical examination of several organs (including the evaluation of extra-cardiac features, i.e., diabetes and deafness) with different criteria for adults or children should initially be performed to determine whether the CM is an isolated pathology or part of a multisystem disease [4,16,121,123,124]. Cardiologists should suspect a mitochondrial dysfunction in patients presenting, in addition to cardiomyopathy, also hearing loss, renal dysfunction, diabetes mellitus, and peripheral myopathy.
A maternal inheritance pattern or family history of mitochondrial disease may support the suspicion of MCM [125].
4.2. Laboratory Investigation
As above reported, the laboratory investigations of suspected MD are very complex, and evaluation of selected biomarkers is necessary to guide genetic investigation. Recently, the Mitochondrial Medicine Society has provided consensus statements on the diagnosis and management of MDs [126,127].
Functional assays on tissue (typically skeletal muscle) based on measurement of RC complexes activity and mitochondrial oxygen consumption have been fundamental in the diagnosis of MDs, especially prior to the recent developments in genomics and remain important procedures to evaluate the mitochondrial function.
Although MDs present with a wide spectrum of clinical manifestations, the skeletal muscle is frequently affected and represents an excellent post-mitotic tissue with distinctive histological and histochemical hallmarks of mitochondrial pathology (in primary mtDNA-related disease). The typical presence of ragged-red fibers (RRF), by using the modified Gomori trichrome stains, represents the histologic hallmark of MDs, resulting from a compensatory response to a RC biochemical defect, although these fibers are typically absent in children and in many adults are present only in late-stage disease [4,8]. Furthermore, the histochemical activities of the partially mtDNA-encoded cytochrome C oxidase complex IV (COX) and the fully nuclear-encoded succinate dehydrogenase complex II (SDH) are measured in the diagnosis of adult-onset MDs [127].
The dual COX/SDH stain is used to determine whether mitochondrial anomalies, observed on Gomori trichrome stains, (and Hematoxylin and Eosin stain) are due to mtDNA or nDNA alterations. Mutations in mtDNA impair mainly synthesis of mtDNA-encoded complex IV subunits, resulting in a decreased or absent complex IV activity and the affected fibers show either partial or complete lack of brown staining (the so-called “COX-negative fibers”).
In contrast, SDH staining depends on the enzymatic activity of the Complex II, which is entirely encoded by nDNA. This complex maintains a normal activity even when mtDNA mutations are present [8,128].
The more recent quadruple immunofluorescence technology, using fluorescently labelled antibodies against subunits of complexes I–IV, provides accurate data on the relative amount of complex I and complex IV and could contribute to the diagnosis of MDs [129].
Furthermore, electron microscopy of the affected muscle biopsy can sometimes highlight an increased number of mitochondria with variable size and shape. In particular enlarged and swollen mitochondria, with irregular cristae and paracrystalline inclusions, are showed [130]. This mitochondrial morphology reflects the perturbation of the rates of fission and fusion, essential processes for the healthy maintenance of mitochondria, leading to either fragmented, punctiform mitochondria, or excessively long or interconnected mitochondria [131].
4.3. Cardiological and Neuroimaging Investigations
As above reported, all the major cardiomyopathy phenotypes may be shown in MDs, but hypertrophic remodeling is the dominant pattern of cardiomyopathy in MDs.
The early stages of MCM are characterized by progressive diastolic dysfunction and heart failure with preserved ejection fraction [125]. The association of LV hypertrophy (with or without apical trabeculation) with systolic dysfunction has been reported as a typical evolution of MCM. T-wave abnormalities are an ECG sign of MD, even in absence of LV hypertrophy. In a cohort of 32 MDs subjects, Limongelli et al. found an abnormal ECG in up to 68% patients [53]. Furthermore, the presence of a short PR interval, various degrees of AV block or Wolff–Parkinson–White syndrome, which is common in patients with mtDNA mutations, also supports the diagnosis.
Regarding cardiac magnetic resonance findings, characteristic patterns of cardiac association might be shown in some MDs, as reported by Florian A. et colleagues [132]. For example, concentric LV hypertrophy, with intramural late gadolinium enhancement (LGE) in the inferolateral wall may be considered a distinctive feature in patients with KSS [25].
5. Molecular Analysis of Mitochondrial Cardiomyopathies
Traditionally, for different pathologies as well as MDs, including MCM, molecular diagnosis is achieved through “candidate gene” studies. This approach is based on recognizable clinical manifestations and family history, followed by molecular testing performed by Sanger sequencing of known genes [133]. Indeed some MD phenotypes are linked to molecular defects in one or few mitochondrial and nuclear genes [12].
To date, for patients suspected of having maternally inherited mtDNA disorders, the molecular diagnosis is still initially based on the mtDNA Sanger sequencing. In adult onset cases, mtDNA aetiology is recurrent and mtDNA sequencing for point mutations or the whole mitochondrial genome, when common point mutations are not detected, remains the first approach [134].
Moreover, Sanger sequencing does not detect either heteroplasmic mutations below 15–20% or allow measuring the degree of heteroplasmy. It also does not show large deletions, which are, conversely, detectable by other different technologies (i.e., array comparative genome hybridization) [135,136,137].
The genetic diagnosis of many MDs, including MCM, is further impaired by numerous nuclear genes whose defects may cause mitochondrial diseases [28].
Nowadays, for the above mentioned reasons, the diagnosis of MCM based on clinical/biochemical and instrumental examinations and sequencing of candidate genes one by one, is not a practical approach because it leaves many undiagnosed cases.
Advances in genomics and high-throughput sequencing technologies, such as NGS, have revolutionized the diagnostic setting of MDs, allowing identification of a large number of nuclear genes simultaneously, but also providing the identification and an accurate measure of heteroplasmy, in case of mtDNA mutations [29,30,138,139,140,141,142,143]. For making diagnosis of many complex diseases, characterized by a wide clinical spectrum and heterogeneous genetic involvement, next generation sequencing based approach is available and sometimes necessary [144,145].
NGS screening encompasses different configurations, ranging from a small number of genes (targeted gene panels: i.e., Complex I genes) to hundred or more genes (targeted exome sequencing: i.e., Mito Exome, that includes about 1500 genes related to mitochondrial structure and function), and even up to Whole Exome Sequencing (WES) [122,146,147]. The choice depends by clinical, laboratory, and instrumental data, based on the clinical indication, family history, biochemical, and histopathological findings and imaging studies. The costs, time, acceptable depth of coverage and the management of off-target findings have to be also evaluated.
Variant Interpretation
The main difficulty in MDs molecular diagnosis, including MCM, is to distinguish rare or novel causative mutations from non-pathogenic polymorphisms in known/unknown genes [29].
Interestingly, rare variants may be also common in isolated genetic groups such as pathogenic mutations, which may show a higher carrier frequency within isolated populations due to the founder effect.
In the last decade, large-scale sequencing projects, integrating data from large population, have improved enormously the variants interpretation. In this context, exome data from different sequenced patients and healthy controls of the same population may contribute to assess the pathogenic role of the variant detected in the patient.
The databases of known nuclear/mitochondrial pathogenic mutations publicly accessible to define the MDs/MCM genetic diagnosis are listed in Supplementary data: online database.
Moreover, the correct classification of novel variants needs functional evidence, conferred by studies often complicated to perform and requiring specific laboratory competences [148,149]. Conversely, in silico evaluations, using several bioinformatics tools, are used to predict the likelihood of pathogenicity of missense and splicing variants. For the interpretation of missense variants the most used bioinformatics tools are: PolyPhen (Polymorphism Phenotyping. Available online: http://coot.embl.de/PolyPhen; accessed on 2 October 2020), SIFT (Sorting Intolerant From Tolerant. Available online: https://sift.bii.a-star.edu.sg/; accessed on 2 October 2020), PMut (Molecular Modeling and Bioinformatics Group. Available online: http://mmb2.pcb.ub.es:8080/PMut/; accessed 2 October 2020). Furthermore, NetGene2 (Center for Biological Sequence Analysis. Available online: http://www.cbs.dtu.dk; accessed on 2 October 2020), ESE finder (Exonic splicing enhancer. Available online: http://rulai.cshl.edu; accessed on 2 October 2020) are investigate to predict splice site mutations.
Guidelines from the American College of Medical Genetics aim to standardize variants interpretation classifying them as “pathogenic”, “likely pathogenic”, “uncertain significance variant” (VUS), “likely benign”, and “benign” considering simultaneously the weight of different criteria, also including in silico analysis [120,150].
However, the significance and the possible consequences of VUS, often detected by NGS technologies, are difficult to establish. Indeed, VUS interpretation and their consequent clinical management is a major challenge following NGS-based molecular testing. In this framework, an important step in the molecular MDs diagnostic approach is to investigate the family segregation of the detected variant in comparison with the clinical phenotype within the family. The detection of the same variant in affected family members, as well as in unrelated individuals with similar phenotype, may provide a strong additional data to support its possible pathogenic role.
Furthermore, interpretation of mtDNA variants for MDs, including MCM, should consider heteroplasmy level, maternal inheritance, and typical clinical manifestations as neuropathy and hearing loss, which provide accurate information for genetic counselling.
6. Literature Review
We reviewed and collected the literature data from July 1990 to October 2020, (one papers published in January 2021) by searching publications including genetic data from patient’s cohort showing mitochondrial cardiomyopathy or mitochondrial disorder with cardiac involvement. Database searching and Inclusion/Exclusion Criteria are reported in Supplementary data: online database. The objective was to investigate the genotype/phenotype correlation in several studies, including patients with syndromic and non-syndromic features of mitochondrial cardiomyopathy. For this aim we investigated (accessed on 2 October 2020) the most known databases, available online.
Furthermore, in recent years, especially since the introduction of NGS technology, several studies, using extensive genetic analysis, highlighted the main disease-genes associated with MCM and MDs showing cardiomyopathy; this has contributed to increase our knowledge on the molecular background of these diseases.
The second aim of this review was to identify disease-genes, by searching publications of the last ten years, in order to prove the utility of genetic tests in MCM diagnosis in patients with syndromic or non-syndromic MDs and to identify a targeted gene panel for the molecular diagnosis.
Summary of Evidence
We found twelve case reports, four case-control studies, eleven retrospective studies, and two prospective studies for a total of twenty-nine papers (Table 2).
Table 2.
Summary of literature data concerning the evaluation of cardiac manifestations in mitochondrial diseases/disorders.
Due to the number of studies identified, it was not possible to describe each of them individually. However, in the Table 2, we show the type study (case-control, case report, retrospective study, prospective study) and briefly the aim of study for each paper. Although with different inclusion criteria, ranging from genotype data (i.e., presence of specific mutation) or phenotype manifestations (i.e., mitochondrial disease, cardiomyopathy, respiratory chain disease) all the selected studies report the number of patients with cardiac phenotype and phenotype/genotype correlation.
Before the advent of NGS technology, few of the genes and mutations were known to be associated with mitochondrial diseases. For example, (as also showed in Table 2) the MT-TL1 m.3243A>G was one of the main mutations known to cause MDs (i.e., MELAS and MIDD phenotype). The use of next generation sequencing technology has revolutionized the MDs diagnosis, leading to the discovery of about half of the overall three hundred currently known genes. Thus, by searching publications regarding the most recent advances in understanding the molecular genetic basis of mitochondrial cardiomyopathy during the last ten years [7,16,19,48,151,152,154,155,174,175,176] and investigating the above mentioned databases, we identified the main nuclear and mitochondrial genes reported in association with mitochondrial cardiomyopathy and MDs showing cardiac manifestations or conduction defects.
Table 3 shows the 130 genes (mtDNA and nDNA genes) collected according to their function showed by colored squares, as reported in Figure 1. This Table can thus represent a gene panel, from literature and database search, performed for targeted genes selection and could be used for the molecular screening of patients with MCM suspect.
Table 3.
List of major mitochondrial and nuclear (mtDNA and nDNA) genes collected through literature and database research over the past ten years in association with mitochondrial heart disease.
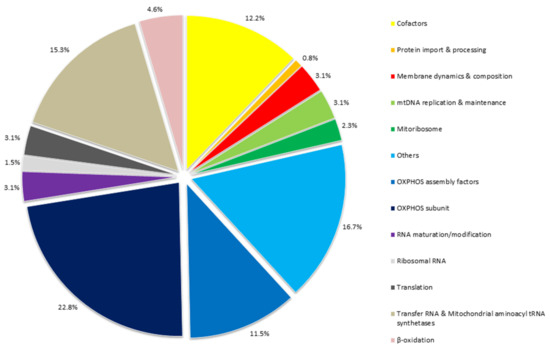
Figure 1.
Rates of protein functions and RNA involved in mitochondrial cardiomyopathies. The figure groups all the 130 genes, reported in Table 3 associated with mitochondrial heart disease, according to their function. A large proportion of these genes (34.3%) encode for key proteins in oxidative phosphorylation system, either as directly OXPHOS subunits (22.8%) and as OXPHOS assembly factors (11.5%). Furthermore, mutations in several mitochondrial tRNA genes have been reported with multi-organ or isolated MCM (15.3%). Mainly HCM, but also DCM or histiocytoid cardiomyopathy, are the main cardiomyopathies associated with pathogenic variants in genes encoding mitochondrial tRNAs. Mutations in mt-tRNA may damage overall mitochondrial translation by alteration of functional mt-tRNAs.
7. Discussion
CMs are reported by the European Society of Cardiology (ESC) Working Group on Myocardial and Pericardial Diseases as structurally and functionally myocardial disorders, in the absence of coronary artery disease, hypertension, valvular heart disease, and congenital heart disease explaining the myocardial defect [177].
The contribution of clinical molecular biology in the evaluation and prevention of cardiological risk, in particular sudden cardiac death, and in the therapeutic approach [178], both in patients and in healthy subjects and athletes, is an important goal that involves many areas of the medical profession [179,180,181,182,183].
MCM, mainly HCM followed by DCM and LVNC or, more rarely, RCM and HICMP, are common cardiac symptoms of MDs and should be considered in patients with multi-organ involvement. Cardiac abnormalities, such as cardiomyopathy, arrhythmias, and conduction defects are, indeed, present in up to 60% of patients with MD and represent often a major determinant of morbidity and mortality [53]. The wide genetic heterogeneity associated with mitochondrial cardiomyopathies, supports the opinion that the “cardiological phenotype” should often be considered as a common feature of many mitochondrial disorders and more rarely may be the first, or even the only, clinical manifestation.
Moreover, because MCM may also occur in non-syndromic MDs or in patients with only mild multisystem involvement and the differential diagnosis with non-mitochondrial cardiomyopathy through cardiological and imaging investigations is often difficult a considerable number of MCM remain undiagnosed or delayed [125]. An early recognition of MCM is crucial to initiate therapeutic procedures avoiding heart failure and arrhythmias as well as mitochondrial crisis.
Currently, the molecular screening for MCM, is a fundamental part of the management of MDs that, through an integrated multidisciplinary diagnostic approach allows getting to the diagnosis.
Recently, the Mitochondrial Medicine Society has provided consensus statements on the diagnosis and management of MDs, including measurements of mitochondrial biomarkers in plasma, urine, and spinal fluid [126].
Moreover, in patients with suspected of MD, biochemical screening, as well as histopathological studies and detailed clinical/cardiological evaluation should be followed by genetic investigation to make a definitive diagnosis.
Molecular analysis of mtDNA may be, initially performed when a clear maternally inheritance is observed, while both the mtDNA and nDNA should be considered if the inheritance pattern is uncertain.
The first objective of this review was to identify the major genes frequently associated with mitochondrial cardiomyopathies, in order to propose, by using Next Generation Sequencing technology, a novel targeted gene panel for the molecular diagnosis of the diseases. For this purpose, we explored literature and public data concerning the cardiac manifestations in mitochondrial diseases/disorders, in order to investigate the genotype/phenotype correlation in patients with MCM. This data collection allowed us to extract twenty-nine papers showing patients with mitochondrial cardiomyopathy and mtDNA and/or nDNA mutations.
The most of these papers report mtDNA mutations in association with the cardiological phenotype, in agreement with the higher frequency, in adults, of mutations in the mitochondrial genome. Moreover, many of these studies are not carried out with high-throughput genetic technologies that are able to highlight several disease-genes associated with MCM and MDs showing cardiomyopathy.
In this context, the next-generation sequencing technology has allowed, for several diseases, a huge expansion of the diagnostic screening, increasing the spectrum of involved genes and suggesting new genotype/phenotype correlations [184]. This correlation is particularly important in the diagnosis of MDs, including MCM and for the identification of pre-symptomatic family members to take in follow up.
The actual NGS strategies allow screening patients with suspected MD through a wide range of approaches, including targeted gene panel sequencing, mitochondrial gene panel, clinical exome panel and Whole Exome Sequencing (WES). The choice of the technique depends on several factors, such as funding, bioinformatics, and laboratory expertise.
WES or clinical exome panel may be considered for group of genetic disorders, which may be examined following the same pipeline. Then, the use of virtually assembled gene panel (i.e., Mitochondrial Cardiomyopathy gene panel, metabolic gene panel, etc.) allows to select only data in genes of interest and filtering for further genes or exome analysis, in a subsequent bioinformatics re-evaluation (i.e., if, in the selected panels, no mutations are found and the clinical suspicion is further deepened and confirmed). This is very important because the MD phenotype overlapped with other metabolic and neuromuscular disorders.
Although coverage for WES is constantly increasing and may be generally comparable to that obtained by targeted gene panel analysis, challenges in obtaining sufficient coverage are due to the need, for economic reasons, to collect a greater number of samples in the same analytical session.
Furthermore, the most challenging aspect of this approach is given by the large amount of unknown identified variants or novel disease genes, which need further investigation both by in silico analysis and through functional studies [185]. In this regards, the role of molecular biology research is fundamental to expand the list of pathogenic variants and increase our knowledge about MCM.
Conversely, smaller or targeted gene panels, such as the one proposed in this study, achieve a high diagnostic yield, at lower operating costs, in particular when they are designed to target the high yield genetic content, or most relevant and clinically associated genes.
This is an ideal NGS approach for the diagnosis of heterogeneous disorders, like MDs, involving dual genome.
In the post-genomic era, the benefits of NGS in molecular diagnosis of rare diseases are well established. However, the use of NGS-based approach in diagnostics is increasing the yield of VUS. The significance and possible consequences of these variants are difficult to establish. The possibility to study a specific VUS, in the context of a large pedigree by analyzing the variant’s segregation, as compared with the clinical phenotype may be useful; however, it is often unfeasible. Additionally, when clinical features suggest an underlying disease, the presence of a VUS may deserve reinterpretation regarding its pathogenicity during follow-up according to accumulating literature knowledge. We believe that the interpretation of variants should be verified approximately every 6 months. Indeed, it is not uncommon that the interpretation of variants changes after further studies are published over time. Moreover, we are fully aware that in silico predictions are not enough to establish the variants’ pathogenicity and in vitro functional studies would be needed to have more conclusive information on each identified variant.
Mitochondrial cardiomyopathies are very rare disorders; in this setting, despite laboratory capacity to generate sequence data has greatly increased, with the advent of high-throughput next generation sequencing, the capacity to interpret genomic data and to identify disease genes can be difficult. The literature review highlighted about thirty high-confidence genes known to cause mitochondrial diseases in large cohorts of MCM patients. At the same time, we selected many other genes rarely reported associated with MCM (<1% of cases, each), which are reported as potential disease-genes based on known gene function, segregation with disease in families, computational analysis, etc., although more information is needed so that they can be classified as clinically actionable genes. Consequently, the probability of finding pathogenic variants is higher in high confidence genes than in low confidence ones, using the updated guidelines for the clinical interpretation of sequence variants developed by the American College of Medical Genetics and Genomics (ACMG).
Employment, in a cohort of patients showing clinical features of MCM, of the genes panel designed by carefully selecting genes previously associated with MCM, will yield rapidly actionable results (variants in high-confidence genes) and also identify potentially causal variants (in low-confidence genes) that will require further information to be clinically actionable.
Although a lot of work is still needed to easily and quickly diagnose a Mitochondrial Cardiomyopathy, a significant number of undiagnosed patients may be identified by NGS technology.
In this review, we identified 130 disease-genes associated with MCM, by searching molecular data from papers published in the last ten years, reporting new sequencing technologies. These genes could be included as target genes to build a novel gene panel for the molecular diagnosis of patients with clinical suspect of MCM.
8. Conclusions
Mitochondrial cardiomyopathy is a challenging diagnosis, requiring wide medical, neuroimaging, and laboratory examinations. The cardiologists have to keep the degree of suspicion high, looking also for signs from other tissues involved and the family history. Echocardiography and neuroimaging findings are variable, but the hypertrophic cardiomyopathy is the most common phenotype. Moreover, the increasing use of Next Generation Sequencing technologies allows collecting more genetic data, continually improving the diagnostic success. This review collects the major genes associated with mitochondrial cardiomyopathy in recent decades, underlining how the link between research and diagnosis is crucial to expand the list of pathogenic variants and concomitantly enhance our knowledge of mitochondrial cardiomyopathy.
9. Limitation
This review has some limitations concerning both the possibility that the filters utilized for the literature and database research, the keywords used, and their association may not have been enough to avoid losing papers. Furthermore, we did not differentiate pediatric and adult studies. Finally, the panel of genes selected from such a large literature will have to be validated to know the diagnostic yield in mitochondrial cardiomyopathies.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/ijms22115742/s1, Supplementary data text: Mitochondrial disease, Mitochondrial genome (mtDNA), online database, Methodology: Database Searching, and Inclusion/Exclusion Criteria.
Author Contributions
Conceived and designed the study, G.F. and C.M.; performed the data, N.T., B.L., and O.S.; collection reviewed the cardiological data, G.L. and M.C.; analyzed the data, B.M., F.B., G.F., and C.M.; wrote the paper, B.M., G.F., and C.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Research Projects of National Interest (PRIN) from the Italian Ministry of University and Research grant (20173ZWACS).
Data Availability Statement
Data is contained within the article or Supplementary material.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AV | Atrioventricular |
| CM | Cardiomyopathies |
| COX | Cytochrome C Oxidase |
| CPEO | Chronic Progressive External Ophthalmoplegia |
| DCM | Dilated Cardiomyopathy |
| DOAJ | Directory Of Open Access Journals |
| HCM | Hypertrophic Cardiomyopathy |
| HICMP | Histiocytoid Cardiomyopathy |
| HMG | High Mobility Group |
| KSS | Kearns–Sayre Syndrome |
| LD | Linear Dichroism |
| LGE | Late Gadolinium Enhancement |
| LHON | Leber Hereditary Optic Neuropathy |
| LV | LeftVentricular |
| LVH | Left Ventricular Hypertrophy |
| LVNC | Left Ventricular Non Compaction |
| MCM | Mitochondrial Cardiomyopathy |
| MDPI | Multidisciplinary Digital Publishing Institute |
| MDs | Mitochondrial Diseases |
| MELAS | Mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes |
| MERRF | Myoclonic Epilepsy With Ragged-Red Fibers |
| MIDD | Maternally Inherited Diabetes And Deafness |
| MNGIE | Mitochondrial Neurogastrointestinal Encephalopathy |
| mtDNA | Mitochondrial DNA |
| NARP | Neurogenic Weakness With Ataxia And Retinitis Pigmentosa |
| nDNA | Nuclear DNA |
| NGS | Next-Generation Sequencing |
| NO | Nitric Oxide |
| OXPHOS | Oxidative Phosphorylation |
| PAH | Pulmonary Arterial Hypertension |
| PMD | Primary Mitochondrial Diseases |
| RC | Respiratory Chain |
| RCD | Respiratory chain disease |
| RCM | Restrictive Cardiomyopathy |
| ROS SCD | Reactive Oxygen Species Sudden cardiac death |
| SDH | Succinate Dehydrogenase |
| SMD | Secondary Mitochondrial Dysfunction |
| TFAM | Mitochondrial Transcription Factor A |
| TLA | Three Letter Acronym |
| TTS | Takotsubo Syndrome |
| VUS | Uncertain Significance Variant |
| WES | Whole Exome Sequencing |
References
- Lee, S.R.; Kim, N.; Noh, Y.H.; Xu, Z.; Ko, K.S.; Rhee, B.D.; Han, J. Mitochondrial DNA, mitochondrial dysfunction, and cardiac manifestations. Front. Biosci. 2017, 22, 1177–1194. [Google Scholar] [CrossRef]
- Towbin, J.A.; Jefferies, J.L. Cardiomyopathies Due to Left Ventricular Noncompaction, Mitochondrial and Storage Diseases, and Inborn Errors of Metabolism. Circ. Res. 2017, 121, 838–854. [Google Scholar] [CrossRef] [PubMed]
- Limongelli, G.; Masarone, D.; D’Alessandro, R.; Elliott, P.M. Mitochondrial diseases and the heart: An overview of molecular basis, diagnosis, treatment and clinical course. Future Cardiol. 2012, 8, 71–88. [Google Scholar] [CrossRef]
- Meyers, D.E.; Basha, H.I.; Koenig, M.K. Mitochondrial cardiomyopathy: Pathophysiology, diagnosis, and management. Tex. Heart Inst. J. 2013, 40, 385–394. [Google Scholar]
- Mazzaccara, C.; Limongelli, G.; Petretta, M.; Vastarella, R.; Pacileo, G.; Bonaduce, D.; Salvatore, F.; Frisso, G. A common polymorphism in the SCN5A gene is associated with dilated cardiomyopathy. J. Cardiovasc. Med. 2018, 19, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Limongelli, G.; Monda, E.; Tramonte, S.; Gragnano, F.; Masarone, D.; Frisso, G.; Esposito, A.; Gravino, R.; Ammendola, E.; Salerno, G.; et al. Prevalence and clinical significance of red flags in patients with hypertrophic cardiomyopathy. Int. J. Cardiol. 2020, 299, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Bates, M.G.; Bourke, J.P.; Giordano, C.; d’Amati, G.; Turnbull, D.M.; Taylor, R.W. Cardiac involvement in mitochondrial DNA disease: Clinical spectrum, diagnosis, and management. Eur. Heart J. 2012, 33, 3023–3033. [Google Scholar] [CrossRef] [PubMed]
- Alston, C.L.; Rocha, M.C.; Lax, N.Z.; Turnbull, D.M.; Taylor, R.W. The genetics and pathology of mitochondrial disease. J. Pathol. 2017, 241, 236–250. [Google Scholar] [CrossRef]
- Schapira, A.H. Mitochondrial diseases. Lancet 2012, 379, 1825–1834. [Google Scholar] [CrossRef]
- Liguori, R.; Mazzaccara, C.; Pasanisi, F.; Buono, P.; Oriani, G.; Finelli, C.; Contaldo, F.; Sacchetti, L. The mtDNA 15497 G/A polymorphism in cytochrome b in severe obese subjects from Southern Italy. Nutr. Metab. Cardiovasc. 2006, 16, 466–470. [Google Scholar] [CrossRef]
- Chinnery, P.F.; Hudson, G. Mitochondrial genetics. Br. Med. Bull. 2013, 106, 135–159. [Google Scholar] [CrossRef] [PubMed]
- Ng, Y.S.; Turnbull, D.M. Mitochondrial disease: Genetics and management. J. Neurol. 2016, 263, 179–191. [Google Scholar] [CrossRef]
- Naing, A.; Kenchaiah, M.; Krishnan, B.; Mir, F.; Charnley, A.; Egan, C.; Bano, G. Maternally inherited diabetes and deafness (MIDD): Diagnosis and management. J. Diabetes Its Complicat. 2014, 28, 542–546. [Google Scholar] [CrossRef]
- Jameson, E.; Morris, A.A.M. Mitochondrial disease—A review. Paediatr. Child. Health 2011, 21, 80–83. [Google Scholar] [CrossRef]
- Schaefer, A.M.; Walker, M.; Turnbull, D.M.; Taylor, R.W. Endocrine disorders in mitochondrial disease. Mol. Cell. Endocrinol. 2013, 379, 2–11. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Scaglia, F. Mitochondrial Cardiomyopathies. Front. Cardiovasc. Med. 2016, 3, 25. [Google Scholar] [CrossRef]
- Mazzaccara, C.; Iafusco, D.; Liguori, R.; Ferrigno, M.; Galderisi, A.; Vitale, D.; Simonelli, F.; Landolfo, P.; Prisco, F.; Masullo, M.; et al. Mitochondrial diabetes in children: Seek and you will find it. PLoS ONE 2012, 7, e34956. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.; Turnbull, D.M.; Walker, M.; Hattersley, A.T. Clinical features, diagnosis and management of maternally inherited diabetes and deafness (MIDD) associated with the 3243A>G mitochondrial point mutation. Diabet. Med. A J. Br. Diabet. Assoc. 2008, 25, 383–399. [Google Scholar] [CrossRef] [PubMed]
- Brunel-Guitton, C.; Levtova, A.; Sasarman, F. Mitochondrial Diseases and Cardiomyopathies. Can. J. Cardiol. 2015, 31, 1360–1376. [Google Scholar] [CrossRef]
- Popov, L.D. Mitochondrial networking in diabetic left ventricle cardiomyocytes. Mitochondrion 2017, 34, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Calvo, S.E.; Mootha, V.K. The Mitochondrial Proteome and Human Disease. Annu. Rev. Genom. Hum. G 2010, 11, 25–44. [Google Scholar] [CrossRef]
- Scaglia, F.; Towbin, J.A.; Craigen, W.J.; Belmont, J.W.; Smith, E.O.; Neish, S.R.; Ware, S.M.; Hunter, J.V.; Fernbach, S.D.; Vladutiu, G.D.; et al. Clinical spectrum, morbidity, and mortality in 113 pediatric patients with mitochondrial disease. Pediatrics 2004, 114, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Wahbi, K.; Bougouin, W.; Behin, A.; Stojkovic, T.; Becane, H.M.; Jardel, C.; Berber, N.; Mochel, F.; Lombes, A.; Eymard, B.; et al. Long-term cardiac prognosis and risk stratification in 260 adults presenting with mitochondrial diseases. Eur. Heart J. 2015, 36, 2886–2893. [Google Scholar] [CrossRef]
- Sebastiani, M.; Giordano, C.; Nediani, C.; Travaglini, C.; Borchi, E.; Zani, M.; Feccia, M.; Mancini, M.; Petrozza, V.; Cossarizza, A.; et al. Induction of mitochondrial biogenesis is a maladaptive mechanism in mitochondrial cardiomyopathies. J. Am. Coll. Cardiol. 2007, 50, 1362–1369. [Google Scholar] [CrossRef] [PubMed]
- Limongelli, G.; Masarone, D.; Pacileo, G. Mitochondrial disease and the heart. Heart 2017, 103, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Legati, A.; Reyes, A.; Nasca, A.; Invernizzi, F.; Lamantea, E.; Tiranti, V.; Garavaglia, B.; Lamperti, C.; Ardissone, A.; Moroni, I.; et al. New genes and pathomechanisms in mitochondrial disorders unraveled by NGS technologies. Biochim. Biophys. Acta 2016, 1857, 1326–1335. [Google Scholar] [CrossRef]
- Falkenberg, M.; Larsson, N.G.; Gustafsson, C.M. DNA replication and transcription in mammalian mitochondria. Annu. Rev. Biochem. 2007, 76, 679–699. [Google Scholar] [CrossRef]
- Wong, L.J.C. Next generation molecular diagnosis of mitochondrial disorders. Mitochondrion 2013, 13, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Carroll, C.J.; Brilhante, V.; Suomalainen, A. Next-generation sequencing for mitochondrial disorders. Br. J. Pharmacol. 2014, 171, 1837–1853. [Google Scholar] [CrossRef]
- Dames, S.; Chou, L.S.; Xiao, Y.; Wayman, T.; Stocks, J.; Singleton, M.; Eilbeck, K.; Mao, R. The development of next-generation sequencing assays for the mitochondrial genome and 108 nuclear genes associated with mitochondrial disorders. J. Mol. Diagn. JMD 2013, 15, 526–534. [Google Scholar] [CrossRef]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016, 2, 16080. [Google Scholar] [CrossRef] [PubMed]
- Petruzzella, V.; Papa, S. Mutations in human nuclear genes encoding for subunits of mitochondrial respiratory complex I: The NDUFS4 gene. Gene 2002, 286, 149–154. [Google Scholar] [CrossRef]
- Sallevelt, S.C.; de Die-Smulders, C.E.; Hendrickx, A.T.; Hellebrekers, D.M.; de Coo, I.F.; Alston, C.L.; Knowles, C.; Taylor, R.W.; McFarland, R.; Smeets, H.J. De novo mtDNA point mutations are common and have a low recurrence risk. J. Med. Genet. 2017, 54, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Gorman, G.S.; Schaefer, A.M.; Ng, Y.; Gomez, N.; Blakely, E.L.; Alston, C.L.; Feeney, C.; Horvath, R.; Yu-Wai-Man, P.; Chinnery, P.F.; et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 2015, 77, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, P.F.; DiMauro, S.; Shanske, S.; Schon, E.A.; Zeviani, M.; Mariotti, C.; Carrara, F.; Lombes, A.; Laforet, P.; Ogier, H.; et al. Risk of developing a mitochondrial DNA deletion disorder. Lancet 2004, 364, 592–596. [Google Scholar] [CrossRef]
- Damas, J.; Samuels, D.C.; Carneiro, J.; Amorim, A.; Pereira, F. Mitochondrial DNA Rearrangements in Health and Disease-A Comprehensive Study. Hum. Mutat. 2014, 35, 1–14. [Google Scholar] [CrossRef]
- Viscomi, C.; Zeviani, M. MtDNA-maintenance defects: Syndromes and genes. J. Inherit. Metab. Dis. 2017, 40, 587–599. [Google Scholar] [CrossRef]
- Niyazov, D.M.; Kahler, S.G.; Frye, R.E. Primary Mitochondrial Disease and Secondary Mitochondrial Dysfunction: Importance of Distinction for Diagnosis and Treatment. Mol. Syndromol. 2016, 7, 122–137. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Scaglia, F. Mitochondrial cytopathies. Cell Calcium 2016, 60, 199–206. [Google Scholar] [CrossRef]
- Schwarz, K.; Siddiqi, N.; Singh, S.; Neil, C.J.; Dawson, D.K.; Frenneaux, M.P. The breathing heart—Mitochondrial respiratory chain dysfunction in cardiac disease. Int. J. Cardiol. 2014, 171, 134–143. [Google Scholar] [CrossRef]
- Dorn, G., II. Mitochondrial fission/fusion and cardiomyopathy. Curr. Opin. Genet. Dev. 2016, 38, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Vasquez-Trincado, C.; Garcia-Carvajal, I.; Pennanen, C.; Parra, V.; Hill, J.A.; Rothermel, B.A.; Lavandero, S. Mitochondrial dynamics, mitophagy and cardiovascular disease. J. Physiol. 2016, 594, 509–525. [Google Scholar] [CrossRef]
- Protasoni, M.; Zeviani, M. Mitochondrial Structure and Bioenergetics in Normal and Disease Conditions. Int. J. Mol. Sci. 2021, 22, 586. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, T.; Tran, A.; Lu, X.; Tomilov, A.A.; Davies, V.; Cortopassi, G.; Chiamvimonvat, N.; Bers, D.M.; Votruba, M.; et al. OPA1 mutation and late-onset cardiomyopathy: Mitochondrial dysfunction and mtDNA instability. J. Am. Heart Assoc. 2012, 1, e003012. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Han, J. Mitochondrial Mutations in Cardiac Disorders. Adv. Exp. Med. Biol. 2017, 982, 81–111. [Google Scholar] [CrossRef] [PubMed]
- Rotig, A. Human diseases with impaired mitochondrial protein synthesis. BBA Bioenergy 2011, 1807, 1198–1205. [Google Scholar] [CrossRef]
- Debray, F.G.; Lambert, M.; Chevalier, I.; Robitaille, Y.; Decarie, J.C.; Shoubridge, E.A.; Robinson, B.H.; Mitchell, G.A. Long-term outcome and clinical spectrum of 73 pediatric patients with mitochondrial diseases. Pediatrics 2007, 119, 722–733. [Google Scholar] [CrossRef]
- Imai-Okazaki, A.; Kishita, Y.; Kohda, M.; Mizuno, Y.; Fushimi, T.; Matsunaga, A.; Yatsuka, Y.; Hirata, T.; Harashima, H.; Takeda, A.; et al. Cardiomyopathy in children with mitochondrial disease: Prognosis and genetic background. Int. J. Cardiol. 2019, 279, 115–121. [Google Scholar] [CrossRef]
- Brignole, M.; Auricchio, A.; Baron-Esquivias, G.; Bordachar, P.; Boriani, G.; Breithardt, O.A.; Cleland, J.; Deharo, J.C.; Delgado, V.; Elliott, P.M.; et al. 2013 ESC Guidelines on cardiac pacing and cardiac resynchronization therapy: The Task Force on cardiac pacing and resynchronization therapy of the European Society of Cardiology (ESC). Developed in collaboration with the European Heart Rhythm Association (EHRA). Eur. Heart J. 2013, 34, 2281–2329. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.C.; Lin, Y.; Xu, X.F.; Lin, S.Y.; Chen, X.M.; Wang, S.S. The alterations of mitochondrial DNA in coronary heart disease. Exp. Mol. Pathol 2020, 114. [Google Scholar] [CrossRef]
- Choi, Y.; Lee, J.H.; Cui, M.N.; Lee, Y.S.; Jung, M.H.; Yi, J.E.; Jung, H.O.; Youn, H.J. Hypertrophic Cardiomyopathy Attributable to Mitochondrial DNA Mutation Diagnosed by Pathology and Gene Sequencing. Circulation 2016, 133, 1297–1299. [Google Scholar] [CrossRef][Green Version]
- Govindaraj, P.; Khan, N.A.; Rani, B.; Rani, D.S.; Selvaraj, P.; Jyothi, V.; Bahl, A.; Narasimhan, C.; Rakshak, D.; Premkumar, K.; et al. Mitochondrial DNA variations associated with hypertrophic cardiomyopathy. Mitochondrion 2014, 16, 65–72. [Google Scholar] [CrossRef]
- Limongelli, G.; Tome-Esteban, M.; Dejthevaporn, C.; Rahman, S.; Hanna, M.G.; Elliott, P.M. Prevalence and natural history of heart disease in adults with primary mitochondrial respiratory chain disease. Eur. J. Heart Fail. 2010, 12, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Brecht, M.; Richardson, M.; Taranath, A.; Grist, S.; Thorburn, D.; Bratkovic, D. Leigh Syndrome Caused by the MT-ND5 m.13513G>A Mutation: A Case Presenting with WPW-Like Conduction Defect, Cardiomyopathy, Hypertension and Hyponatraemia. JIMD Rep. 2015, 19, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Fassone, E.; Rahman, S. Complex I deficiency: Clinical features, biochemistry and molecular genetics. J. Med. Genet. 2012, 49, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Abdulhag, U.N.; Soiferman, D.; Schueler-Furman, O.; Miller, C.; Shaag, A.; Elpeleg, O.; Edvardson, S.; Saada, A. Mitochondrial complex IV deficiency, caused by mutated COX6B1, is associated with encephalomyopathy, hydrocephalus and cardiomyopathy. Eur. J. Hum. Genet. 2015, 23, 159–164. [Google Scholar] [CrossRef]
- Marin-Garcia, J.; Goldenthal, M.J.; Ananthakrishnan, R.; Pierpont, M.E. The complete sequence of mtDNA genes in idiopathic dilated cardiomyopathy shows novel missense and tRNA mutations. J. Card. Fail. 2000, 6, 321–329. [Google Scholar] [CrossRef]
- Alston, C.; Ceccatelli Berti, C.; Blakely, E.; Olahova, M.; He, L.P.; McMahon, C.; Olpin, S.; Hargreaves, I.; Nolli, C.; McFarland, R.; et al. A recessive homozygous p.Asp92Gly SDHD mutation causes prenatal cardiomyopathy and a severe mitochondrial complex II deficiency. Hum. Genet. 2015, 134, 869–879. [Google Scholar] [CrossRef]
- Ahola, S.; Isohanni, P.; Euro, L.; Brilhante, V.; Palotie, A.; Pihko, H.; Lonnqvist, T.; Lehtonen, T.; Laine, J.; Tyynismaa, H.; et al. Mitochondrial EFTs defects in juvenile-onset Leigh disease, ataxia, neuropathy, and optic atrophy. Neurology 2014, 83, 743–751. [Google Scholar] [CrossRef]
- Wahbi, K.; Larue, S.; Jardel, C.; Meune, C.; Stojkovic, T.; Ziegler, F.; Lombes, A.; Eymard, B.; Duboc, D.; Laforet, P. Cardiac involvement is frequent in patients with the m.8344A > G mutation of mitochondrial DNA. Neurology 2010, 74, 674–677. [Google Scholar] [CrossRef]
- Ng, Y.S.; Grady, J.P.; Lax, N.Z.; Bourke, J.P.; Alston, C.L.; Hardy, S.A.; Falkous, G.; Schaefer, A.G.; Radunovic, A.; Mohiddin, S.A.; et al. Sudden adult death syndrome in m.3243A > G-related mitochondrial disease: An unrecognized clinical entity in young, asymptomatic adults. Eur. Heart J. 2016, 37, 2552–2559. [Google Scholar] [CrossRef] [PubMed]
- Distelmaier, F.; Haack, T.B.; Catarino, C.B.; Gallenmuller, C.; Rodenburg, R.J.; Strom, T.M.; Baertling, F.; Meitinger, T.; Mayatepek, E.; Prokisch, H.; et al. MRPL44 mutations cause a slowly progressive multisystem disease with childhood-onset hypertrophic cardiomyopathy. Neurogenetics 2015, 16, 319–323. [Google Scholar] [CrossRef]
- Galmiche, L.; Serre, V.; Beinat, M.; Assouline, Z.; Lebre, A.S.; Chretien, D.; Nietschke, P.; Benes, V.; Boddaert, N.; Sidi, D.; et al. Exome sequencing identifies MRPL3 mutation in mitochondrial cardiomyopathy. Hum. Mutat. 2011, 32, 1225–1231. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Song, Y.; Li, D.; He, X.; Li, S.; Wu, B.; Wang, W.; Gu, S.; Zhu, X.; Wang, X.; et al. The novel mitochondrial 16S rRNA 2336T>C mutation is associated with hypertrophic cardiomyopathy. J. Med. Genet. 2014, 51, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.W.; Giordano, C.; Davidson, M.M.; d’Amati, G.; Bain, H.; Hayes, C.M.; Leonard, H.; Barron, M.J.; Casali, C.; Santorelli, F.M.; et al. A homoplasmic mitochondrial transfer ribonucleic acid mutation as a cause of maternally inherited hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2003, 41, 1786–1796. [Google Scholar] [CrossRef]
- Govindaraj, P.; Rani, B.; Sundaravadivel, P.; Vanniarajan, A.; Indumathi, K.P.; Khan, N.A.; Dhandapany, P.S.; Rani, D.S.; Tamang, R.; Bahl, A.; et al. Mitochondrial genome variations in idiopathic dilated cardiomyopathy. Mitochondrion 2019, 48, 51–59. [Google Scholar] [CrossRef]
- Bates, M.G.; Nesbitt, V.; Kirk, R.; He, L.; Blakely, E.L.; Alston, C.L.; Brodlie, M.; Hasan, A.; Taylor, R.W.; McFarland, R. Mitochondrial respiratory chain disease in children undergoing cardiac transplantation: A prospective study. Int. J. Cardiol. 2012, 155, 305–306. [Google Scholar] [CrossRef] [PubMed]
- Terasaki, F.; Tanaka, M.; Kawamura, K.; Kanzaki, Y.; Okabe, M.; Hayashi, T.; Shimomura, H.; Ito, T.; Suwa, M.; Gong, J.S.; et al. A case of cardiomyopathy showing progression from the hypertrophic to the dilated form: Association of Mt8348A-->G mutation in the mitochondrial tRNA(Lys) gene with severe ultrastructural alterations of mitochondria in cardiomyocytes. Jpn. Circ. J. 2001, 65, 691–694. [Google Scholar] [CrossRef]
- Khogali, S.S.; Mayosi, B.M.; Beattie, J.M.; McKenna, W.J.; Watkins, H.; Poulton, J. A common mitochondrial DNA variant associated with susceptibility to dilated cardiomyopathy in two different populations. Lancet 2001, 357, 1265–1267. [Google Scholar] [CrossRef]
- Thebault, C.; Ollivier, R.; Leurent, G.; Marcorelles, P.; Langella, B.; Donal, E. Mitochondriopathy: A rare aetiology of restrictive cardiomyopathy. Eur. J. Echocardiogr. J. Work. Group Echocardiogr. Eur. Soc. Cardiol. 2008, 9, 840–845. [Google Scholar] [CrossRef]
- Santorelli, F.M.; Tanji, K.; Manta, P.; Casali, C.; Krishna, S.; Hays, A.P.; Mancini, D.M.; DiMauro, S.; Hirano, M. Maternally inherited cardiomyopathy: An atypical presentation of the mtDNA 12S rRNA gene A1555G mutation. Am. J. Hum. Genet. 1999, 64, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Batra, A.; Zhang, Y.; Ebenroth, E.S.; Huang, T.S. Left ventricular noncompaction is associated with mutations in the mitochondrial genome. Mitochondrion 2010, 10, 350–357. [Google Scholar] [CrossRef]
- Ye, Z.; Gu, X. A mitochondrial mutation A8701G is associated with maternally inherited hypertension and dilated cardiomyopathy in a Chinese pedigree of a consanguineous marriage. J. Am. Coll. Cardiol. 2016, 68, C132. [Google Scholar] [CrossRef]
- Alila, O.F.; Rebai, E.M.; Tabebi, M.; Tej, A.; Chamkha, I.; Tlili, A.; Bouguila, J.; Tilouche, S.; Soyah, N.; Boughamoura, L.; et al. Whole mitochondrial genome analysis in two families with dilated mitochondrial cardiomyopathy: Detection of mutations in MT-ND2 and MT-TL1 genes. Mitochondrial DNA A 2016, 27, 2873–2880. [Google Scholar] [CrossRef] [PubMed]
- Gilbert-Barness, E. Review: Metabolic cardiomyopathy and conduction system defects in children. Ann. Clin. Lab. Sci. 2004, 34, 15–34. [Google Scholar] [PubMed]
- Vallance, H.D.; Jeven, G.; Wallace, D.C.; Brown, M.D. A case of sporadic infantile histiocytoid cardiomyopathy caused by the A8344G (MERRF) mitochondrial DNA mutation. Pediatr. Cardiol. 2004, 25, 538–540. [Google Scholar] [CrossRef]
- Andreu, A.L.; Checcarelli, N.; Iwata, S.; Shanske, S.; DiMauro, S. A missense mutation in the mitochondrial cytochrome b gene in a revisited case with histiocytoid cardiomyopathy. Pediatr. Res. 2000, 48, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J. Histiocytoid cardiomyopathy: A mitochondrial disorder. Clin. Cardiol. 2008, 31, 225–227. [Google Scholar] [CrossRef]
- Zhou, L.F.; Solhjoo, S.; Millare, B.; Plank, G.; Abraham, M.R.; Cortassa, S.; Trayanova, N.; O’Rourke, B. Effects of Regional Mitochondrial Depolarization on Electrical Propagation Implications for Arrhythmogenesis. Circ. Arrhythmia Elec. 2014, 7, 143–151. [Google Scholar] [CrossRef]
- Majamaa-Voltti, K.; Peuhkurinen, K.; Kortelainen, M.L.; Hassinen, I.E.; Majamaa, K. Cardiac abnormalities in patients with mitochondrial DNA mutation 3243A>G. BMC Cardiovasc. Disord. 2002, 2, 12. [Google Scholar] [CrossRef]
- Lev, D.; Nissenkorn, A.; Leshinsky-Silver, E.; Sadeh, M.; Zeharia, A.; Garty, B.Z.; Blieden, L.; Barash, V.; Lerman-Sagie, T. Clinical presentations of mitochondrial cardiomyopathies. Pediatr. Cardiol. 2004, 25, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Montaigne, D.; Pentiah, A.D. Mitochondrial cardiomyopathy and related arrhythmias. Card. Electrophysiol. Clin. 2015, 7, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.F.; Hu, H.C.; Lin, Y.; Huang, F.Z.; Ji, H.H.; Yin, L.; Lin, S.Y.; Chen, X.M.; Duan, S.W. Differences in Leukocyte Telomere Length between Coronary Heart Disease and Normal Population: A Multipopulation Meta-Analysis. BioMed Res. Int. 2019, 2019. [Google Scholar] [CrossRef]
- Andreassi, M.G. Coronary atherosclerosis and somatic mutations: An overview of the contributive factors for oxidative DNA damage. Mutat Res. Rev. Mutat 2003, 543, 67–86. [Google Scholar] [CrossRef]
- Mueller, E.E.; Brunner, S.M.; Mayr, J.A.; Stanger, O.; Sperl, W.; Kofler, B. Functional Differences between Mitochondrial Haplogroup T and Haplogroup H in HEK293 Cybrid Cells. PLoS ONE 2012, 7, e52367. [Google Scholar] [CrossRef] [PubMed]
- Kofler, B.; Mueller, E.E.; Eder, W.; Stanger, O.; Maier, R.; Weger, M.; Haas, A.; Winker, R.; Schmut, O.; Paulweber, B.; et al. Mitochondrial DNA haplogroup T is associated with coronary artery disease and diabetic retinopathy: A case control study. BMC Med. Genet. 2009, 10, 35. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.Y.; Sun, L.; Chen, X.P.; Zhang, D.S. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural. Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef]
- Cenini, G.; Lloret, A.; Cascella, R. Oxidative Stress and Mitochondrial Damage in Neurodegenerative Diseases: From Molecular Mechanisms to Targeted Therapies. Oxid. Med. Cell Longev. 2020, 2020. [Google Scholar] [CrossRef]
- Ait-Aissa, K.; Blaszak, S.C.; Beutner, G.; Tsaih, S.W.; Morgan, G.; Santos, J.H.; Flister, M.J.; Joyce, D.L.; Camara, A.K.S.; Gutterman, D.D.; et al. Mitochondrial Oxidative Phosphorylation defect in the Heart of Subjects with Coronary Artery Disease. Sci. Rep. 2019, 9, 7623. [Google Scholar] [CrossRef]
- Sazonova, M.A.; Sinyov, V.V.; Barinova, V.A.; Ryzhkova, A.I.; Zhelankin, A.V.; Postnov, A.Y.; Sobenin, I.A.; Bobryshev, Y.V.; Orekhov, A.N. Mosaicism of mitochondrial genetic variation in atherosclerotic lesions of the human aorta. Biomed. Res. Int. 2015, 2015, 825468. [Google Scholar] [CrossRef]
- Mitrofanov, K.Y.; Zhelankin, A.V.; Shiganova, G.M.; Sazonova, M.A.; Bobryshev, Y.V.; Postnov, A.Y.; Sobenin, I.A.; Orekhov, A.N. Analysis of mitochondrial DNA heteroplasmic mutations A1555G, C3256T, T3336C, C5178A, G12315A, G13513A, G14459A, G14846A and G15059A in CHD patients with the history of myocardial infarction. Exp. Mol. Pathol. 2016, 100, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Jefferies, J.L. Barth syndrome. Am. J. Med. Genet. Part. C Semin. Med. Genet. 2013, 163C, 198–205. [Google Scholar] [CrossRef]
- Mazurova, S.; Tesarova, M.; Magner, M.; Houstkova, H.; Hansikova, H.; Augustinova, J.; Tomek, V.; Vondrackova, A.; Zeman, J.; Honzik, T. Novel mutations in the TAZ gene in patients with Barth syndrome. Prague Med. Rep. 2013, 114, 139–153. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Guleray, N.; Kosukcu, C.; Taskiran, Z.E.; Simsek Kiper, P.O.; Utine, G.E.; Gucer, S.; Tokatli, A.; Boduroglu, K.; Alikasifoglu, M. Atypical Presentation of Sengers Syndrome: A Novel Mutation Revealed with Postmortem Genetic Testing. Fetal Pediatric Pathol. 2020, 39, 163–171. [Google Scholar] [CrossRef]
- Sanchez-Caballero, L.; Elurbe, D.M.; Baertling, F.; Guerrero-Castillo, S.; van den Brand, M.; van Strien, J.; van Dam, T.J.P.; Rodenburg, R.; Brandt, U.; Huynen, M.A.; et al. TMEM70 functions in the assembly of complexes I and V. Biochim. Biophys. Acta. Bioenerg. 2020, 1861, 148202. [Google Scholar] [CrossRef]
- Cizkova, A.; Stranecky, V.; Mayr, J.A.; Tesarova, M.; Havlickova, V.; Paul, J.; Ivanek, R.; Kuss, A.W.; Hansikova, H.; Kaplanova, V.; et al. TMEM70 mutations cause isolated ATP synthase deficiency and neonatal mitochondrial encephalocardiomyopathy. Nat. Genet. 2008, 40, 1288–1290. [Google Scholar] [CrossRef]
- Cameron, J.M.; Levandovskiy, V.; Mackay, N.; Ackerley, C.; Chitayat, D.; Raiman, J.; Halliday, W.H.; Schulze, A.; Robinson, B.H. Complex V TMEM70 deficiency results in mitochondrial nucleoid disorganization. Mitochondrion 2011, 11, 191–199. [Google Scholar] [CrossRef]
- Koeppen, A.H.; Ramirez, R.L.; Becker, A.B.; Bjork, S.T.; Levi, S.; Santambrogio, P.; Parsons, P.J.; Kruger, P.C.; Yang, K.X.; Feustel, P.J.; et al. The Pathogenesis of Cardiomyopathy in Friedreich Ataxia. PLoS ONE 2015, 10, 0116396. [Google Scholar] [CrossRef]
- Weidemann, F.; Stork, S.; Liu, D.; Hu, K.; Herrmann, S.; Ertl, G.; Niemann, M. Cardiomyopathy of Friedreich Ataxia. J. Neurochem. 2013, 126, 88–93. [Google Scholar] [CrossRef]
- Cook, A.; Giunti, P. Friedreich’s ataxia: Clinical features, pathogenesis and management. Brit. Med. Bull. 2017, 124, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Haack, T.B.; Danhauser, K.; Haberberger, B.; Hoser, J.; Strecker, V.; Boehm, D.; Uziel, G.; Lamantea, E.; Invernizzi, F.; Poulton, J.; et al. Exome sequencing identifies ACAD9 mutations as a cause of complex I deficiency. Nat. Genet. 2010, 42, 1131–1134. [Google Scholar] [CrossRef]
- Mayr, J.A.; Haack, T.B.; Graf, E.; Zimmermann, F.A.; Wieland, T.; Haberberger, B.; Superti-Furga, A.; Kirschner, J.; Steinmann, B.; Baumgartner, M.R.; et al. Lack of the Mitochondrial Protein Acylglycerol Kinase Causes Sengers Syndrome. Am. J. Hum. Genet. 2012, 90, 314–320. [Google Scholar] [CrossRef]
- Carroll, C.J.; Isohanni, P.; Poyhonen, R.; Euro, L.; Richter, U.; Brilhante, V.; Gotz, A.; Lahtinen, T.; Paetau, A.; Pihko, H.; et al. Whole-exome sequencing identifies a mutation in the mitochondrial ribosome protein MRPL44 to underlie mitochondrial infantile cardiomyopathy. J. Med. Genet. 2013, 50, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Schuelke, M.; Krude, H.; Finckh, B.; Mayatepek, E.; Janssen, A.; Schmelz, M.; Trefz, F.; Trijbels, F.; Smeitink, J. Septo-optic dysplasia associated with a new mitochondrial cytochrome b mutation. Ann. Neurol. 2002, 51, 388–392. [Google Scholar] [CrossRef]
- Moslemi, A.R.; Darin, N.; Tulinius, M.; Wiklund, L.M.; Holme, E.; Oldfors, A. Progressive encephalopathy and complex I deficiency associated with mutations in MTND1. Neuropediatrics 2008, 39, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Song, Y.R.; Gu, S.L.; He, X.Y.; Zhu, X.Y.; Shen, Y.Y.; Wu, B.F.; Wang, W.; Li, S.S.; Jiang, P.P.; et al. Mitochondrial ND5 12338T > C variant is associated with maternally inherited hypertrophic cardiomyopathy in a Chinese pedigree. Gene 2012, 506, 339–343. [Google Scholar] [CrossRef]
- Li, D.; Sun, Y.P.; Zhuang, Q.Q.; Song, Y.R.; Wu, B.F.; Jia, Z.X.; Pan, H.Y.; Zhou, H.; Hu, S.Y.; Zhang, B.T.; et al. Mitochondrial dysfunction caused by m.2336T > C mutation with hypertrophic cardiomyopathy in cybrid cell lines. Mitochondrion 2019, 46, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Loeffen, J.; Elpeleg, O.; Smeitink, J.; Smeets, R.; Stockler-Ipsiroglu, S.; Mandel, H.; Sengers, R.; Trijbels, F.; van den Heuvel, L. Mutations in the complex I NDUFS2 gene of patients with cardiomyopathy and encephalomyopathy. Ann. Neurol. 2001, 49, 195–201. [Google Scholar] [CrossRef]
- Cameron, J.M.; MacKay, N.; Feigenbaum, A.; Tarnopolsky, M.; Blaser, S.; Robinson, B.H.; Schulze, A. Exome sequencing identifies complex I NDUFV2 mutations as a novel cause of Leigh syndrome. Eur. J. Paediatr. Neuro. 2015, 19, 525–532. [Google Scholar] [CrossRef]
- Hoefs, S.J.G.; Dieteren, C.E.J.; Distelmaier, F.; Janssen, R.J.R.J.; Epplen, A.; Swarts, H.G.P.; Forkink, M.; Rodenburg, R.J.; Nijtmans, L.G.; Willems, P.H.; et al. NDUFA2 complex I mutation leads to Leigh disease. Am. J. Hum. Genet. 2008, 82, 1306–1315. [Google Scholar] [CrossRef]
- Fassone, E.; Taanman, J.W.; Hargreaves, I.P.; Sebire, N.J.; Cleary, M.A.; Burch, M.; Rahman, S. Mutations in the mitochondrial complex I assembly factor NDUFAF1 cause fatal infantile hypertrophic cardiomyopathy. J. Med. Genet. 2011, 48, 691–697. [Google Scholar] [CrossRef]
- Hallas, T.; Eisen, B.; Shemer, Y.; Ben Jehuda, R.; Mekies, L.N.; Naor, S.; Schick, R.; Eliyahu, S.; Reiter, I.; Vlodavsky, E.; et al. Investigating the cardiac pathology of SCO2-mediated hypertrophic cardiomyopathy using patients induced pluripotent stem cell-derived cardiomyocytes. J. Cell. Mol. Med. 2018, 22, 913–925. [Google Scholar] [CrossRef]
- Piekutowska-Abramczuk, D.; Magner, M.; Popowska, E.; Pronicki, M.; Karczmarewicz, E.; Sykut-Cegielska, J.; Kmiec, T.; Jurkiewicz, E.; Szymanska-Debinska, T.; Bielecka, L.; et al. SURF1 missense mutations promote a mild Leigh phenotype. Clin. Genet. 2009, 76, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Courage, C.; Jackson, C.B.; Hahn, D.; Euro, L.; Nuoffer, J.M.; Gallati, S.; Schaller, A. SDHA Mutation with Dominant Transmission Results in Complex II Deficiency with Ocular, Cardiac, and Neurologic Involvement. Am. J. Med. Genet. A 2017, 173, 225–230. [Google Scholar] [CrossRef]
- Diodato, D.; Invernizzi, F.; Lamantea, E.; Fagiolari, G.; Parini, R.; Menni, F.; Parenti, G.; Bollani, L.; Pasquini, E.; Donati, M.A.; et al. Common and Novel TMEM70 Mutations in a Cohort of Italian Patients with Mitochondrial Encephalocardiomyopathy. JIMD Rep. 2015, 15, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, R.; Khayat, M.; Shalev, S.A.; Horovitz, Y.; Mandel, H.; Hershkovitz, E.; Barghuti, F.; Shaag, A.; Saada, A.; Korman, S.H.; et al. TMEM70 mutations are a common cause of nuclear encoded ATP synthase assembly defect: Further delineation of a new syndrome. J. Med. Genet. 2011, 48, 177–182. [Google Scholar] [CrossRef]
- Smeitink, J.A.M.; Elpeleg, O.; Antonicka, H.; Diepstra, H.; Saada, A.; Smits, P.; Sasarman, F.; Vriend, G.; Jacob-Hirsch, J.; Shaag, A.; et al. Distinct clinical phenotypes associated with a mutation in the mitochondrial translation elongation factor EFTs. Am. J. Hum. Genet. 2006, 79, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Guo, Y.; Huang, M.R.; Zhang, Z.; Zhu, J.X.; Liu, T.L.; Shi, L.; Li, F.; Huang, H.M.; Fu, L.J. Identification of TAZ mutations in pediatric patients with cardiomyopathy by targeted next-generation sequencing in a Chinese cohort. Orphanet J. Rare Dis. 2017, 12. [Google Scholar] [CrossRef]
- Perli, E.; Pisano, A.; Glasgow, R.I.C.; Carbo, M.; Hardy, S.A.; Falkous, G.; He, L.; Cerbelli, B.; Pignataro, M.G.; Zacara, E.; et al. Novel compound mutations in the mitochondrial translation elongation factor (TSFM) gene cause severe cardiomyopathy with myocardial fibro-adipose replacement. Sci. Rep. 2019, 9, 5108. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Finsterer, J.; Kothari, S. Cardiac manifestations of primary mitochondrial disorders. Int. J. Cardiol. 2014, 177, 754–763. [Google Scholar] [CrossRef]
- Wortmann, S.B.; Koolen, D.; Smeitink, J.A.; van den Heuvel, L.; Rodenburg, R.J. Whole exome sequencing of suspected mitochondrial patients in clinical practice. J. Inherit. Metab. Dis. 2015, 38, 437–443. [Google Scholar] [CrossRef]
- Morava, E.; van den Heuvel, L.; Hol, F.; de Vries, M.C.; Hogeveen, M.; Rodenburg, R.J.; Smeitink, J.A.M. Mitochondrial disease criteria—Diagnostic applications in children. Neurology 2006, 67, 1823–1826. [Google Scholar] [CrossRef] [PubMed]
- Bernier, F.P.; Boneh, A.; Dennett, X.; Chow, C.W.; Cleary, M.A.; Thorburn, D.R. Diagnostic criteria for respiratory chain disorders in adults and children. Neurology 2002, 59, 1406–1411. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, G.; Steinberg, C.; Dubois, M.; Senechal, M. What the Cardiologist Should Know About Mitochondrial Cardiomyopathy? Can. J. Cardiol. 2019, 35, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.; Goldstein, A.; Koenig, M.K.; Scaglia, F.; Enns, G.M.; Saneto, R.; Anselm, I.; Cohen, B.H.; Falk, M.J.; Greene, C.; et al. Diagnosis and management of mitochondrial disease: A consensus statement from the Mitochondrial Medicine Society. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Greaves, L.C.; Reeve, A.K.; Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA and disease. J. Pathol. 2012, 226, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.H.G.; Margeta, M. Educational Case: Mitochondrial Myopathy. Acad Pathol. 2019, 6. [Google Scholar] [CrossRef] [PubMed]
- Rocha, M.C.; Grady, J.P.; Grunewald, A.; Vincent, A.; Dobson, P.F.; Taylor, R.W.; Turnbull, D.M.; Rygiel, K.A. A novel immunofluorescent assay to investigate oxidative phosphorylation deficiency in mitochondrial myopathy: Understanding mechanisms and improving diagnosis. Sci Rep. Uk 2015, 5. [Google Scholar] [CrossRef]
- Tashiro, R.; Onoue, N.; Rikimaru, H.; Tsukita, K.; Fujita, H.; Yamaguchi, N.; Ishizuka, T.; Suzuki, Y.; Suzuki, H.; Shinozaki, T. Mitochondrial Cardiomyopathy with a Unique (99m)Tc-MIBI/(123)I-BMIPP Mismatch Pattern. Intern. Med. 2017, 56, 321–325. [Google Scholar] [CrossRef][Green Version]
- Murphy, E.; Ardehali, H.; Balaban, R.S.; DiLisa, F.; Dorn, G.W., 2nd; Kitsis, R.N.; Otsu, K.; Ping, P.; Rizzuto, R.; Sack, M.N.; et al. Mitochondrial Function, Biology, and Role in Disease: A Scientific Statement From the American Heart Association. Circ. Res. 2016, 118, 1960–1991. [Google Scholar] [CrossRef]
- Florian, A.; Ludwig, A.; Stubbe-Drager, B.; Boentert, M.; Young, P.; Waltenberger, J.; Rosch, S.; Sechtem, U.; Yilmaz, A. Characteristic cardiac phenotypes are detected by cardiovascular magnetic resonance in patients with different clinical phenotypes and genotypes of mitochondrial myopathy. J. Cardiovasc. Magn. Reson. Off. J. Soc. Cardiovasc. Magn. Reson. 2015, 17, 40. [Google Scholar] [CrossRef] [PubMed]
- Delvecchio, M.; Salzano, G.; Bonura, C.; Cauvin, V.; Cherubini, V.; d’Annunzio, G.; Franzese, A.; Giglio, S.; Grasso, V.; Graziani, V.; et al. Can HbA1c combined with fasting plasma glucose help to assess priority for GCK-MODY vs HNF1A-MODY genetic testing? Acta Diabetol. 2018, 55, 981–983. [Google Scholar] [CrossRef] [PubMed]
- Tuppen, H.A.; Blakely, E.L.; Turnbull, D.M.; Taylor, R.W. Mitochondrial DNA mutations and human disease. Biochim. Biophys. Acta 2010, 1797, 113–128. [Google Scholar] [CrossRef]
- Chinault, A.C.; Shaw, C.A.; Brundage, E.K.; Tang, L.Y.; Wong, L.J. Application of dual-genome oligonucleotide array-based comparative genomic hybridization to the molecular diagnosis of mitochondrial DNA deletion and depletion syndromes. Genet. Med. Off. J. Am. Coll. Med. Genet. 2009, 11, 518–526. [Google Scholar] [CrossRef]
- Wang, J.; Zhan, H.L.; Li, F.Y.; Pursley, A.N.; Schmitt, E.S.; Wong, L.J. Targeted array CGH as a valuable molecular diagnostic approach: Experience in the diagnosis of mitochondrial and metabolic disorders. Mol. Genet. Metab. 2012, 106, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.J.; Dimmock, D.; Geraghty, M.T.; Quan, R.; Lichter-Konecki, U.; Wang, J.; Brundage, E.K.; Scaglia, F.; Chinault, A.C. Utility of oligonucleotide array-based comparative genomic hybridization for detection of target gene deletions. Clin. Chem. 2008, 54, 1141–1148. [Google Scholar] [CrossRef] [PubMed]
- Dinwiddie, D.L.; Smith, L.D.; Miller, N.A.; Atherton, A.M.; Farrow, E.G.; Strenk, M.E.; Soden, S.E.; Saunders, C.J.; Kingsmore, S.F. Diagnosis of mitochondrial disorders by concomitant next-generation sequencing of the exome and mitochondrial genome. Genomics 2013, 102, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Hahn, S.H. Targeted next-generation sequencing expands the spectrum of mitochondrial disorders. Genome Med. 2012, 4, 22. [Google Scholar] [CrossRef]
- Liu, Z.; Fang, F.; Ding, C.; Zhang, W.; Li, J.; Yang, X.; Wang, X.; Wu, Y.; Wang, H.; Liu, L.; et al. [Diagnosis of mitochondrial disorders in children with next generation sequencing]. Zhonghua Er Ke Za Zhi Chin. J. Pediatr. 2015, 53, 747–753. [Google Scholar]
- Zhang, W.; Cui, H.; Wong, L. Development and Validation of Next Generation Sequencing for Clinical Molecular Diagnosis of Mitochondrial Disorders. J. Mol. Diagn. 2012, 14, 637–638. [Google Scholar]
- Zhang, V.W.; Cui, H.; Wong, L.J. Implementation of next generation sequencing for clinical molecular diagnosis of mitochondrial disorders. Mitochondrion 2012, 12, 555–556. [Google Scholar] [CrossRef]
- Vasta, V.; Ng, S.B.; Turner, E.H.; Shendure, J.; Hahn, S.H. Next generation sequence analysis for mitochondrial disorders. Genome Med. 2009, 1. [Google Scholar] [CrossRef]
- Matthijs, G.; Souche, E.; Alders, M.; Corveleyn, A.; Eck, S.; Feenstra, I.; Race, V.; Sistermans, E.; Sturm, M.; Weiss, M.; et al. Guidelines for diagnostic next-generation sequencing. Eur. J. Hum. Genet. 2016, 24, 1515. [Google Scholar] [CrossRef]
- Tang, S.; Wang, J.; Zhang, V.W.; Li, F.Y.; Landsverk, M.; Cui, H.; Truong, C.K.; Wang, G.; Chen, L.C.; Graham, B.; et al. Transition to next generation analysis of the whole mitochondrial genome: A summary of molecular defects. Hum. Mutat 2013, 34, 882–893. [Google Scholar] [CrossRef]
- Pronicka, E.; Piekutowska-Abramczuk, D.; Ciara, E.; Trubicka, J.; Rokicki, D.; Karkucinska-Wieckowska, A.; Pajdowska, M.; Jurkiewicz, E.; Halat, P.; Kosinska, J.; et al. New perspective in diagnostics of mitochondrial disorders: Two years’ experience with whole-exome sequencing at a national paediatric centre. J. Transl. Med. 2016, 14, 174. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, B.; D’Argenio, V.; Monda, E.; Vitale, A.; Caiazza, M.; Sacchetti, L.; Pastore, L.; Limongelli, G.; Frisso, G.; Mazzaccara, C. Genetic analysis resolves differential diagnosis of a familial syndromic dilated cardiomyopathy: A new case of Alstrom syndrome. Mol. Genet. Genom. Med. 2020, 8, e1260. [Google Scholar] [CrossRef]
- Frisso, G.; Detta, N.; Coppola, P.; Mazzaccara, C.; Pricolo, M.R.; D’Onofrio, A.; Limongelli, G.; Calabro, R.; Salvatore, F. Functional Studies and In Silico Analyses to Evaluate Non-Coding Variants in Inherited Cardiomyopathies. Int. J. Mol. Sci. 2016, 17, 1883. [Google Scholar] [CrossRef] [PubMed]
- Suay-Corredera, C.; Pricolo, M.R.; Herrero-Galán, E.; Velázquez-Carreras, D.; Sánchez-Ortiz, D.; García-Giustiniani, D.; Delgado, J.; Galano-Frutos, J.J.; García-Cebollada, H.; Vilches, S.; et al. Protein haploinsufficiency drivers identify MYBPC3 mutations that cause hypertrophic cardiomyopathy. MedRxiv 2020. [Google Scholar] [CrossRef]
- Girolami, F.; Frisso, G.; Benelli, M.; Crotti, L.; Iascone, M.; Mango, R.; Mazzaccara, C.; Pilichou, K.; Arbustini, E.; Tomberli, B.; et al. Contemporary genetic testing in inherited cardiac disease: Tools, ethical issues, and clinical applications. J. Cardiovasc. Med. 2018, 19, 1–11. [Google Scholar] [CrossRef]
- Correia, S.P.; Moedas, M.F.; Naess, K.; Bruhn, H.; Maffezzini, C.; Calvo-Garrido, J.; Lesko, N.; Wibom, R.; Schober, F.A.; Jemt, A.; et al. Severe congenital lactic acidosis and hypertrophic cardiomyopathy caused by an intronic variant in NDUFB7. Hum. Mutat. 2021. [Google Scholar] [CrossRef]
- Chung, H.; Kim, Y.; Cho, S.M.; Lee, H.J.; Park, C.H.; Kim, J.Y.; Lee, S.H.; Min, P.K.; Yoon, Y.W.; Lee, B.K.; et al. Differential contributions of sarcomere and mitochondria-related multigene variants to the endophenotype of hypertrophic cardiomyopathy. Mitochondrion 2020, 53, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Chau, E.M.C.; Ma, E.S.K.; Chan, A.O.O.; Tsio, T.H.; Law, W.L. Mitochondrial cardiomyopathy due to m.3243A > G mitochondrial DNA mutation presenting in late adulthood: A case report. Hong Kong Med. J. 2020, 26, 240–242. [Google Scholar] [CrossRef]
- Kamps, R.; Szklarczyk, R.; Theunissen, T.E.; Hellebrekers, D.; Sallevelt, S.; Boesten, I.B.; de Koning, B.; van den Bosch, B.J.; Salomons, G.S.; Simas-Mendes, M.; et al. Genetic defects in mtDNA-encoded protein translation cause pediatric, mitochondrial cardiomyopathy with early-onset brain disease. Eur. J. Hum. Genet. 2018, 26, 537–551. [Google Scholar] [CrossRef]
- Feichtinger, R.G.; Olahova, M.; Kishita, Y.; Garone, C.; Kremer, L.S.; Yagi, M.; Uchiumi, T.; Jourdain, A.A.; Thompson, K.; D’Souza, A.R.; et al. Biallelic C1QBP Mutations Cause Severe Neonatal-, Childhood-, or Later-Onset Cardiomyopathy Associated with Combined Respiratory-Chain Deficiencies. Am. J. Hum. Genet. 2017, 101, 525–538. [Google Scholar] [CrossRef]
- Haack, T.B.; Kopajtich, R.; Freisinger, P.; Wieland, T.; Rorbach, J.; Nicholls, T.J.; Baruffini, E.; Walther, A.; Danhauser, K.; Zimmermann, F.A.; et al. ELAC2 Mutations Cause a Mitochondrial RNA Processing Defect Associated with Hypertrophic Cardiomyopathy. Am. J. Hum. Genet. 2013, 93, 211–223. [Google Scholar] [CrossRef]
- Malfatti, E.; Laforet, P.; Jardel, C.; Stojkovic, T.; Behin, A.; Eymard, B.; Lombes, A.; Benmalek, A.; Becane, H.M.; Berber, N.; et al. High risk of severe cardiac adverse events in patients with mitochondrial m.3243A > G mutation. Neurology 2013, 80, 100–105. [Google Scholar] [CrossRef]
- Hollingsworth, K.G.; Gorman, G.S.; Trenell, M.I.; McFarland, R.; Taylor, R.W.; Turnbull, D.M.; MacGowan, G.A.; Blamire, A.M.; Chinnery, P.F. Cardiomyopathy is common in patients with the mitochondrial DNA m.3243A > G mutation and correlates with mutation load. Neuromuscul. Disord. 2012, 22, 592–596. [Google Scholar] [CrossRef]
- Kaufmann, P.; Engelstad, K.; Wei, Y.; Kulikova, R.; Oskoui, M.; Sproule, D.M.; Battista, V.; Koenigsberger, D.Y.; Pascual, J.M.; Shanske, S.; et al. Natural history of MELAS associated with mitochondrial DNA m.3243A > G genotype. Neurology 2011, 77, 1965–1971. [Google Scholar] [CrossRef] [PubMed]
- Nozieres, C.; Quillasi, V.; Mouly-Bertin, C.; Thomson, V.; Gachon-Lanier, E.; Lantelme, P. [Diabetes and hypokinetic cardiopathy: When to consider mitochondrial disease?]. Ann. De Cardiol. Et D’angeiologie 2011, 60, 176–178. [Google Scholar] [CrossRef]
- Yajima, N.; Yazaki, Y.; Yoshida, K.; Sano, K.; Takahashi, W.; Sasaki, Y.; Ikeda, U. A case of mitochondrial cardiomyopathy with pericardial effusion evaluated by Tc-99m-MIBI myocardial scintigraphy. J. Nucl. Cardiol. 2009, 16, 989–994. [Google Scholar] [CrossRef]
- Majamaa-Voltti, K.A.M.; Winqvist, S.; Remes, A.M.; Tolonen, U.; Pyhtinen, J.; Uimonen, S.; Karppa, M.; Sorri, M.; Peuhkurinen, K.; Majamaa, K. A 3-year clinical follow-up of adult patients with 3243A > G in mitochondrial DNA. Neurology 2006, 66, 1470–1475. [Google Scholar] [CrossRef]
- Chae, J.H.; Hwang, H.; Lim, B.C.; Cheong, H.I.; Hwang, Y.S.; Kim, K.J. Clinical features of A3243G mitochondrial tRNA mutation. Brain Dev. 2004, 26, 459–462. [Google Scholar] [CrossRef]
- Holmgren, D.; Wahlander, H.; Eriksson, B.O.; Oldfors, A.; Holme, E.; Tulinius, M. Cardiomyopathy in children with mitochondrial disease—Clinical course and cardiological findings. Eur. Heart J. 2003, 24, 280–288. [Google Scholar] [CrossRef]
- Momiyama, Y.; Furutani, M.; Suzuki, Y.; Ohmori, R.; Imamura, S.; Mokubo, A.; Asahina, T.; Murata, C.; Kato, K.; Anazawa, S.; et al. A mitochondrial DNA variant associated with left ventricular hypertrophy in diabetes. Biochem. Bioph Res. Co 2003, 312, 858–864. [Google Scholar] [CrossRef]
- Shin, W.S.; Tanaka, M.; Suzuki, J.; Hemmi, C.; Toyo-oka, T. A novel homoplasmic mutation in mtDNA with a single evolutionary origin as a risk factor for cardiomyopathy. Am. J. Hum. Genet. 2000, 67, 1617–1620. [Google Scholar] [CrossRef] [PubMed]
- Okajima, Y.; Tanabe, Y.; Takayanagi, M.; Aotsuka, H. A follow up study of myocardial involvement in patients with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS). Heart 1998, 80, 292–295. [Google Scholar] [CrossRef]
- Chinnery, P.F.; Howell, N.; Lightowlers, R.N.; Turnbull, D.M. Molecular pathology of MELAS and MERRF - The relationship between mutation load and clinical phenotypes. Brain 1997, 120, 1713–1721. [Google Scholar] [CrossRef]
- Anan, R.; Nakagawa, M.; Miyata, M.; Higuchi, I.; Nakao, S.; Suehara, M.; Osame, M.; Tanaka, H. Cardiac Involvement in Mitochondrial Diseases—A Study on 17 Patients with Documented Mitochondrial-DNA Defects. Circulation 1995, 91, 955–961. [Google Scholar] [CrossRef]
- Guenthard, J.; Wyler, F.; Fowler, B.; Baumgartner, R. Cardiomyopathy in Respiratory-Chain Disorders. Arch. Dis. Child. 1995, 72, 223–226. [Google Scholar] [CrossRef]
- Ciafaloni, E.; Ricci, E.; Shanske, S.; Moraes, C.T.; Silvestri, G.; Hirano, M.; Simonetti, S.; Angelini, C.; Donati, M.A.; Garcia, C.; et al. Melas—Clinical-Features, Biochemistry, and Molecular-Genetics. Ann. Neurol. 1992, 31, 391–398. [Google Scholar] [CrossRef]
- Zeviani, M.; Gellera, C.; Antozzi, C.; Rimoldi, M.; Morandi, L.; Villani, F.; Tiranti, V.; Didonato, S. Maternally Inherited Myopathy and Cardiomyopathy—Association with Mutation in Mitochondrial-DNA Transfer Rnaleu(Uur). Lancet 1991, 338, 143–147. [Google Scholar] [CrossRef]
- Ozawa, T.; Tanaka, M.; Sugiyama, S.; Hattori, K.; Ito, T.; Ohno, K.; Takahashi, A.; Sato, W.; Takada, G.; Mayumi, B.; et al. Multiple Mitochondrial-DNA Deletions Exist in Cardiomyocytes of Patients with Hypertrophic or Dilated Cardiomyopathy. Biochem. Bioph. Res. Commun. 1990, 170, 830–836. [Google Scholar] [CrossRef]
- Thompson, K.; Collier, J.J.; Glasgow, R.I.C.; Robertson, F.M.; Pyle, A.; Blakely, E.L.; Alston, C.L.; Olahova, M.; McFarland, R.; Taylor, R.W. Recent advances in understanding the molecular genetic basis of mitochondrial disease. J. Inherit. Metab. Dis. 2020, 43, 36–50. [Google Scholar] [CrossRef]
- Bayona-Bafaluy, M.P.; Iglesias, E.; Lopez-Gallardo, E.; Emperador, S.; Pacheu-Grau, D.; Labarta, L.; Montoya, J.; Ruiz-Pesini, E. Genetic aspects of the oxidative phosphorylation dysfunction in dilated cardiomyopathy. Mutat. Res. 2020, 786, 108334. [Google Scholar] [CrossRef]
- Rafiq, M.A.; Chaudhry, A.; Care, M.; Spears, D.A.; Morel, C.F.; Hamilton, R.M. Whole exome sequencing identified 1 base pair novel deletion in BCL2-associated athanogene 3 (BAG3) gene associated with severe dilated cardiomyopathy (DCM) requiring heart transplant in multiple family members. Am. J. Med. Genet. Part. A 2017, 173, 699–705. [Google Scholar] [CrossRef]
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kuhl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the cardiomyopathies: A position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2008, 29, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Danese, E.; Raimondi, S.; Montagnana, M.; Tagetti, A.; Langaee, T.; Borgiani, P.; Ciccacci, C.; Carcas, A.J.; Borobia, A.M.; Tong, H.Y.; et al. Effect of CYP4F2, VKORC1, and CYP2C9 in Influencing Coumarin Dose: A Single-Patient Data Meta-Analysis in More Than 15,000 Individuals. Clin. Pharmacol. Ther. 2019, 105, 1477–1491. [Google Scholar] [CrossRef]
- Lombardo, B.; Izzo, V.; Terracciano, D.; Ranieri, A.; Mazzaccara, C.; Fimiani, F.; Cesaro, A.; Gentile, L.; Leggiero, E.; Pero, R.; et al. Laboratory medicine: Health evaluation in elite athletes. Clin. Chem. Lab. Med. 2019, 57, 1450–1473. [Google Scholar] [CrossRef]
- Mazzaccara, C.; Redi, A.; Lemme, E.; Pelliccia, A.; Salvatore, F.; Frisso, G. Impact of molecular diagnostics in an asymptomatic amateur athlete found to be affected by hypertrophic cardiomyopathy. Med. Sport 2018, 71, 405–412. [Google Scholar] [CrossRef]
- Barretta, F.; Mirra, B.; Monda, E.; Caiazza, M.; Lombardo, B.; Tinto, N.; Scudiero, O.; Frisso, G.; Mazzaccara, C. The Hidden Fragility in the Heart of the Athletes: A Review of Genetic Biomarkers. Int. J. Mol. Sci. 2020, 21, 6682. [Google Scholar] [CrossRef]
- Mazzaccara, C.; D’Argenio, V.; Nunziato, M.; Esposito, M.V.; Salvatore, F.; Frisso, G. Clinical molecular biology in the assessment and prevention of cardiological risk in case of participation in sports activity and intense physical activity. Biochim. Clin. 2019, 43, 24–43. [Google Scholar] [CrossRef]
- Limongelli, G.; Nunziato, M.; D’Argenio, V.; Esposito, M.V.; Monda, E.; Mazzaccara, C.; Caiazza, M.; D’Aponte, A.; D’Andrea, A.; Bossone, E.; et al. Yield and clinical significance of genetic screening in elite and amateur athletes. Eur. J. Prev. Cardiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Limongelli, G.; Nunziato, M.; Mazzaccara, C.; Intrieri, M.; D’Argenio, V.; Esposito, M.V.; Monda, E.; Di Maggio, F.; Frisso, G.; Salvatore, F. Genotype-Phenotype Correlation: A Triple DNA Mutational Event in a Boy Entering Sport Conveys an Additional Pathogenicity Risk. Genes 2020, 11, 524. [Google Scholar] [CrossRef] [PubMed]
- Pricolo, M.R.; Herrero-Galan, E.; Mazzaccara, C.; Losi, M.A.; Alegre-Cebollada, J.; Frisso, G. Protein Thermodynamic Destabilization in the Assessment of Pathogenicity of a Variant of Uncertain Significance in Cardiac Myosin Binding Protein C. J. Cardiovasc. Transl. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).