
Intracellular Na+ Modulates Pacemaking Activity in Murine Sinoatrial Node Myocytes: An In Silico Analysis

 , , , and
, , , and
Abstract
1. Introduction
2. Results
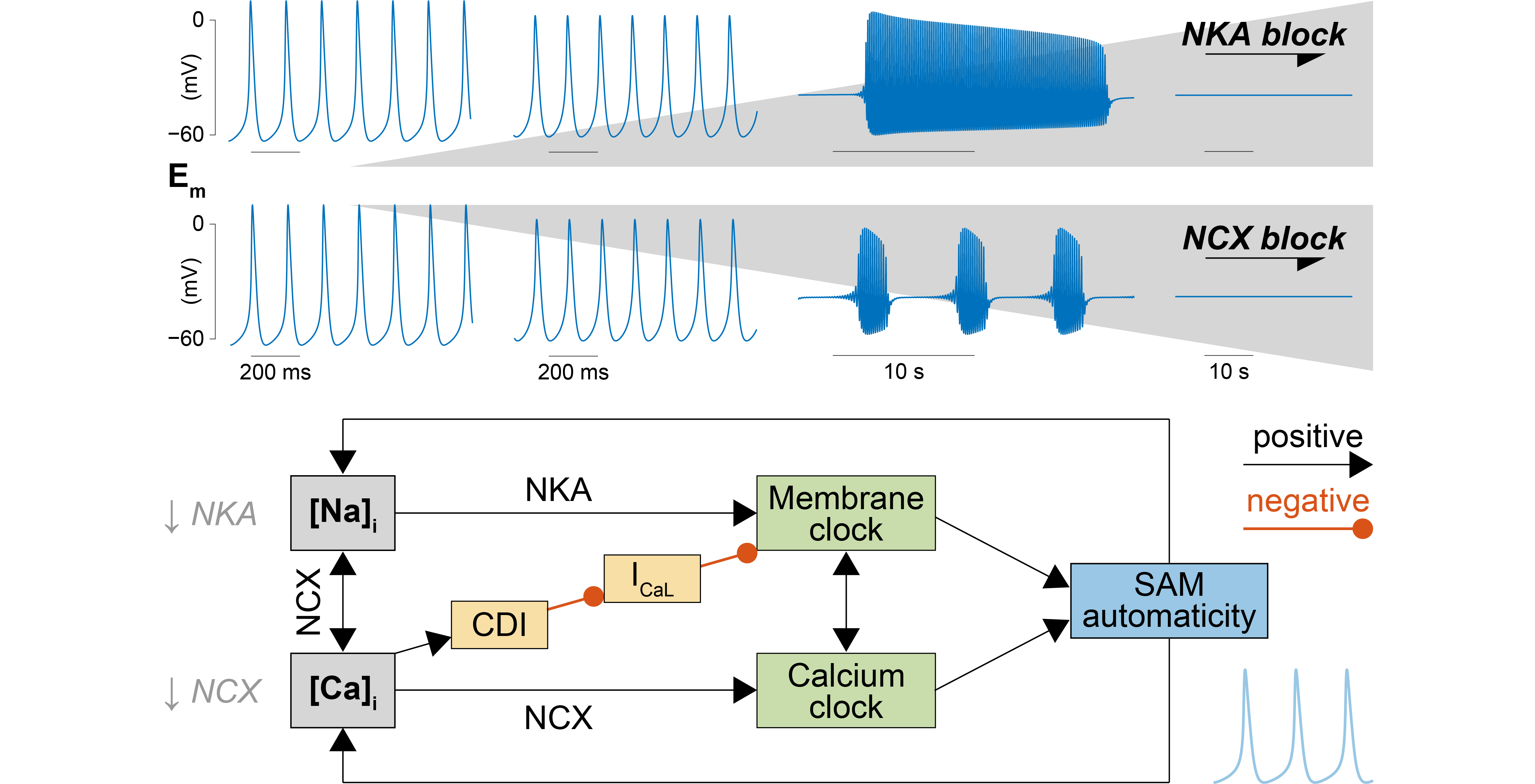
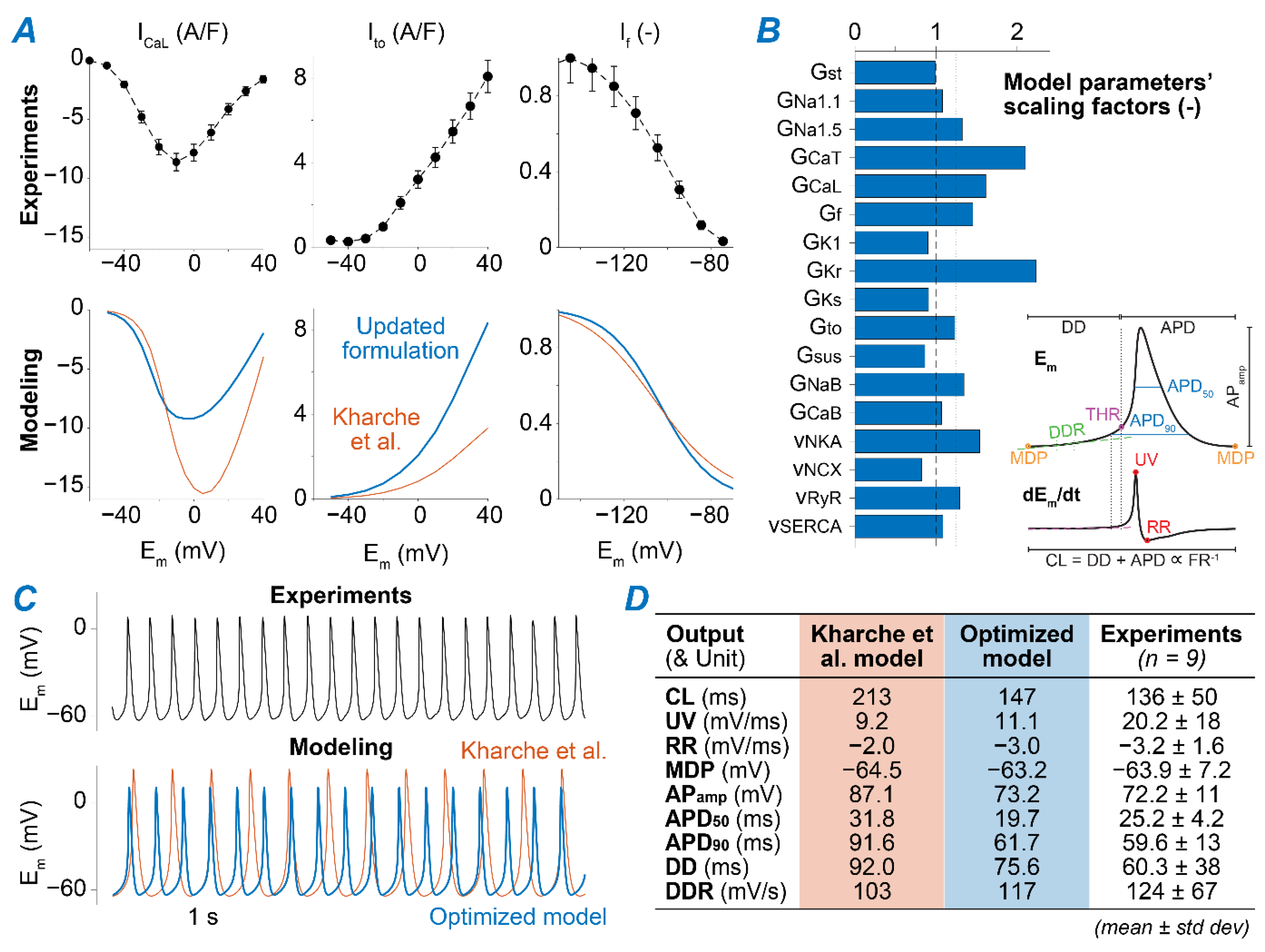
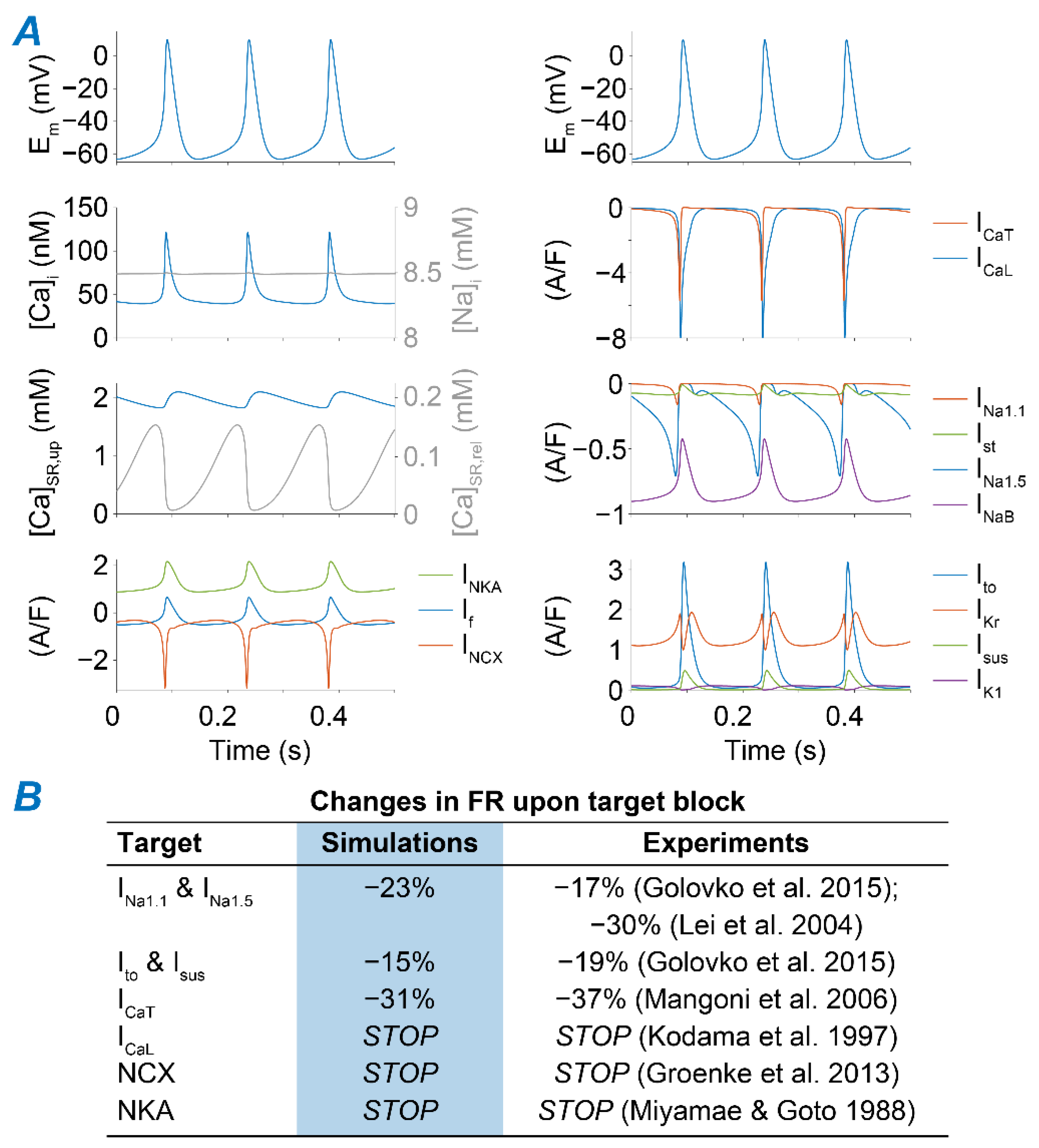
2.1. A Computational Model of Murine SAMs Well Recapitulates a Broad Experimental Dataset
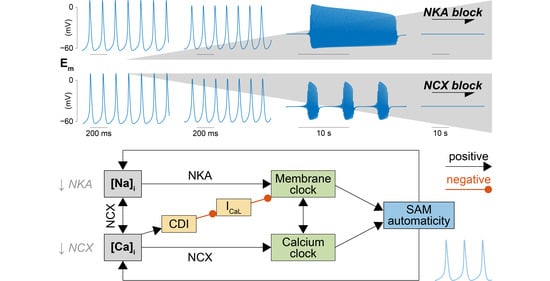
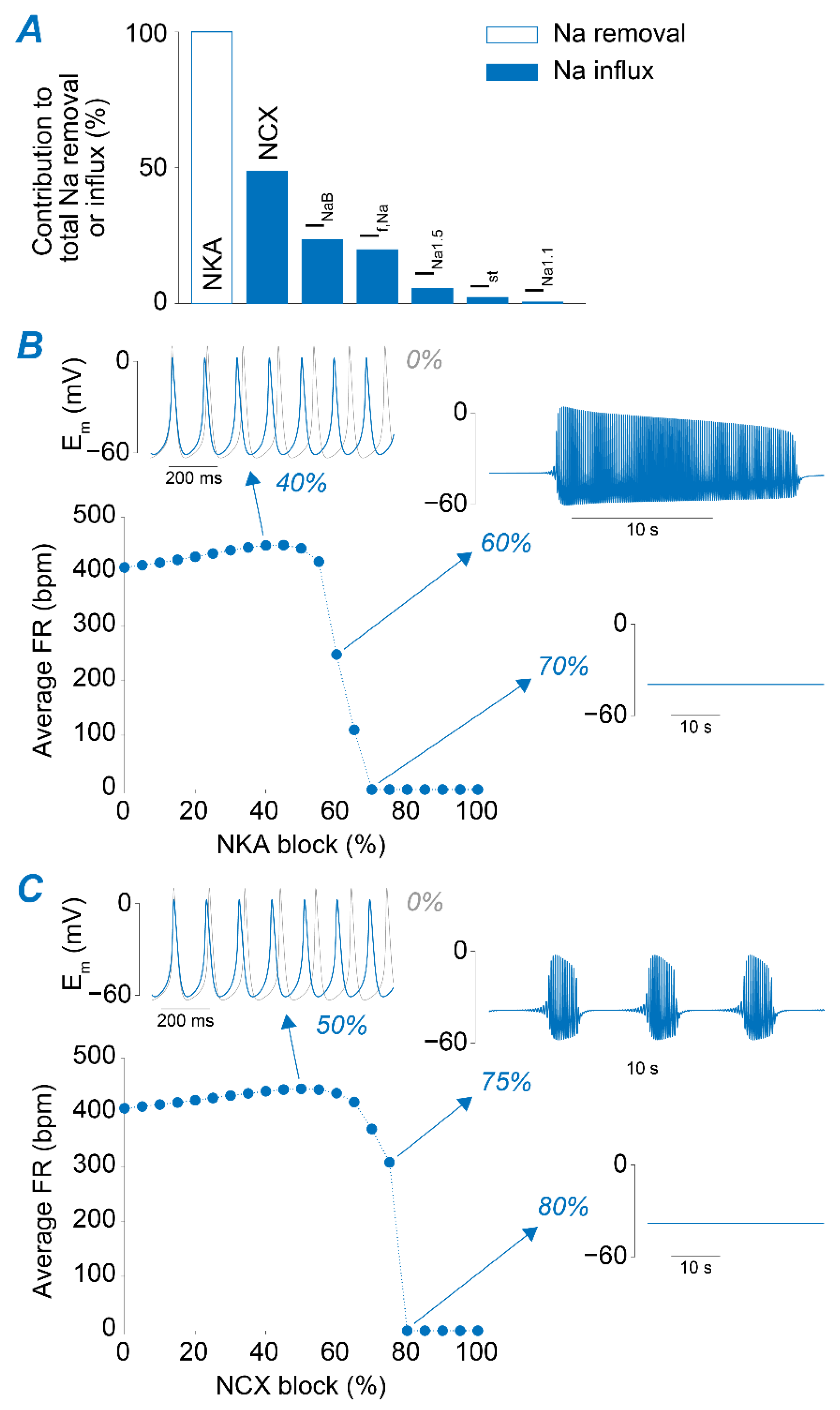
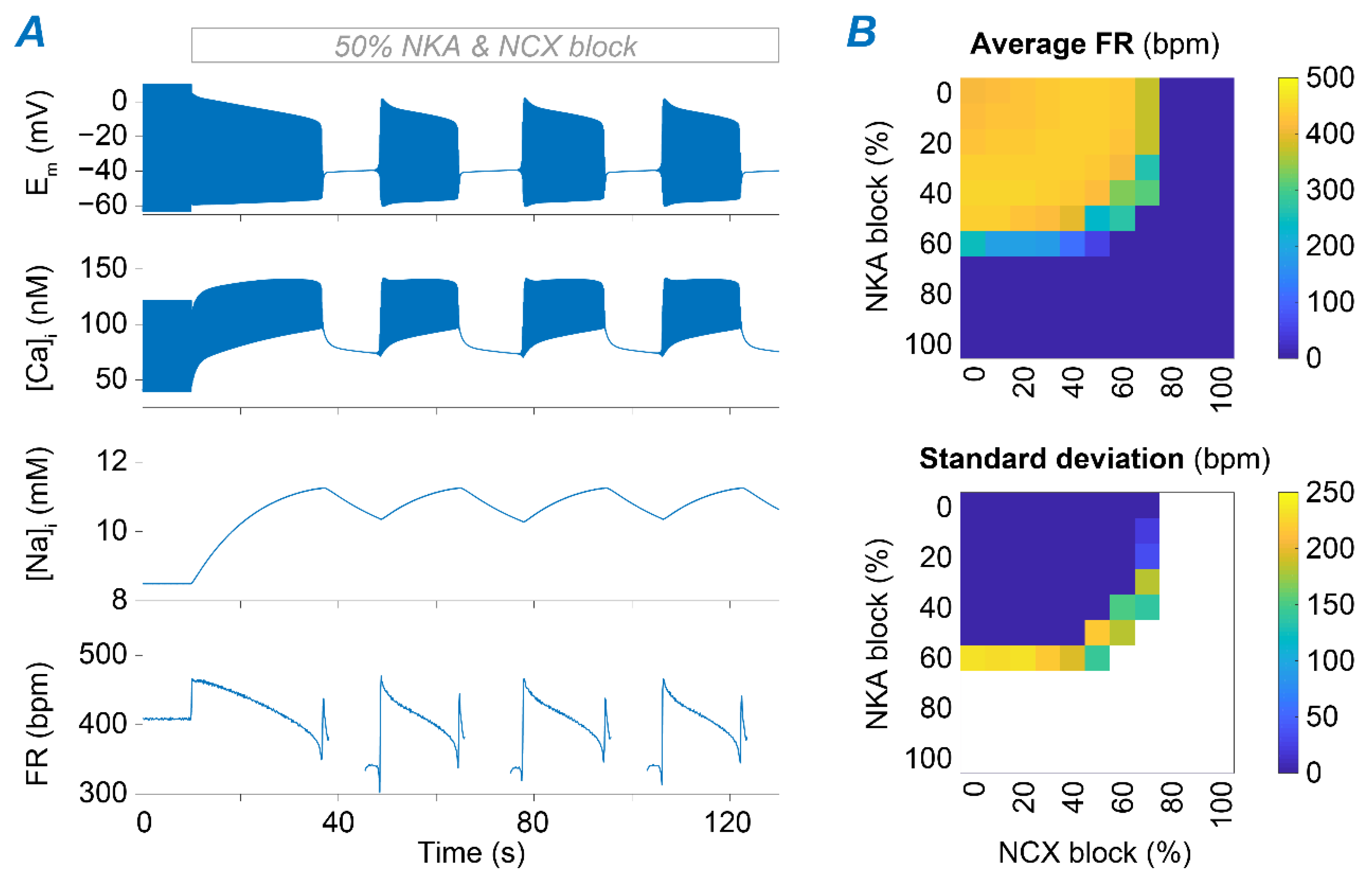
2.2. NKA and NCX Inhibition Can Both Boost and Disrupt SAM Automaticity
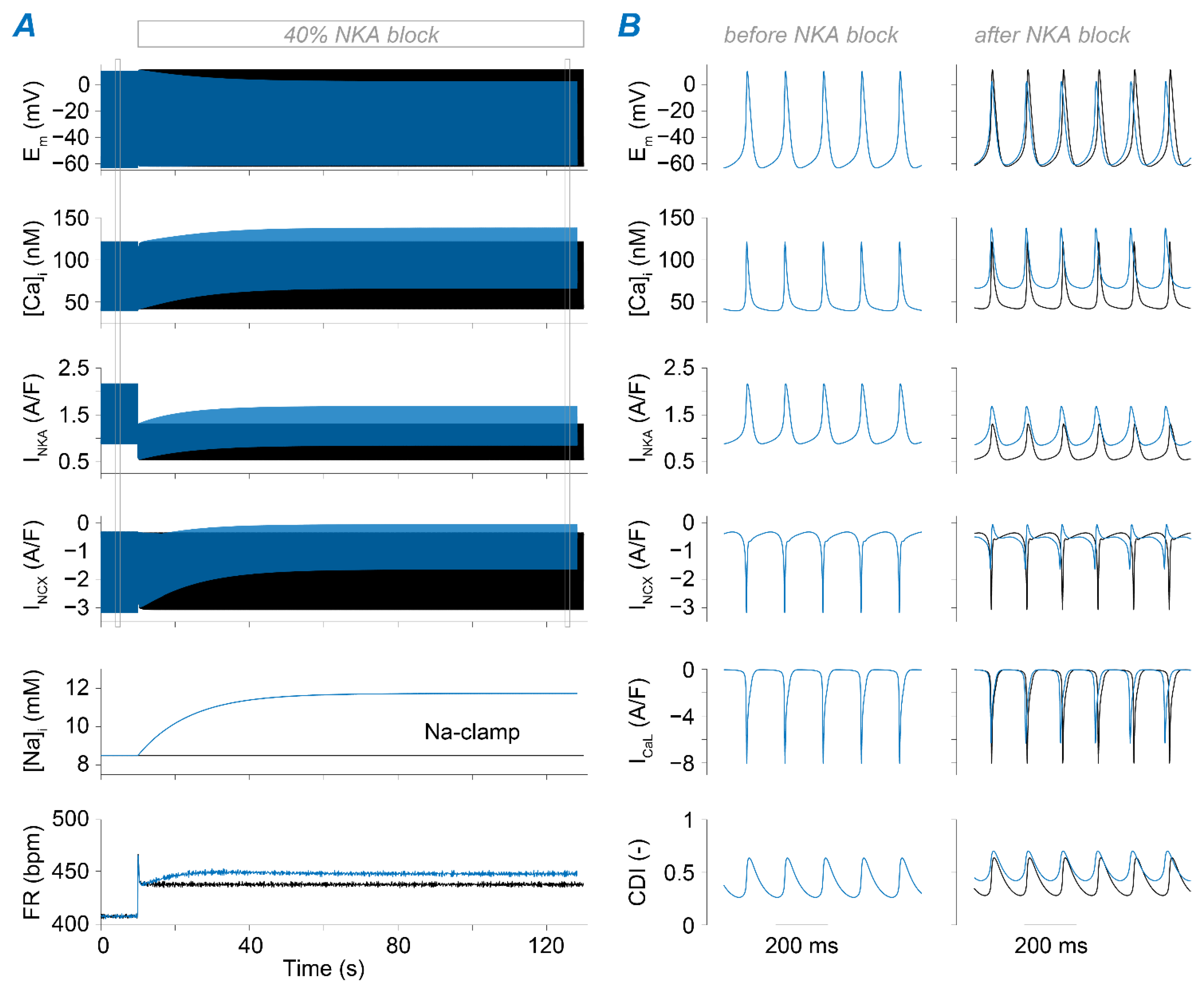
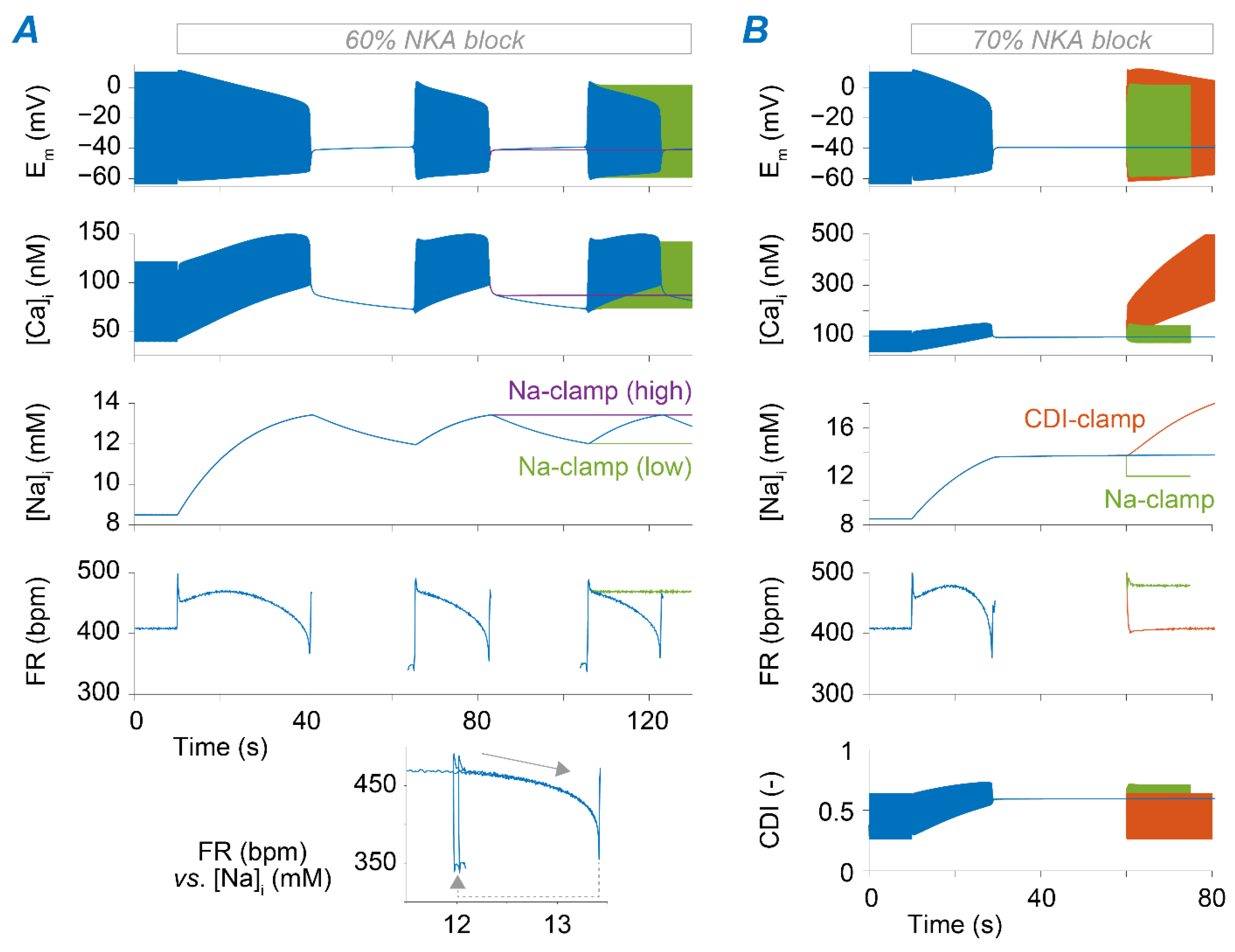
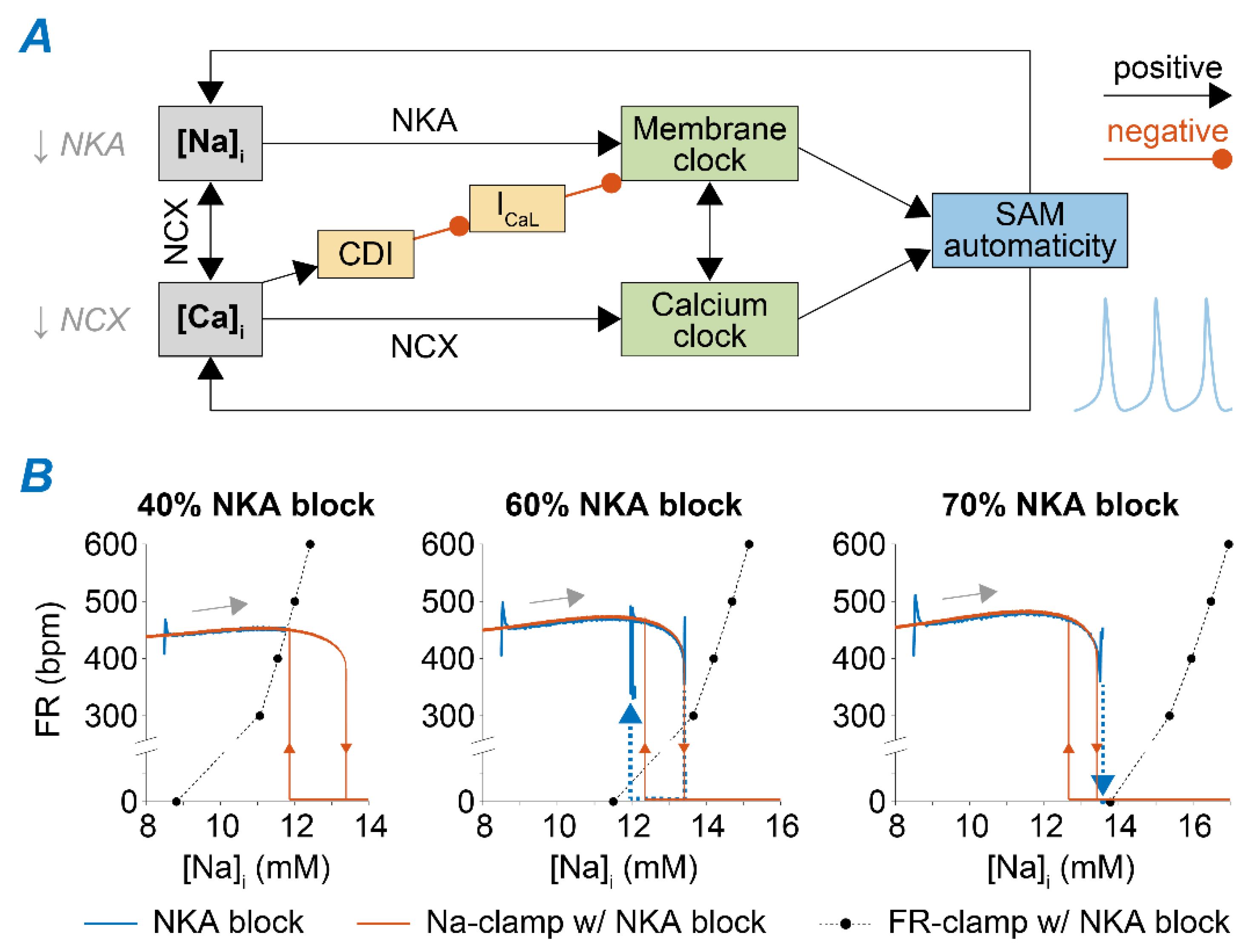
2.3. Na+ Accumulation Induced by NKA Impairment Reduces SAM Automaticity via Ca2+ Overload and Excessive Ca2+-Dependent ICaL Inactivation
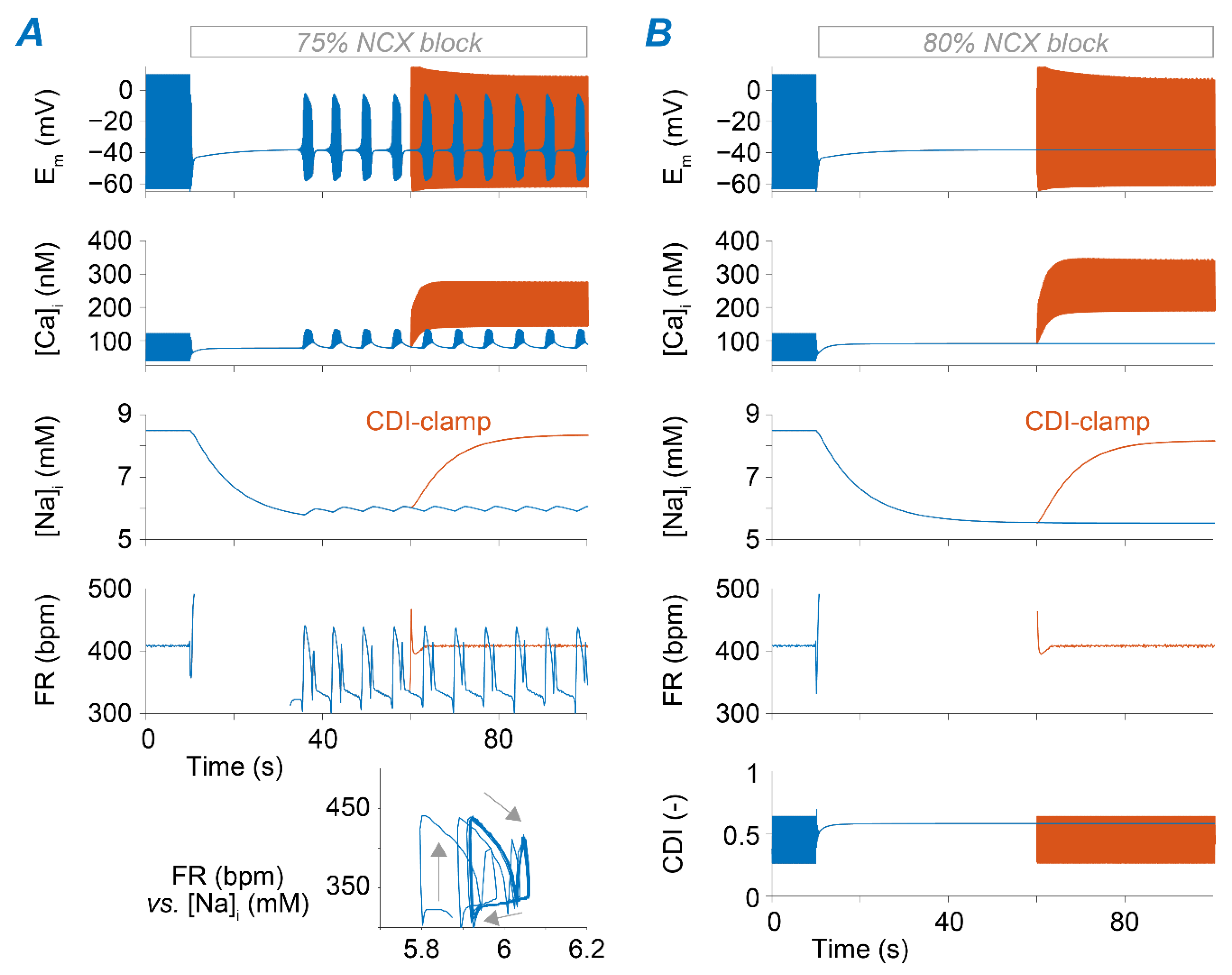
2.4. Ca2+ Accumulation Induced by NCX Impairment Reduces SAM Automaticity via Excessive Ca2+-Dependent ICaL Inactivation
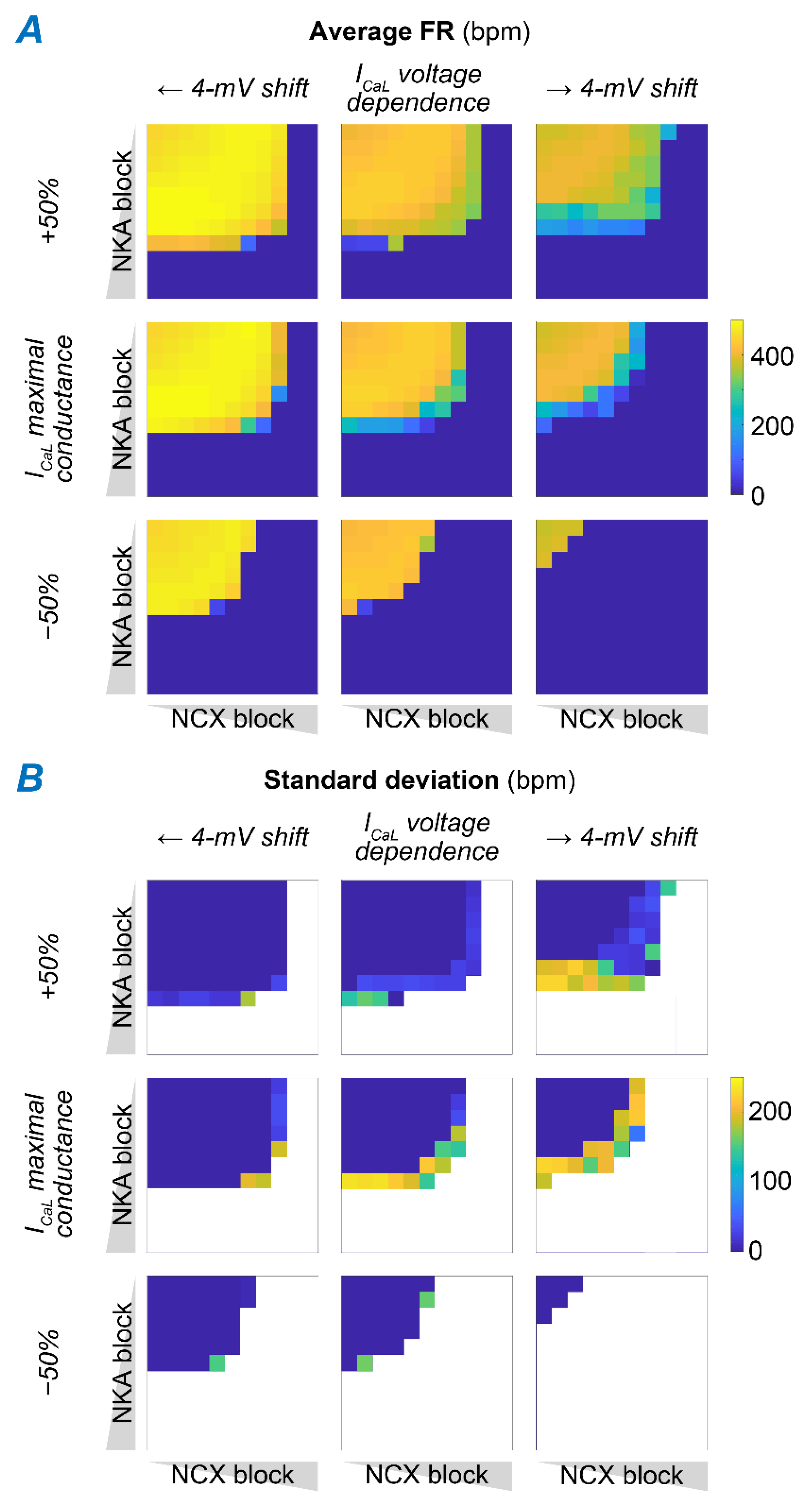
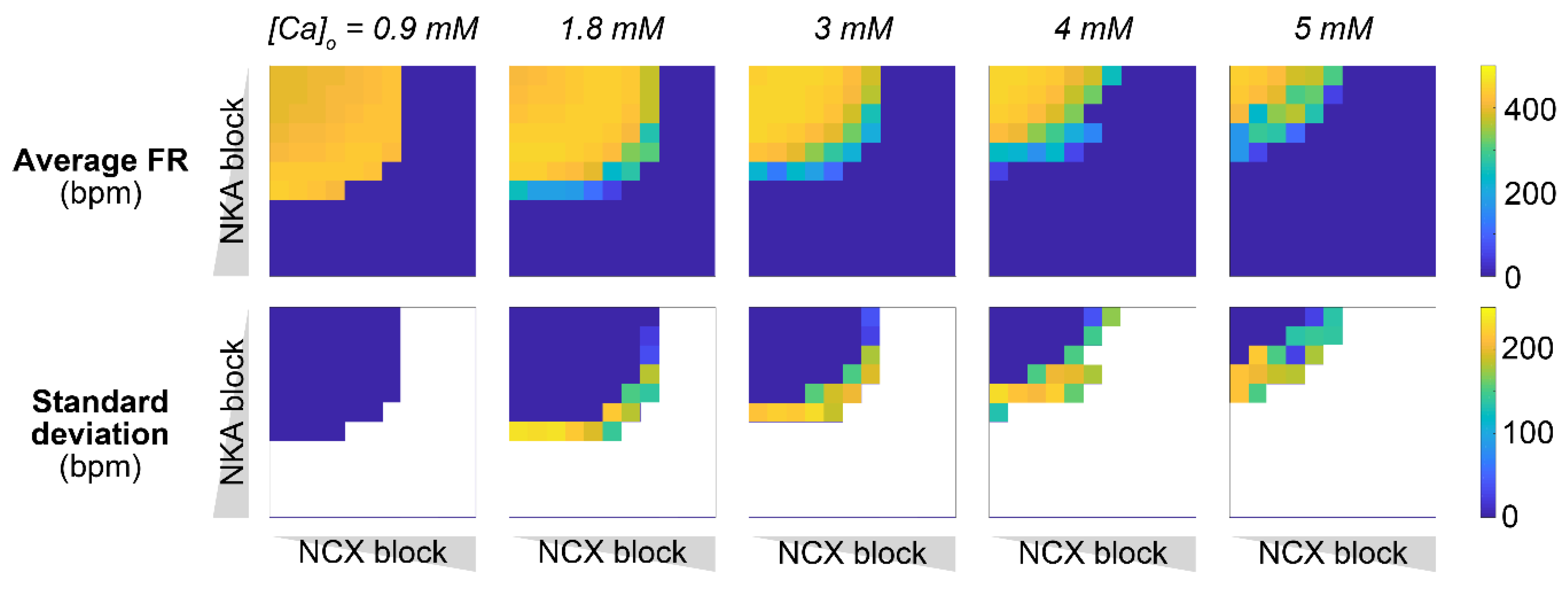
2.5. Concomitant NKA, NCX, and ICaL Block Increases the Susceptibility to Pacemaking Dysfunction
3. Discussion
3.1. Summary of the Results
3.2. Feedback of Na+ and Ca2+ Signals and Membrane Clock Dynamically Modulates SAM Automaticity
3.3. Disruption of Na+ Homeostatic Processes Contributes to SAN Dysfunction in Animal Models and Patients
3.4. Limitations and Future Directions
3.5. Conclusions
4. Methods
4.1. Experimental Data
4.2. Model Development
- (i)
- ICaL. The original Kharche et al. model includes isoform-specific formulations for ICaL1.2 and ICaL1.3 based on data obtained from genetically modified mice lacking either isoform. Given the lack of pharmacological agents that unequivocally distinguish between Cav1.2 and Cav1.3 when both expressed in wild-type mice, we eliminated ICaL1.2 and re-parameterized the formulation of ICaL1.3 to reproduce our experimental peak ICaL-voltage relationship [35]. This was obtained by negatively shifting (−7 mV) the voltage-dependence of activation and inactivation and decreasing the maximal conductance by one-third.
- (ii)
- Ito and Isus. Maximal conductances of both components of the outward K+ currents were increased by 2.5-fold [36].
- (iii)
- If. The original If the formulation of the Kharche et al. model was replaced with our recently updated version [37]. Briefly, we modified and extended the Hodgkin-Huxley type If model originally present in the Kharche et al. framework based on our novel data in murine SAMs describing voltage-dependence of If availability and activation/deactivation kinetics. Notably, in our novel formulation, activation and deactivation of If exhibit both fast and slow kinetics [37].
4.3. Simulation Protocols and Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Maximal Conductances | |
| Gst | Sustained inward Na+ current (Ist) |
| GNa1.1 | TTX-sensitive Na+ current (INa1.1) |
| GNa1.5 | TTX-resistant Na+ current (INa1.5) |
| GCaT | T-type Ca2+ current (ICaT) |
| GCaL | L-type Ca2+ current (ICaL) |
| Gf | Hyperpolarization-activated (funny) current (If) |
| GK1 | Time-independent K+ current (IK1) |
| GKr | Rapid delayed rectifying K+ current (IKr) |
| GKs | Slow delayed rectifying K+ current (IKs) |
| Gto | Transient component of the 4-AP-sensitive K+ current (Ito) |
| Gsus | Sustained component of the 4-AP-sensitive K+ current (Isus) |
| GNaB | Background Na+ current (INaB) |
| GCaB | Background Ca2+ current (ICaB) |
| Maximal Transport Rates | |
| vNKA | Na+/K+ ATPase (NKA) |
| vNCX | Na+/Ca2+ exchanger (NCX) |
| vRyR | Ca2+ release via ryanodine receptor |
| vSERCA | Sarcoplasmic reticulum (SR) Ca2+ pump |
| Output | Unit | |
|---|---|---|
| FR | Firing rate | bpm |
| CL | Cycle length | ms |
| UV | Upstroke velocity | mV/ms |
| RR | Repolarization rate | mV/ms |
| DD | Diastolic duration | ms |
| APD | Action potential (AP) duration | ms |
| APD90 | APD at 90% of repolarization | ms |
| APD50 | APD at 50% of repolarization | ms |
| MDP | Maximum diastolic potential | mV |
| APamp | AP amplitude | mV |
| THR | Threshold potential | mV |
| DDR | Diastolic depolarization rate | mV/s |
References
- Monfredi, O.; Dobrzynski, H.; Mondal, T.; Boyett, M.R.; Morris, G.M. The anatomy and physiology of the sinoatrial node-a contemporary review: Sinoatrial nodal anatomy and physiology. Pacing Clin. Electrophysiol. 2010, 33, 1392–1406. [Google Scholar] [CrossRef] [PubMed]
- Boyett, M.R.; Honjo, H.; Kodama, I. The sinoatrial node, a heterogeneous pacemaker structure. Cardiovasc. Res. 2000, 47, 658–687. [Google Scholar] [CrossRef]
- Lakatta, E.G.; Maltsev, V.A.; Vinogradova, T.M. A Coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart’s pacemaker. Circ. Res. 2010, 106, 659–673. [Google Scholar] [CrossRef] [PubMed]
- Yaniv, Y.; Lakatta, E.G.; Maltsev, V.A. From two competing oscillators to one coupled-clock pacemaker cell system. Front. Physiol. 2015, 6. [Google Scholar] [CrossRef]
- DiFrancesco, D. The role of the funny current in pacemaker activity. Circ. Res. 2010, 106, 434–446. [Google Scholar] [CrossRef] [PubMed]
- Torrente, A.G.; Mesirca, P.; Bidaud, I.; Mangoni, M.E. Channelopathies of Voltage-Gated L-Type Cav1.3/A1D and T-Type Cav3.1/A1G Ca2+ Channels in dysfunction of heart automaticity. Pflugers Arch. Eur. J Physiol. 2020, 472, 817–830. [Google Scholar] [CrossRef]
- Bers, D.M.; Morotti, S. Ca(2+) Current facilitation Is CaMKII-dependent and has arrhythmogenic consequences. Front. Pharmacol. 2014, 5, 144. [Google Scholar] [CrossRef] [PubMed]
- Mesirca, P.; Fedorov, V.V.; Hund, T.J.; Torrente, A.G.; Bidaud, I.; Mohler, P.J.; Mangoni, M.E. Pharmacologic approach to sinoatrial node dysfunction. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 757–778. [Google Scholar] [CrossRef] [PubMed]
- Milanesi, R.; Bucchi, A.; Baruscotti, M. The genetic basis for inherited forms of sinoatrial dysfunction and atrioventricular node dysfunction. J. Interv. Card. Electrophysiol. 2015, 43, 121–134. [Google Scholar] [CrossRef]
- Wallace, M.J.; El Refaey, M.; Mesirca, P.; Hund, T.J.; Mangoni, M.E.; Mohler, P.J. Genetic complexity of sinoatrial node dysfunction. Front. Genet. 2021, 12, 654925. [Google Scholar] [CrossRef] [PubMed]
- Sanders, P.; Kistler, P.M.; Morton, J.B.; Spence, S.J.; Kalman, J.M. Remodeling of sinus node function in patients with congestive heart failure: Reduction in sinus node reserve. Circulation 2004, 110, 897–903. [Google Scholar] [CrossRef]
- John, R.M.; Kumar, S. Sinus node and atrial arrhythmias. Circulation 2016, 133, 1892–1900. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.H.; Sharpe, E.J.; Proenza, C. Cardiac pacemaker activity and aging. Annu. Rev. Physiol. 2020, 82, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Despa, S.; Bers, D.M. Na+ Transport in the normal and failing heart—Remember the balance. J. Mol. Cell. Cardiol. 2013, 61, 2–10. [Google Scholar] [CrossRef]
- Grandi, E.; Herren, A.W. CaMKII-dependent regulation of cardiac Na+ homeostasis. Front. Pharmacol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Despa, S. Myocyte [Na+]i dysregulation in heart failure and diabetic cardiomyopathy. Front. Physiol. 2018, 9, 1303. [Google Scholar] [CrossRef]
- Horvath, B.; Banyasz, T.; Jian, Z.; Hegyi, B.; Kistamas, K.; Nanasi, P.P.; Izu, L.T.; Chen-Izu, Y. Dynamics of the late Na+ current during cardiac action potential and its contribution to afterdepolarizations. J. Mol. Cell. Cardiol. 2013, 64, 59–68. [Google Scholar] [CrossRef]
- Hegyi, B.; Bányász, T.; Shannon, T.R.; Chen-Izu, Y.; Izu, L.T. Electrophysiological determination of submembrane Na+ concentration in cardiac myocytes. Biophys. J. 2016, 111, 1304–1315. [Google Scholar] [CrossRef]
- Despa, S.; Islam, M.A.; Pogwizd, S.M.; Bers, D.M. Intracellular [Na+] and Na+ pump rate in rat and rabbit ventricular myocytes. J. Physiol. 2002, 539, 133–143. [Google Scholar] [CrossRef]
- Despa, S.; Islam, M.A.; Weber, C.R.; Pogwizd, S.M.; Bers, D.M. Intracellular Na+ concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation 2002, 105, 2543–2548. [Google Scholar] [CrossRef]
- Pieske, B.; Maier, L.S.; Piacentino, V.; Weisser, J.; Hasenfuss, G.; Houser, S. Rate Dependence of [Na+]i and contractility in nonfailing and failing human myocardium. Circulation 2002, 106, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Sossalla, S.; Wagner, S.; Rasenack, E.C.L.; Ruff, H.; Weber, S.L.; Schöndube, F.A.; Tirilomis, T.; Tenderich, G.; Hasenfuss, G.; Belardinelli, L.; et al. Ranolazine improves diastolic dysfunction in isolated myocardium from failing human hearts—role of late sodium current and intracellular ion accumulation. J. Mol. Cell. Cardiol. 2008, 45, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Sossalla, S.; Maurer, U.; Schotola, H.; Hartmann, N.; Didié, M.; Zimmermann, W.-H.; Jacobshagen, C.; Wagner, S.; Maier, L.S. Diastolic dysfunction and arrhythmias caused by overexpression of CaMKIIδC can be reversed by inhibition of late Na+ current. Basic Res. Cardiol. 2011, 106, 263–272. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Morotti, S.; Edwards, A.G.; McCulloch, A.D.; Bers, D.M.; Grandi, E. A novel computational model of mouse myocyte electrophysiology to assess the synergy between Na+ loading and CaMKII. J. Physiol. 2014, 592, 1181–1197. [Google Scholar] [CrossRef]
- Morotti, S.; Grandi, E. Quantitative systems models illuminate arrhythmia mechanisms in heart failure: Role of the Na+–Ca2+–Ca2+/calmodulin-dependent protein kinase ii-reactive oxygen species feedback. Wiley Interdiscip. Rev. Syst. Biol. Med. 2019, 11, e1434. [Google Scholar] [CrossRef]
- Bers, D.M. Excitation-contraction coupling and cardiac contractile force. In Developments in Cardiovascular Medicine; Springer: Dordrecht, The Netherlands, 2001; Volume 237, ISBN 978-0-7923-7158-8. [Google Scholar]
- Xie, Y.; Liao, Z.; Grandi, E.; Shiferaw, Y.; Bers, D.M. Slow [Na]i changes and positive feedback between membrane potential and [Ca]i underlie intermittent early afterdepolarizations and arrhythmias. Circ. Arrhythm. Electrophysiol. 2015, 8, 1472–1480. [Google Scholar] [CrossRef]
- Krogh-Madsen, T.; Christini, D.J. Slow [Na+]i dynamics impacts arrhythmogenesis and spiral wave reentry in cardiac myocyte ionic model. Chaos 2017, 27, 093907. [Google Scholar] [CrossRef] [PubMed]
- Torrente, A.G.; Zhang, R.; Zaini, A.; Giani, J.F.; Kang, J.; Lamp, S.T.; Philipson, K.D.; Goldhaber, J.I. Burst pacemaker activity of the sinoatrial node in sodium–calcium exchanger knockout mice. Proc. Natl. Acad. Sci. USA 2015, 112, 9769–9774. [Google Scholar] [CrossRef]
- Yue, X.; Hazan, A.; Lotteau, S.; Zhang, R.; Torrente, A.G.; Philipson, K.D.; Ottolia, M.; Goldhaber, J.I. Na/Ca exchange in the atrium: Role in sinoatrial node pacemaking and excitation-contraction coupling. Cell Calcium 2020, 87, 102167. [Google Scholar] [CrossRef]
- Mohler, P.J.; Splawski, I.; Napolitano, C.; Bottelli, G.; Sharpe, L.; Timothy, K.; Priori, S.G.; Keating, M.T.; Bennett, V. A Cardiac Arrhythmia syndrome caused by loss of ankyrin-b function. Proc. Natl. Acad. Sci. USA 2004, 101, 9137–9142. [Google Scholar] [CrossRef]
- Le Scouarnec, S.; Bhasin, N.; Vieyres, C.; Hund, T.J.; Cunha, S.R.; Koval, O.; Marionneau, C.; Chen, B.; Wu, Y.; Demolombe, S.; et al. Dysfunction in Ankyrin-B-Dependent ion channel and transporter targeting causes human sinus node disease. Proc. Natl. Acad. Sci. USA 2008, 105, 15617–15622. [Google Scholar] [CrossRef] [PubMed]
- Glukhov, A.V.; Fedorov, V.V.; Anderson, M.E.; Mohler, P.J.; Efimov, I.R. Functional anatomy of the murine sinus node: High-resolution optical mapping of Ankyrin-B Heterozygous Mice. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H482–H491. [Google Scholar] [CrossRef] [PubMed]
- Kharche, S.; Yu, J.; Lei, M.; Zhang, H. A Mathematical Model of Action Potentials of Mouse Sinoatrial Node Cells with Molecular Bases. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H945–H963. [Google Scholar] [CrossRef]
- Larson, E.D.; St Clair, J.R.; Sumner, W.A.; Bannister, R.A.; Proenza, C. Depressed pacemaker activity of sinoatrial node myocytes contributes to the age-dependent decline in maximum heart rate. Proc. Natl. Acad. Sci. USA 2013, 110, 18011–18016. [Google Scholar] [CrossRef]
- St Clair, J.R.; Sharpe, E.J.; Hagar, A.; Garthe, C.; Juchno, J.; Proenza, C. Diabetes slows heart rate via electrical remodeling of K+ currents in sinoatrial node myocytes. Biophys. J. 2015, 108, 195a. [Google Scholar] [CrossRef][Green Version]
- Peters, C.H.; Liu, P.W.; Morotti, S.; Gantz, S.C.; Grandi, E.; Bean, B.P.; Proenza, C. Bi-directional flow of the funny current (If) during the pacemaking cycle in murine sinoatrial node myocytes. bioRxiv 2021. [Google Scholar] [CrossRef]
- Sarkar, A.X.; Sobie, E.A. Regression analysis for constraining free parameters in electrophysiological models of cardiac cells. PLoS Comput. Biol. 2010, 6, e1000914. [Google Scholar] [CrossRef] [PubMed]
- Rickert, C.; Proenza, C. Action potential heterogeneity in murine sinoatrial node myocytes. Biophys. J. 2017, 112, 35a. [Google Scholar] [CrossRef]
- Rickert, C.; Proenza, C. ParamAP: Standardized parameterization of sinoatrial node myocyte action potentials. Biophys. J. 2017, 113, 765–769. [Google Scholar] [CrossRef]
- Lei, M.; Jones, S.A.; Liu, J.; Lancaster, M.K.; Fung, S.S.-M.; Dobrzynski, H.; Camelliti, P.; Maier, S.K.G.; Noble, D.; Boyett, M.R. Requirement of neuronal- and cardiac-type sodium channels for murine sinoatrial node pacemaking: Sodium channels in the SA Node. J. Physiol. 2004, 559, 835–848. [Google Scholar] [CrossRef]
- Golovko, V.; Gonotkov, M.; Lebedeva, E. Effects of 4-aminopyridine on action potentials generation in mouse sinoauricular Node Strips. Physiol. Rep. 2015, 3, e12447. [Google Scholar] [CrossRef]
- Mangoni, M.E.; Traboulsie, A.; Leoni, A.-L.; Couette, B.; Marger, L.; Le Quang, K.; Kupfer, E.; Cohen-Solal, A.; Vilar, J.; Shin, H.-S.; et al. Bradycardia and slowing of the atrioventricular conduction in mice lacking CaV3.1/α1G T-Type calcium channels. Circ. Res. 2006, 98, 1422–1430. [Google Scholar] [CrossRef] [PubMed]
- Kodama, I.; Nikmaram, M.R.; Boyett, M.R.; Suzuki, R.; Honjo, H.; Owen, J.M. Regional differences in the role of the Ca2+ and Na+ currents in pacemaker activity in the sinoatrial node. Am. J. Physiol. Heart Circ. Physiol. 1997, 272, H2793–H2806. [Google Scholar] [CrossRef] [PubMed]
- Groenke, S.; Larson, E.D.; Alber, S.; Zhang, R.; Lamp, S.T.; Ren, X.; Nakano, H.; Jordan, M.C.; Karagueuzian, H.S.; Roos, K.P.; et al. Complete atrial-specific knockout of sodium-calcium exchange eliminates sinoatrial node pacemaker activity. PLoS ONE 2013, 8, e81633. [Google Scholar] [CrossRef] [PubMed]
- Miyamae, S.I.; Goto, K. Effects of Ethylene Glycol-Bis-(Beta-Aminoethyl Ether)-N,N′-Tetraacetic acid on rabbit sinoatrial node cells treated with cardiotonic steroids. J. Pharmacol. Exp. Ther. 1988, 245, 706–717. [Google Scholar]
- Baudot, M.; Torre, E.; Bidaud, I.; Louradour, J.; Torrente, A.G.; Fossier, L.; Talssi, L.; Nargeot, J.; Barrère-Lemaire, S.; Mesirca, P.; et al. Concomitant Genetic Ablation of L-Type Cav1.3 (A1D) and T-Type Cav3.1 (A1G) Ca2+ channels disrupts heart automaticity. Sci. Rep. 2020, 10, 18906. [Google Scholar] [CrossRef]
- Pott, C.; Philipson, K.D.; Goldhaber, J.I. Excitation–Contraction Coupling in Na+–Ca2+ Exchanger Knockout Mice: Reduced Transsarcolemmal Ca2+ Flux. Circ. Res. 2005, 97, 1288–1295. [Google Scholar] [CrossRef]
- Altamirano, J.; Li, Y.; DeSantiago, J.; Piacentino, V.; Houser, S.R.; Bers, D.M. The inotropic effect of cardioactive glycosides in ventricular myocytes requires Na+–Ca2+ Exchanger Function: Na+ and cardioactive glycoside action. J. Physiol. 2006, 575, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Steinbeck, G.; Bonke, F.I.; Allessie, M.A.; Lammers, W.J. The effect of ouabain on the isolated sinus node preparation of the rabbit studied with microelectrodes. Circ. Res. 1980, 46, 406–414. [Google Scholar] [CrossRef]
- Gonzalez, M.D.; Vassalle, M. Role of oscillatory potential and pacemaker shifts in digitalis intoxication of the sinoatrial node. Circulation 1993, 87, 1705–1714. [Google Scholar] [CrossRef]
- El-Sherif, N.; Turitto, G. Electrolyte disorders and arrhythmogenesis. Cardiol. J. 2011, 18, 233–245. [Google Scholar]
- Loewe, A.; Lutz, Y.; Nairn, D.; Fabbri, A.; Nagy, N.; Toth, N.; Ye, X.; Fuertinger, D.H.; Genovesi, S.; Kotanko, P.; et al. Hypocalcemia-induced slowing of human sinus node pacemaking. Biophys. J. 2019, 117, 2244–2254. [Google Scholar] [CrossRef]
- Loewe, A.; Lutz, Y.; Nagy, N.; Fabbri, A.; Schweda, C.; Varro, A.; Severi, S. Inter-species differences in the response of sinus node cellular pacemaking to changes of extracellular calcium. In Proceedings of the 2019 41st Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC), Berlin, Germany, 23–27 July 2019; IEEE: Berlin, Germany, 2019; pp. 1875–1878. [Google Scholar]
- Torrente, A.G.; Fossier, L.; Baudot, M.; Bidaud, I.; Mesirca, P.; Mangoni, M. Hypercalcemia impairs sino-atrial automaticity through excessive Cav1.2-Mediated Ca2+ Influx. Arch. Cardiovasc. Dis. Suppl. 2020, 12, 255. [Google Scholar] [CrossRef]
- Torrente, A.G.; Zhang, R.; Wang, H.; Zaini, A.; Kim, B.; Yue, X.; Philipson, K.D.; Goldhaber, J.I. Contribution of small conductance K+ channels to sinoatrial node pacemaker activity: Insights from atrial-specific Na+/Ca2+ exchange knockout mice: Role of small conductance K+ channels in sinus node activity. J. Physiol. 2017, 595, 3847–3865. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, N.; Tran, A.; Kang, J.; Zhang, R.; Philipson, K.D.; Goldhaber, J.I. Regulation of calcium clock-mediated pacemaking by inositol-1,4,5-trisphosphate receptors in mouse sinoatrial nodal cells: IP3 R-mediated regulation of SAN Pacemaking. J. Physiol. 2015, 593, 2649–2663. [Google Scholar] [CrossRef]
- Cingolani, H.E.; Ennis, I.L. Sodium-hydrogen exchanger, cardiac overload, and myocardial hypertrophy. Circulation 2007, 115, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- van Borren, M.M.G.J.; Vos, M.A.; Houtman, M.J.C.; Antoons, G.; Ravesloot, J.H. Increased Sarcolemmal Na+/H+ Exchange Activity in hypertrophied myocytes from dogs with chronic atrioventricular block. Front. Physiol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Anderson, M.E. CaMKII in sinoatrial node physiology and dysfunction. Front. Pharmacol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Arbel-Ganon, L.; Behar, J.A.; Gómez, A.M.; Yaniv, Y. Distinct mechanisms mediate pacemaker dysfunction associated with catecholaminergic polymorphic ventricular tachycardia mutations: Insights from computational modeling. J. Mol. Cell Cardiol. 2020, 143, 85–95. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, E.A.; Rose, R.A.; Quinn, T.A. Neurohumoral control of sinoatrial node activity and heart rate: Insight from experimental models and findings from humans. Front. Physiol. 2020, 11, 170. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, K.; Monfredi, O.J.; Sirenko-Tagirova, S.G.; Maltseva, L.A.; Bychkov, R.; Kim, M.S.; Ziman, B.D.; Tarasov, K.V.; Tarasova, Y.S.; Zhang, J.; et al. A coupled-clock system drives the automaticity of human sinoatrial nodal pacemaker cells. Sci. Signal. 2018, 11, eaap7608. [Google Scholar] [CrossRef] [PubMed]
- Despa, S.; Bossuyt, J.; Han, F.; Ginsburg, K.S.; Jia, L.-G.; Kutchai, H.; Tucker, A.L.; Bers, D.M. Phospholemman-Phosphorylation Mediates the β-Adrenergic Effects on Na/K pump function in cardiac myocytes. Circ. Res. 2005, 97, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Despa, S.; Tucker, A.L.; Bers, D.M. Phospholemman-mediated activation of Na/K-ATPase Limits [Na]i and inotropic state during β-Adrenergic stimulation in mouse ventricular myocytes. Circulation 2008, 117, 1849–1855. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Maltsev, A.V.; Monfredi, O.; Maltseva, L.A.; Wirth, A.; Florio, M.C.; Tsutsui, K.; Riordon, D.R.; Parsons, S.P.; Tagirova, S.; et al. Heterogeneity of calcium clock functions in dormant, dysrhythmically and rhythmically firing single pacemaker cells isolated from SA Node. Cell Calcium 2018, 74, 168–179. [Google Scholar] [CrossRef]
- Lang, D.; Glukhov, A.V. Functional microdomains in heart’s pacemaker: A step beyond classical electrophysiology and remodeling. Front. Physiol. 2018, 9, 1686. [Google Scholar] [CrossRef] [PubMed]
- Csepe, T.A.; Kalyanasundaram, A.; Hansen, B.J.; Zhao, J.; Fedorov, V.V. Fibrosis: A structural modulator of sinoatrial node physiology and Dysfunction. Front. Physiol. 2015, 6. [Google Scholar] [CrossRef]
- Ni, H.; Morotti, S.; Grandi, E. A heart for diversity: Simulating variability in cardiac arrhythmia research. Front. Physiol. 2018, 9, 958. [Google Scholar] [CrossRef]
- Kurata, Y.; Matsuda, H.; Hisatome, I.; Shibamoto, T. Regional difference in dynamical property of sinoatrial node pacemaking: Role of Na+ Channel Current. Biophys. J. 2008, 95, 951–977. [Google Scholar] [CrossRef]
- Clancy, C.E.; Chen-Izu, Y.; Bers, D.M.; Belardinelli, L.; Boyden, P.A.; Csernoch, L.; Despa, S.; Fermini, B.; Hool, L.C.; Izu, L.; et al. Deranged sodium to sudden death: Deranged sodium to sudden death. J. Physiol. 2015, 593, 1331–1345. [Google Scholar] [CrossRef]
- Cingolani, E.; Goldhaber, J.I.; Marbán, E. Next-generation pacemakers: From small devices to biological pacemakers. Nat. Rev. Cardiol. 2018, 15, 139–150. [Google Scholar] [CrossRef]
- St Clair, J.R.; Sharpe, E.J.; Proenza, C. Culture and adenoviral infection of sinoatrial node myocytes from adult mice. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H490–H498. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, E.J.; St Clair, J.R.; Proenza, C. Methods for the isolation, culture, and functional characterization of sinoatrial node myocytes from adult mice. JoVE 2016, 54555. [Google Scholar] [CrossRef] [PubMed]
- Sobie, E.A. Parameter sensitivity analysis in electrophysiological models using multivariable regression. Biophys. J. 2009, 96, 1264–1274. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morotti, S.; Ni, H.; Peters, C.H.; Rickert, C.; Asgari-Targhi, A.; Sato, D.; Glukhov, A.V.; Proenza, C.; Grandi, E. Intracellular Na+ Modulates Pacemaking Activity in Murine Sinoatrial Node Myocytes: An In Silico Analysis. Int. J. Mol. Sci. 2021, 22, 5645. https://doi.org/10.3390/ijms22115645
Morotti S, Ni H, Peters CH, Rickert C, Asgari-Targhi A, Sato D, Glukhov AV, Proenza C, Grandi E. Intracellular Na+ Modulates Pacemaking Activity in Murine Sinoatrial Node Myocytes: An In Silico Analysis. International Journal of Molecular Sciences. 2021; 22(11):5645. https://doi.org/10.3390/ijms22115645
Chicago/Turabian StyleMorotti, Stefano, Haibo Ni, Colin H. Peters, Christian Rickert, Ameneh Asgari-Targhi, Daisuke Sato, Alexey V. Glukhov, Catherine Proenza, and Eleonora Grandi. 2021. "Intracellular Na+ Modulates Pacemaking Activity in Murine Sinoatrial Node Myocytes: An In Silico Analysis" International Journal of Molecular Sciences 22, no. 11: 5645. https://doi.org/10.3390/ijms22115645
APA StyleMorotti, S., Ni, H., Peters, C. H., Rickert, C., Asgari-Targhi, A., Sato, D., Glukhov, A. V., Proenza, C., & Grandi, E. (2021). Intracellular Na+ Modulates Pacemaking Activity in Murine Sinoatrial Node Myocytes: An In Silico Analysis. International Journal of Molecular Sciences, 22(11), 5645. https://doi.org/10.3390/ijms22115645