
Mutant p53 Mediates Sensitivity to Cancer Treatment Agents in Oesophageal Adenocarcinoma Associated with MicroRNA and SLC7A11 Expression

 , , , , and
, , , , and
Abstract
1. Introduction
2. Results
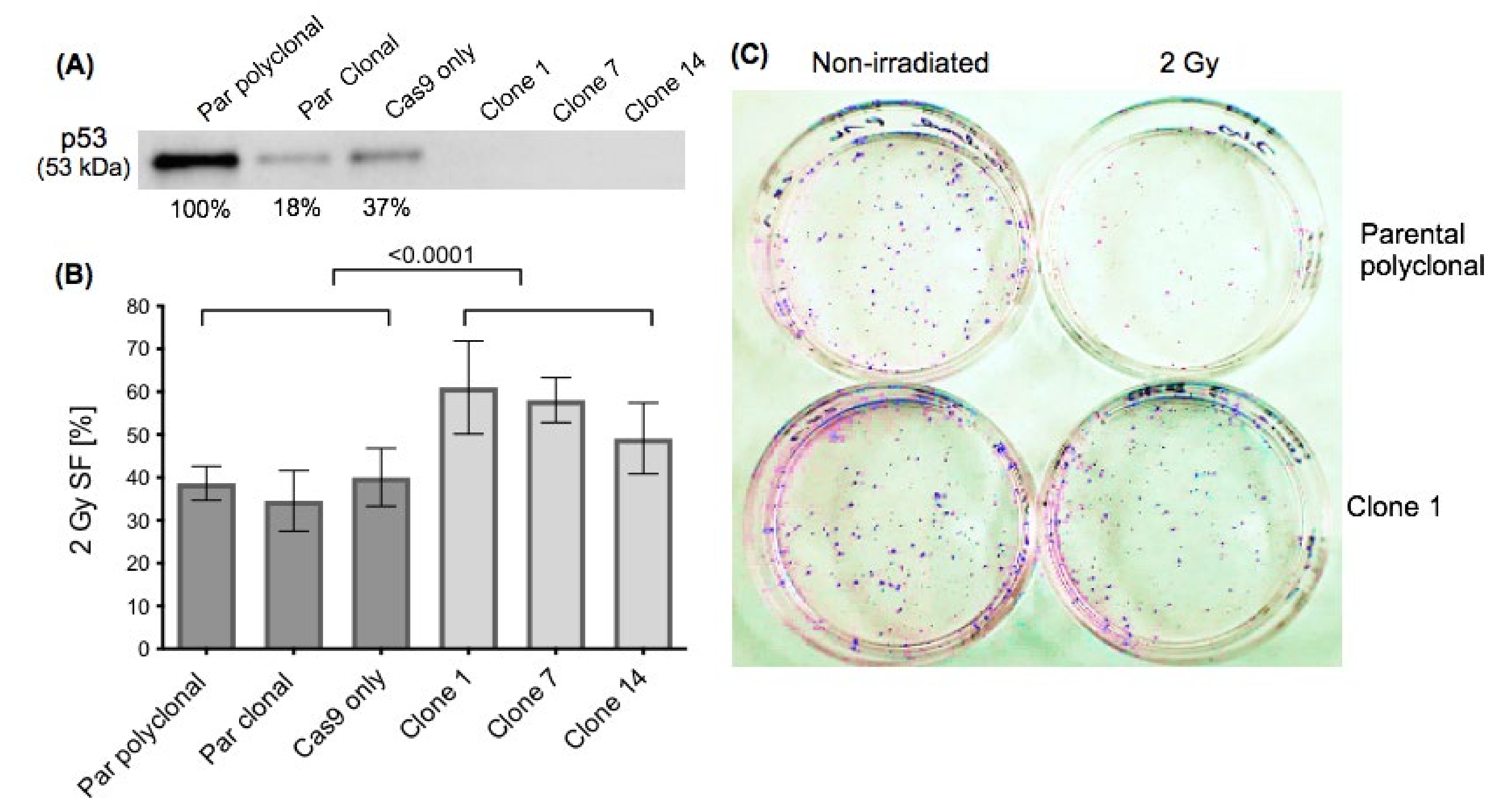
2.1. Effect of p53 Knockout on Cell Survival after Ionising Irradiation
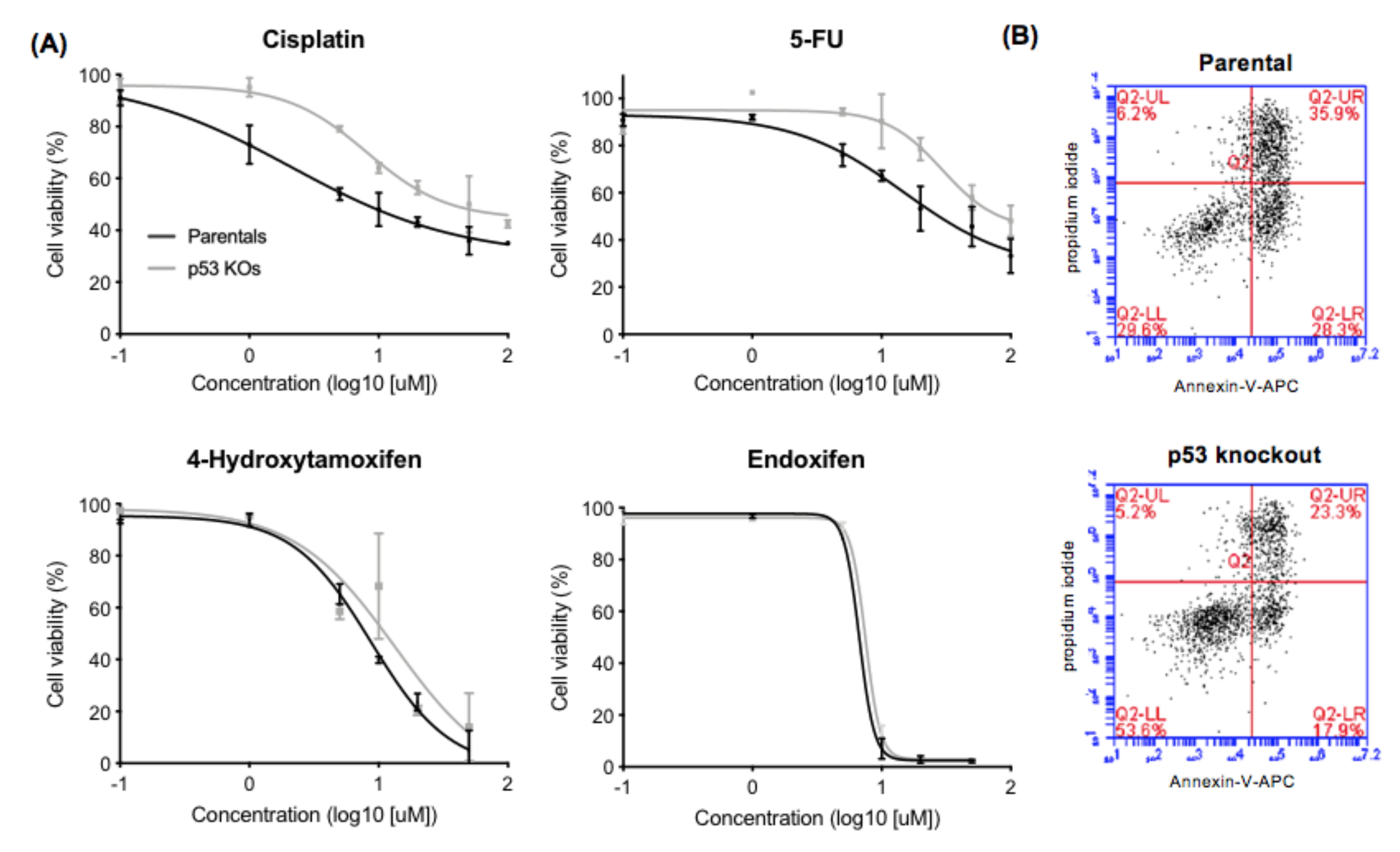
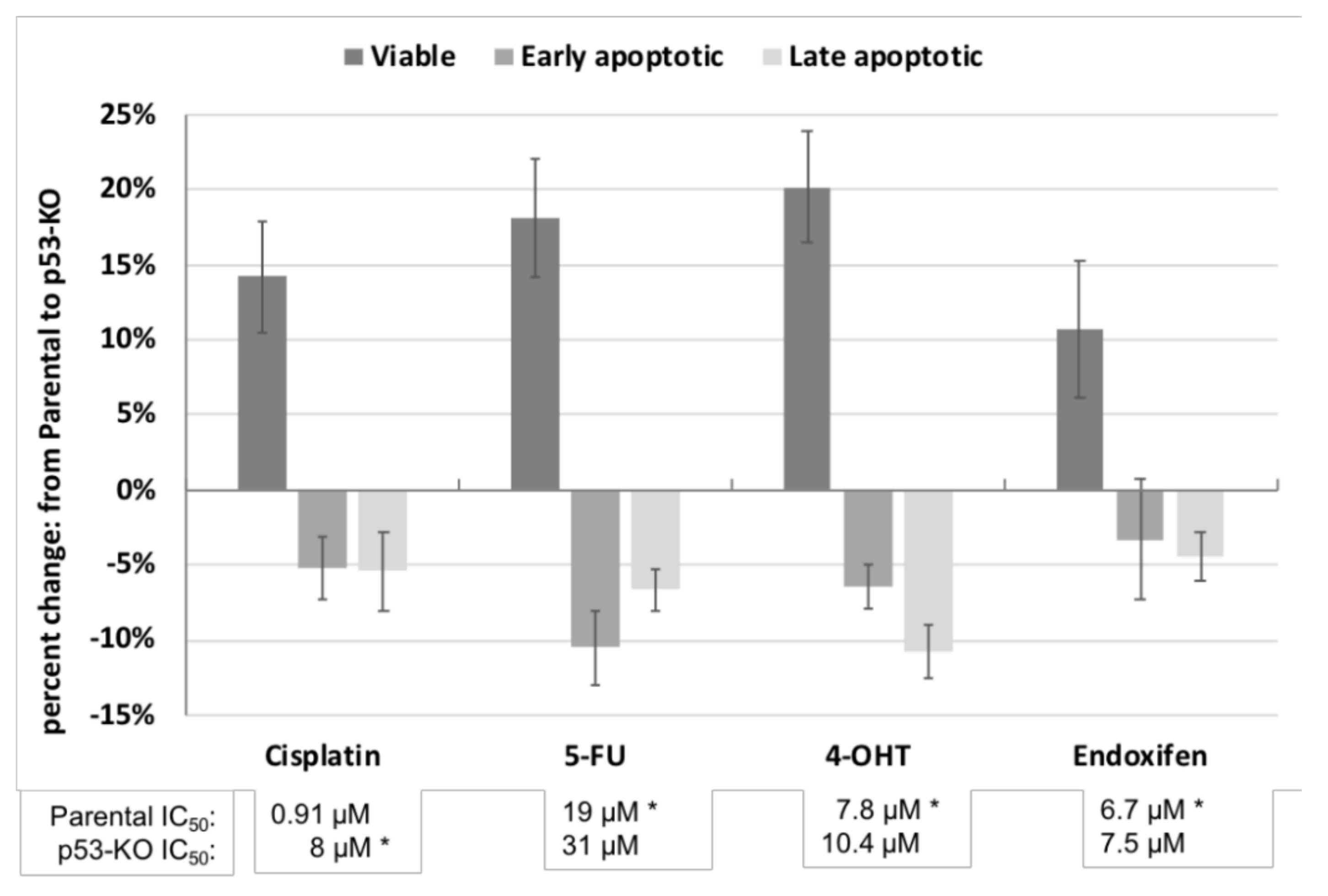
2.2. Effect of p53 Knockout on Cell Survival after Drug Treatment
2.3. Alterations in MiRNA Expression in JH-EsoAd1 Cells after p53 Knockout
2.4. Pathway Analyses
2.4.1. Pathway Analyses of Increased miRNAs
2.4.2. Pathway Analyses of Decreased MiRNAs
2.5. Rationale for Studying SLC7A11, a Key Regulator of the Antioxidant Response
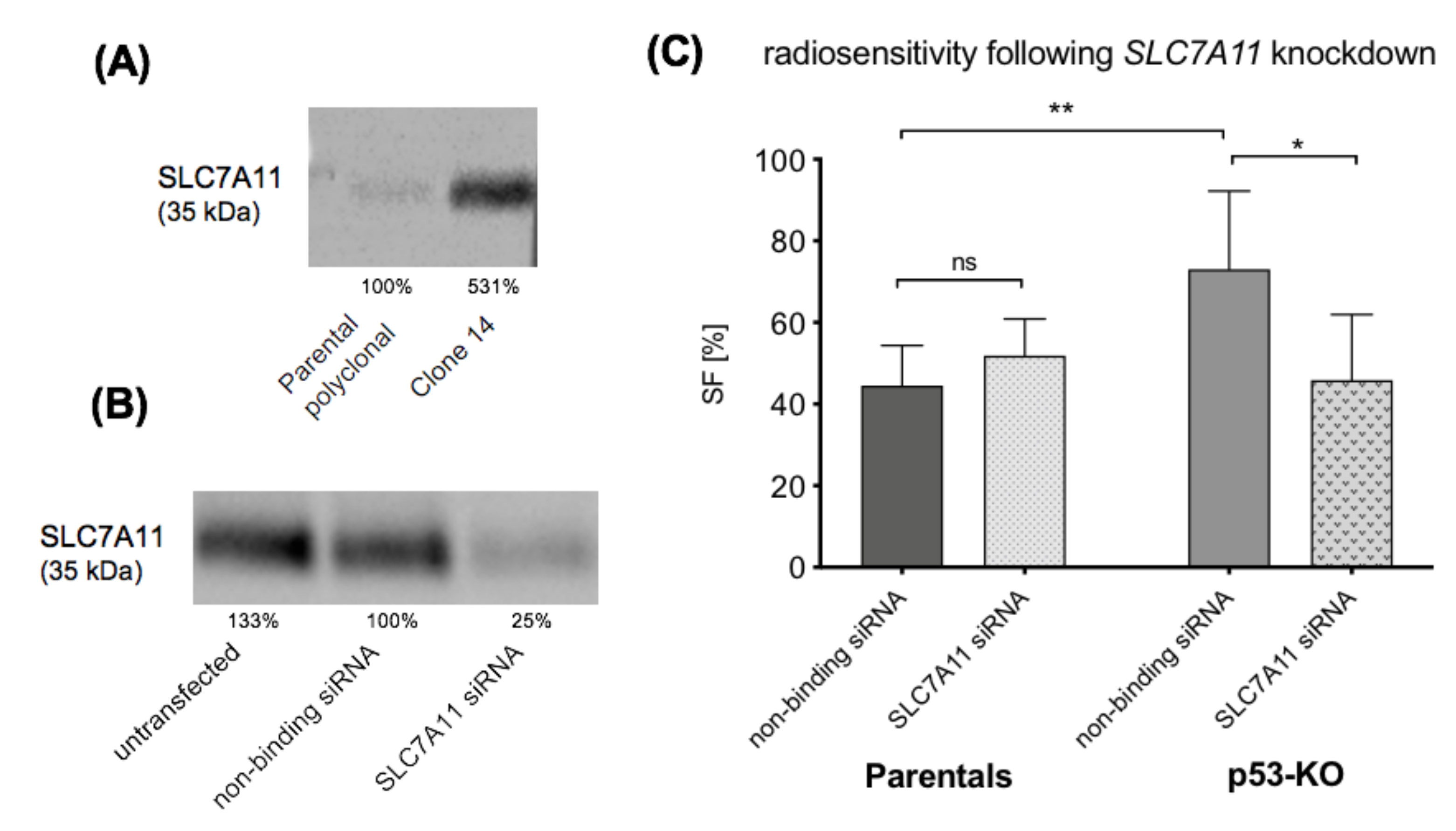
2.6. SLC7A11 Knockdown Restores Radiosensitivity of p53-KO Cells
2.7. Alterations in MiRNA Expression and Predicted Pathways Resulting from SLC7A11 Knockdown
2.8. Interaction of MiRNAs with Oestrogen Signaling
3. Discussion
3.1. Effect of p53 Knockout on Cell Survival after Ionizing Irradiation and Drug Treatment
3.2. Alterations in MiRNA Expression in JH-EsoAd1 Cells after p53 Knockout Followed by Pathway Analysis
3.2.1. Pathway Analyses of Increased MiRNAs
3.2.2. Pathway Analyses of Decreased MiRNAs
3.3. SLC7A11 Knockdown Restores Radiosensitivity of p53-KO JH-EsoAd1 Cells
3.4. Alterations in MiRNA Expression after SLC7A11 Knockdown Followed by Pathway Analysis
3.5. Possible Roles for Oestrogen Receptor Signaling
3.6. Limitations
4. Methods
4.1. Cell Lines and Culture Conditions
4.2. Irradiation and Clonogenic Survival Assay
4.3. Drug Treatment and Apoptosis Assay
4.4. SLC7A11 Knockdown Using siRNA
4.5. Irradiation and Clonogenic Survival Assay Following SLC7A11 Knockdown
4.6. Statistical Analyses
4.7. Western Blot
4.8. RNA Extraction
4.9. TaqMan® OpenArray® MiRNA Profiling
4.10. House Keeping Gene (HKG) Selection
4.11. Sample Comparisons, for Pooling of Cell Lines
4.12. Differential Expression Analyses
4.13. Gene Ontology and Biological Pathway Enrichment Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 5-FU | 5-Fluorouracil |
| CA | California |
| Cat | Catalogue number |
| cDNA | Complementary DNA |
| CSS | Charcoal-stripped bovine serum |
| Ct CT | Cycle threshold Connecticut |
| DB | Database |
| DE | Differential expression |
| ECL | Enhanced chemiluminescence |
| ER | Oestrogen receptor |
| FBS | Fetal bovine serum |
| FDR | False discovery rate |
| g | Gravity |
| Gy | Gray |
| HKG | Housekeeping gene |
| IARC | International Agency for Research on Cancer |
| Inh | Inhibitor |
| IQR | interquartile range |
| IC50 | 50% inhibitory concentration |
| KO | Knockout |
| MA | Massachusetts |
| miRNA | MicroRNA |
| Mut | Mutant |
| NPC OAC PA | Nasopharyngeal carcinoma Oesophageal adenocarcinoma Pennsylvania |
| PCR | Polymerase chain reaction |
| PE | Plating efficiency |
| PI | Propidium iodide |
| PPAR-α | Peroxisome proliferator-activated receptor alpha |
| RACS | Royal Australasian College of Surgeons |
| ROS | Reactive oxidative species |
| RP | Ribosomal protein |
| RT | Reverse transcription |
| SDS | Sodium dodecyl sulfate |
| SF UK | Survival Fraction United Kingdom |
| USA Wa | United States of America Washington |
| Wt | Wild-type |
References
- Domper Arnal, M.J.; Ferrandez Arenas, A.; Lanas Arbeloa, A. Esophageal cancer: Risk factors, screening and endoscopic treatment in Western and Eastern countries. World J. Gastroenterol. 2015, 21, 7933–7943. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Laversanne, M.; Brown, L.M.; Devesa, S.S.; Bray, F. Predicting the Future Burden of Esophageal Cancer by Histological Subtype: International Trends in Incidence up to 2030. Am. J. Gastroenterol. 2017, 112, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Rice, T.W.; Gress, D.M.; Patil, D.T.; Hofstetter, W.L.; Kelsen, D.P.; Blackstone, E.H. Cancer of the esophagus and esophagogastric junction-Major changes in the American Joint Committee on Cancer eighth edition cancer staging manual. CA Cancer J. Clin. 2017, 67, 304–317. [Google Scholar] [CrossRef]
- Sjoquist, K.M.; Burmeister, B.H.; Smithers, B.M.; Zalcberg, J.R.; Simes, R.J.; Barbour, A.; Gebski, V.; Australasian Gastro-Intestinal Trials Group. Survival after neoadjuvant chemotherapy or chemoradiotherapy for resectable oesophageal carcinoma: An updated meta-analysis. Lancet Oncol. 2011, 12, 681–692. [Google Scholar] [CrossRef]
- Haisley, K.R.; Hart, K.D.; Nabavizadeh, N.; Bensch, K.G.; Vaccaro, G.M.; Thomas, C.R.; Schipper, P.H.; Hunter, J.G.; Dolan, J.P. Neoadjuvant chemoradiotherapy with concurrent cisplatin/5-fluorouracil is associated with increased pathologic complete response and improved survival compared to carboplatin/paclitaxel in patients with locally advanced esophageal cancer. Dis. Esophagus 2017, 30. [Google Scholar] [CrossRef] [PubMed]
- Dittrick, G.W.; Weber, J.M.; Shridhar, R.; Hoffe, S.; Melis, M.; Almhanna, K.; Barthel, J.; McLoughlin, J.; Karl, R.C.; Meredith, K.L. Pathologic nonresponders after neoadjuvant chemoradiation for esophageal cancer demonstrate no survival benefit compared with patients treated with primary esophagectomy. Ann. Surg. Oncol. 2012, 19, 1678–1684. [Google Scholar] [CrossRef]
- Meredith, K.L.; Weber, J.M.; Turaga, K.K.; Siegel, E.M.; McLoughlin, J.; Hoffe, S.; Marcovalerio, M.; Shah, N.; Kelley, S.; Karl, R. Pathologic response after neoadjuvant therapy is the major determinant of survival in patients with esophageal cancer. Ann. Surg. Oncol. 2010, 17, 1159–1167. [Google Scholar] [CrossRef]
- Blandino, G.; Di Agostino, S. New therapeutic strategies to treat human cancers expressing mutant p53 proteins. J. Exp. Clin. Cancer Res. 2018, 37, 30. [Google Scholar] [CrossRef]
- Yamasaki, M.; Miyata, H.; Fujiwara, Y.; Takiguchi, S.; Nakajima, K.; Nishida, T.; Yasuda, T.; Matsuyama, J.; Mori, M.; Doki, Y. p53 genotype predicts response to chemotherapy in patients with squamous cell carcinoma of the esophagus. Ann. Surg. Oncol. 2010, 17, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Fisher, O.M.; Lord, S.J.; Falkenback, D.; Clemons, N.J.; Eslick, G.D.; Lord, R.V. The prognostic value of TP53 mutations in oesophageal adenocarcinoma: A systematic review and meta-analysis. Gut 2017, 66, 399–410. [Google Scholar] [CrossRef]
- Secrier, M.; Li, X.; de Silva, N.; Eldridge, M.D.; Contino, G.; Bornschein, J.; MacRae, S.; Grehan, N.; O’Donovan, M.; Miremadi, A.; et al. Mutational signatures in esophageal adenocarcinoma define etiologically distinct subgroups with therapeutic relevance. Nat. Genet. 2016, 48, 1131–1141. [Google Scholar] [CrossRef]
- Zhu, G.; Pan, C.; Bei, J.X.; Li, B.; Liang, C.; Xu, Y.; Fu, X. Mutant p53 in cancer progression and targeted therapies. Front. Oncol. 2020, 10, 595187. [Google Scholar] [CrossRef] [PubMed]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef]
- Fei, P.; El-Deiry, W.S. P53 and radiation responses. Oncogene 2003, 22, 5774–5783. [Google Scholar] [CrossRef] [PubMed]
- Cuddihy, A.R.; Bristow, R.G. The p53 protein family and radiation sensitivity: Yes or no? Cancer Metastasis Rev. 2004, 23, 237–257. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, J.; Xu, D.; Zhang, T.; Hu, W.; Feng, Z. Gain-of-function mutant p53 in cancer progression and therapy. J. Mol. Cell Biol. 2020, 12, 674–687. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.S.; Read, M.; Cullinane, C.; Azar, W.J.; Fennell, C.M.; Montgomery, K.G.; Haupt, S.; Haupt, Y.; Wiman, K.G.; Duong, C.P.; et al. APR-246 potently inhibits tumour growth and overcomes chemoresistance in preclinical models of oesophageal adenocarcinoma. Gut 2015, 64, 1506–1516. [Google Scholar] [CrossRef]
- Zhang, Q.; Bykov, V.J.N.; Wiman, K.G.; Zawacka-Pankau, J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis. 2018, 9, 439. [Google Scholar] [CrossRef]
- Liu, D.S.; Duong, C.P.; Haupt, S.; Montgomery, K.G.; House, C.M.; Azar, W.J.; Pearson, H.B.; Fisher, O.M.; Read, M.; Guerra, G.R.; et al. Inhibiting the system xC(-)/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat. Commun. 2017, 8, 14844. [Google Scholar] [CrossRef]
- Koppula, P.; Zhang, Y.; Zhuang, L.; Gan, B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun. 2018, 38, 12. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.F.; Chen, M.S.; Chou, Y.C.; Ueng, Y.F.; Yin, P.H.; Yeh, T.S.; Lee, H.C. Mitochondrial dysfunction enhances cisplatin resistance in human gastric cancer cells via the ROS-activated GCN2-eIF2α-ATF4-xCT pathway. Oncotarget 2016, 7, 74132–74151. [Google Scholar] [CrossRef]
- Huang, Y.; Dai, Z.; Barbacioru, C.; Sadée, W. Cystine-glutamate transporter SLC7A11 in cancer chemosensitivity and chemoresistance. Cancer Res. 2005, 65, 7446–7454. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Zhao, Y.; Feng, Z. MicroRNA Control of p53. J. Cell. Biochem. 2017, 118, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Mueller, A.K.; Lindner, K.; Hummel, R.; Haier, J.; Watson, D.I.; Hussey, D.J. MicroRNAs and Their Impact on Radiotherapy for Cancer. Radiat. Res. 2016, 185, 668–677. [Google Scholar] [CrossRef]
- Donzelli, S.; Mori, F.; Biagioni, F.; Bellissimo, T.; Pulito, C.; Muti, P.; Strano, S.; Blandino, G. MicroRNAs: Short non-coding players in cancer chemoresistance. Mol. Cell 2014, 2, 16. [Google Scholar] [CrossRef]
- Lindner, K.; Eichelmann, A.K.; Matuszcak, C.; Hussey, D.J.; Haier, J.; Hummel, R. Complex Epigenetic Regulation of Chemotherapy Resistance and Biohlogy in Esophageal Squamous Cell Carcinoma via MicroRNAs. Int. J. Mol. Sci. 2018, 19, 499. [Google Scholar] [CrossRef] [PubMed]
- Due, S.L.; Watson, D.I.; Bastian, I.; Ding, G.Q.; Sukocheva, O.A.; Astill, D.S.; Vat, L.; Hussey, D.J. Tamoxifen enhances the cytotoxicity of conventional chemotherapy in esophageal adenocarcinoma cells. Surg. Oncol. 2016, 25, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Sukocheva, O.A.; Wee, C.; Ansar, A.; Hussey, D.J.; Watson, D.I. Effect of estrogen on growth and apoptosis in esophageal adenocarcinoma cells. Dis. Esophagus 2013, 26, 628–635. [Google Scholar] [CrossRef]
- Bouaoun, L.; Sonkin, D.; Ardin, M.; Hollstein, M.; Byrnes, G.; Zavadil, J.; Olivier, M. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum. Mutat. 2016, 37, 865–876. [Google Scholar] [CrossRef]
- Alvarez, H.; Koorstra, J.B.; Hong, S.M.; Boonstra, J.J.; Dinjens, W.N.; Foratiere, A.A.; Wu, T.T.; Montgomery, E.; Eshleman, J.R.; Maitra, A. Establishment and characterization of a bona fide Barrett esophagus-associated adenocarcinoma cell line. Cancer Biol. Ther. 2008, 7, 1753–1755. [Google Scholar] [CrossRef][Green Version]
- Towers, C.G.; Guarnieri, A.L.; Micalizzi, D.S.; Harrell, J.C.; Gillen, A.E.; Kim, J.; Wang, C.A.; Oliphant, M.U.J.; Drasin, D.J.; Guney, M.A.; et al. The Six1 oncoprotein downregulates p53 via concomitant regulation of RPL26 and microRNA-27a-3p. Nat. Commun. 2015, 6, 10077. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Luo, L.; Chen, W.; Xu, H.X.; Chen, F.; Chen, L.Z.; Zeng, W.T.; Chen, J.S.; Huang, X.H. MicroRNA-24 increases hepatocellular carcinoma cell metastasis and invasion by targeting p53: miR-24 targeted p53. Biomed. Pharm. 2016, 84, 1113–1118. [Google Scholar] [CrossRef]
- Dong, P.; Karaayvaz, M.; Jia, N.; Kaneuchi, M.; Hamada, J.; Watari, H.; Sudo, S.; Ju, J.; Sakuragi, N. Mutant p53 gain-of-function induces epithelial-mesenchymal transition through modulation of the miR-130b-ZEB1 axis. Oncogene 2013, 32, 3286–3295. [Google Scholar] [CrossRef] [PubMed]
- Raut, S.K.; Singh, G.B.; Rastogi, B.; Saikia, U.N.; Mittal, A.; Dogra, N.; Singh, S.; Prasad, R.; Khullar, M. miR-30c and miR-181a synergistically modulate p53-p21 pathway in diabetes induced cardiac hypertrophy. Mol. Cell Biochem. 2016, 417, 191–203. [Google Scholar] [CrossRef]
- Mutharasan, R.K.; Nagpal, V.; Ichikawa, Y.; Ardehali, H. microRNA-210 is upregulated in hypoxic cardiomyocytes through Akt- and p53-dependent pathways and exerts cytoprotective effects. Am. J. Physiol. Heart 2011, 301, H1519–H1530. [Google Scholar] [CrossRef]
- Gao, X.M.; Zhu, Y.; Li, J.H.; Wang, X.Y.; Zhang, X.F.; Yi, C.H.; Yang, X. microRNA-26a induces a mitochondrial apoptosis mediated by p53 through targeting to inhibit Mcl1 in human hepatocellular carcinoma. Oncotargets 2018, 11, 2227–2239. [Google Scholar] [CrossRef]
- Helwak, A.; Kudla, G.; Dudnakova, T.; Tollervey, D. Mapping the Human miRNA Interactome by CLASH Reveals Frequent Noncanonical Binding. Cell 2013, 153, 654–665. [Google Scholar] [CrossRef]
- Costa, C.; Indovina, P.; Mattioli, E.; Forte, I.M.; Iannuzzi, C.A.; Luzzi, L.; Bellan, C.; De Summa, S.; Bucci, E.; Di Marzo, D.; et al. P53-regulated miR-320a targets PDL1 and is downregulated in malignant mesothelioma. Cell Death Dis. 2020, 11, 748. [Google Scholar]
- Hock, J.; Meister, G. The Argonaute protein family. Genome Biol. 2008, 9, 210. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.Y.; Wang, C.Q.; Liu, G.P.; Bi, C.; Wang, X.; Zhou, Q.Y.; Jin, H.C. SLC7A11/xCT in cancer: Biological functions and therapeutic implications. Am. J. Cancer Res. 2020, 10, 3106–3126. [Google Scholar] [PubMed]
- Calabrese, E.J.; Kozumbo, W.J. The hormetic dose-response mechanism: Nrf2 activation. Pharm. Res. 2021, 167, 105526. [Google Scholar] [CrossRef] [PubMed]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Kong, D.; Hou, X.; Liang, B.; He, M.; Liang, N.; Ma, S.; Liu, X. The tumor suppressor, p53, contributes to radiosensitivity of lung cancer cells by regulating autophagy and apoptosis. Cancer Biother. Radiopharm. 2013, 28, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Stein, Y.; Rotter, V.; Aloni-Grinstein, R. Gain-of-Function Mutant p53: All the Roads Lead to Tumorigenesis. Int. J. Mol. Sci. 2019, 20, 6197. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zhai, H.; Wang, X.; Han, Z.; Liu, C.; Lan, M.; Du, J.; Guo, C.; Zhang, Y.; Wu, K.; et al. Ribosomal proteins S13 and L23 promote multidrug resistance in gastric cancer cells by suppressing drug-induced apoptosis. Exp. Cell Res. 2004, 296, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Gregorio, A.; Martinez-Ramirez, I.; Pedraza-Chaverri, J.; Lizano, M. Reprogramming of Energy Metabolism in Response to Radiotherapy in Head and Neck Squamous Cell Carcinoma. Cancers 2019, 11, 182. [Google Scholar] [CrossRef]
- Richardson, R.B.; Harper, M.E. Mitochondrial stress controls the radiosensitivity of the oxygen effect: Implications for radiotherapy. Oncotarget 2016, 7, 21469–21483. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Butz, F.; Eichelmann, A.K.; Mayne, G.C.; Wang, T.; Bastian, I.; Chiam, K.; Marri, S.; Sykes, P.J.; Wijnhoven, B.P.; Toxopeus, E.; et al. MicroRNA Profiling in Oesophageal Adenocarcinoma Cell Lines and Patient Serum Samples Reveals a Role for miR-451a in Radiation Resistance. Int. J. Mol. Sci. 2020, 21, 8898. [Google Scholar] [CrossRef]
- El-Deiry, W.S. The role of p53 in chemosensitivity and radiosensitivity. Oncogene 2003, 22, 7486–7495. [Google Scholar] [CrossRef] [PubMed]
- Lynam-Lennon, N.; Bibby, B.A.; Mongan, A.M.; Marignol, L.; Paxton, C.N.; Geiersbach, K.; Bronner, M.P.; O’Sullivan, J.; Reynolds, J.; Maher, S.G. Low miR-187 expression promotes resistance to chemoradiation therapy in vitro and correlates with treatment failure in patients with esophageal adenocarcinoma. Mol. Med. 2016, 22, 388–397. [Google Scholar] [CrossRef]
- Lynam-Lennon, N.; Heavey, S.; Sommerville, G.; Bibby, B.A.; Ffrench, B.; Quinn, J.; Gasch, C.; O’Leary, J.J.; Gallagher, M.F.; Reynolds, J.V.; et al. MicroRNA-17 is downregulated in esophageal adenocarcinoma cancer stem-like cells and promotes a radioresistant phenotype. Oncotarget 2017, 8, 11400–11413. [Google Scholar] [CrossRef]
- Lynam-Lennon, N.; Reynolds, J.V.; Marignol, L.; Sheils, O.M.; Pidgeon, G.P.; Maher, S.G. MicroRNA-31 modulates tumour sensitivity to radiation in oesophageal adenocarcinoma. J. Mol. Med. 2012, 90, 1449–1458. [Google Scholar] [CrossRef]
- Matuszcak, C.; Lindner, K.; Eichelmann, A.K.; Hussey, D.J.; Haier, J.; Hummel, R. microRNAs: Key regulators of chemotherapy response and metastatic potential via complex control of target pathways in esophageal adenocarcinoma. Surg. Oncol. 2018, 27, 392–401. [Google Scholar] [CrossRef]
- Zhang, H.W.; Li, M.B.; Han, Y.; Hong, L.; Gong, T.Q.; Sun, L.; Zheng, X.S. Down-Regulation of miR-27a Might Reverse Multidrug Resistance of Esophageal Squamous Cell Carcinoma. Dig. Dis. Sci. 2010, 55, 2545–2551. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.Z.; Duan, Y.T.; Zhou, H.B. MicroRNA-27a directly targets KRAS to inhibit cell proliferation in esophageal squamous cell carcinoma. Oncol. Lett. 2015, 9, 471–477. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, S.; Shi, R.; Zhao, G. miR-27 promotes human gastric cancer cell metastasis by inducing epithelial-to-mesenchymal transition. Cancer Genet. 2011, 204, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Tang, H.; Lang, Y.Y.; Liu, M.; Li, X. MicroRNA-27a functions as an oncogene in gastric adenocarcinoma by targeting prohibitin. Cancer Lett. 2009, 273, 233–242. [Google Scholar] [CrossRef]
- Xu, L.L.; Xiang, J.Y.; Shen, J.; Zou, X.; Zhai, S.L.; Yin, Y.X.; Li, P.; Wang, X.R.; Sun, Q.M. Oncogenic MicroRNA-27a is a Target for Genistein in Ovarian Cancer Cells. Anti-Cancer Agent Med. Chem. 2013, 13, 1126–1132. [Google Scholar] [CrossRef] [PubMed]
- Mertens-Talcott, S.U.; Chintharlapalli, S.; Li, M.R.; Safe, S. The oncogenic microRNA-27a targets genes that regulate specificity protein transcription factors and the G(2)-M checkpoint in MDA-MB-231 breast cancer cells. Cancer Res. 2007, 67, 11001–11011. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.M.; Huang, Q.D.; Zheng, S.R.; Lin, K.L.; You, J.; Zhang, X.H. miR-27a regulates the sensitivity of breast cancer cells to cisplatin treatment via BAK-SMAC/DIABLO-XIAP axis. Tumor Biol. 2016, 37, 6837–6845. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.A.; Wang, Z.J.; Fan, Q.X.; Wang, R.L.; Sun, Y. microRNA-27a functions as a tumor suppressor in esophageal squamous cell carcinoma by targeting KRAS. Oncol. Rep. 2014, 31, 280–286. [Google Scholar] [CrossRef]
- Li, Y.Y.; Li, J.; Sun, X.L.; Chen, J.C.; Sun, X.Q.; Zheng, J.N.; Chen, R.F. MicroRNA-27a functions as a tumor suppressor in renal cell carcinoma by targeting epidermal growth factor receptor. Oncol. Lett. 2016, 11, 4217–4223. [Google Scholar] [CrossRef]
- Wu, Y.; Sun, X.; Song, B.; Qiu, X.; Zhao, J. MiR-375/SLC7A11 axis regulates oral squamous cell carcinoma proliferation and invasion. Cancer Med. 2017, 6, 1686–1697. [Google Scholar] [CrossRef]
- Li, X.W.; Xu, M.; Ding, L.; Tang, J.H. MiR-27a: A Novel Biomarker and Potential Therapeutic Target in Tumors. J. Cancer 2019, 10, 2836–2848. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.C.; Cao, Z.; Yang, G.; You, L.; Zhang, T.P.; Zhao, Y.P. MicroRNA-27a (miR-27a) in Solid Tumors: A Review Based on Mechanisms and Clinical Observations. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef]
- Cheng, C.M.; Geng, F.; Cheng, X.; Guo, D.L. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun. 2018, 38. [Google Scholar] [CrossRef] [PubMed]
- Lynam-Lennon, N.; Maher, S.G.; Maguire, A.; Phelan, J.; Muldoon, C.; Reynolds, J.V.; O’Sullivan, J. Altered Mitochondrial Function and Energy Metabolism Is Associated with a Radioresistant Phenotype in Oesophageal Adenocarcinoma. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Acunzo, M.; Romano, G.; Palmieri, D.; Lagana, A.; Garofalo, M.; Balatti, V.; Drusco, A.; Chiariello, M.; Nana-Sinkam, P.; Croce, C.M. Cross-talk between MET and EGFR in non-small cell lung cancer involves miR-27a and Sprouty2. Proc. Natl. Acad. Sci. USA 2013, 110, 8573–8578. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.Z.; Li, J.; Shao, D.; Pan, Y.; Chen, Y.J.; Li, S.; Yao, X.X.; Li, H.; Liu, W.W.; Zhang, M.; et al. Adipose tissue-secreted miR-27a promotes liver cancer by targeting FOXO1 in obese individuals. Oncotargets 2015, 8, 735–744. [Google Scholar] [CrossRef]
- Harris, S.L.; Levine, A.J. The p53 pathway: Positive and negative feedback loops. Oncogene 2005, 24, 2899–2908. [Google Scholar] [CrossRef]
- Wang, W.; Cheng, B.; Miao, L.; Mei, Y.; Wu, M. Mutant p53-R273H gains new function in sustained activation of EGFR signaling via suppressing miR-27a expression. Cell Death Dis. 2013, 4. [Google Scholar] [CrossRef]
- Li, G.; Liu, Y.; Su, Z.; Ren, S.; Zhu, G.; Tian, Y.; Qiu, Y. MicroRNA-324-3p regulates nasopharyngeal carcinoma radioresistance by directly targeting WNT2B. Eur. J. Cancer 2013, 49, 2596–2607. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Hu, Y.; Yan, K.; Qi, Y.; Zhang, C.; Zhu, D.; Liu, D.; Zhao, S. microRNA-10b confers cisplatin resistance by activating AKT/mTOR/P70S6K signaling via targeting PPARgamma in esophageal cancer. J. Cell Physiol. 2020, 235, 1247–1258. [Google Scholar] [CrossRef] [PubMed]
- Tao, S.; Liu, M.; Shen, D.; Zhang, W.; Wang, T.; Bai, Y. TGF-beta/Smads Signaling Affects Radiation Response and Prolongs Survival by Regulating DNA Repair Genes in Malignant Glioma. DNA Cell Biol. 2018, 37, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Ai, Q.; Cao, H.; Liu, Q. MiR-185-3p and miR-324-3p Predict Radiosensitivity of Nasopharyngeal Carcinoma and Modulate Cancer Cell Growth and Apoptosis by Targeting SMAD7. Med. Sci. Monit. 2015, 21, 2828–2836. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Zhou, H.; Wei, X.; Meng, Y.; Guo, Q. Downregulation of microRNA-324-3p inhibits lung cancer by blocking the NCAM1-MAPK axis through ALX4. Cancer Gene 2020. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.H.; Chen, Y.Z.; Lee, M.Y.; Weng, K.P.; Chang, H.T.; Yu, S.Y.; Dong, B.J.; Kuo, F.R.; Hung, L.T.; Liu, L.F.; et al. Comprehensive identification of microRNA arm selection preference in lung cancer: miR-324-5p and -3p serve oncogenic functions in lung cancer. Oncol. Lett. 2018, 15, 9818–9826. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Yuan, Q.; Liu, G.; Zhang, Y.; Yan, M.; Sun, Q.; Zhu, C. Long non-coding RNA MALAT1 regulates oxaliplatin-resistance via miR-324-3p/ADAM17 axis in colorectal cancer cells. Cancer Cell Int. 2020, 20, 473. [Google Scholar] [CrossRef] [PubMed]
- Reza, A.; Yuan, Y.G. microRNAs Mediated Regulation of the Ribosomal Proteins and its Consequences on the Global Translation of Proteins. Cells 2021, 10, 110. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Xiong, X.; Sun, Y. The role of ribosomal proteins in the regulation of cell proliferation, tumorigenesis, and genomic integrity. Sci. China Life Sci. 2016, 59, 656–672. [Google Scholar] [CrossRef] [PubMed]
- Boominathan, L. The tumor suppressors p53, p63, and p73 are regulators of microRNA processing complex. PLoS ONE 2010, 5, e10615. [Google Scholar] [CrossRef]
- Jiang, L.H.; Zhang, H.D.; Tang, J.H. MiR-30a: A Novel Biomarker and Potential Therapeutic Target for Cancer. J. Oncol. 2018, 2018, 5167829. [Google Scholar] [CrossRef] [PubMed]
- Saad, M.A.; Kuo, S.Z.; Rahimy, E.; Zou, A.E.; Korrapati, A.; Rahimy, M.; Kim, E.; Zheng, H.; Yu, M.A.; Wang-Rodriguez, J.; et al. Alcohol-dysregulated miR-30a and miR-934 in head and neck squamous cell carcinoma. Mol. Cancer 2015, 14. [Google Scholar] [CrossRef] [PubMed]
- Teramoto, K.; Katoh, H. The cystine/glutamate antiporter xCT is a key regulator of EphA2 S897 phosphorylation under glucose-limited conditions. Cell Signal. 2019, 62. [Google Scholar] [CrossRef] [PubMed]
- Schopman, N.C.; Heynen, S.; Haasnoot, J.; Berkhout, B. A miRNA-tRNA mix-up: tRNA origin of proposed miRNA. RNA Biol. 2010, 7, 573–576. [Google Scholar] [CrossRef]
- Wang, G.J.; Liu, G.H.; Ye, Y.W.; Fu, Y.; Zhang, X.F. The role of microRNA-1274a in the tumorigenesis of gastric cancer: Accelerating cancer cell proliferation and migration via directly targeting FOXO4. Biochem. Bioph. Res. Commun. 2015, 459, 629–635. [Google Scholar] [CrossRef]
- Yoshino, H.; Yonezawa, T.; Yonemori, M.; Miyamoto, K.; Sakaguchi, T.; Sugita, S.; Osako, Y.; Tatarano, S.; Nakagawa, M.; Enokida, H. Downregulation of microRNA-1274a induces cell apoptosis through regulation of BMPR1B in clear cell renal cell carcinoma. Oncol. Rep. 2018, 39, 173–181. [Google Scholar] [CrossRef]
- Guzman, N.; Agarwal, K.; Asthagiri, D.; Yu, L.; Saji, M.; Ringel, M.D.; Paulaitis, M.E. Breast Cancer-Specific miR Signature Unique to Extracellular Vesicles Includes “microRNA-like” tRNA Fragments. Mol. Cancer Res. 2015, 13, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Haussecker, D.; Huang, Y.; Lau, A.; Parameswaran, P.; Fire, A.Z.; Kay, M.A. Human tRNA-derived small RNAs in the global regulation of RNA silencing. RNA 2010, 16, 673–695. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.X.; Merchant, M.L.; Burke, T.J.; Ritzenthaler, J.D.; Li, M.; Gaweda, A.E.; Benz, F.W.; Roman, J.; Watson, W.H. Redox States of Protein Cysteines in Pathways of Protein Turnover and Cytoskeleton Dynamics Are Changed with Aging and Reversed by Slc7a11 Restoration in Mouse Lung Fibroblasts. Oxid. Med. Cell Longev. 2020, 2020. [Google Scholar] [CrossRef] [PubMed]
- Akgun, H.; Lechago, J.; Younes, M. Estrogen receptor-beta is expressed in Barrett’s metaplasia and associated adenocarcinoma of the esophagus. Anticancer Res. 2002, 22, 1459–1461. [Google Scholar]
- Kalayarasan, R.; Ananthakrishnan, N.; Kate, V.; Basu, D. Estrogen and progesterone receptors in esophageal carcinoma. Dis. Esophagus 2008, 21, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Al-Khyatt, W.; Tufarelli, C.; Khan, R.; Iftikhar, S.Y. Selective oestrogen receptor antagonists inhibit oesophageal cancer cell proliferation in vitro. BMC Cancer 2018, 18. [Google Scholar] [CrossRef]
- Rong, C.; Meinert, E.; Hess, J. Estrogen Receptor Signaling in Radiotherapy: From Molecular Mechanisms to Clinical Studies. Int. J. Mol. Sci. 2018, 19, 713. [Google Scholar] [CrossRef]
- Thomas, C.; Gustafsson, J.A. The different roles of ER subtypes in cancer biology and therapy. Nat. Rev. Cancer 2011, 11, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Konduri, S.D.; Bansal, S.; Nayak, B.K.; Rajasekaran, S.A.; Karuppayil, S.M.; Rajasekaran, A.K.; Das, G.M. Estrogen receptor-alpha binds p53 tumor suppressor protein directly and represses its function. J. Biol. Chem. 2006, 281, 9837–9840. [Google Scholar] [CrossRef]
- Schiff, R.; Reddy, P.; Ahotupa, M.; Coronado-Heinsohn, E.; Grim, M.; Hilsenbeck, S.G.; Lawrence, R.; Deneke, S.; Herrera, R.; Chamness, G.C.; et al. Oxidative stress and AP-1 activity in tamoxifen-resistant breast tumors in vivo. J. Natl. Cancer Inst. 2000, 92, 1926–1934. [Google Scholar] [CrossRef]
- Bekele, R.T.; Venkatraman, G.; Liu, R.Z.; Tang, X.; Mi, S.; Benesch, M.G.; Mackey, J.R.; Godbout, R.; Curtis, J.M.; McMullen, T.P.; et al. Oxidative stress contributes to the tamoxifen-induced killing of breast cancer cells: Implications for tamoxifen therapy and resistance. Sci. Rep. 2016, 6, 21164. [Google Scholar] [CrossRef]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef] [PubMed]
- Rafehi, H.; Orlowski, C.; Georgiadis, G.T.; Ververis, K.; El-Osta, A.; Karagiannis, T.C. Clonogenic Assay: Adherent Cells. Jove J. Vis. Exp. 2011. [Google Scholar] [CrossRef]
- Cao, Z.; West, C.; Norton-Wenzel, C.S.; Rej, R.; Davis, F.B.; Davis, P.J. Effects of resin or charcoal treatment on fetal bovine serum and bovine calf serum. Endocr. Res. 2009, 34, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Colella, A.D.; Chegenii, N.; Tea, M.N.; Gibbins, I.L.; Williams, K.A.; Chataway, T.K. Comparison of Stain-Free gels with traditional immunoblot loading control methodology. Anal. Biochem. 2012, 430, 108–110. [Google Scholar] [CrossRef]
- Bianchi, F.; Nicassio, F.; Marzi, M.; Belloni, E.; Dall’olio, V.; Bernard, L.; Pelosi, G.; Maisonneuve, P.; Veronesi, G.; Di Fiore, P.P. A serum circulating miRNA diagnostic test to identify asymptomatic high-risk individuals with early stage lung cancer. EMBO Mol. Med. 2011, 3, 495–503. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Storey, J.D. A direct approach to false discovery rates. J. R. Stat. Soc. 2002, 64, 479–498. [Google Scholar] [CrossRef]
- Chou, C.H.; Shrestha, S.; Yang, C.D.; Chang, N.W.; Lin, Y.L.; Liao, K.W.; Huang, W.C.; Sun, T.H.; Tu, S.J.; Lee, W.H.; et al. miRTarBase update 2018: A resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018, 46, D296–D302. [Google Scholar] [CrossRef]
- Huang, H.Y.; Lin, Y.C.D.; Li, J.; Huang, K.Y.; Shrestha, S.; Hong, H.C.; Tang, Y.; Chen, Y.G.; Jin, C.N.; Yu, Y.; et al. miRTarBase 2020: Updates to the experimentally validated microRNA-target interaction database. Nucleic Acids Res. 2020, 48, D148–D154. [Google Scholar] [CrossRef]
- Breuer, K.; Foroushani, A.K.; Laird, M.R.; Chen, C.; Sribnaia, A.; Lo, R.; Winsor, G.L.; Hancock, R.E.; Brinkman, F.S.; Lynn, D.J. InnateDB: Systems biology of innate immunity and beyond--recent updates and continuing curation. Nucleic Acids Res. 2013, 41, D1228–D1233. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Increased or Decreased in p53-KO | MiRNAs | Differential Expression in p53-KO (Fold Change) | p-Value (Limma; FDR = 10%) | Reference for Interaction with p53 |
|---|---|---|---|---|
| increased | hsa-miR-27a-3p * | 1.37 | 0.0003 | [31] |
| hsa-miR-24-3p * | 1.24 | 0.0043 | [32] | |
| hsa-miR-185-5p * | 1.25 | 0.0070 | ||
| hsa-miR-130b-3p ^ | 1.31 | 0.0103 | [33] | |
| hsa-miR-181a-5p * | 1.16 | 0.0304 | [34] | |
| decreased | hsa-miR-324-3p | 0.73 | 0.0001 | |
| hsa-miR-345-5p | 0.71 | 0.0057 | ||
| hsa-miR-328-3p | 0.62 | 0.0068 | ||
| hsa-miR-146b-5p | 0.64 | 0.0084 | ||
| hsa-miR-210-3p ^ | 0.55 | 0.0101 | [35] | |
| hsa-miR-140-3p | 0.48 | 0.0117 | ||
| hsa-miR-26a-5p ^ | 0.86 | 0.0144 | [36] | |
| hsa-miR-324-5p * | 0.73 | 0.0225 | [37] | |
| hsa-miR-26b-5p | 0.86 | 0.0312 | ||
| hsa-miR-320a-3p ^ | 0.79 | 0.0461 | [38] |
| Pathway Name | Pathway Uploaded Gene Count | Genes in InnateDB for This Entity | Pathway p-Value (Corrected) |
|---|---|---|---|
| Cellular responses to stress | 81 | 240 | 1.93 × 10−12 |
| Pathways in cancer | 94 | 329 | 8.30 × 10−10 |
| p53 pathway * | 25 | 47 | 4.20 × 10−8 |
| p53 signalling pathway | 31 | 68 | 4.45 × 10−8 |
| EGFR1 * | 114 | 472 | 1.46 × 10−7 |
| Gene expression * | 223 | 1118 | 4.13 × 10−7 |
| Cell cycle * | 42 | 126 | 1.30 × 10−6 |
| Cyclins and cell cycle regulation | 14 | 23 | 8.72 × 10−6 |
| Direct p53 effectors * | 40 | 129 | 1.61 × 10−5 |
| Rb tumour suppressor/checkpoint signaling in response to DNA damage * | 10 | 13 | 1.75 × 10−5 |
| WNT signaling pathway * | 42 | 140 | 2.08 × 10−5 |
| Cellular response to hypoxia | 13 | 25 | 1.72 × 10−4 |
| Fatty acid, triacylglycerol and ketone body metabolism* | 46 | 176 | 2.61 × 10−4 |
| mTOR signaling pathway | 22 | 62 | 3.03 × 10−4 |
| Regulation of lipid metabolism by PPARalpha * | 32 | 109 | 3.92 × 10−4 |
| VEGF signaling pathway | 46 | 183 | 6.06 × 10−4 |
| TGF-beta signaling pathway | 25 | 80 | 8.39 × 10−4 |
| Signal Transduction | 4 | 4 | 0.004 |
| Nuclear receptor transcription pathway * | 14 | 39 | 0.004 |
| Intrinsic pathway for apoptosis | 13 | 38 | 0.008 |
| PTEN dependent cell cycle arrest and apoptosis | 7 | 15 | 0.013 |
| Apoptosis | 35 | 155 | 0.013 |
| Plasma membrane oestrogen receptor signaling | 9 | 24 | 0.019 |
| Signaling to RAS | 10 | 31 | 0.032 |
| Apoptotic signaling in response to DNA damage | 6 | 14 | 0.034 |
| Metabolism of lipids and lipoproteins * | 97 | 554 | 0.040 |
| p53-Dependent G1 DNA damage response | 15 | 57 | 0.040 |
| p53-Dependent G1/S DNA damage checkpoint | 15 | 57 | 0.040 |
| Pyruvate metabolism * | 8 | 24 | 0.050 |
| Pathway Name | Pathway Uploaded Gene Count | Genes in InnateDB for This Entity | Pathway p-Value (Corrected) |
|---|---|---|---|
| Gene expression | 330 | 1118 | 2.85 × 10−9 |
| Translation | 68 | 145 | 3.11 × 10−9 |
| Cell cycle | 172 | 523 | 3.80 × 10−8 |
| SRP-dependent cotranslational protein targeting to membrane | 51 | 107 | 1.57 × 10−7 |
| Cellular responses to stress | 91 | 240 | 2.18 × 10−7 |
| Apoptosis | 65 | 155 | 4.26 × 10−7 |
| p53 signaling pathway | 34 | 68 | 1.04 × 10−5 |
| DNA Replication | 44 | 105 | 5.86 × 10−5 |
| p53-Dependent G1 DNA damage response | 28 | 57 | 1.08 × 10−4 |
| p53-Dependent G1/S DNA damage checkpoint | 28 | 57 | 1.08 × 10−4 |
| Direct p53 effectors | 50 | 129 | 1.44 × 10−4 |
| Metabolism of proteins | 191 | 678 | 1.77 × 10−4 |
| Stabilization of p53 | 24 | 52 | 0.001 |
| Metabolism | 382 | 1535 | 0.002 |
| Ribosome | 48 | 137 | 0.002 |
| Cell cycle checkpoints | 42 | 117 | 0.003 |
| Regulation of apoptosis | 25 | 60 | 0.004 |
| Intrinsic pathway for apoptosis | 18 | 38 | 0.004 |
| WNT signaling pathway | 33 | 91 | 0.008 |
| p53 pathway | 20 | 47 | 0.009 |
| Alpha-linolenic (omega3) and linoleic (omega6) acid metabolism | 7 | 10 | 0.013 |
| Alpha-linolenic acid metabolism | 7 | 10 | 0.013 |
| Caspase cascade in apoptosis | 21 | 53 | 0.016 |
| Linoleic acid metabolism | 5 | 6 | 0.018 |
| Fatty acid, triacylglycerol and ketone body metabolism | 53 | 176 | 0.024 |
| Apoptotic signaling in response to DNA damage | 8 | 14 | 0.026 |
| Regulation of lipid metabolism by PPARalpha | 35 | 109 | 0.032 |
| Internal ribosome entry pathway | 9 | 18 | 0.038 |
| Validated nuclear oestrogen receptor alpha network | 22 | 64 | 0.051 |
| Increased or Decreased after SLC7A11 Knockdown | MiRNAs | Differential Expression after SLC7A11 Knockdown (Fold Change) | p-Value (Empirical Bayes; FDR = 10%) |
|---|---|---|---|
| decreased | hsa-miR-331-3p | 0.93 | 0.019 |
| increased | hsa-miR-30a-5p | 1.27 | 0.025 |
| hsa-miR-1274A | 1.37 | 0.041 | |
| hsa-miR-1274B | 1.37 | 0.047 |
| Pathway Name | Pathway Uploaded Gene Count | Genes in InnateDB for This Entity | Pathway p-Value (Corrected) |
|---|---|---|---|
| Direct p53 effectors | 19 | 129 | 0.003 |
| Intrinsic pathway for apoptosis | 9 | 38 | 0.008 |
| Cellular responses to stress | 25 | 240 | 0.011 |
| ATM signalling pathway | 6 | 18 | 0.012 |
| Apoptosis | 13 | 88 | 0.016 |
| Apoptotic signalling in response to DNA damage | 5 | 14 | 0.020 |
| Plasma membrane oestrogen receptor signalling | 6 | 24 | 0.028 |
| p53 signalling pathway | 10 | 68 | 0.034 |
| Hypoxia and p53 in the cardiovascular system | 5 | 20 | 0.041 |
| Cell cycle | 14 | 126 | 0.043 |
| Gene expression | 70 | 1118 | 0.046 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eichelmann, A.-K.; Mayne, G.C.; Chiam, K.; Due, S.L.; Bastian, I.; Butz, F.; Wang, T.; Sykes, P.J.; Clemons, N.J.; Liu, D.S.; et al. Mutant p53 Mediates Sensitivity to Cancer Treatment Agents in Oesophageal Adenocarcinoma Associated with MicroRNA and SLC7A11 Expression. Int. J. Mol. Sci. 2021, 22, 5547. https://doi.org/10.3390/ijms22115547
Eichelmann A-K, Mayne GC, Chiam K, Due SL, Bastian I, Butz F, Wang T, Sykes PJ, Clemons NJ, Liu DS, et al. Mutant p53 Mediates Sensitivity to Cancer Treatment Agents in Oesophageal Adenocarcinoma Associated with MicroRNA and SLC7A11 Expression. International Journal of Molecular Sciences. 2021; 22(11):5547. https://doi.org/10.3390/ijms22115547
Chicago/Turabian StyleEichelmann, Ann-Kathrin, George C. Mayne, Karen Chiam, Steven L. Due, Isabell Bastian, Frederike Butz, Tingting Wang, Pamela J. Sykes, Nicholas J. Clemons, David S. Liu, and et al. 2021. "Mutant p53 Mediates Sensitivity to Cancer Treatment Agents in Oesophageal Adenocarcinoma Associated with MicroRNA and SLC7A11 Expression" International Journal of Molecular Sciences 22, no. 11: 5547. https://doi.org/10.3390/ijms22115547
APA StyleEichelmann, A.-K., Mayne, G. C., Chiam, K., Due, S. L., Bastian, I., Butz, F., Wang, T., Sykes, P. J., Clemons, N. J., Liu, D. S., Michael, M. Z., Karapetis, C. S., Hummel, R., Watson, D. I., & Hussey, D. J. (2021). Mutant p53 Mediates Sensitivity to Cancer Treatment Agents in Oesophageal Adenocarcinoma Associated with MicroRNA and SLC7A11 Expression. International Journal of Molecular Sciences, 22(11), 5547. https://doi.org/10.3390/ijms22115547