
Type IIa RPTPs and Glycans: Roles in Axon Regeneration and Synaptogenesis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Roles of Type IIa RPTPs and Glycans in Axon Regeneration and Synaptogenesis
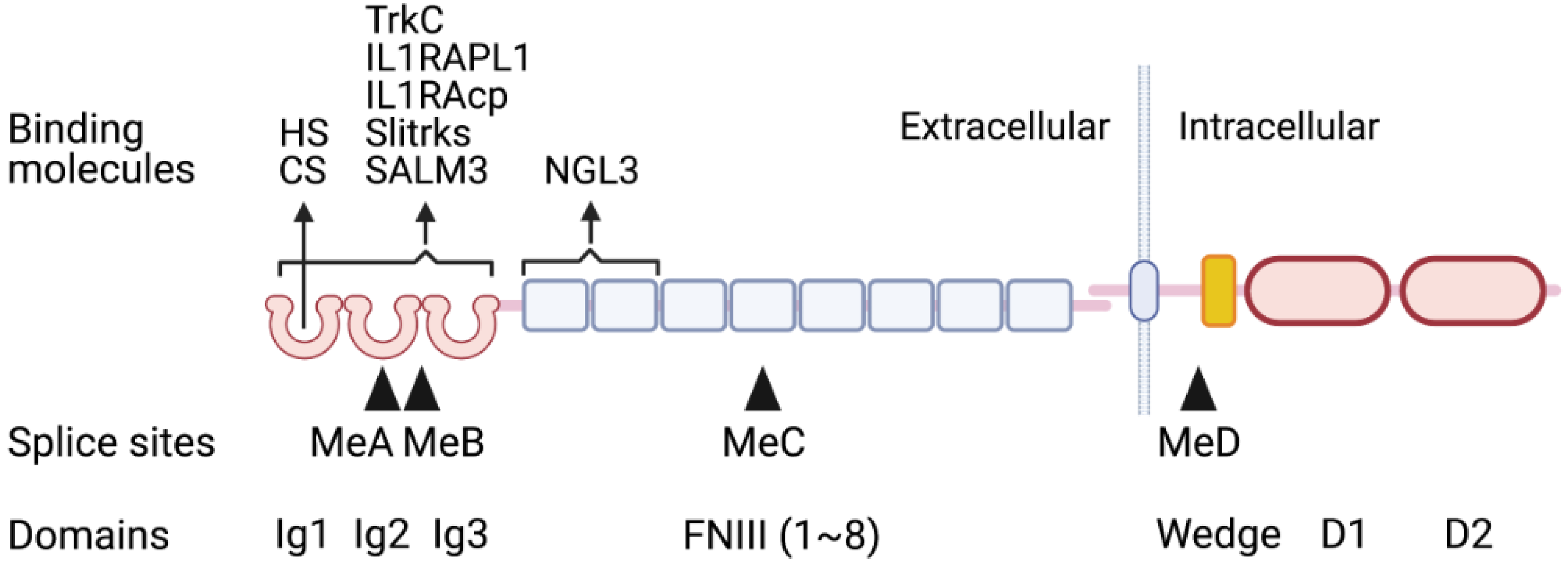
2.1. Structures of Type IIa RPTPs
2.2. Axon Regeneration
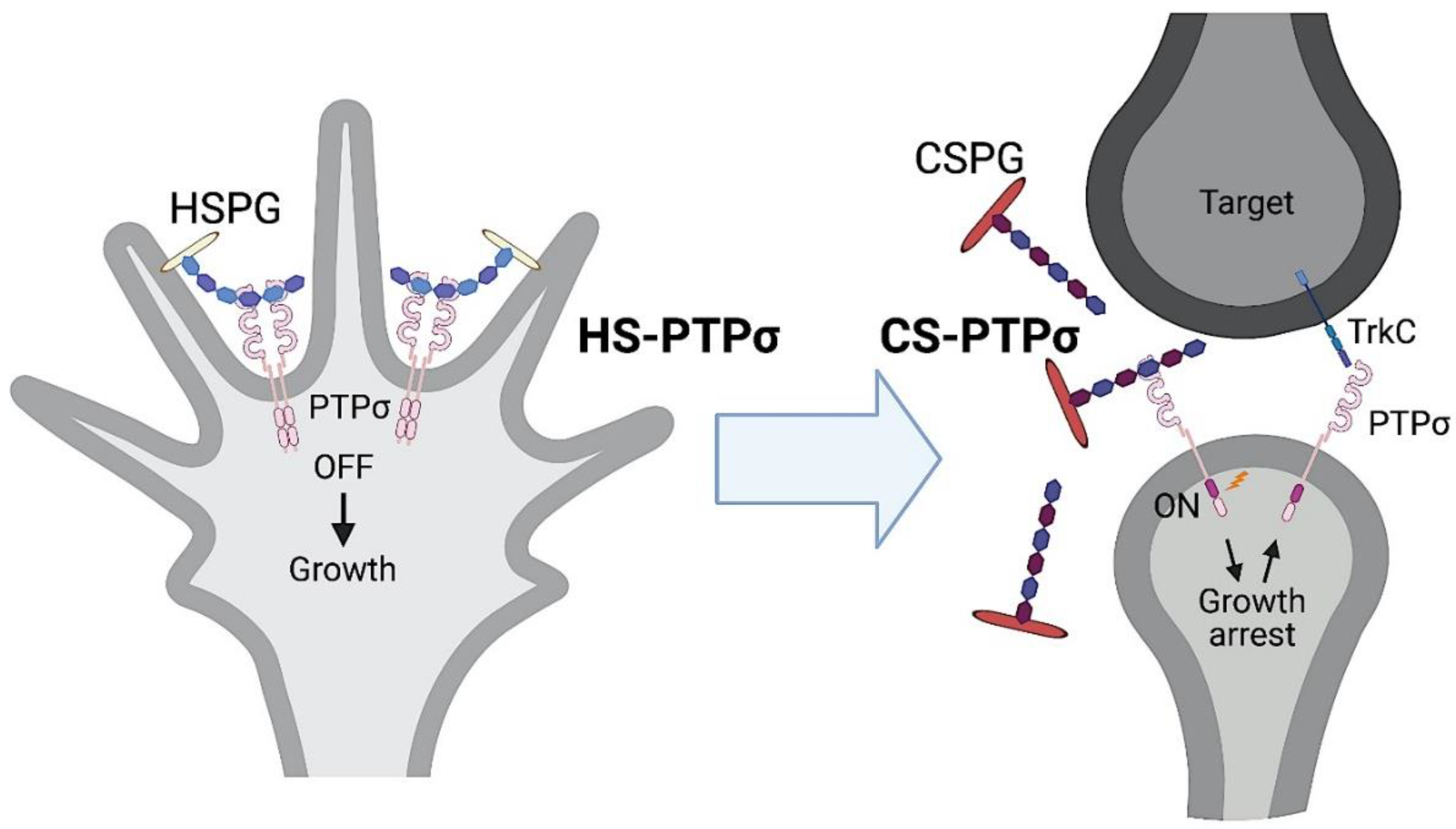
2.2.1. Type IIa RPTPs and Glycosaminoglycans in Axon Regeneration
2.2.2. Substrates of Type IIa RPTPs
2.2.3. Autophagy in Neurodegenerative Diseases
2.2.4. Axon Extension vs. Synapse Formation
2.3. Synaptogenesis
2.3.1. Synapse Organizers
2.3.2. HS Involved in Synapse Formation
3. Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coles, C.H.; Shen, Y.; Tenney, A.P.; Siebold, C.; Sutton, G.C.; Lu, W.; Gallagher, J.T.; Jones, E.Y.; Flanagan, J.G.; Aricescu, A.R. Proteoglycan-specific molecular switch for RPTPσ clustering and neuronal extension. Science 2011, 332, 484–488. [Google Scholar] [CrossRef]
- Takahashi, H.; Arstikaitis, P.; Prasad, T.; Bartlett, T.E.; Wang, Y.T.; Murphy, T.H.; Craig, A.M. Postsynaptic TrkC and presynaptic PTPσ function as a bidirectional excitatory synaptic organizing complex. Neuron 2011, 69, 287–303. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Yasumura, M.; Uemura, T.; Lee, S.J.; Ra, M.; Taguchi, R.; Iwakura, Y.; Mishina, M. IL-1 receptor accessory protein-like 1 associated with mental retardation and autism mediates synapse formation by trans-synaptic interaction with protein tyrosine phosphataseδ. J. Neurosci. 2011, 31, 13485–13499. [Google Scholar] [CrossRef] [PubMed]
- Valnegri, P.; Montrasio, C.; Brambilla, D.; Ko, J.; Passafaro, M.; Sala, C. The X-linked intellectual disability protein IL1RAPL1 regulates excitatory synapse formation by binding PTPδ and RhoGAP2. Hum. Mol. Genet. 2011, 20, 4797–4809. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Shiroshima, T.; Lee, S.J.; Yasumura, M.; Uemura, T.; Chen, X.; Iwakura, Y.; Mishina, M. Interleukin-1 receptor accessory protein organizes neuronal synaptogenesis as a cell adhesion molecule. J. Neurosci. 2012, 32, 2588–2600. [Google Scholar] [CrossRef]
- Um, J.W.; Kim, K.H.; Park, B.S.; Choi, Y.; Kim, D.; Kim, C.Y.; Kim, S.J.; Kim, M.; Ko, J.S.; Lee, S.G.; et al. Structural basis for LAR-RPTP/Slitrk complex-mediated synaptic adhesion. Nat. Commun. 2014, 5, 5423. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, P.; Choi, T.Y.; Park, S.K.; Park, H.; Lee, E.J.; Lee, D.; Roh, J.D.; Mah, W.; Kim, R.; et al. Splicing-dependent trans-synaptic SALM3-LAR-RPTP interactions regulate excitatory synapse development and locomotion. Cell Rep. 2015, 12, 1618–1630. [Google Scholar] [CrossRef]
- Ko, J.S.; Pramanik, G.; Um, J.W.; Shim, J.S.; Lee, D.; Kim, K.H.; Chung, G.Y.; Condomitti, G.; Kim, H.M.; Kim, H.; et al. PTPσ functions as a presynaptic receptor for the glypican-4/LRRTM4 complex and is essential for excitatory synaptic transmission. Proc. Natl. Acad. Sci. USA 2015, 112, 1874–1879. [Google Scholar] [CrossRef]
- Woo, J.; Kwon, S.K.; Choi, S.; Kim, S.; Lee, J.R.; Dunah, A.W.; Sheng, M.; Kim, E. Trans-synaptic adhesion between NGL-3 and LAR regulates the formation of excitatory synapses. Nat. Neurosci. 2009, 12, 428–437. [Google Scholar] [CrossRef]
- Kwon, S.K.; Woo, J.; Kim, S.Y.; Kim, H.; Kim, E. Trans-synaptic adhesions between netrin-G ligand-3 (NGL-3) and receptor tyrosine phosphatases LAR, protein-tyrosine phosphataseδ (PTPδ), and PTPσ via specific domains regulate excitatory synapse formation. J. Biol. Chem. 2010, 285, 13966–13978. [Google Scholar] [CrossRef]
- Bilwes, A.M.; den Hertog, J.; Hunter, T.; Noel, J.P. Structural basis for inhibition of receptor protein-tyrosine phosphatase-α by dimerization. Nature 1996, 382, 555–559. [Google Scholar] [CrossRef]
- Jiang, G.; den Hertog, J.; Su, J.; Noel, J.; Sap, J.; Hunter, T. Dimerization inhibits the activity of receptor-like protein-tyrosine phosphatase-α. Nature 1999, 401, 606–610. [Google Scholar] [CrossRef] [PubMed]
- Barr, A.J.; Ugochukwu, E.; Lee, W.H.; King, O.N.F.; Filippakopoulos, P.; Alfano, I.; Savitsky, P.; Burgess-Brown, N.A.; Müller, S.; Knapp, S. Large-scale structural analysis of the classical human protein tyrosine phosphatome. Cell 2009, 136, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Massa, S.M.; Ensslen-Craig, S.E.; Major, D.L.; Yang, T.; Tisi, M.A.; Derevyanny, V.D.; Runge, W.O.; Mehta, B.P.; Moore, L.A.; et al. Protein-tyrosine phosphatase (PTP) wedge domain peptides: A novel approach for inhibition of PTP function and augmentation of protein-tyrosine kinase function. J. Biol. Chem. 2006, 281, 16482–16492. [Google Scholar] [CrossRef] [PubMed]
- Lang, B.T.; Cregg, J.M.; DePaul, M.A.; Tran, A.P.; Xu, K.; Dyck, S.M.; Madalena, K.M.; Brown, B.P.; Weng, Y.-L.; Li, S.; et al. Modulation of the proteoglycan receptor PTPσ promotes recovery after spinal cord injury. Nature 2015, 518, 404–408. [Google Scholar] [CrossRef]
- Schwab, M.E.; Strittmatter, S.M. Nogo limits neural plasticity and recovery from injury. Curr. Opin. Neurobiol. 2014, 27, 53–60. [Google Scholar] [CrossRef]
- Harel, N.Y.; Strittmatter, S.M. Can regenerating axons recapitulate developmental guidance during recovery from spinal cord injury? Nat. Rev. Neurosci. 2006, 7, 603–616. [Google Scholar] [CrossRef]
- Rauvala, H.; Paveliev, M.; Kuja-Panula, J.; Kulesskaya, N. Inhibition and enhancement of neural regeneration by chondroitin sulfate proteoglycans. Neural. Regen. Res. 2017, 12, 687–691. [Google Scholar] [CrossRef]
- Tom, V.J.; Steinmetz, M.P.; Miller, J.H.; Doller, C.M.; Silver, J. Studies on the development and behavior of the dystrophic growth cone, the hallmark of regeneration failure, in an in vitro model of the glial scar and after spinal cord injury. J. Neurosci. 2004, 24, 6531–6539. [Google Scholar] [CrossRef]
- Asher, R.A.; Morgenstern, D.A.; Fidler, P.S.; Adcock, K.H.; Oohira, A.; Braistead, J.E.; Levine, J.M.; Margolis, R.U.; Rogers, J.H.; Fawcett, J.W. Neurocan is upregulated in injured brain and in cytokine-treated astrocytes. J. Neurosci. 2000, 20, 2427–2438. [Google Scholar] [CrossRef] [PubMed]
- Yamada, H.; Fredette, B.; Shitara, K.; Hagihara, K.; Miura, R.; Ranscht, B.; Stallcup, W.B.; Yamaguchi, Y. The brain chondroitin sulfate proteoglycan brevican associates with astrocytes ensheathing cerebellar glomeruli and inhibits neurite outgrowth from granule neurons. J. Neurosci. 1997, 17, 7784–7795. [Google Scholar] [CrossRef] [PubMed]
- Snow, D.M.; Lemmon, V.; Carrino, D.A.; Caplan, A.I.; Silver, J. Sulfated proteoglycans in astroglial barriers inhibit neurite outgrowth in vitro. Exp. Neurol. 1990, 109, 111–130. [Google Scholar] [CrossRef]
- Moon, L.D.; Asher, R.A.; Rhodes, K.E.; Fawcett, J.W. Regeneration of CNS axons back to their target following treatment of adult rat brain with chondroitinase ABC. Nat. Neurosci. 2001, 4, 465–466. [Google Scholar] [CrossRef]
- Bradbury, E.J.; Moon, L.D.; Popat, R.J.; King, V.R.; Bennett, G.S.; Patel, P.N.; Fawcett, J.W.; McMahon, S.B. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature 2002, 416, 636–640. [Google Scholar] [CrossRef]
- Zhang, H.; Muramatsu, T.; Murase, A.; Yuasa, S.; Uchimura, K.; Kadomatsu, K. N-Acetylglucosamine 6-O-sulfotransferase-1 is required for brain keratan sulfate biosynthesis and glial scar formation after brain injury. Glycobiology 2006, 16, 702–710. [Google Scholar] [CrossRef][Green Version]
- Ito, Z.; Sakamoto, K.; Imagama, S.; Matsuyama, Y.; Zhang, H.; Hirano, K.; Ando, K.; Yamashita, T.; Ishiguro, N.; Kadomatsu, K. N-acetylglucosamine 6-O-sulfotransferase-1-deficient mice show better functional recovery after spinal cord injury. J. Neurosci. 2010, 30, 5937–5947. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Imagama, S.; Sakamoto, K.; Tauchi, R.; Shinjo, R.; Ohgomori, T.; Ito, Z.; Zhang, H.; Nishida, Y.; Asami, N.; Takeshita, S.; et al. Keratan sulfate restricts neural plasticity after spinal cord injury. J. Neurosci. 2011, 31, 17091–17102. [Google Scholar] [CrossRef] [PubMed]
- Frischknecht, R.; Heine, M.; Perrais, D.; Seidenbecher, C.I.; Choquet, D.; Gundelfinger, E.D. Brain extracellular matrix affects AMPA receptor lateral mobility and short-term synaptic plasticity. Nat. Neurosci. 2009, 12, 897–904. [Google Scholar] [CrossRef]
- Orlando, C.; Ster, J.; Gerber, U.; Fawcett, J.W.; Raineteau, O. Perisynaptic chondroitin sulfate proteoglycans restrict structural plasticity in an integrin-dependent manner. J. Neurosci. 2012, 32, 18009–18017. [Google Scholar] [CrossRef]
- Pizzorusso, T.; Medini, P.; Berardi, N.; Chierzi, S.; Fawcett, J.W.; Maffei, L. Reactivation of ocular dominance plasticity in the adult visual cortex. Science 2002, 298, 1248–1251. [Google Scholar] [CrossRef]
- Gogolla, N.; Caroni, P.; Lüthi, A.; Herry, C. Perineuronal nets protect fear memories from erasure. Science 2009, 325, 1258–1261. [Google Scholar] [CrossRef] [PubMed]
- Miyata, S.; Komatsu, Y.; Yoshimura, Y.; Taya, C.; Kitagawa, H. Persistent cortical plasticity by upregulation of chondroitin 6-sulfation. Nat. Neurosci. 2012, 15, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.Z.; Duan, M.R.; Lin, N.; Zhao, W.J. The emerging role of the chondroitin sulfate proteoglycan family in neurodegenerative diseases. Rev. Neurosci. 2021. [Google Scholar] [CrossRef] [PubMed]
- Cajal, S.R.Y. Degeneration & Regeneration of the Nervous System; Oxford University Press, Humphrey Milford: London, UK, 1928. [Google Scholar]
- Ruschel, J.; Hellal, F.; Flynn, K.C.; Norenberg, M.D.; Blesch, A.; Bradke, F. Systemic administration of epothilone B promotes axon regeneration after spinal cord injury. Science 2015, 348, 347–352. [Google Scholar] [CrossRef]
- Shen, Y.; Tenney, A.P.; Busch, S.A.; Horn, K.P.; Cuascut, F.X.; Liu, K.; He, Z.; Silver, J.; Flanagan, J.G. PTPσ is a receptor for chondroitin sulfate proteoglycan, an inhibitor of neural regeneration. Science 2009, 326, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Ozaki, T.; Ko, Y.C.; Tsai, C.F.; Gong, Y.; Morozumi, M.; Ishikawa, Y.; Uchimura, K.; Nadanaka, S.; Kitagawa, H.; et al. Glycan sulfation patterns define autophagy flux at axon tip via PTPRσ-cortactin axis. Nat. Chem. Biol. 2019, 15, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, J.; Iwamoto, R.; Otomo, T.; Nezu, A.; Hamasaki, M.; Yoshimori, T. Autophagosome-lysosome fusion in neurons requires INPP5E, a protein associated with Joubert syndrome. EMBO J. 2016, 35, 1853–1867. [Google Scholar] [CrossRef]
- Takeuchi, K.; Yoshioka, N.; Higa Onaga, S.; Watanabe, Y.; Miyata, S.; Wada, Y.; Kudo, C.; Okada, M.; Ohko, K.; Oda, K.; et al. Chondroitin sulphate N-acetylgalactosaminyl-transferase-1 inhibits recovery from neural injury. Nat. Commun. 2013, 4, 2740. [Google Scholar] [CrossRef]
- Properzi, F.; Carulli, D.; Asher, R.A.; Muir, E.; Camargo, L.M.; van Kuppevelt, T.H.; ten Dam, G.B.; Furukawa, Y.; Mikami, T.; Sugahara, K.; et al. Chondroitin 6-sulphate synthesis is up-regulated in injured CNS, induced by injury-related cytokines and enhanced in axon-growth inhibitory glia. Eur. J. Neurosci. 2005, 21, 378–390. [Google Scholar] [CrossRef]
- Properzi, F.; Lin, R.; Kwok, J.; Naidu, M.; van Kuppevelt, T.H.; Ten Dam, G.B.; Camargo, L.M.; Raha-Chowdhury, R.; Furukawa, Y.; Mikami, T.; et al. Heparan sulphate proteoglycans in glia and in the normal and injured CNS: Expression of sulphotransferases and changes in sulphation. Eur. J. Neurosci. 2008, 27, 593–604. [Google Scholar] [CrossRef]
- Ito, S.; Ozaki, T.; Morozumi, M.; Imagama, S.; Kadomatsu, K.; Sakamoto, K. Enoxaparin promotes functional recovery after spinal cord injury by antagonizing PTPRσ. Exp. Neurol. 2021, 340, 113679. [Google Scholar] [CrossRef]
- Kantor, D.B.; Chivatakarn, O.; Peer, K.L.; Oster, S.F.; Inatani, M.; Hansen, M.J.; Flanagan, J.G.; Yamaguchi, Y.; Sretavan, D.W.; Giger, R.J.; et al. Semaphorin 5A is a bifunctional axon guidance cue regulated by heparan and chondroitin sulfate proteoglycans. Neuron 2004, 44, 961–975. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.; Xing, B.; Dill, J.; Li, H.; Hoang, H.H.; Zhao, Z.; Yang, X.L.; Bachoo, R.; Cannon, S.; Longo, F.M.; et al. Leukocyte common antigen-related phosphatase is a functional receptor for chondroitin sulfate proteoglycan axon growth inhibitors. J. Neurosci. 2011, 31, 14051–14066. [Google Scholar] [CrossRef] [PubMed]
- Wills, Z.; Bateman, J.; Korey, C.A.; Comer, A.; Van Vactor, D. The tyrosine kinase Abl and its substrate enabled collaborate with the receptor phosphatase Dlar to control motor axon guidance. Neuron 1999, 22, 301–312. [Google Scholar] [CrossRef]
- Chagnon, M.J.; Wu, C.L.; Nakazawa, T.; Yamamoto, T.; Noda, M.; Blanchetot, C.; Tremblay, M.L. Receptor tyrosine phosphatase sigma (RPTPσ) regulates, p250GAP, a novel substrate that attenuates Rac signaling. Cell Signal. 2010, 22, 1626–1633. [Google Scholar] [CrossRef]
- Debant, A.; Serra-Pagès, C.; Seipel, K.; O’Brien, S.; Tang, M.; Park, S.H.; Streuli, M. The multidomain protein Trio binds the LAR transmembrane tyrosine phosphatase, contains a protein kinase domain, and has separate rac-specific and rho-specific guanine nucleotide exchange factor domains. Proc. Natl. Acad. Sci. USA 1996, 93, 5466–5471. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.; Choidas, A.; Reichmann, E.; Ullrich, A. Phosphorylation and free pool of beta-catenin are regulated by tyrosine kinases and tyrosine phosphatases during epithelial cell migration. J. Biol. Chem. 1999, 274, 10173–10183. [Google Scholar] [CrossRef]
- Siu, R.; Fladd, C.; Rotin, D. N-cadherin is an in vivo substrate for protein tyrosine phosphatase sigma (PTPsigma) and participates in PTPsigma-mediated inhibition of axon growth. Mol. Cell Biol. 2007, 27, 208–219. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kaufmann, N.; DeProto, J.; Ranjan, R.; Wan, H.; Van Vactor, D. Drosophila liprin-alpha and the receptor phosphatase Dlar control synapse morphogenesis. Neuron 2002, 34, 27–38. [Google Scholar] [CrossRef]
- Pulido, R.; Serra-Pagès, C.; Tang, M.; Streuli, M. The LAR/PTP delta/PTP sigma subfamily of transmembrane protein-tyrosine-phosphatases: Multiple human LAR, PTP delta, and PTP sigma isoforms are expressed in a tissue-specific manner and associate with the LAR-interacting protein LIP.1. Proc. Natl. Acad. Sci. USA 1995, 92, 11686–11690. [Google Scholar] [CrossRef]
- Gong, Y.; Abudureyimu, S.; Kadomatsu, K.; Sakamoto, K. Identification of PTPRsigma-interacting proteins by proximity-labelling assay. J. Biochem. 2021, 169, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Geisler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Bento, C.F.; Ricketts, T.; Vicinanza, M.; Siddiqi, F.; Pavel, M.; Squitieri, F.; Hardenberg, M.C.; Imarisio, S.; Menzies, F.M.; et al. Polyglutamine tracts regulate beclin 1-dependent autophagy. Nature 2017, 545, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef]
- DeWitt, D.A.; Silver, J.; Canning, D.R.; Perry, G. Chondroitin sulfate proteoglycans are associated with the lesions of Alzheimer’s disease. Exp. Neurol. 1993, 121, 149–152. [Google Scholar] [CrossRef]
- Mizuno, H.; Warita, H.; Aoki, M.; Itoyama, Y. Accumulation of chondroitin sulfate proteoglycans in the microenvironment of spinal motor neurons in amyotrophic lateral sclerosis transgenic rats. J. Neurosci. Res. 2008, 86, 2512–2523. [Google Scholar] [CrossRef]
- Tran, A.P.; Warren, P.M.; Silver, J. Regulation of autophagy by inhibitory CSPG interactions with receptor PTPσ and its impact on plasticity and regeneration after spinal cord injury. Exp. Neurol. 2020, 328, 113276. [Google Scholar] [CrossRef]
- Clark, E.S.; Whigham, A.S.; Yarbrough, W.G.; Weaver, A.M. Cortactin is an essential regulator of matrix metalloproteinase secretion and extracellular matrix degradation in invadopodia. Cancer Res. 2007, 67, 4227–4235. [Google Scholar] [CrossRef]
- Tran, A.P.; Sundar, S.; Yu, M.; Lang, B.T.; Silver, J. Modulation of receptor protein tyrosine phosphatase sigma increases chondroitin sulfate proteoglycan degradation through Cathepsin B secretion to enhance axon outgrowth. J. Neurosci. 2018, 38, 5399–5414. [Google Scholar] [CrossRef]
- Filous, A.R.; Tran, A.; Howell, C.J.; Busch, S.A.; Evans, T.A.; Stallcup, W.B.; Kang, S.H.; Bergles, D.E.; Lee, S.-I.; Levine, J.M.; et al. Entrapment via synaptic-like connections between NG2 proteoglycan+ cells and dystrophic axons in the lesion plays a role in regeneration failure after spinal cord injury. J. Neurosci. 2014, 34, 16369–16384. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.-J. Synapsing with NG2 cells (polydendrocytes), unappreciated barrier to axon regeneration? Neural Regen. Res. 2015, 10, 346. [Google Scholar] [CrossRef] [PubMed]
- Busch, S.A.; Horn, K.P.; Cuascut, F.X.; Hawthorne, A.L.; Bai, L.; Miller, R.H.; Silver, J. Adult NG2+ cells are permissive to neurite outgrowth and stabilize sensory axons during macrophage-induced axonal dieback after spinal cord injury. J. Neurosci. 2010, 30, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Südhof, T.C. Synaptic Neurexin Complexes: A Molecular Code for the Logic of Neural Circuits. Cell 2017, 171, 745–769. [Google Scholar] [CrossRef]
- Südhof, T.C. Towards an Understanding of Synapse Formation. Neuron 2018, 100, 276–293. [Google Scholar] [CrossRef]
- Han, K.A.; Jeon, S.; Um, J.W.; Ko, J. Emergent Synapse Organizers: LAR-RPTPs and Their Companions. Int. Rev. Cell Mol. Biol. 2016, 324, 39–65. [Google Scholar] [CrossRef] [PubMed]
- Joo, W.; Hippenmeyer, S.; Luo, L. Dendrite morphogenesis depends on relative levels of NT-3/TrkC signaling. Science 2014, 346, 626–629. [Google Scholar] [CrossRef]
- Ammendrup-Johnsen, I.; Naito, Y.; Craig, A.M.; Takahashi, H. Neurotrophin-3 enhances the synaptic organizing function of TrkC-protein tyrosine phosphatase σ in rat hippocampal neurons. J. Neurosci. 2015, 35, 12425–12431. [Google Scholar] [CrossRef]
- Yasumura, M.; Yoshida, T.; Yamazaki, M.; Abe, M.; Natsume, R.; Kanno, K.; Uemura, T.; Takao, K.; Sakimura, K.; Kikusui, T.; et al. IL1RAPL1 knockout mice show spine density decrease, learning deficiency, hyperactivity and reduced anxiety-like behaviours. Sci. Rep. 2014, 4, 6613. [Google Scholar] [CrossRef]
- Tabolacci, E.; Pomponi, M.G.; Pietrobono, R.; Terracciano, A.; Chiurazzi, P.; Neri, G. A truncating mutation in the IL1RAPL1 gene is responsible for X-linked mental retardation in the MRX21 family. Am. J. Med. Genet. A 2006, 140, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Dinopoulos, A.; Stefanou, M.I.; Attilakos, A.; Tsirouda, M.; Papaevangelou, V. A case of startle epilepsy associated with IL1RAPL1 gene deletion. Pediatr. Neurol. 2014, 51, 271–274. [Google Scholar] [CrossRef]
- Takahashi, H.; Katayama, K.; Sohya, K.; Miyamoto, H.; Prasad, T.; Matsumoto, Y.; Ota, M.; Yasuda, H.; Tsumoto, T.; Aruga, J.; et al. Selective control of inhibitory synapse development by Slitrk3-PTPδ trans-synaptic interaction. Nat. Neurosci. 2012, 15, 389–398. [Google Scholar] [CrossRef]
- Yim, Y.S.; Kwon, Y.; Nam, J.; Yoon, H.I.; Lee, K.; Kim, D.G.; Kim, E.; Kim, C.H.; Ko, J. Slitrks control excitatory and inhibitory synapse formation with LAR receptor protein tyrosine phosphatases. Proc. Natl. Acad. Sci. USA 2013, 110, 4057–4062. [Google Scholar] [CrossRef]
- Mah, W.; Ko, J.; Nam, J.; Han, K.; Chung, W.S.; Kim, E. Selected SALM (synaptic adhesion-like molecule) family proteins regulate synapse formation. J. Neurosci. 2010, 30, 5559–5568. [Google Scholar] [CrossRef]
- Munezane, H.; Oizumi, H.; Wakabayashi, T.; Nishio, S.; Hirasawa, T.; Sato, T.; Harada, A.; Yoshida, T.; Eguchi, T.; Yamanashi, Y.; et al. Roles of Collagen XXV and Its Putative Receptors PTPσ/δ in Intramuscular Motor Innervation and Congenital Cranial Dysinnervation Disorder. Cell Rep. 2019, 29, 4362–4376. [Google Scholar] [CrossRef]
- Han, K.A.; Ko, J.S.; Pramanik, G.; Kim, J.Y.; Tabuchi, K.; Um, J.W.; Ko, J. PTPσ Drives Excitatory Presynaptic Assembly via Various Extracellular and Intracellular Mechanisms. J. Neurosci. 2018, 38, 6700–6721. [Google Scholar] [CrossRef]
- Coles, C.H.; Mitakidis, N.; Zhang, P.; Elegheert, J.; Lu, W.; Stoker, A.W.; Nakagawa, T.; Craig, A.M.; Jones, E.Y.; Aricescu, A.R. Structural basis for extracellular cis and trans RPTPσ signal competition in synaptogenesis. Nat. Commun. 2014, 5, 5209. [Google Scholar] [CrossRef]
- Farhy-Tselnicker, I.; van Casteren, A.C.M.; Lee, A.; Chang, V.T.; Aricescu, A.R.; Allen, N.J. Astrocyte-Secreted Glypican 4 Regulates Release of Neuronal Pentraxin 1 from Axons to Induce Functional Synapse Formation. Neuron 2017, 96, 428–445. [Google Scholar] [CrossRef]
- Allen, N.J.; Bennett, M.L.; Foo, L.C.; Wang, G.X.; Chakraborty, C.; Smith, S.J.; Barres, B.A. Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature 2012, 486, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.G.; Tenney, A.P.; Ghose, A.; Duckworth, A.M.; Higashi, M.E.; Parfitt, K.; Marcu, O.; Heslip, T.R.; Marsh, J.L.; Schwarz, T.L.; et al. The HSPGs Syndecan and Dallylike bind the receptor phosphatase LAR and exert distinct effects on synaptic development. Neuron 2006, 49, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Fox, A.N.; Zinn, K. The heparan sulfate proteoglycan syndecan is an in vivo ligand for the Drosophila LAR receptor tyrosine phosphatase. Curr. Biol. 2005, 15, 1701–1711. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.U.; Kwong, J.; Chang, J.; Gillet, V.G.; Lee, R.M.; Johnson, K.G. The Extracellular and Cytoplasmic Domains of Syndecan Cooperate Postsynaptically to Promote Synapse Growth at the Drosophila Neuromuscular Junction. PLoS ONE 2016, 11, e0151621. [Google Scholar] [CrossRef]
- Zhang, P.; Lu, H.; Peixoto, R.T.; Pines, M.K.; Ge, Y.; Oku, S.; Siddiqui, T.J.; Xie, Y.; Wu, W.; Archer-Hartmann, S.; et al. Heparan Sulfate Organizes Neuronal Synapses through Neurexin Partnerships. Cell 2018, 174, 1450–1464. [Google Scholar] [CrossRef] [PubMed]
- Roppongi, R.T.; Dhume, S.H.; Padmanabhan, N.; Silwal, P.; Zahra, N.; Karimi, B.; Bomkamp, C.; Patil, C.S.; Champagne-Jorgensen, K.; Twilley, R.E.; et al. LRRTMs Organize Synapses through Differential Engagement of Neurexin and PTPσ. Neuron 2020, 106, 108–125. [Google Scholar] [CrossRef]
- Li, H.; Yamagata, T.; Mori, M.; Momoi, M.Y. Association of autism in two patients with hereditary multiple exostoses caused by novel deletion mutations of EXT1. J. Hum. Genet. 2002, 47, 262–265. [Google Scholar] [CrossRef]
- Wang, K.; Zhang, H.; Ma, D.; Bucan, M.; Glessner, J.T.; Abrahams, B.S.; Salyakina, D.; Imielinski, M.; Bradfield, J.P.; Sleiman, P.M.; et al. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature 2009, 459, 528–533. [Google Scholar] [CrossRef] [PubMed]
- Pearson, B.L.; Corley, M.J.; Vasconcellos, A.; Blanchard, D.C.; Blanchard, R.J. Heparan sulfate deficiency in autistic postmortem brain tissue from the subventricular zone of the lateral ventricles. Behav. Brain Res. 2013, 243, 138–145. [Google Scholar] [CrossRef]
- Irie, F.; Badie-Mahdavi, H.; Yamaguchi, Y. Autism-like socio-communicative deficits and stereotypies in mice lacking heparan sulfate. Proc. Natl. Acad. Sci. USA 2012, 109, 5052–5056. [Google Scholar] [CrossRef]
- Martin, K.R.; Xu, Y.; Looyenga, B.D.; Davis, R.J.; Wu, C.L.; Tremblay, M.L.; Xu, H.E.; MacKeigan, J.P. Identification of PTPsigma as an autophagic phosphatase. J. Cell Sci. 2011, 124, 812–819. [Google Scholar] [CrossRef]
- Zhang, Y.; Roos, M.; Himburg, H.; Termini, C.M.; Quarmyne, M.; Li, M.; Zhao, L.; Kan, J.; Fang, T.; Yan, X.; et al. PTPσ inhibitors promote hematopoietic stem cell regeneration. Nat. Commun. 2019, 10, 3667. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sakamoto, K.; Ozaki, T.; Suzuki, Y.; Kadomatsu, K. Type IIa RPTPs and Glycans: Roles in Axon Regeneration and Synaptogenesis. Int. J. Mol. Sci. 2021, 22, 5524. https://doi.org/10.3390/ijms22115524
Sakamoto K, Ozaki T, Suzuki Y, Kadomatsu K. Type IIa RPTPs and Glycans: Roles in Axon Regeneration and Synaptogenesis. International Journal of Molecular Sciences. 2021; 22(11):5524. https://doi.org/10.3390/ijms22115524
Chicago/Turabian StyleSakamoto, Kazuma, Tomoya Ozaki, Yuji Suzuki, and Kenji Kadomatsu. 2021. "Type IIa RPTPs and Glycans: Roles in Axon Regeneration and Synaptogenesis" International Journal of Molecular Sciences 22, no. 11: 5524. https://doi.org/10.3390/ijms22115524
APA StyleSakamoto, K., Ozaki, T., Suzuki, Y., & Kadomatsu, K. (2021). Type IIa RPTPs and Glycans: Roles in Axon Regeneration and Synaptogenesis. International Journal of Molecular Sciences, 22(11), 5524. https://doi.org/10.3390/ijms22115524