
Analysis and Interpretation of the Impact of Missense Variants in Cancer

 , , ,
, , ,
Abstract
1. Introduction
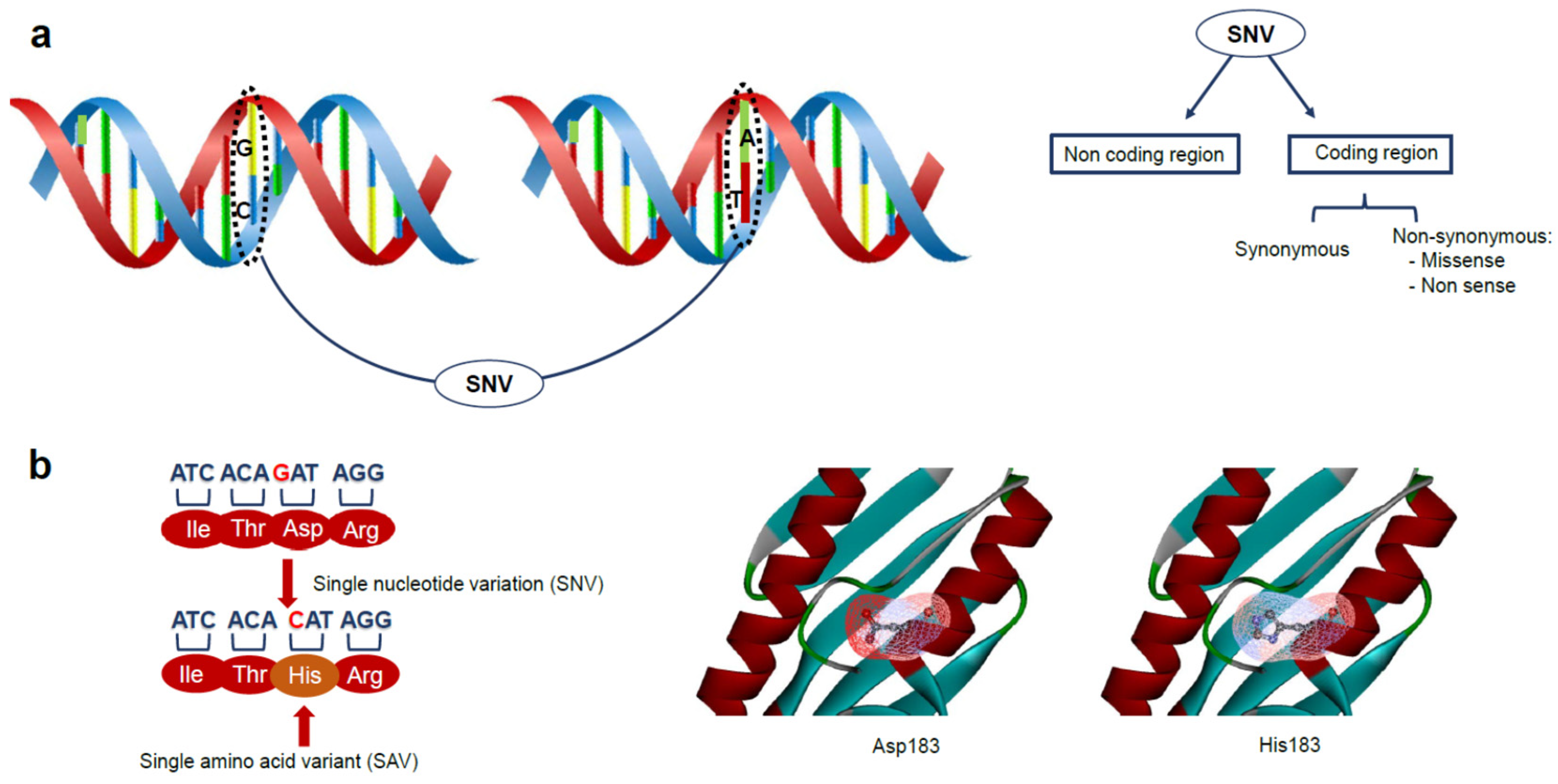
2. Genetic Variations and Disease: The Role of Protein Stability in Cancer
3. Experimental Analysis of Protein Variants in Cancer-Related Genes
3.1. Effect on Protein Stability
3.2. Effect on Protein Conformation
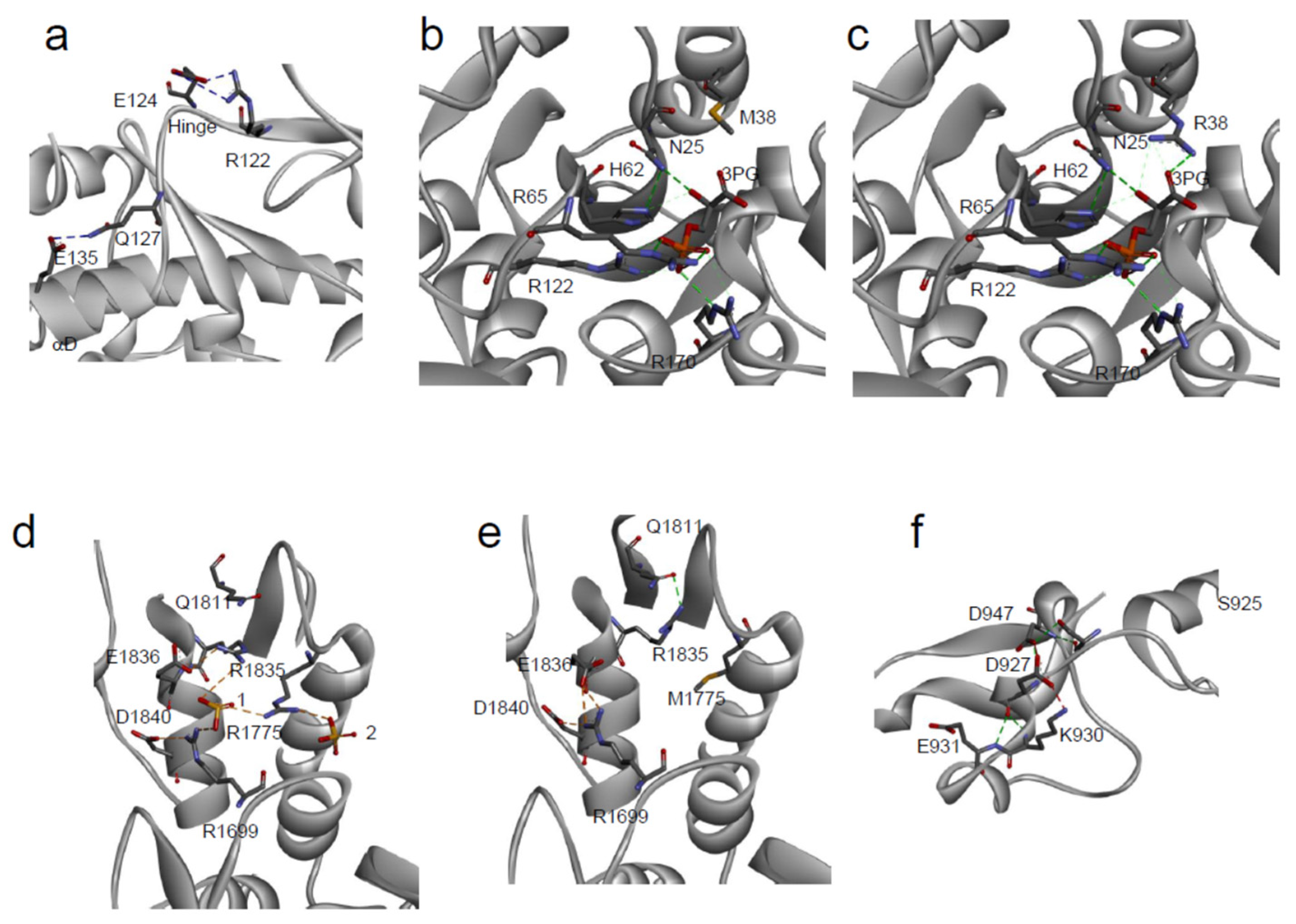
3.3. Effect on Protein Interactions
3.4. Effect on Protein Catalytic Activity
4. Computational Analysis of Protein Variants in Cancer-Related Genes
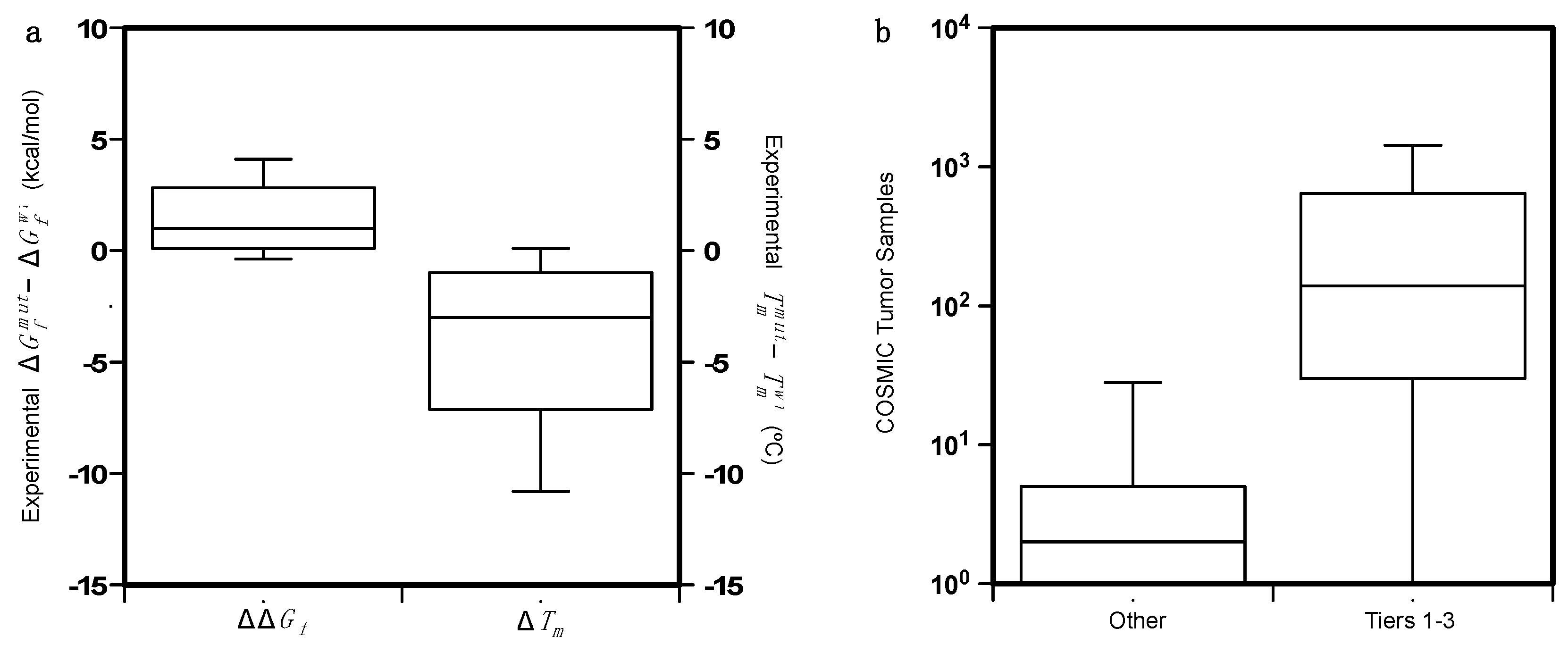
4.1. Collection of the Protein Variant Datasets
4.2. Analysis of the Protein Variants
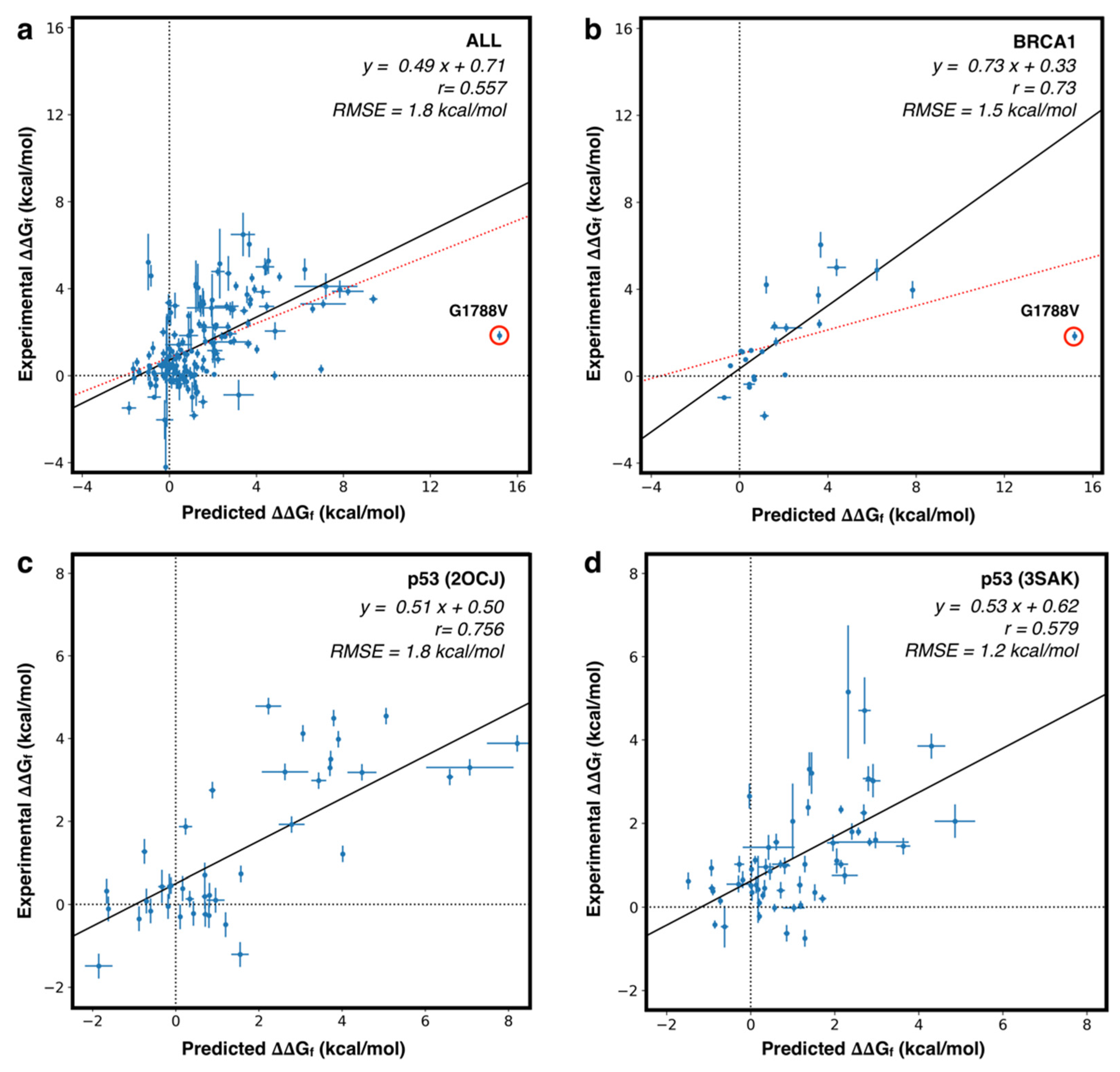
4.3. Predicting the Folding Free Energy Change of the Protein Variants
4.4. Predicting the Pathogenicity of Protein Variants
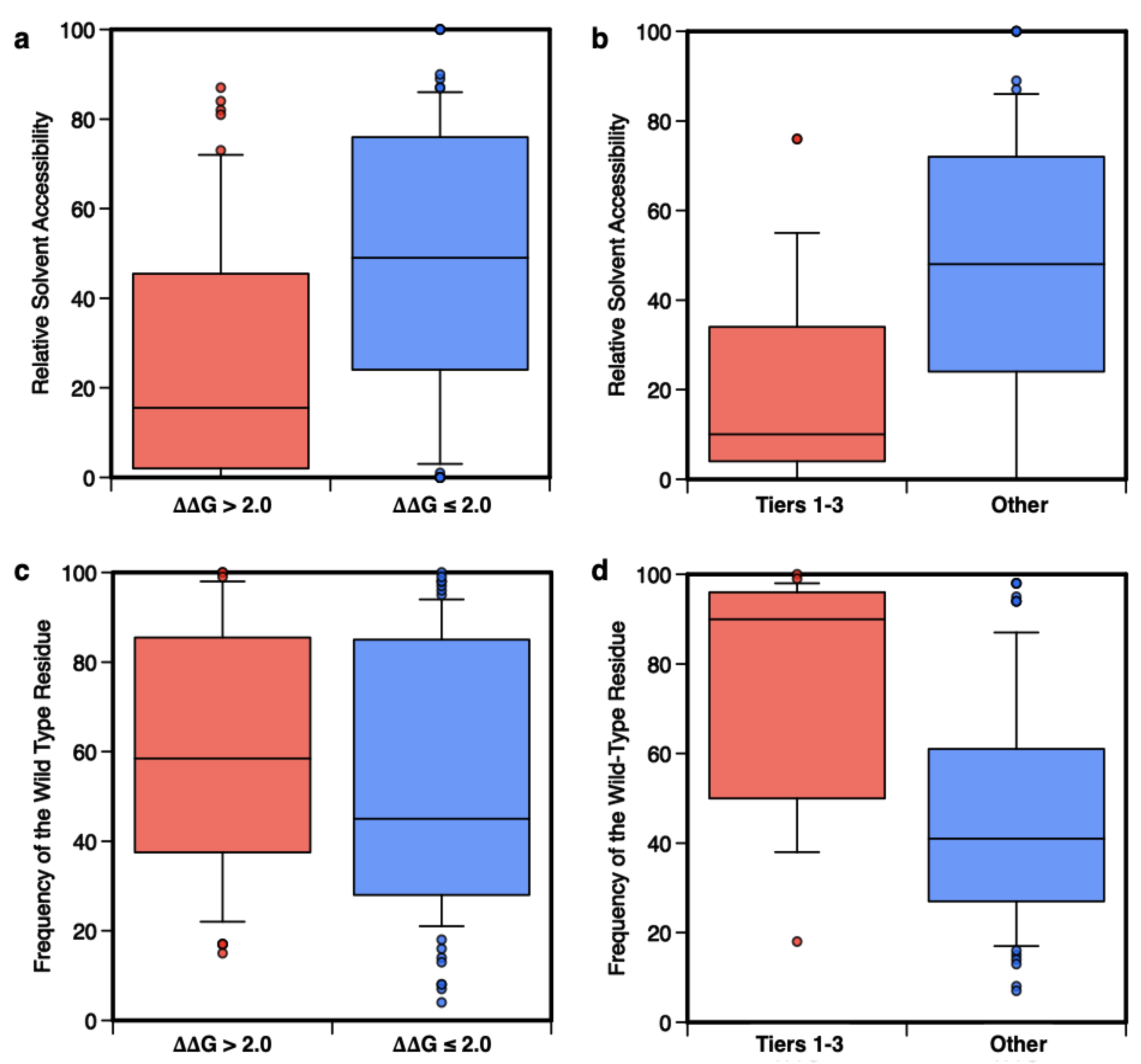
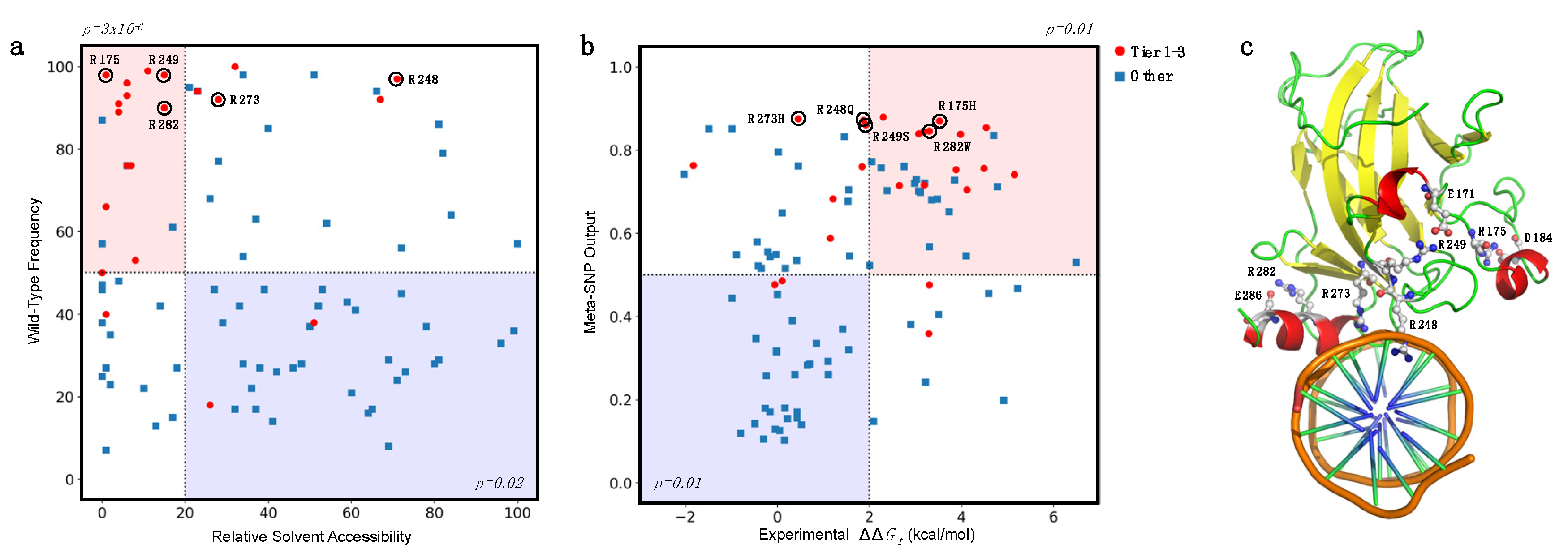
4.5. Analysis of the Prediction on the Basis of the Amino Acid Accessibility and Conservation
5. Conclusions and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public Archive of Interpretations of Clinically Relevant Variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- 1000 Genomes Project Consortium; Abecasis, G.R.; Auton, A.; Brooks, L.D.; DePristo, M.A.; Durbin, R.M.; Handsaker, R.E.; Kang, H.M.; Marth, G.T.; McVean, G.A. An Integrated Map of Genetic Variation from 1092 Human Genomes. Nature 2012, 491, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Collins, F.S.; Guyer, M.S.; Charkravarti, A. Variations on a Theme: Cataloging Human DNA Sequence Variation. Science 1997, 278, 1580–1581. [Google Scholar] [CrossRef] [PubMed]
- HapMap Consortium. The International HapMap Project. Nature 2003, 426, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Kruglyak, L.; Nickerson, D.A. Variation Is the Spice of Life. Nat. Genet. 2001, 27, 234–236. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.; Li, M.; Zhao, L.; Zhang, B.; Yang, Y.; Zheng, C.H.; Xia, J. Comparison and Integration of Computational Methods for Deleterious Synonymous Mutation Prediction. Brief. Bioinform. 2019, 21, 970–981. [Google Scholar] [CrossRef]
- Zhou, T.; Zhang, Y.; Macchiarulo, A.; Yang, Z.; Cellanetti, M.; Coto, E.; Xu, P.; Pellicciari, R.; Wang, L. Novel Polymorphisms of Nuclear Receptor SHP Associated with Functional and Structural Changes. J. Biol. Chem. 2010, 285, 24871–24881. [Google Scholar] [CrossRef] [PubMed]
- Ancien, F.; Pucci, F.; Godfroid, M.; Rooman, M. Prediction and Interpretation of Deleterious Coding Variants in Terms of Protein Structural Stability. Sci. Rep. 2018, 8, 4480. [Google Scholar] [CrossRef]
- Malhotra, S.; Alsulami, A.F.; Heiyun, Y.; Ochoa, B.M.; Jubb, H.; Forbes, S.; Blundell, T.L. Understanding the Impacts of Missense Mutations on Structures and Functions of Human Cancer-Related Genes: A Preliminary Computational Analysis of the COSMIC Cancer Gene Census. PLoS ONE 2019, 14, e0219935. [Google Scholar] [CrossRef]
- MacArthur, D.G.; Manolio, T.A.; Dimmock, D.P.; Rehm, H.L.; Shendure, J.; Abecasis, G.R.; Adams, D.R.; Altman, R.B.; Antonarakis, S.E.; Ashley, E.A.; et al. Guidelines for Investigating Causality of Sequence Variants in Human Disease. Nature 2014, 508, 469–476. [Google Scholar] [CrossRef]
- Lappalainen, T.; Scott, A.J.; Brandt, M.; Hall, I.M. Genomic Analysis in the Age of Human Genome Sequencing. Cell 2019, 177, 70–84. [Google Scholar] [CrossRef]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of Published Genome-Wide Association Studies, Targeted Arrays and Summary Statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef] [PubMed]
- Flannick, J.; Florez, J.C. Type 2 Diabetes: Genetic Data Sharing to Advance Complex Disease Research. Nat. Rev. Genet. 2016, 17, 535–549. [Google Scholar] [CrossRef]
- Liu, Y.; Easton, J.; Shao, Y.; Maciaszek, J.; Wang, Z.; Wilkinson, M.R.; McCastlain, K.; Edmonson, M.; Pounds, S.B.; Shi, L.; et al. The Genomic Landscape of Pediatric and Young Adult T-Lineage Acute Lymphoblastic Leukemia. Nat. Genet. 2017, 49, 1211–1218. [Google Scholar] [CrossRef]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385.e18. [Google Scholar] [CrossRef]
- ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium Pan-Cancer Analysis of Whole Genomes. Nature 2020, 578, 82–93. [CrossRef]
- Nussinov, R.; Tsai, C.-J.; Jang, H. Why Are Some Driver Mutations Rare? Trends Pharmacol. Sci. 2019, 40, 919–929. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. DbSNP: The NCBI Database of Genetic Variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef]
- Mottaz, A.; David, F.P.; Veuthey, A.L.; Yip, Y.L. Easy Retrieval of Single Amino-Acid Polymorphisms and Phenotype Information Using SwissVar. Bioinformatics 2010, 26, 851–852. [Google Scholar] [CrossRef]
- Zhang, J.; Baran, J.; Cros, A.; Guberman, J.M.; Haider, S.; Hsu, J.; Liang, Y.; Rivkin, E.; Wang, J.; Whitty, B.; et al. International Cancer Genome Consortium Data Portal--a One-Stop Shop for Cancer Genomics Data. Database Oxf. 2011, 2011, bar026. [Google Scholar] [CrossRef] [PubMed]
- Amberger, J.S.; Bocchini, C.A.; Scott, A.F.; Hamosh, A. OMIM.Org: Leveraging Knowledge across Phenotype-Gene Relationships. Nucleic Acids Res. 2019, 47, D1038–D1043. [Google Scholar] [CrossRef] [PubMed]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a Shared Vision for Cancer Genomic Data. N. Engl. J. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Stein, A.; Fowler, D.M.; Hartmann-Petersen, R.; Lindorff-Larsen, K. Biophysical and Mechanistic Models for Disease-Causing Protein Variants. Trends Biochem. Sci. 2019, 44, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Sanavia, T.; Birolo, G.; Montanucci, L.; Turina, P.; Capriotti, E.; Fariselli, P. Limitations and Challenges in Protein Stability Prediction upon Genome Variations: Towards Future Applications in Precision Medicine. Comput. Struct. Biotechnol. J. 2020, 18, 1968–1979. [Google Scholar] [CrossRef]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX Web Server: An Online Force Field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef]
- Capriotti, E.; Altman, R.B.; Bromberg, Y. Collective Judgment Predicts Disease-Associated Single Nucleotide Variants. BMC Genom. 2013, 14 (Suppl. S3), S2. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Brown, J. Mutations, Evolution and the Central Role of a Self-Defined Fitness Function in the Initiation and Progression of Cancer. Biochim. Biophys. Acta Rev. Cancer 2017, 1867, 162–166. [Google Scholar] [CrossRef]
- Sackton, T.B.; Lazzaro, B.P.; Schlenke, T.A.; Evans, J.D.; Hultmark, D.; Clark, A.G. Dynamic Evolution of the Innate Immune System in Drosophila. Nat. Genet. 2007, 39, 1461–1468. [Google Scholar] [CrossRef]
- Cairns, J. Mutation Selection and the Natural History of Cancer. Nature 1975, 255, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.; Maley, C.C. Clonal Evolution in Cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef]
- Nowell, P.C. The Clonal Evolution of Tumor Cell Populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Tran, B.; Hsu, A.L.; Taylor, G.R.; Fox, S.B.; Fellowes, A.; Marquis, R.; Mooi, J.; Desai, J.; Doig, K.; et al. Exploring the Feasibility and Utility of Exome-Scale Tumour Sequencing in a Clinical Setting. Intern. Med. J. 2018, 48, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Manjunath, M.; Yan, J.; Baur, B.A.; Zhang, S.; Roy, S.; Song, J.S. The Cancer-Associated Genetic Variant Rs3903072 Modulates Immune Cells in the Tumor Microenvironment. Front. Genet. 2019, 10, 754. [Google Scholar] [CrossRef] [PubMed]
- Milanese, J.-S.; Wang, E. Germline Mutations and Their Clinical Applications in Cancer. Breast Cancer Manag. 2019, 8, BMT23. [Google Scholar] [CrossRef]
- Chan, S.H.; Lim, W.K.; Ishak, N.D.B.; Li, S.-T.; Goh, W.L.; Tan, G.S.; Lim, K.H.; Teo, M.; Young, C.N.C.; Malik, S.; et al. Germline Mutations in Cancer Predisposition Genes Are Frequent in Sporadic Sarcomas. Sci. Rep. 2017, 7, 10660. [Google Scholar] [CrossRef]
- Hu, C.; Hart, S.N.; Polley, E.C.; Gnanaolivu, R.; Shimelis, H.; Lee, K.Y.; Lilyquist, J.; Na, J.; Moore, R.; Antwi, S.O.; et al. Association Between Inherited Germline Mutations in Cancer Predisposition Genes and Risk of Pancreatic Cancer. JAMA 2018, 319, 2401–2409. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Brusco, L.; Daniels, M.; Wathoo, C.; Bailey, A.M.; Strong, L.; Shaw, K.; Lu, K.; Qi, Y.; Zhao, H.; et al. Incidental Germline Variants in 1000 Advanced Cancers on a Prospective Somatic Genomic Profiling Protocol. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016, 27, 795–800. [Google Scholar] [CrossRef]
- Schrader, K.A.; Cheng, D.T.; Joseph, V.; Prasad, M.; Walsh, M.; Zehir, A.; Ni, A.; Thomas, T.; Benayed, R.; Ashraf, A.; et al. Germline Variants in Targeted Tumor Sequencing Using Matched Normal DNA. JAMA Oncol. 2016, 2, 104–111. [Google Scholar] [CrossRef]
- Seifert, B.A.; O’Daniel, J.M.; Amin, K.; Marchuk, D.S.; Patel, N.M.; Parker, J.S.; Hoyle, A.P.; Mose, L.E.; Marron, A.; Hayward, M.C.; et al. Germline Analysis from Tumor-Germline Sequencing Dyads to Identify Clinically Actionable Secondary Findings. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 4087–4094. [Google Scholar] [CrossRef] [PubMed]
- Mandelker, D.; Zhang, L.; Kemel, Y.; Stadler, Z.K.; Joseph, V.; Zehir, A.; Pradhan, N.; Arnold, A.; Walsh, M.F.; Li, Y.; et al. Mutation Detection in Patients with Advanced Cancer by Universal Sequencing of Cancer-Related Genes in Tumor and Normal DNA vs Guideline-Based Germline Testing. JAMA 2017, 318, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Bertelsen, B.; Tuxen, I.V.; Yde, C.W.; Gabrielaite, M.; Torp, M.H.; Kinalis, S.; Oestrup, O.; Rohrberg, K.; Spangaard, I.; Santoni-Rugiu, E.; et al. High Frequency of Pathogenic Germline Variants within Homologous Recombination Repair in Patients with Advanced Cancer. NPJ Genom. Med. 2019, 4, 13. [Google Scholar] [CrossRef]
- Qing, T.; Mohsen, H.; Marczyk, M.; Ye, Y.; O’Meara, T.; Zhao, H.; Townsend, J.P.; Gerstein, M.; Hatzis, C.; Kluger, Y.; et al. Germline Variant Burden in Cancer Genes Correlates with Age at Diagnosis and Somatic Mutation Burden. Nat. Commun. 2020, 11, 2438. [Google Scholar] [CrossRef]
- Kampmeyer, C.; Nielsen, S.V.; Clausen, L.; Stein, A.; Gerdes, A.-M.; Lindorff-Larsen, K.; Hartmann-Petersen, R. Blocking Protein Quality Control to Counter Hereditary Cancers. Genes. Chromosomes Cancer 2017, 56, 823–831. [Google Scholar] [CrossRef]
- Nielsen, S.V.; Poulsen, E.G.; Rebula, C.A.; Hartmann-Petersen, R. Protein Quality Control in the Nucleus. Biomolecules 2014, 4, 646–661. [Google Scholar] [CrossRef]
- Kriegenburg, F.; Jakopec, V.; Poulsen, E.G.; Nielsen, S.V.; Roguev, A.; Krogan, N.; Gordon, C.; Fleig, U.; Hartmann-Petersen, R. A Chaperone-Assisted Degradation Pathway Targets Kinetochore Proteins to Ensure Genome Stability. PLoS Genet. 2014, 10, e1004140. [Google Scholar] [CrossRef] [PubMed]
- Casadio, R.; Vassura, M.; Tiwari, S.; Fariselli, P.; Luigi Martelli, P. Correlating Disease-Related Mutations to Their Effect on Protein Stability: A Large-Scale Analysis of the Human Proteome. Hum. Mutat. 2011, 32, 1161–1170. [Google Scholar] [CrossRef]
- Matreyek, K.A.; Starita, L.M.; Stephany, J.J.; Martin, B.; Chiasson, M.A.; Gray, V.E.; Kircher, M.; Khechaduri, A.; Dines, J.N.; Hause, R.J.; et al. Multiplex Assessment of Protein Variant Abundance by Massively Parallel Sequencing. Nat. Genet. 2018, 50, 874–882. [Google Scholar] [CrossRef]
- Nielsen, S.V.; Stein, A.; Dinitzen, A.B.; Papaleo, E.; Tatham, M.H.; Poulsen, E.G.; Kassem, M.M.; Rasmussen, L.J.; Lindorff-Larsen, K.; Hartmann-Petersen, R. Predicting the Impact of Lynch Syndrome-Causing Missense Mutations from Structural Calculations. PLoS Genet. 2017, 13, e1006739. [Google Scholar] [CrossRef]
- Ahner, A.; Nakatsukasa, K.; Zhang, H.; Frizzell, R.A.; Brodsky, J.L. Small Heat-Shock Proteins Select DeltaF508-CFTR for Endoplasmic Reticulum-Associated Degradation. Mol. Biol. Cell 2007, 18, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting Mutant P53 for Efficient Cancer Therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef]
- Chen, S.; Wu, J.-L.; Liang, Y.; Tang, Y.-G.; Song, H.-X.; Wu, L.-L.; Xing, Y.-F.; Yan, N.; Li, Y.-T.; Wang, Z.-Y.; et al. Arsenic Trioxide Rescues Structural P53 Mutations through a Cryptic Allosteric Site. Cancer Cell 2021, 39, 225–239.e8. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Y.; Li, X.; Yi, S.; Xu, J. Gain-of-Function Mutations: An Emerging Advantage for Cancer Biology. Trends Biochem. Sci. 2019, 44, 659–674. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-H.; Babu, M.M. Human Diseases from Gain-of-Function Mutations in Disordered Protein Regions. Cell 2018, 175, 40–42. [Google Scholar] [CrossRef] [PubMed]
- Stein, Y.; Rotter, V.; Aloni-Grinstein, R. Gain-of-Function Mutant P53: All the Roads Lead to Tumorigenesis. Int. J. Mol. Sci. 2019, 20, 6197. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W. A Strong Candidate for the Breast and Ovarian Cancer Susceptibility Gene BRCA1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational Landscape and Significance across 12 Major Cancer Types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Guccini, I.; Serio, D.; Condò, I.; Rufini, A.; Tomassini, B.; Mangiola, A.; Maira, G.; Anile, C.; Fina, D.; Pallone, F.; et al. Frataxin Participates to the Hypoxia-Induced Response in Tumors. Cell Death Dis. 2011, 2, e123. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Knapp, S. Targeting Bromodomains: Epigenetic Readers of Lysine Acetylation. Nat. Rev. Drug Discov. 2014, 13, 337–356. [Google Scholar] [CrossRef]
- Ruvolo, P.P. Role of Protein Phosphatases in the Cancer Microenvironment. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Sauer, S. Ligands for the Nuclear Peroxisome Proliferator-Activated Receptor Gamma. Trends Pharmacol. Sci. 2015, 36, 688–704. [Google Scholar] [CrossRef] [PubMed]
- Savojardo, C.; Manfredi, M.; Martelli, P.L.; Casadio, R. Solvent Accessibility of Residues Undergoing Pathogenic Variations in Humans: From Protein Structures to Protein Sequences. Front. Mol. Biosci. 2021, 7. [Google Scholar] [CrossRef]
- Gilis, D.; Rooman, M. Stability Changes upon Mutation of Solvent-Accessible Residues in Proteins Evaluated by Database-Derived Potentials. J. Mol. Biol. 1996, 257, 1112–1126. [Google Scholar] [CrossRef]
- Wei, Q.; Xu, Q.; Dunbrack, R.L. Prediction of Phenotypes of Missense Mutations in Human Proteins from Biological Assemblies. Proteins 2013, 81, 199–213. [Google Scholar] [CrossRef]
- Duning, K.; Wennmann, D.O.; Bokemeyer, A.; Reissner, C.; Wersching, H.; Thomas, C.; Buschert, J.; Guske, K.; Franzke, V.; Flöel, A.; et al. Common Exonic Missense Variants in the C2 Domain of the Human KIBRA Protein Modify Lipid Binding and Cognitive Performance. Transl. Psychiatry 2013, 3, e272. [Google Scholar] [CrossRef][Green Version]
- Feinberg, H.; Rowntree, T.J.W.; Tan, S.L.W.; Drickamer, K.; Weis, W.I.; Taylor, M.E. Common Polymorphisms in Human Langerin Change Specificity for Glycan Ligands. J. Biol. Chem. 2013, 288, 36762–36771. [Google Scholar] [CrossRef]
- Haraksingh, R.R.; Snyder, M.P. Impacts of Variation in the Human Genome on Gene Regulation. J. Mol. Biol. 2013, 425, 3970–3977. [Google Scholar] [CrossRef]
- Silva, J.L.; De Moura Gallo, C.V.; Costa, D.C.F.; Rangel, L.P. Prion-like Aggregation of Mutant P53 in Cancer. Trends Biochem. Sci. 2014, 39, 260–267. [Google Scholar] [CrossRef]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2.0: Predicting Stability Changes upon Mutation from the Protein Sequence or Structure. Nucleic Acids Res. 2005, 33, W306–W310. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, L.; Gao, Y.; Zhang, J.; Zhenirovskyy, M.; Alexov, E. Predicting Folding Free Energy Changes upon Single Point Mutations. Bioinforma. Oxf. Engl. 2012, 28, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Moult, J. SNPs, Protein Structure, and Disease. Hum. Mutat. 2001, 17, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Yue, P.; Li, Z.; Moult, J. Loss of Protein Structure Stability as a Major Causative Factor in Monogenic Disease. J. Mol. Biol. 2005, 353, 459–473. [Google Scholar] [CrossRef] [PubMed]
- Martelli, P.L.; Fariselli, P.; Savojardo, C.; Babbi, G.; Aggazio, F.; Casadio, R. Large Scale Analysis of Protein Stability in OMIM Disease Related Human Protein Variants. BMC Genom. 2016, 17 (Suppl. S2), 397. [Google Scholar] [CrossRef]
- Soragni, A.; Janzen, D.M.; Johnson, L.M.; Lindgren, A.G.; Thai-Quynh Nguyen, A.; Tiourin, E.; Soriaga, A.B.; Lu, J.; Jiang, L.; Faull, K.F.; et al. A Designed Inhibitor of P53 Aggregation Rescues P53 Tumor Suppression in Ovarian Carcinomas. Cancer Cell 2016, 29, 90–103. [Google Scholar] [CrossRef]
- Yue, P.; Moult, J. Identification and Analysis of Deleterious Human SNPs. J. Mol. Biol. 2006, 356, 1263–1274. [Google Scholar] [CrossRef] [PubMed]
- Pasquo, A.; Consalvi, V.; Knapp, S.; Alfano, I.; Ardini, M.; Stefanini, S.; Chiaraluce, R. Structural Stability of Human Protein Tyrosine Phosphatase ρ Catalytic Domain: Effect of Point Mutations. PLoS ONE 2012, 7, e32555. [Google Scholar] [CrossRef] [PubMed]
- Grothe, H.L.; Little, M.R.; Sjogren, P.P.; Chang, A.A.; Nelson, E.F.; Yuan, C. Altered Protein Conformation and Lower Stability of the Dystrophic Transforming Growth Factor Beta-Induced Protein Mutants. Mol. Vis. 2013, 19, 593–603. [Google Scholar]
- Khan, S.; Vihinen, M. Performance of Protein Stability Predictors. Hum. Mutat. 2010, 31, 675–684. [Google Scholar] [CrossRef]
- Waters, P.J. Degradation of Mutant Proteins, Underlying “Loss of Function” Phenotypes, Plays a Major Role in Genetic Disease. Curr. Issues Mol. Biol. 2001, 3, 57–65. [Google Scholar]
- Gummlich, L. ATO Stabilizes Structural P53 Mutants. Nat. Rev. Cancer 2021, 21, 141. [Google Scholar] [CrossRef]
- Bullock, A.N.; Henckel, J.; Fersht, A.R. Quantitative Analysis of Residual Folding and DNA Binding in Mutant P53 Core Domain: Definition of Mutant States for Rescue in Cancer Therapy. Oncogene 2000, 19, 1245–1256. [Google Scholar] [CrossRef]
- Williams, R.S.; Chasman, D.I.; Hau, D.D.; Hui, B.; Lau, A.Y.; Glover, J.N.M. Detection of Protein Folding Defects Caused by BRCA1-BRCT Truncation and Missense Mutations. J. Biol. Chem. 2003, 278, 53007–53016. [Google Scholar] [CrossRef] [PubMed]
- Rowling, P.J.E.; Cook, R.; Itzhaki, L.S. Toward Classification of BRCA1 Missense Variants Using a Biophysical Approach. J. Biol. Chem. 2010, 285, 20080–20087. [Google Scholar] [CrossRef] [PubMed]
- Petrosino, M.; Pasquo, A.; Novak, L.; Toto, A.; Gianni, S.; Mantuano, E.; Veneziano, L.; Minicozzi, V.; Pastore, A.; Puglisi, R.; et al. Characterization of Human Frataxin Missense Variants in Cancer Tissues. Hum. Mutat. 2019, 40, 1400–1413. [Google Scholar] [CrossRef] [PubMed]
- Lori, C.; Lantella, A.; Pasquo, A.; Alexander, L.T.; Knapp, S.; Chiaraluce, R.; Consalvi, V. Effect of Single Amino Acid Substitution Observed in Cancer on Pim-1 Kinase Thermodynamic Stability and Structure. PLoS ONE 2013, 8, e64824. [Google Scholar] [CrossRef] [PubMed]
- Fiorillo, A.; Petrosino, M.; Ilari, A.; Pasquo, A.; Cipollone, A.; Maggi, M.; Chiaraluce, R.; Consalvi, V. The Phosphoglycerate Kinase 1 Variants Found in Carcinoma Cells Display Different Catalytic Activity and Conformational Stability Compared to the Native Enzyme. PLoS ONE 2018, 13, e0199191. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.S.; Glover, J.N.M. Structural Consequences of a Cancer-Causing BRCA1-BRCT Missense Mutation. J. Biol. Chem. 2003, 278, 2630–2635. [Google Scholar] [CrossRef]
- Venkitaraman, A.R. Cancer Susceptibility and the Functions of BRCA1 and BRCA2. Cell 2002, 108, 171–182. [Google Scholar] [CrossRef]
- Yarden, R.I.; Brody, L.C. BRCA1 Interacts with Components of the Histone Deacetylase Complex. Proc. Natl. Acad. Sci. USA 1999, 96, 4983–4988. [Google Scholar] [CrossRef]
- Cantor, S.B.; Bell, D.W.; Ganesan, S.; Kass, E.M.; Drapkin, R.; Grossman, S.; Wahrer, D.C.; Sgroi, D.C.; Lane, W.S.; Haber, D.A.; et al. BACH1, a Novel Helicase-like Protein, Interacts Directly with BRCA1 and Contributes to Its DNA Repair Function. Cell 2001, 105, 149–160. [Google Scholar] [CrossRef]
- Yu, X.; Wu, L.C.; Bowcock, A.M.; Aronheim, A.; Baer, R. The C-Terminal (BRCT) Domains of BRCA1 Interact in Vivo with CtIP, a Protein Implicated in the CtBP Pathway of Transcriptional Repression. J. Biol. Chem. 1998, 273, 25388–25392. [Google Scholar] [CrossRef]
- Li, S.; Chen, P.L.; Subramanian, T.; Chinnadurai, G.; Tomlinson, G.; Osborne, C.K.; Sharp, Z.D.; Lee, W.H. Binding of CtIP to the BRCT Repeats of BRCA1 Involved in the Transcription Regulation of P21 Is Disrupted upon DNA Damage. J. Biol. Chem. 1999, 274, 11334–11338. [Google Scholar] [CrossRef]
- Schuster-Böckler, B.; Bateman, A. Protein Interactions in Human Genetic Diseases. Genome Biol. 2008, 9, R9. [Google Scholar] [CrossRef] [PubMed]
- Teng, S.; Madej, T.; Panchenko, A.; Alexov, E. Modeling Effects of Human Single Nucleotide Polymorphisms on Protein-Protein Interactions. Biophys. J. 2009, 96, 2178–2188. [Google Scholar] [CrossRef] [PubMed]
- David, A.; Sternberg, M.J.E. The Contribution of Missense Mutations in Core and Rim Residues of Protein-Protein Interfaces to Human Disease. J. Mol. Biol. 2015, 427, 2886–2898. [Google Scholar] [CrossRef] [PubMed]
- Dixit, A.; Verkhivker, G.M. Hierarchical Modeling of Activation Mechanisms in the ABL and EGFR Kinase Domains: Thermodynamic and Mechanistic Catalysts of Kinase Activation by Cancer Mutations. PLoS Comput. Biol. 2009, 5, e1000487. [Google Scholar] [CrossRef]
- Dixit, A.; Yi, L.; Gowthaman, R.; Torkamani, A.; Schork, N.J.; Verkhivker, G.M. Sequence and Structure Signatures of Cancer Mutation Hotspots in Protein Kinases. PLoS ONE 2009, 4, e7485. [Google Scholar] [CrossRef]
- Dixit, A.; Torkamani, A.; Schork, N.J.; Verkhivker, G. Computational Modeling of Structurally Conserved Cancer Mutations in the RET and MET Kinases: The Impact on Protein Structure, Dynamics, and Stability. Biophys. J. 2009, 96, 858–874. [Google Scholar] [CrossRef] [PubMed]
- Acuner Ozbabacan, S.E.; Gursoy, A.; Keskin, O.; Nussinov, R. Conformational Ensembles, Signal Transduction and Residue Hot Spots: Application to Drug Discovery. Curr. Opin. Drug Discov. Devel. 2010, 13, 527–537. [Google Scholar] [PubMed]
- Bauer-Mehren, A.; Furlong, L.I.; Rautschka, M.; Sanz, F. From SNPs to Pathways: Integration of Functional Effect of Sequence Variations on Models of Cell Signalling Pathways. BMC Bioinform. 2009, 10 (Suppl. S6). [Google Scholar] [CrossRef] [PubMed]
- Vanunu, O.; Magger, O.; Ruppin, E.; Shlomi, T.; Sharan, R. Associating Genes and Protein Complexes with Disease via Network Propagation. PLoS Comput. Biol. 2010, 6, e1000641. [Google Scholar] [CrossRef]
- Barabasi, A.L.; Gulbahce, N.; Loscalzo, J. Network Medicine: A Network-Based Approach to Human Disease. Nat. Rev. Genet. 2011, 12, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Akhavan, S.; Miteva, M.A.; Villoutreix, B.O.; Venisse, L.; Peyvandi, F.; Mannucci, P.M.; Guillin, M.C.; Bezeaud, A. A Critical Role for Gly25 in the B Chain of Human Thrombin. J. Thromb. Haemost. JTH 2005, 3, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Gallione, C.; Aylsworth, A.S.; Beis, J.; Berk, T.; Bernhardt, B.; Clark, R.D.; Clericuzio, C.; Danesino, C.; Drautz, J.; Fahl, J.; et al. Overlapping Spectra of SMAD4 Mutations in Juvenile Polyposis (JP) and JP-HHT Syndrome. Am. J. Med. Genet. A 2010, 152A, 333–339. [Google Scholar] [CrossRef]
- Sayed, M.G.; Ahmed, A.F.; Ringold, J.R.; Anderson, M.E.; Bair, J.L.; Mitros, F.A.; Lynch, H.T.; Tinley, S.T.; Petersen, G.M.; Giardiello, F.M.; et al. Germline SMAD4 or BMPR1A Mutations and Phenotype of Juvenile Polyposis. Ann. Surg. Oncol. 2002, 9, 901–906. [Google Scholar] [CrossRef]
- Jung, B.; Staudacher, J.J.; Beauchamp, D. Transforming Growth Factor β Superfamily Signaling in Development of Colorectal Cancer. Gastroenterology 2017, 152, 36–52. [Google Scholar] [CrossRef]
- Massagué, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef]
- Lori, L.; Pasquo, A.; Lori, C.; Petrosino, M.; Chiaraluce, R.; Tallant, C.; Knapp, S.; Consalvi, V. Effect of BET Missense Mutations on Bromodomain Function, Inhibitor Binding and Stability. PLoS ONE 2016, 11, e0159180. [Google Scholar] [CrossRef]
- Stevanin, G.; Hahn, V.; Lohmann, E.; Bouslam, N.; Gouttard, M.; Soumphonphakdy, C.; Welter, M.-L.; Ollagnon-Roman, E.; Lemainque, A.; Ruberg, M.; et al. Mutation in the Catalytic Domain of Protein Kinase C Gamma and Extension of the Phenotype Associated with Spinocerebellar Ataxia Type 14. Arch. Neurol. 2004, 61, 1242–1248. [Google Scholar] [CrossRef]
- Dehal, P.; Satou, Y.; Campbell, R.K.; Chapman, J.; Degnan, B.; De Tomaso, A.; Davidson, B.; Di Gregorio, A.; Gelpke, M.; Goodstein, D.M.; et al. The Draft Genome of Ciona Intestinalis: Insights into Chordate and Vertebrate Origins. Science 2002, 298, 2157–2167. [Google Scholar] [CrossRef] [PubMed]
- Takamiya, O.; Seta, M.; Tanaka, K.; Ishida, F. Human Factor VII Deficiency Caused by S339C Mutation Located Adjacent to the Specificity Pocket of the Catalytic Domain. Clin. Lab. Haematol. 2002, 24, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Teng, S.; Wang, L.; Schwartz, C.E.; Alexov, E. Computational Analysis of Missense Mutations Causing Snyder-Robinson Syndrome. Hum. Mutat. 2010, 31, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Norris, J.; Schwartz, C.; Alexov, E. In Silico and in Vitro Investigations of the Mutability of Disease-Causing Missense Mutation Sites in Spermine Synthase. PLoS ONE 2011, 6, e20373. [Google Scholar] [CrossRef]
- Wang, Z.; Shen, D.; Parsons, D.W.; Bardelli, A.; Sager, J.; Szabo, S.; Ptak, J.; Silliman, N.; Peters, B.A.; van der Heijden, M.S.; et al. Mutational Analysis of the Tyrosine Phosphatome in Colorectal Cancers. Science 2004, 304, 1164–1166. [Google Scholar] [CrossRef]
- Compiani, M.; Capriotti, E. Computational and Theoretical Methods for Protein Folding. Biochemistry 2013, 52, 8601–8624. [Google Scholar] [CrossRef] [PubMed]
- Marabotti, A.; Scafuri, B.; Facchiano, A. Predicting the Stability of Mutant Proteins by Computational Approaches: An Overview. Brief. Bioinform. 2020, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.D.; Bava, K.A.; Gromiha, M.M.; Prabakaran, P.; Kitajima, K.; Uedaira, H.; Sarai, A. ProTherm and ProNIT: Thermodynamic Databases for Proteins and Protein-Nucleic Acid Interactions. Nucleic Acids Res. 2006, 34, D204–D206. [Google Scholar] [CrossRef]
- Fernald, G.H.; Capriotti, E.; Daneshjou, R.; Karczewski, K.J.; Altman, R.B. Bioinformatics Challenges for Personalized Medicine. Bioinformatics 2011, 27, 1741–1748. [Google Scholar] [CrossRef]
- Niroula, A.; Vihinen, M. Variation Interpretation Predictors: Principles, Types, Performance, and Choice. Hum. Mutat. 2016, 37, 579–597. [Google Scholar] [CrossRef]
- Capriotti, E.; Ozturk, K.; Carter, H. Integrating Molecular Networks with Genetic Variant Interpretation for Precision Medicine. Wiley Interdiscip. Rev. Syst. Biol. Med. 2019, 11, e1443. [Google Scholar] [CrossRef] [PubMed]
- Capriotti, E.; Calabrese, R.; Casadio, R. Predicting the Insurgence of Human Genetic Diseases Associated to Single Point Protein Mutations with Support Vector Machines and Evolutionary Information. Bioinformatics 2006, 22, 2729–2734. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.D.; Kejariwal, A. Coding Single-Nucleotide Polymorphisms Associated with Complex vs. Mendelian Disease: Evolutionary Evidence for Differences in Molecular Effects. Proc. Natl. Acad. Sci. USA 2004, 101, 15398–15403. [Google Scholar] [CrossRef] [PubMed]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT Web Server: Predicting Effects of Amino Acid Substitutions on Proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, Y.; Rost, B. SNAP: Predict Effect of Non-Synonymous Polymorphisms on Function. Nucleic Acids Res. 2007, 35, 3823–3835. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Sander, C. Dictionary of Protein Secondary Structure: Pattern Recognition of Hydrogen-Bonded and Geometrical Features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, R.A.; Chan, C.S. Why Are There Hotspot Mutations in the TP53 Gene in Human Cancers? Cell Death Differ. 2018, 25, 154–160. [Google Scholar] [CrossRef]
- Kim, M.P.; Lozano, G. Mutant P53 Partners in Crime. Cell Death Differ. 2018, 25, 161–168. [Google Scholar] [CrossRef]
- Bloom, J.D.; Labthavikul, S.T.; Otey, C.R.; Arnold, F.H. Protein Stability Promotes Evolvability. Proc. Natl. Acad. Sci. USA 2006, 103, 5869–5874. [Google Scholar] [CrossRef]
- Tokuriki, N.; Tawfik, D.S. Stability Effects of Mutations and Protein Evolvability. Curr. Opin. Struct. Biol. 2009, 19, 596–604. [Google Scholar] [CrossRef]
- Vihinen, M. Functional Effects of Protein Variants. Biochimie 2021, 180, 104–120. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, S.; Pérez-Palma, E.; Jespersen, J.B.; May, P.; Hoksza, D.; Heyne, H.O.; Ahmed, S.S.; Rifat, Z.T.; Rahman, M.S.; Lage, K.; et al. Comprehensive Characterization of Amino Acid Positions in Protein Structures Reveals Molecular Effect of Missense Variants. Proc. Natl. Acad. Sci. USA 2020, 117, 28201–28211. [Google Scholar] [CrossRef] [PubMed]
- Andreoletti, G.; Pal, L.R.; Moult, J.; Brenner, S.E. Reports from the Fifth Edition of CAGI: The Critical Assessment of Genome Interpretation. Hum. Mutat. 2019, 40, 1197–1201. [Google Scholar] [CrossRef]
- Savojardo, C.; Petrosino, M.; Babbi, G.; Bovo, S.; Corbi-Verge, C.; Casadio, R.; Fariselli, P.; Folkman, L.; Garg, A.; Karimi, M.; et al. Evaluating the Predictions of the Protein Stability Change upon Single Amino Acid Substitutions for the FXN CAGI5 Challenge. Hum. Mutat. 2019, 40, 1392–1399. [Google Scholar] [CrossRef]
- Chandonia, J.-M.; Adhikari, A.; Carraro, M.; Chhibber, A.; Cutting, G.R.; Fu, Y.; Gasparini, A.; Jones, D.T.; Kramer, A.; Kundu, K.; et al. Lessons from the CAGI-4 Hopkins Clinical Panel Challenge. Hum. Mutat. 2017, 38, 1155–1168. [Google Scholar] [CrossRef]
- Pal, L.R.; Kundu, K.; Yin, Y.; Moult, J. CAGI4 SickKids Clinical Genomes Challenge: A Pipeline for Identifying Pathogenic Variants. Hum. Mutat. 2017, 38, 1169–1181. [Google Scholar] [CrossRef] [PubMed]
- Stein, Y.; Aloni-Grinstein, R.; Rotter, V. Mutant P53 Oncogenicity: Dominant-Negative or Gain-of-Function? Carcinogenesis 2020, 41, 1635–1647. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. P53 Proteoforms and Intrinsic Disorder: An Illustration of the Protein Structure–Function Continuum Concept. Int. J. Mol. Sci. 2016, 17, 1874. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, R.C.; Sandrini, F.; Figueiredo, B.; Zambetti, G.P.; Michalkiewicz, E.; Lafferty, A.R.; DeLacerda, L.; Rabin, M.; Cadwell, C.; Sampaio, G.; et al. An Inherited P53 Mutation That Contributes in a Tissue-Specific Manner to Pediatric Adrenal Cortical Carcinoma. Proc. Natl. Acad. Sci. USA 2001, 98, 9330–9335. [Google Scholar] [CrossRef]
- Latronico, A.C.; Pinto, E.M.; Domenice, S.; Fragoso, M.C.; Martin, R.M.; Zerbini, M.C.; Lucon, A.M.; Mendonca, B.B. An Inherited Mutation Outside the Highly Conserved DNA-Binding Domain of the P53 Tumor Suppressor Protein in Children and Adults with Sporadic Adrenocortical Tumors. J. Clin. Endocrinol. Metab. 2001, 86, 4970–4973. [Google Scholar] [CrossRef]
- Bhattacharya, R.; Rose, P.W.; Burley, S.K.; Prlić, A. Impact of Genetic Variation on Three Dimensional Structure and Function of Proteins. PLoS ONE 2017, 12, e0171355. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.J.; Sottoriva, A.; Graham, T.A. Measuring Clonal Evolution in Cancer with Genomics. Annu. Rev. Genomics Hum. Genet. 2019, 20, 309–329. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant Database | Database Description | Web Site | Reference |
|---|---|---|---|
| ClinVar | ClinVar aggregates information about genomic variation and its relationship to human health. | http://www.ncbi.nlm.nih.gov/clinvar (accessed on 1 May 2021) | [1] |
| COSMIC | Manually curated resource of somatic mutation of human cancers. | https://cancer.sanger.ac.uk/cosmic (accessed on 1 May 2021) | [19] |
| dbSNP | Database of human single nucleotide variations, microsatellites, and small insertions and deletions for both common variations and clinical mutations. | http://www.ncbi.nlm.nih.gov/snp (accessed on 1 May 2021) | [20] |
| UniProt humsvar | Lists of missense variants annotated in UniProtKB/Swiss-Prot human entries. The variant classification should not be considered for clinical and diagnostic use. | https://www.uniprot.org/docs/humsavar (accessed on 1 May 2021) | [21] |
| ICGC Data Portal | The ICGC Data Portal provides many tools for visualizing, querying, and downloading cancer data. | https://dcc.icgc.org/ (accessed on 1 May 2021) | [22] |
| OMIM | Online Catalog of Human Genes and Genetic Disorders. | http://www.omim.org (accessed on 1 May 2021) | [23] |
| GDC | The GDC Data Portal provides access to GDC harmonized data as well as an archive of legacy data from TCGA and other NCI programs. | https://portal.gdc.cancer.gov/ (accessed on 1 May 2021) | [24] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrosino, M.; Novak, L.; Pasquo, A.; Chiaraluce, R.; Turina, P.; Capriotti, E.; Consalvi, V. Analysis and Interpretation of the Impact of Missense Variants in Cancer. Int. J. Mol. Sci. 2021, 22, 5416. https://doi.org/10.3390/ijms22115416
Petrosino M, Novak L, Pasquo A, Chiaraluce R, Turina P, Capriotti E, Consalvi V. Analysis and Interpretation of the Impact of Missense Variants in Cancer. International Journal of Molecular Sciences. 2021; 22(11):5416. https://doi.org/10.3390/ijms22115416
Chicago/Turabian StylePetrosino, Maria, Leonore Novak, Alessandra Pasquo, Roberta Chiaraluce, Paola Turina, Emidio Capriotti, and Valerio Consalvi. 2021. "Analysis and Interpretation of the Impact of Missense Variants in Cancer" International Journal of Molecular Sciences 22, no. 11: 5416. https://doi.org/10.3390/ijms22115416
APA StylePetrosino, M., Novak, L., Pasquo, A., Chiaraluce, R., Turina, P., Capriotti, E., & Consalvi, V. (2021). Analysis and Interpretation of the Impact of Missense Variants in Cancer. International Journal of Molecular Sciences, 22(11), 5416. https://doi.org/10.3390/ijms22115416