
An Integrated Transcriptomic Approach to Identify Molecular Markers of Calcineurin Inhibitor Nephrotoxicity in Pediatric Kidney Transplant Recipients


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
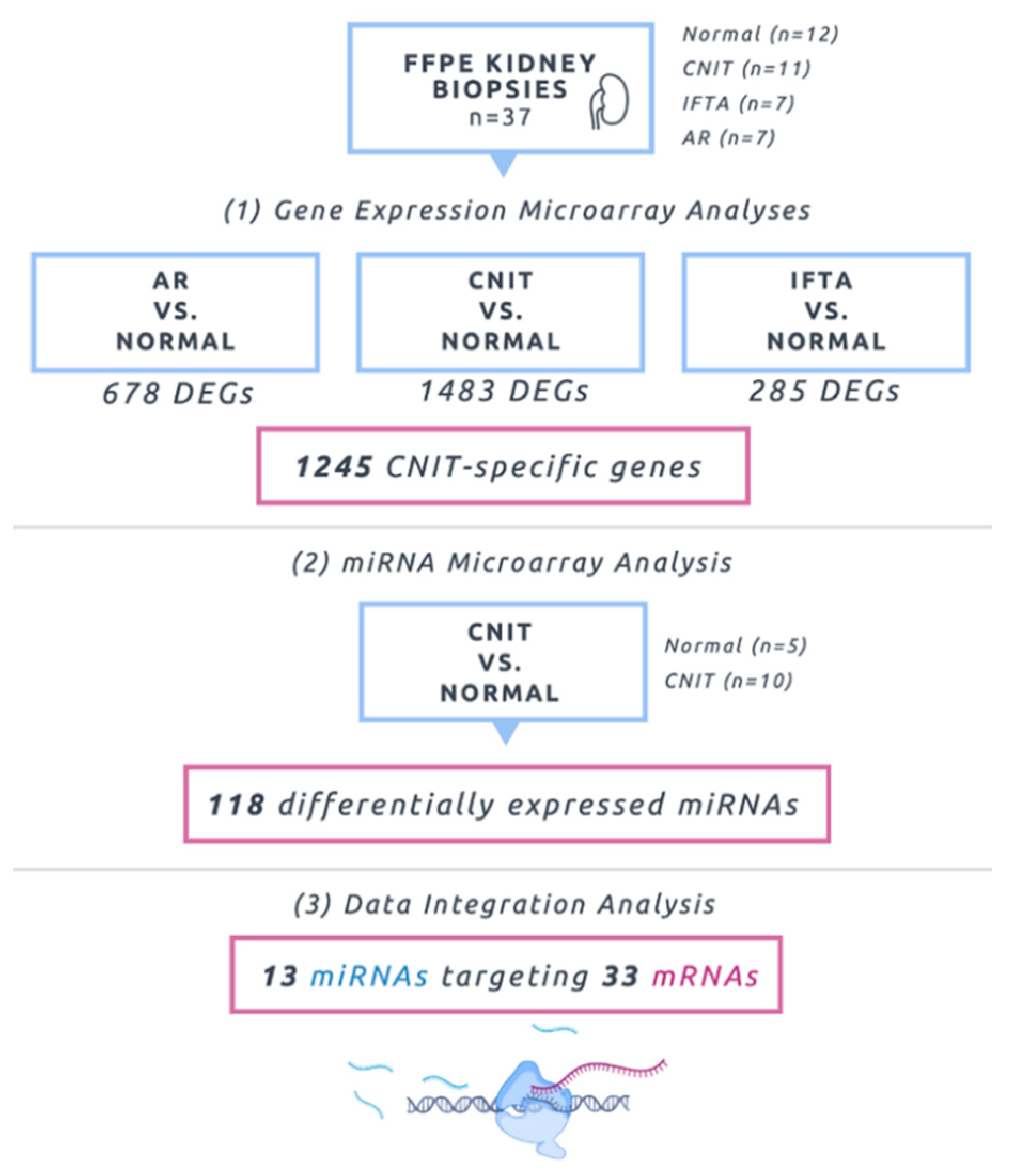
2.1. Molecular Profiling Using Microarrays
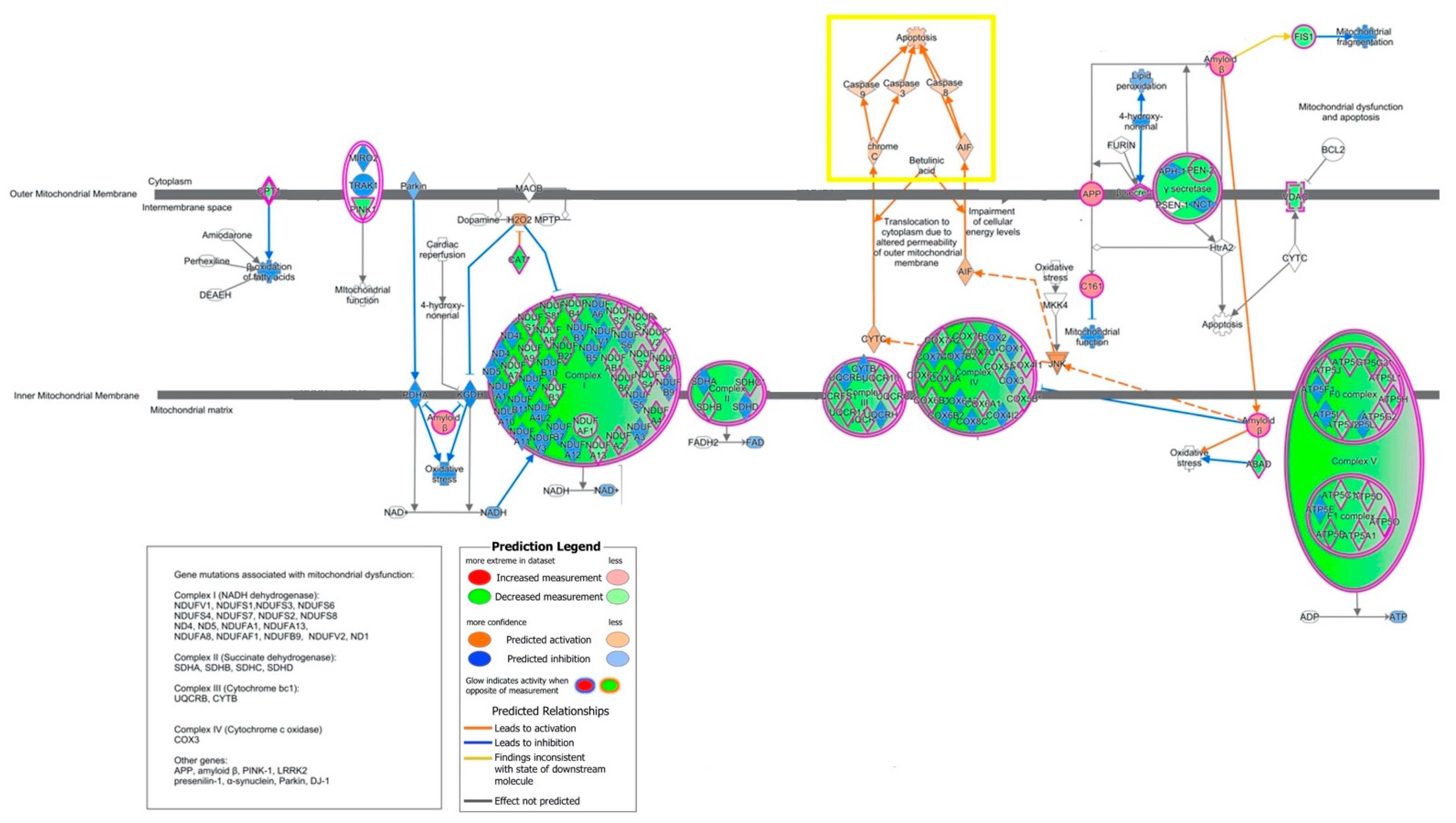
2.2. Canonical Pathways and Upstream Regulators
2.3. Biological Characterization
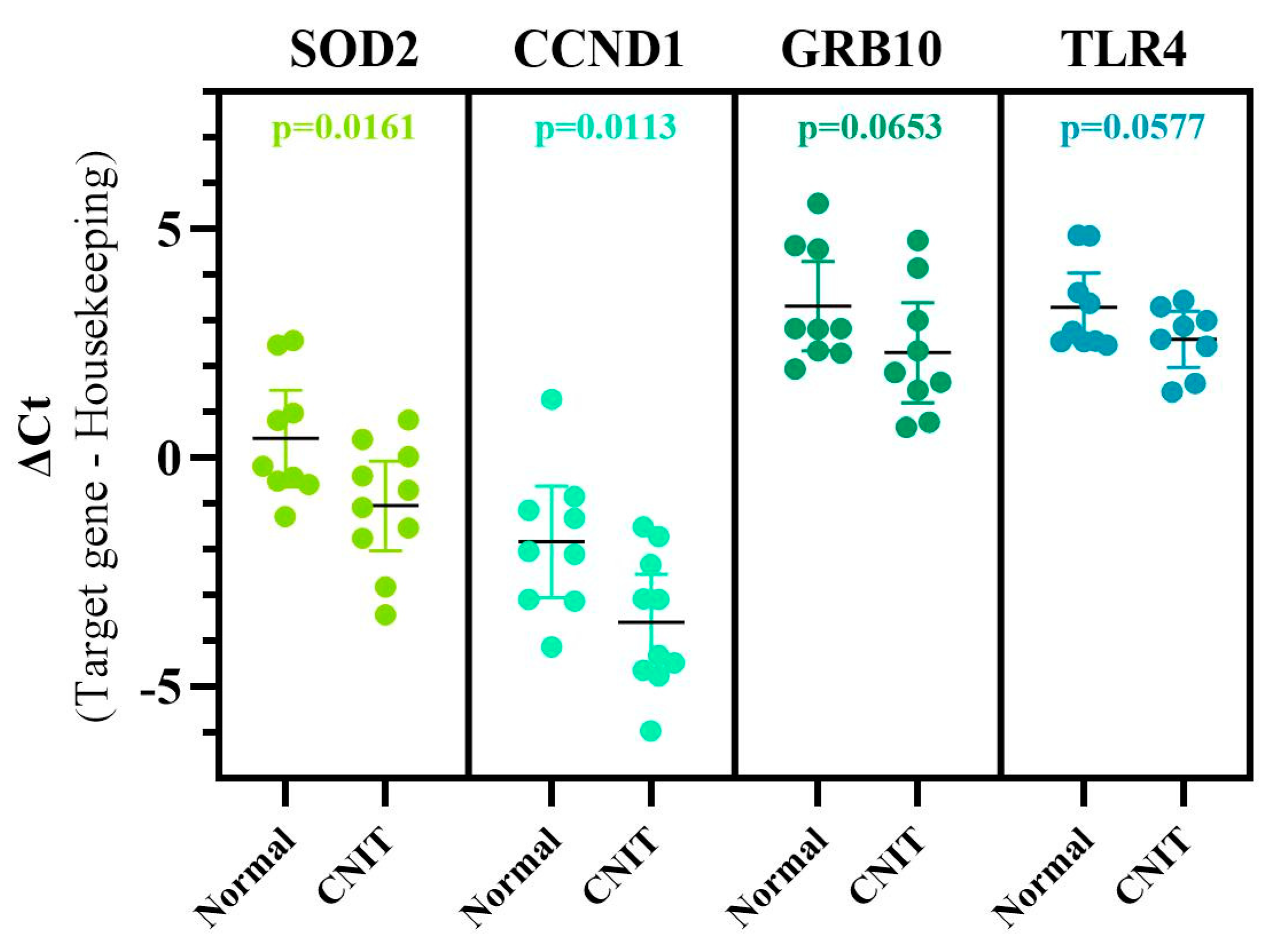
2.4. Marker Specificity, Unique Genes Associated with CNIT, and Comparison Analyses
2.5. Integrative Analysis (miRNA–mRNA Interactions)
3. Discussion
4. Materials and Methods
4.1. Study Cohort
4.2. RNA Isolation and Microarray Hybridization
4.3. Gene Expression/miRNA Microarrays
4.4. Gene Expression/miRNA Microarray Analysis and Quality Control
4.5. Interaction Networks, Functional/Pathway Analysis, Upstream Regulators
4.6. Data Integration Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McDonald, S.P.; Craig, J.C. Australian and New Zealand Paediatric Nephrology Association. Long-term survival of children with end-stage renal disease. N. Engl. J. Med. 2004, 350, 2654–2662. [Google Scholar] [CrossRef]
- Shroff, R.; Ledermann, S. Long-term outcome of chronic dialysis in children. Pediatr. Nephrol. 2009, 24, 463–474. [Google Scholar] [CrossRef]
- Gillen, D.L.; Stehman-Breen, C.O.; Smith, J.M.; McDonald, R.A.; Warady, B.A.; Brandt, J.R.; Wong, C.S. Survival Advantage of Pediatric Recipients of a First Kidney Transplant Among Children Awaiting Kidney Transplantation. Arab. Archaeol. Epigr. 2008, 8, 2600–2606. [Google Scholar] [CrossRef]
- Tonelli, M.; Wiebe, N.; Knoll, G.; Bello, A.; Browne, S.; Jadhav, D.; Klarenbach, S.; Gill, J. Systematic Review: Kidney Transplantation Compared With Dialysis in Clinically Relevant Outcomes. Arab. Archaeol. Epigr. 2011, 11, 2093–2109. [Google Scholar] [CrossRef]
- Meier-Kriesche, H.-U.; Schold, J.D.; Srinivas, T.R.; Kaplan, B. Lack of Improvement in Renal Allograft Survival Despite a Marked Decrease in Acute Rejection Rates Over the Most Recent Era. Arab. Archaeol. Epigr. 2004, 4, 378–383. [Google Scholar] [CrossRef]
- Marcén, R.; Fernández-Rodriguez, A.; Rodríguez-Mendiola, N.; Ponte, B.; Galeano, C.; Villafruela, J.; Teruel, J.; Burgos, F.; Ortuño, J. Evolution of Rejection Rates and Kidney Graft Survival: A Historical Analysis. Transplant. Proc. 2009, 41, 2357–2359. [Google Scholar] [CrossRef]
- Van Arendonk, K.J.; Boyarsky, B.J.; Orandi, B.J.; James, N.T.; Smith, J.M.; Colombani, P.M.; Segev, D.L. National trends over 25 years in pediatric kidney transplant outcomes. Pediatrics 2014, 133, 594–601. [Google Scholar] [CrossRef]
- Hart, A.; Lentine, K.L.; Smith, J.M.; Miller, J.M.; Skeans, M.A.; Prentice, M.; Robinson, A.; Foutz, J.; Booker, S.E.; Israni, A.K.; et al. OPTN/SRTR 2019 Annual Data Report: Kidney. Am. J. Transplant 2021, 21 (Suppl. S2), 21–137. [Google Scholar] [CrossRef] [PubMed]
- Laskin, B.L.; Mitsnefes, M.M.; Dahhou, M.; Zhang, X.; Foster, B.J. The mortality risk with graft function has decreased among children receiving a first kidney transplant in the United States. Kidney Int. 2015, 87, 575–583. [Google Scholar] [CrossRef]
- Sayegh, M.H.; Carpenter, C.B. Transplantation 50 years later: Progress, challenges, and promises. N. Engl. J. Med. 2004, 351, 2761–2766. [Google Scholar] [CrossRef]
- Chapman, J.R.; O’Connell, P.J.; Nankivell, B.J. Chronic renal allograft dysfunction. J. Am. Soc. Nephrol. 2005, 16, 3015–3026. [Google Scholar] [CrossRef]
- Langewisch, E.; Mannon, R.B. Chronic Allograft Injury. Clin. J. Am. Soc. Nephrol. 2021, CJN.15590920. [Google Scholar] [CrossRef] [PubMed]
- Nankivell, B.J.; Kuypers, D.R. Diagnosis and prevention of chronic kidney allograft loss. Lancet 2011, 378, 1428–1437. [Google Scholar] [CrossRef]
- Farouk, S.S.; Rein, J.L. The Many Faces of Calcineurin Inhibitor Toxicity-What the FK? Adv. Chronic Kidney Dis. 2020, 27, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Nankivell, B.J.; Borrows, R.J.; Fung, C.L.-S.; O’Connell, P.J.; Allen, R.D.; Chapman, J.R. The Natural History of Chronic Allograft Nephropathy. N. Engl. J. Med. 2003, 349, 2326–2333. [Google Scholar] [CrossRef]
- Myers, B.D.; Ross, J.; Newton, L.; Luetscher, J.; Perlroth, M. Cyclosporine-Associated Chronic Nephropathy. N. Engl. J. Med. 1984, 311, 699–705. [Google Scholar] [CrossRef]
- Calne, R.Y.; White, D.J.; Thiru, S.; Evans, D.B.; McMaster, P.; Dunn, D.C.; Pentlow, B.D.; Rolles, K. Cyclosporin A in patients receiving renal allograft from cadaveric donors. Lancet 1978, 2, 1323–1327. [Google Scholar] [CrossRef]
- Naesens, M.; Kuypers, D.R.J.; Sarwal, M. Calcineurin Inhibitor Nephrotoxicity. Clin. J. Am. Soc. Nephrol. 2009, 4, 481–508. [Google Scholar] [CrossRef]
- Kambham, N.; Nagarajan, S.; Shah, S.; Li, L.; Salvatierra, O.; Sarwal, M.M. A Novel, Semiquantitative, Clinically Correlated Calcineurin Inhibitor Toxicity Score for Renal Allograft Biopsies. Clin. J. Am. Soc. Nephrol. 2007, 2, 135–142. [Google Scholar] [CrossRef]
- Liptak, P.; Ivanyi, B. Primer: Histopathology of calcineurin-inhibitor toxicity in renal allografts. Nat. Clin. Pract. Nephrol. 2006, 2, 398–404. [Google Scholar] [CrossRef]
- Devarajan, P. Biomarkers for the early detection of acute kidney injury. Curr. Opin. Pediatr. 2011, 23, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Snanoudj, R.; Royal, V.; Elie, C.; Rabant, M.; Girardin, C.; Morelon, E.; Kreis, H.; Fournet, J.-C.; Noël, L.-H.; Legendre, C. Specificity of histological markers of long-term CNI nephrotoxicity in kidney-transplant recipients under low-dose cyclosporine therapy. Am. J. Transpl. 2011, 11, 2635–2646. [Google Scholar] [CrossRef]
- Huang, Y.; Farkash, E. Protocol Biopsies: Utility and Limitations. Adv. Chronic Kidney Dis. 2016, 23, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Furness, P.N.; Taub, N. International variation in the interpretation of renal transplant biopsies: Report of the CERTPAP Project. Kidney Int. 2001, 60, 1998–2012. [Google Scholar] [CrossRef]
- Serón, D.; Moreso, F.; Fulladosa, X.; Hueso, M.; Carrera, M.; Grinyó, J.M. Reliability of chronic allograft nephropathy diagnosis in sequential protocol biopsies. Kidney Int. 2002, 61, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Scian, M.J.; Maluf, D.G.; Mas, V.R. MiRNAs in kidney transplantation: Potential role as new biomarkers. Expert Rev. Mol. Diagn. 2013, 13, 93–104. [Google Scholar] [CrossRef]
- Fan, Y.; Chen, H.; Huang, Z.; Zheng, H.; Zhou, J. Emerging role of miRNAs in renal fibrosis. RNA Biol. 2020, 17, 1–12. [Google Scholar] [CrossRef]
- Maluf, D.G.; Dumur, C.I.; Suh, J.L.; Lee, J.K.; Cathro, H.P.; King, A.L.; Gallon, L.; Brayman, K.L.; Mas, V.R. Evaluation of Molecular Profiles in Calcineurin Inhibitor Toxicity Post-Kidney Transplant: Input to Chronic Allograft Dysfunction. Arab. Archaeol. Epigr. 2014, 14, 1152–1163. [Google Scholar] [CrossRef]
- Goldman, J.; Becker, M.L.; Jones, B.; Clements, M.; Leeder, J.S. Development of biomarkers to optimize pediatric patient management: What makes children different? Biomark. Med. 2011, 5, 781–794. [Google Scholar] [CrossRef]
- Burckart, G.J.; Green, D.J. The personalized medicine revolution: Getting it right for children. Pediatr. Transplant. 2012, 16, 530–532. [Google Scholar] [CrossRef]
- Savage, W.J.; Everett, A.D. Biomarkers in pediatrics: Children as biomarker orphans. Proteom. Clin. Appl. 2010, 4, 915–921. [Google Scholar] [CrossRef]
- Van Driest, S.L.; McGregor, T.L. Pharmacogenetics in clinical pediatrics: Challenges and strategies. PerMed 2013, 10, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Dharnidharka, V.R.; Fiorina, P.; Harmon, W.E. Kidney transplantation in children. N. Engl. J. Med. 2014, 371, 549–558. [Google Scholar] [CrossRef]
- Leeder, J.S. Developmental and pediatric pharmacogenomics. Pharmacogenomics 2003, 4, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Lipshultz, S.E.; Chandar, J.J.; Rusconi, P.G.; Fornoni, A.; Abitbol, C.L.; Burke, G.W.; Zilleruelo, G.E.; Pham, S.M.; Perez, E.E.; Karnik, R.; et al. Issues in solid-organ transplantation in children: Translational research from bench to bedside. Clinics 2014, 69, 55–72. [Google Scholar] [CrossRef]
- Ettenger, R.B. Age and the immune response in pediatric renal transplantation. Eur. J. Nucl. Med. Mol. Imaging 1992, 151, S7–S8. [Google Scholar] [CrossRef]
- Przepiorka, D.; Blamble, D.; Hilsenbeck, S.; Danielson, M.; Krance, R.; Chan, K.W. Tacrolimus clearance is age-dependent within the pediatric population. Bone Marrow Transplant. 2000, 26, 601–605. [Google Scholar] [CrossRef]
- Montini, G.; Ujka, F.; Varagnolo, C.; Ghio, L.; Ginevri, F.; Murer, L.; Thafam, B.S.; Carasi, C.; Zacchello, G.; Plebani, M. The pharmacokinetics and immunosuppressive response of tacrolimus in paediatric renal transplant recipients. Pediatr. Nephrol. 2006, 21, 719–724. [Google Scholar] [CrossRef]
- Naesens, M.; Salvatierra, O.; Li, L.; Kambham, N.; Concepcion, W.; Sarwal, M. Maturation of Dose-Corrected Tacrolimus Predose Trough Levels in Pediatric Kidney Allograft Recipients. Transplant 2008, 85, 1139–1145. [Google Scholar] [CrossRef]
- Marfo, K.; Altshuler, J.; Lu, A. Tacrolimus Pharmacokinetic and Pharmacogenomic Differences between Adults and Pediatric Solid Organ Transplant Recipients. Pharmaceutics 2010, 2, 291–299. [Google Scholar] [CrossRef]
- Birk, P.E. Surveillance biopsies in children post-kidney transplant. Pediatr. Nephrol. 2011, 27, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Ettenger, R.B.; Blifeld, C.; Prince, H.; Gradus, D.B.-E.; Cho, S.; Sekiya, N.; Salusky, I.B.; Fine, R.N. The pediatric nephrologist’s dilemma: Growth after renal transplantation and its interaction with age as a possible immunologic variable. J. Pediatr. 1987, 111, 1022–1025. [Google Scholar] [CrossRef]
- Holcik, M.; Sonenberg, N. Translational control in stress and apoptosis. Nat. Rev. Mol. Cell Biol. 2005, 6, 318–327. [Google Scholar] [CrossRef] [PubMed]
- DuRose, J.B.; Scheuner, D.; Kaufman, R.J.; Rothblum, L.I.; Niwa, M. Phosphorylation of Eukaryotic Translation Initiation Factor 2α Coordinates rRNA Transcription and Translation Inhibition during Endoplasmic Reticulum Stress. Mol. Cell. Biol. 2009, 29, 4295–4307. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Ishii, J.; Ota, Y.; Sasaki, E.; Shibagaki, Y.; Hattori, S. Mammalian target of rapamycin (mTOR) complex 2 regulates filamin A-dependent focal adhesion dynamics and cell migration. Genes Cells 2016, 21, 579–593. [Google Scholar] [CrossRef]
- Yasuda, K.; Takahashi, M.; Mori, N. Mdm20 Modulates Actin Remodeling through the mTORC2 Pathway via Its Effect on Rictor Expression. PLoS ONE 2015, 10, e0142943. [Google Scholar] [CrossRef]
- Lamouille, S.; Connolly, E.; Smyth, J.W.; Akhurst, R.J.; Derynck, R. TGF-ß-induced activation of MTOR complex 2 drives epithelial-mesenchymal transition and cell invasion. J. Cell Sci. 2012, 125, 1259–1273. [Google Scholar] [CrossRef]
- Li, J.; Ren, J.; Liu, X.; Jiang, L.; He, W.; Yuan, W.; Yang, J.; Dai, C. RICTOR/mTORC2 signaling mediates TGFß1-induced fibroblast activation and kidney fibrosis. Kidney Int. 2015, 88, 515–527. [Google Scholar] [CrossRef]
- Betz, C.; Stracka, D.; Prescianotto-Baschong, C.; Frieden, M.; Demaurex, N.; Hall, M.N. mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc. Natl. Acad. Sci. USA 2013, 110, 12526–12534. [Google Scholar] [CrossRef] [PubMed]
- Appenzeller-Herzog, C.; Hall, M.N. Bidirectional crosstalk between endoplasmic reticulum stress and MTOR signaling. Trends Cell Biol. 2012, 22, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Arciuch, V.G.A.; Galli, S.; Franco, M.C.; Lam, P.Y.; Cadenas, E.; Carreras, M.C.; Poderoso, J.J. Akt1 intramitochondrial cycling is a crucial step in the redox modulation of cell cycle progression. PLoS ONE 2009, 4, e7523. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef]
- Los, M.J.; Maddika, S.; Erb, B.; Schulze-Osthoff, K. Switching Akt: From survival signaling to deadly response. BioEssays 2009, 31, 492–495. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, V.; Park, Y.; Chen, C.-C.; Xu, P.-Z.; Chen, M.-L.; Tonic, I.; Unterman, T.; Hay, N. Akt Determines Replicative Senescence and Oxidative or Oncogenic Premature Senescence and Sensitizes Cells to Oxidative Apoptosis. Cancer Cell 2008, 14, 458–470. [Google Scholar] [CrossRef]
- Venner, J.M.; Famulski, K.S.; Reeve, J.; Chang, J.; Halloran, P.F. Relationships among injury, fibrosis, and time in human kidney transplants. JCI Insight 2016, 1, e85323. [Google Scholar] [CrossRef]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.-A.; Han, S.H.; Chinga, F.; Park, A.S.D.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Zhao, L.; Dong, M.; Liao, S.; Du, Y.; Zhou, Q.; Zheng, H.; Chen, M.; Ji, J.; Gao, H. Identification of key metabolic changes in renal interstitial fibrosis rats using metabolomics and pharmacology. Sci. Rep. 2016, 6, 27194. [Google Scholar] [CrossRef]
- Gamboa, J.L.; Billings, F.T.; Bojanowski, M.T.; Gilliam, L.A.; Yu, C.; Roshanravan, B.; Roberts, L.J.; Himmelfarb, J.; Ikizler, T.A.; Brown, N.J. Mitochondrial dysfunction and oxidative stress in patients with chronic kidney disease. Physiol. Rep. 2016, 4, e12780. [Google Scholar] [CrossRef]
- Ishimoto, Y.; Inagi, R. Mitochondria: A therapeutic target in acute kidney injury. Nephrol. Dial. Transplant. 2016, 31, 1062–1069. [Google Scholar] [CrossRef]
- Zhan, M.; Brooks, C.; Liu, F.; Sun, L.; Dong, Z. Mitochondrial dynamics: Regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int. 2013, 83, 568–581. [Google Scholar] [CrossRef] [PubMed]
- Simon, N.; Morin, C.; Urien, S.; Tillement, J.P.; Bruguerolle, B. Tacrolimus and sirolimus decrease oxidative phosphorylation of isolated rat kidney mitochondria. Br. J. Pharmacol. 2003, 138, 369–376. [Google Scholar] [CrossRef]
- Servais, H.; Ortiz, A.; Devuyst, O.; Denamur, S.; Tulkens, P.M.; Mingeot-Leclercq, M.-P.; Arduan, A.O. Renal cell apoptosis induced by nephrotoxic drugs: Cellular and molecular mechanisms and potential approaches to modulation. Apoptosis 2007, 13, 11–32. [Google Scholar] [CrossRef] [PubMed]
- Palacín, M.; Coto, E.; Llobet, L.; Pacheu-Grau, D.; Montoya, J.; Ruiz-Pesini, E. FK506 affects mitochondrial protein synthesis and oxygen consumption in human cells. Cell Biol. Toxicol. 2013, 29, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Hortelano, S.; Castilla, M.; Torres, A.M.; Tejedor, A.; Boscá, L. Potentiation by nitric oxide of cyclosporin A and FK506-induced apoptosis in renal proximal tubule cells. J. Am. Soc. Nephrol. 2000, 11, 2315–2323. [Google Scholar] [CrossRef]
- Henke, W.; Jung, K. Comparison of the effects of the immunosuppressive agents FK 506 and cyclosporin a on rat kidney mitochondria. Biochem. Pharmacol. 1993, 46, 829–832. [Google Scholar] [CrossRef]
- Ruiz-Cabello, J.; Buss, W.C.; Collier, S.W.; I Glazer, R.; Cohen, J.S. Changes in ATP after cyclosporin A treatment in a renal epithelial cell line in the rat studied by 31P-NMR spectroscopy. Res. Commun. Mol. Pathol. Pharmacol. 1994, 86, 3–13. [Google Scholar]
- Ortiz, A.; Tejedor, A.; Caramelo, C. Nephrotoxicity. In Drug-Induced Mitochondrial Dysfunction; Dykens, J.A., Will, Y., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008; pp. 291–310. [Google Scholar]
- Forbes, J.M. Mitochondria-Power players in kidney function? Trends Endocrinol. Metab. 2016, 27, 441–442. [Google Scholar] [CrossRef]
- Higgins, G.C.; Coughlan, M.T. Mitochondrial dysfunction and mitophagy: The beginning and end to diabetic nephropathy? Br. J. Pharmacol. 2014, 171, 1917–1942. [Google Scholar] [CrossRef]
- Balaban, R.S.; Mandel, L.J. Metabolic substrate utilization by rabbit proximal tubule. An NADH fluorescence study. Am. J. Physiol. 1988, 254, F407–F416. [Google Scholar] [CrossRef]
- Rodrigues-Diez, R.; González-Guerrero, C.; Ocaña-Salceda, C.; Rodrigues-Diez, R.R.; Egido, J.; Ortiz, A.; Ruiz-Ortega, M.; Ramos, A.M. Calcineurin inhibitors cyclosporine A and tacrolimus induce vascular inflammation and endothelial activation through TLR4 signaling. Sci. Rep. 2016, 6, 27915. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Jiao, J.; Gao, G.; Prabhakar, B.S. Control of mitochondrial activity by miRNAs. J. Cell. Biochem. 2011, 113, 1104–1110. [Google Scholar] [CrossRef]
- Sripada, L.; Tomar, D.; Singh, R. Mitochondria: One of the destinations of miRNAs. Mitochondrion 2012, 12, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, M.; Neuzil, J.; Dong, L. MicroRNAs as regulators of mitochondrial function: Role in cancer suppression. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 1441–1453. [Google Scholar] [CrossRef] [PubMed]
- Duarte, F.V.; Palmeira, C.M.; Rolo, A.P. The Role of microRNAs in Mitochondria: Small Players Acting Wide. Genes 2014, 5, 865–886. [Google Scholar] [CrossRef]
- Bontha, S.V.; Maluf, D.G.; Mueller, T.F.; Mas, V.R. Systems Biology in Kidney Transplantation: The Application of Multi-Omics to a Complex Model. Arab. Archaeol. Epigr. 2016, 17, 11–21. [Google Scholar] [CrossRef]
- Liang, M.; Cowley, A.W., Jr.; Hessner, M.J.; Lazar, J.; Basile, D.P.; Pietrusz, J.L. Transcriptome analysis and kidney research: Toward systems biology. Kidney Int. 2005, 67, 2114–2122. [Google Scholar] [CrossRef]
- Li, Z.; Rana, T.M. Therapeutic targeting of microRNAs: Current status and future challenges. Nat. Rev. Drug Discov. 2014, 13, 622–638. [Google Scholar] [CrossRef]
- Mattes, J.; Yang, M.; Foster, P.S. Regulation of microRNA by antagomirs: A new class of pharmacological antagonists for the specific regulation of gene function? Am. J. Respir. Cell Mol. Biol. 2007, 36, 8–12. [Google Scholar] [CrossRef]
- Belder, N.; Coşkun, Ö.; Erdoğan, B.D.; Savaş, B.; Ensari, A.; Özdağ, H. Optimization of gene expression microarray protocol for formalin-fixed paraffin-embedded tissues. Genom. Data 2016, 7, 303–306. [Google Scholar] [CrossRef]
- Gehrau, R.C.; Mas, V.R.; Villamil, F.G.; Dumur, C.I.; Suh, J.L.; Maluf, D.G.; Mehta, N.K. MicroRNA Signature at the Time of Clinical HCV Recurrence Associates With Aggressive Fibrosis Progression Post-Liver Transplantation. Am. J. Transplant. 2013, 13, 729–737. [Google Scholar] [CrossRef]
- April, C.; Klotzle, B.; Royce, T.; Wickham-Garcia, E.; Boyaniwsky, T.; Izzo, J.; Cox, D.; Jones, W.; Rubio, R.; Holton, K.; et al. Whole-Genome Gene Expression Profiling of Formalin-Fixed, Paraffin-Embedded Tissue Samples. PLoS ONE 2009, 4, e8162. [Google Scholar] [CrossRef]
- Klopfleisch, R.; Weiss, A.T.; Gruber, A.D. Excavation of a buried treasure—DNA, mRNA, miRNA and protein analysis in formalin-fixed, paraffin-embedded tissues. Histol. Histolpathol. 2011, 26, 797–810. [Google Scholar]
- Solez, K.; Colvin, R.B.; Racusen, L.C.; Haas, M.; Sis, B.; Mengel, M.; Halloran, P.F.; Baldwin, W.; Banfi, G.; Collins, A.B.; et al. Banff 07 Classification of Renal Allograft Pathology: Updates and Future Directions. Arab. Archaeol. Epigr. 2008, 8, 753–760. [Google Scholar] [CrossRef]
- Solez, K.; Racusen, L.C. The Banff classification revisited. Kidney Int. 2013, 83, 201–216. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.; Sis, B.; Racusen, L.C.; Solez, K.; Glotz, D.; Colvin, R.B.; Castro, M.C.R.; David, D.S.R.; David-Neto, E.; Bagnasco, S.M.; et al. Banff 2013 meeting report: Inclusion of c4d-negative antibody-mediated rejection and antibody-associated arterial lesions. Am. J. Transplant. 2014, 14, 272–283. [Google Scholar] [CrossRef]
- Barash, Y.; Dehan, E.; Krupsky, M.; Franklin, W.; Geraci, M.; Friedman, N.; Kaminski, N. Comparative analysis of algorithms for signal quantitation from oligonucleotide microarrays. Bioinformatics 2004, 20, 839–846. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef]
- Carvalho, B.S.; Irizarry, R.A. A framework for oligonucleotide microarray preprocessing. Bioinformatics 2010, 26, 2363–2367. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Benjamini, Y.; Yekutieli, D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 2001, 29, 1165–1188. [Google Scholar] [CrossRef]
- Dumur, C.I.; Nasim, S.; Best, A.M.; Archer, K.J.; Ladd, A.C.; Mas, V.R.; Wilkinson, D.S.; Garrett, C.T.; Ferreira-Gonzalez, A. Evaluation of Quality-Control Criteria for Microarray Gene Expression Analysis. Clin. Chem. 2004, 50, 1994–2002. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rhone, E.T.; Bardhi, E.; Bontha, S.V.; Walker, P.D.; Almenara, J.A.; Dumur, C.I.; Cathro, H.; Maluf, D.; Mas, V. An Integrated Transcriptomic Approach to Identify Molecular Markers of Calcineurin Inhibitor Nephrotoxicity in Pediatric Kidney Transplant Recipients. Int. J. Mol. Sci. 2021, 22, 5414. https://doi.org/10.3390/ijms22115414
Rhone ET, Bardhi E, Bontha SV, Walker PD, Almenara JA, Dumur CI, Cathro H, Maluf D, Mas V. An Integrated Transcriptomic Approach to Identify Molecular Markers of Calcineurin Inhibitor Nephrotoxicity in Pediatric Kidney Transplant Recipients. International Journal of Molecular Sciences. 2021; 22(11):5414. https://doi.org/10.3390/ijms22115414
Chicago/Turabian StyleRhone, Erika T., Elissa Bardhi, Sai Vineela Bontha, Patrick D. Walker, Jorge A. Almenara, Catherine I. Dumur, Helen Cathro, Daniel Maluf, and Valeria Mas. 2021. "An Integrated Transcriptomic Approach to Identify Molecular Markers of Calcineurin Inhibitor Nephrotoxicity in Pediatric Kidney Transplant Recipients" International Journal of Molecular Sciences 22, no. 11: 5414. https://doi.org/10.3390/ijms22115414
APA StyleRhone, E. T., Bardhi, E., Bontha, S. V., Walker, P. D., Almenara, J. A., Dumur, C. I., Cathro, H., Maluf, D., & Mas, V. (2021). An Integrated Transcriptomic Approach to Identify Molecular Markers of Calcineurin Inhibitor Nephrotoxicity in Pediatric Kidney Transplant Recipients. International Journal of Molecular Sciences, 22(11), 5414. https://doi.org/10.3390/ijms22115414