
Mechanisms of TP53 Pathway Inactivation in Embryonic and Somatic Cells—Relevance for Understanding (Germ Cell) Tumorigenesis




Abstract
1. Introduction
1.1. Embryonic Stem Cells and Germ Cell Tumors Versus Somatic Cells
1.2. Protecting Genomic Integrity
1.3. P53 Pathway
1.3.1. Induction of Cell Cycle Arrest
1.3.2. Senescence or Apoptosis as a Final Measure to Protect Genomic Integrity
1.3.3. Autophagy
2. Inhibitory Mechanisms of the P53 Pathway
2.1. TP53 Modifications
2.1.1. TP53 Mutations
2.1.2. P53 Isoforms
2.1.3. P53 Post-Translational Modifications and Regulatory Elements
2.1.4. Chromosomal Number Alterations
2.2. Negative Regulators of the P53 Pathway
2.2.1. MDM2
2.2.2. Elevated Levels of MDM2
2.2.3. Isoforms of MDM2
2.2.4. Post-Translational Modifications of MDM2
2.2.5. Ribosomal Proteins Influencing MDM2
2.2.6. MDM4
2.2.7. Elevated Levels of MDM4
2.2.8. SNPs and Overexpression of Transcriptional and Translational Regulators of MDM4
2.2.9. Overexpression of MDM4-S
2.2.10. Post-Translational Modifications of MDM4
2.3. Alternative Mechanisms to Inactivate the P53 Pathway
2.3.1. P21
2.3.2. ARF
2.3.3. TRIM
2.3.4. Long Non-Coding RNAs
2.3.5. MicroRNA
2.3.6. CTCF
3. Summarizing Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Juan, H.-C.; Lin, Y.; Chen, H.-R.; Fann, M.-J. Cdk12 is essential for embryonic development and the maintenance of genomic stability. Cell Death Differ. 2016, 23, 1038–1048. [Google Scholar] [CrossRef]
- Kobayashi, T.; Surani, M.A. On the origin of the human germline. Development 2018, 145. [Google Scholar] [CrossRef] [PubMed]
- Oosterhuis, J.W.; Looijenga, L.H.J. Human germ cell tumours from a developmental perspective. Nat. Rev. Cancer 2019, 19, 522–537. [Google Scholar] [CrossRef] [PubMed]
- Dvash, T.; Ben-Yosef, D.; Eiges, R. Human Embryonic Stem Cells as a Powerful Tool for Studying Human Embryogenesis. Pediatr. Res. 2006, 60. [Google Scholar] [CrossRef]
- Vazin, T.; Freed, W.J. Human embryonic stem cells: Derivation, culture, and differentiation: A review. Restor. Neurol. Neurosci. 2010, 28, 589–603. [Google Scholar] [CrossRef]
- Wright, W.E.; Piatyszek, M.A.; Rainey, W.E.; Byrd, W.; Shay, J.W. Telomerase activity in human germline and embryonic tissues and cells. Dev. Genet. 1996, 18, 173–179. [Google Scholar] [CrossRef]
- Liu, L.; Bailey, S.M.; Okuka, M.; Muñoz, P.; Li, C.; Zhou, L.; Wu, C.; Czerwiec, E.; Sandler, L.; Seyfang, A.; et al. Telomere lengthening early in development. Nat. Cell Biol. 2007, 9. [Google Scholar] [CrossRef]
- Liu, L.; Michowski, W.; Kolodziejczyk, A.; Sicinski, P. The cell cycle in stem cell proliferation, pluripotency and differentiation. Nat. Cell Biol. 2019, 21, 1060–1067. [Google Scholar] [CrossRef]
- Bloom, J.C.; Loehr, A.R.; Schimenti, J.C.; Weiss, R.S. Germline genome protection: Implications for gamete quality and germ cell tumorigenesis. Andrology 2019, 7, 516–526. [Google Scholar] [CrossRef]
- Filion, T.M.; Qiao, M.; Ghule, P.N.; Mandeville, M.; Van Wijnen, A.J.; Stein, J.L.; Lian, J.B.; Altieri, D.C.; Stein, G.S. Survival Responses of Human Embryonic Stem Cells to DNA Damage. J. Cell. Physiol. 2009, 220, 586–592. [Google Scholar] [CrossRef]
- Magnúsdóttir, E.; Gillich, A.; Grabole, N.; Surani, M.A. Combinatorial control of cell fate and reprogramming in the mammalian germline. Curr. Opin. Genet. Dev. 2012, 22, 466–474. [Google Scholar] [CrossRef]
- Greenberg, M.V.C.; Bourchis, D. The diverse roles of DNA methylation in mammalian development and disease. Nature 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- González, F.; Boué, S.; Carlos Izpisúa Belmonte, J. Methods for making induced pluripotent stem cells: Reprogramming à la carte. Nat. Rev. Genet. 2011. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Fazal, Z.; Freemantle, S.J.; Spinella, M.J. Mechanisms of cisplatin sensitivity and resistance in testicular germ cell tumors HHS Public Access. Cancer Drug Resist. 2019, 2, 580–594. [Google Scholar] [CrossRef] [PubMed]
- Van Echten, J.; Sleijfer, D.T.; Wiersema, J.; Schraffordt Koops, H.; Oosterhuis, J.W.; De Jong, B. Cytogenetics of Primary Testicular Nonseminoma, Residual Mature Teratoma, and Growing Teratoma Lesion in Individual Patients. Cancer Genet. Cytogenet. 1997, 96, 1–6. [Google Scholar] [CrossRef]
- Van Echten, J.; Van Der Vloedt, W.S.; Sleijfer, D.T.; Oosterhuis, J.W.; De Jong, B. Comparison of the Chromosomal Pattern of Primary Testicular Nonseminomas and Residual Mature Teratomas after Chemotherapy. Cancer Genet. Cytogenet. 1997, 99, 59–67. [Google Scholar] [CrossRef]
- Jacobsen, C.; Honecker, F. Cisplatin resistance in germ cell tumours: Models and mechanisms. Andrology 2015, 3, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Bártková, J.; Meyts, E.R.-D.; Skakkebaek, N.E.; Lukas, J.; Bartek, J.; Meyts, R.-D.E. Deregulation of the G1/S-phase control in human testicular germ cell tumours. APMIS 2003, 111, 252–266. [Google Scholar] [CrossRef] [PubMed]
- Bártková, J.; Thullberg, M.; Rajpert-De Meyts, E.; Skakkebaek, N.E.; Bartek, J. Cell cycle regulators in resticular cancer: Loss of P18INK4c marks progression from carcinoma in situ to invasive germ cell tumours. Int. J. Cancer 2000, 85, 370–375. [Google Scholar] [CrossRef]
- Romano, F.J.; Rossetti, S.; Conteduca, V.; Schepisi, G.; Cavaliere, C.; Di Franco, R.; La Mantia, E.; Castaldo, L.; Nocerino, F.; Ametrano, G.; et al. Role of DNA repair machinery and p53 in the testicular germ cell cancer: A review. Oncotarget 2016, 7, 85641–85649. [Google Scholar] [CrossRef] [PubMed]
- Spierings, D.C.; Ge De Vries, E.; Stel, A.J.; Te Rietstap, N.; Vellenga, E.; De Jong, S. Low p21 Waf1/Cip1 protein level sensitizes testicular germ cell tumor cells to Fas-mediated apoptosis. Oncogene 2004, 23, 4862–4872. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Cervantes, R.B.; Tichy, E.; Tischfield, J.A.; Stambrook, P.J. Protecting genomic integrity in somatic cells and embryonic stem cells. Mutat. Res. 2007, 614, 48–55. [Google Scholar] [CrossRef]
- Datta, M.W.; Macri, E.; Signoretti, S.; Renshaw, A.A.; Loda, M. Transition from In Situ to Invasive Testicular Germ Cell Neoplasia is Associated with the Loss of p21 and Gain of mdm-2 Expression. Mod. Pathol. 2001, 14, 437–442. [Google Scholar] [CrossRef]
- Kerley-Hamilton, J.S.; Pike, A.M.; Li, N.; Direnzo, J.; Spinella, M.J. A p53-dominant transcriptional response to cisplatin in testicular germ cell tumor-derived human embyronal carcinoma. Oncogene 2005, 24, 6090–6100. [Google Scholar] [CrossRef] [PubMed]
- Spierings, D.; De Vries, E.; Vellenga, E.; De Jong, S. Loss of drug-induced activation of the CD95 apoptotic pathway in a cisplatin-resistant testicular germ cell tumor cell line. Cell Death Differ. 2003, 10, 808–822. [Google Scholar] [CrossRef] [PubMed]
- Spierings, D.C.; Ge De Vries, E.; Vellenga, E.; De Jong, S. The attractive Achilles heel of germ cell tumours: An inherent sensitivity to apoptosis-inducing stimuli. J. Pathol. 2003, 200, 137–148. [Google Scholar] [CrossRef]
- Gutekunst, M.; Oren, M.; Weilbacher, A.; Dengler, M.A.; Markwardt, C.; Thomale, J.; Aulitzky, W.E.; van der Kuip, H. P53 hypersensitivity is the predominant mechanism of the unique responsiveness of testicular germ cell tumor (TGCT) cells to Cisplatin. PLoS ONE 2011, 6, e19198. [Google Scholar] [CrossRef]
- Bártková, J.; Bártek, J.; Lukáŝ, J.; Vojtêŝek, B.; Staŝková, Z.; Rejthar, A.; Kovarík, J.; Midgley, C.A.; Lane, D.P. p53 Protein alterations in human testicular cancer including pre-invasive intratubular germ-cell neoplasia. Int. J. Cancer 1991, 49, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Burger, H.; Nooter, K.; Boersma, A.W.M.; Kortland, C.J.; Stoter, G. Lack of correlation between cisplatin-induced apoptosis, p53 status and expression of Bcl-2 family proteins in testicular germ cell tumour cell lines. Int. J. Cancer 1997, 73, 592–599. [Google Scholar] [CrossRef]
- Burger, H.; Nooter, K.; Boersma, A.W.M.; van Wingerden, K.E.; Looijenga, L.H.J.; Jochemsen, A.G.; Stoter, G. Distinct p53-independent apoptotic cell death signalling pathways in testicular germ cell tumour cell lines. Int. J. Cancer 1999, 628, 620–628. [Google Scholar] [CrossRef]
- Kersemaekers, A.M.F.; Mayer, F.; Molier, M.; Van Weeren, P.C.; Oosterhuis, J.W.; Bokemeyer, C.; Looijenga, L.H.J. Role of P53 and MDM2 in treatment response of human germ cell tumors. J. Clin. Oncol. 2002, 20, 1551–1561. [Google Scholar] [CrossRef] [PubMed]
- Ulbright, T.M.; Orazi, A.; de Riese, W.; de Riese, C.; Messemer, J.E.; Foster, R.S.; Donohue, J.P.; Eble, J.N. The correlation of P53 protein expression with proliferative activity and occult metastases in clinical stage I non-seminomatous germ cell tumors of the testis. Mod. Pathol. 1994, 7, 64–68. [Google Scholar] [PubMed]
- Riou, G.; Barrois, M.; Prost, S.; Terrier, M.J.; Theodore, C.; Levine, A.J. The p53 and mdm-2 genes in human testicular germ-cell tumors. Mol. Carcinog. 1995, 12, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Schenkman, N.S.; Sesterhenn, I.A.; Washington, L.; Weghorst, C.M.; Buzard, G.S.; Srnastava, S.; Moul, J.W.; PRIIDyn, B. Increased p53 protein does not correlate to p53 gene mutations in microdissected human testicular germ cell tumors. J. Urol. 1995, 154, 617–621. [Google Scholar] [CrossRef]
- Guillou, L.; Estreicher, A.; Chaubert, P.; Hurlimann, J.; Kurt, A.-M.; Metthez, G.; Iggo, R.; Gray, A.C.; Jichlinski, P.; Leisinger, H.-J.; et al. Germ cell tumors of the testis overexpress Wild-Type P53. Am. J. Pathol. 1996, 149, 1221–1228. [Google Scholar]
- Lutzker, S.G. P53 tumour suppressor gene and germ cell neoplasia. APMIS 1998, 106, 85–89. [Google Scholar] [CrossRef]
- Bagrodia, A.; Lee, B.H.; Lee, W.; Cha, E.K.; Sfakianos, J.P.; Iyer, G.; Pietzak, E.J.; Gao, S.P.; Zabor, E.C.; Ostrovnaya, I.; et al. Genetic determinants of cisplatin resistance in patients with advanced germ cell tumors. J. Clin. Oncol. 2016, 34, 4000–4007. [Google Scholar] [CrossRef]
- Koster, R.; Di Pietro, A.; Timmer-Bosscha, H.; Gibcus, J.H.; Van Den Berg, A.; Suurmeijer, A.J.; Bischoff, R.; Gietema, J.A.; De Jong, S. Cytoplasmic p21 expression levels determine cisplatin resistance in human testicular cancer. J. Clin. Investig. 2010, 120. [Google Scholar] [CrossRef]
- Koster, R.; van Vugt, M.A.; Timmer-Bosscha, H.; Gietema, J.A.; de Jong, S. Unravelling mechanisms of cisplatin sensitivity and resistance in testicular cancer. Expert Rev. Mol. Med. 2013, 15, 2013. [Google Scholar] [CrossRef]
- Koster, R.; Timmer-Bosscha, H.; Bischoff, R.; Gietema, J.A.; De Jong, S. Disruption of the MDM2-p53 interaction strongly potentiates p53-dependent apoptosis in cisplatin-resistant human testicular carcinoma cells via the Fas/FasL pathway. Cell Death Dis. 2011, 2. [Google Scholar] [CrossRef]
- Lobo, J.; Alzamora, M.A.; Guimarães, R.; Cantante, M.; Lopes, P.; Braga, I.; Maurício, J.; Jerónimo, C.; Henrique, R. p53 and MDM2 expression in primary and metastatic testicular germ cell tumors: Association with clinical outcome. Andrology 2020, 8, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- Ménézo, Y.; Dale, B.; Cohen, M. DNA damage and repair in human oocytes and embryos: A review. Zygote 2010, 18, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y. A New Role for p53 in Maintaining Genetic Stability in Embryonic Stem Cells. Cell Cycle 2005, 363, 363–364. [Google Scholar] [CrossRef]
- Lin, T.; Chao, C.; Saito, I.; JMazur, S.; Murphy, M.E.; Appella, E.; Xu, Y. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat. Cell Biol. 2005, 7, 165. [Google Scholar] [CrossRef]
- Maltzman, W.; Czyzyk, L. UV Irradiation Stimulates Levels of p53 Cellular Tumor Antigen in Nontransformed Mouse Cells. Mol. Cell. Biol. 1984, 4, 1689–1694. [Google Scholar] [CrossRef] [PubMed]
- Kastan, M.B.; Onyekwere, O.; Sidransky, D.; Vogelstein, B.; Craig, R.W. Participation of p53 Protein in the Cellular Response to DNA Damage1. Cancer Res. 1991, 51, 6304–6311. [Google Scholar] [PubMed]
- Baker, S.J.; Markowitz, S.; Fearon, E.R.; Willson, J.K.V.; Vogelstein, B. Suppression of Human Colorectal Carcinoma Cell Growth by Wild-Type p53. Science 1990, 249, 912–915. [Google Scholar] [CrossRef]
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 661–663. [Google Scholar] [CrossRef]
- Greenblatt, M.S.; Bennett, W.P.; Hollstein, M.; Harris, C.C. Mutations in the p53 Tumor Suppressor Gene: Clues to Cancer Etiology and Molecular Pathogenesis. Cancer Res. 1994, 54, 4855–4878. [Google Scholar]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Schvartzman, J.M.; Duijf, P.H.G.; Sotillo, R.; Coker, C.; Benezra, R. Mad2 is a critical mediator of the chromosome instability observed upon Rb and p53 pathway inhibition. Cancer Cell 2011, 19, 701–714. [Google Scholar] [CrossRef] [PubMed]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170. [Google Scholar] [CrossRef] [PubMed]
- Beauséjour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO J. 2003, 22, 4212–4222. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28. [Google Scholar] [CrossRef]
- Voorhoeve, P.M.; Le Sage, C.; Schrier, M.; Gillis, A.J.M.; Stoop, H.; Nagel, R.; Liu, Y.-P.; Van Duijse, J.; Drost, J.; Griekspoor, A.; et al. A Genetic Screen Implicates miRNA-372 and miRNA-373 As Oncogenes in Testicular Germ Cell Tumors. Cell 2006, 124, 1169–1181. [Google Scholar] [CrossRef]
- Dieckmann, K.-P.; Spiekermann, M.; Balks, T.; Flor, I.; Lö Ning, T.; Bullerdiek, J.; Belge, G. MicroRNAs miR-371-3 in serum as diagnostic tools in the management of testicular germ cell tumours. Br. J. Cancer 2012, 107. [Google Scholar] [CrossRef] [PubMed]
- Dieckmann, K.P.; Radtke, A.; Spiekermann, M.; Balks, T.; Matthies, C.; Becker, P.; Ruf, C.; Oing, C.; Oechsle, K.; Bokemeyer, C.; et al. Serum Levels of MicroRNA miR-371a-3p: A Sensitive and Specific New Biomarker for Germ Cell Tumours. Eur. Urol. 2017, 71, 213–220. [Google Scholar] [CrossRef]
- Dieckmann, K.-P.; Radtke, A.; Geczi, L.; Matthies, C.; Anheuser, P.; Eckardt, U.; Sommer, J.; Zengerling, F.; Trenti, E.; Pichler, R.; et al. Serum Levels of MicroRNA-371a-3p (M371 Test) as a New Biomarker of Testicular Germ Cell Tumors: Results of a Prospective Multicentric Study. J. Clin. Oncol. 2019, 37, 1412–1423. [Google Scholar] [CrossRef]
- Helton, E.S.; Chen, X. p53 Modulation of the DNA Damage Response. J. Cell. Biochem. 2007, 100, 883–896. [Google Scholar] [CrossRef]
- Harris, S.L.; Levine, A.J. The p53 pathway: Positive and negative feedback loops. Oncogene 2005, 24, 2899–2908. [Google Scholar] [CrossRef]
- Marine, J.-C.; Francoz, S.; Maetens, M.; Wahl, G.; Toledo, F.; Lozano, G. Keeping p53 in check: Essential and synergistic functions of Mdm2 and Mdm4. Cell Death Differ. 2006, 13, 927–934. [Google Scholar] [CrossRef]
- Harms, K.L.; Chen, X. The functional domains in p53 family proteins exhibit both common and distinct properties. Cell Death Differ. 2006, 13, 890–897. [Google Scholar] [CrossRef]
- Toledo, F.; Wahl, G.M. Regulating the p53 pathway: In vitro hypotheses, in vivo veritas. Nat. Rev. Cancer 2006, 6. [Google Scholar] [CrossRef]
- Butler, J.S.; Loh, S.N. Structure, Function, and Aggregation of the Zinc-Free Form of the p53 DNA Binding Domain. Biochemistry 2003, 42, 2396–2403. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal Structure of a p53 Tumor Suppressor-DNA Complex: Understanding Tumorigenic. Science 1994, 265, 346–355. [Google Scholar] [CrossRef]
- Stracker, T.H.; Roig, I.; Knobel, P.A.; Marjanoví, M. The ATM signaling network in development and disease. Front. Genet. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Engeland, K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 2018, 25, 114–132. [Google Scholar] [CrossRef] [PubMed]
- El-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Edward Mercer, W.; Kinzler, K.W.; Vogelstein, B. WAF1, a Potential Mediator of p53 Tumor Suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Harper, J.W.; Adami, G.R.; Wei, N.; Keyomarsi, K.; Elledge, S.J. The p21 Cdk-Interacting Protein Cip1 Is a Potent Inhibitor of G1 Cyclin-Dependent Kinases. Cell 1993, 75, 805–816. [Google Scholar] [CrossRef]
- Chen, J. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.-K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef]
- Choudhury, S.; Kolukula, V.K.; Preet, A.; Albanese, C.; Avantaggiati, M.L. Dissecting the pathways that destabilize mutant p53: The proteasome or autophagy? Cell Cycle 2013, 12, 1022–1029. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hu, W.; De Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.; Levine, A.J. The Regulation of AMPK B1, TSC2, and PTEN Expression by p53: Stress, Cell and Tissue Specificity, and the Role of These Gene Products in Modulating the IGF-1-AKT-mTOR Pathways. Cancer Res. 2007, 67, 3043–3053. [Google Scholar] [CrossRef] [PubMed]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta 2013, 1833, 3448–3459. [Google Scholar] [CrossRef] [PubMed]
- Shamas-Din, A.; Kale, J.; Leber, B.; Andrews, D.W. Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, 1–21. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Le Toumelin, G.; Criollo, A.; Rain, J.C.; Gautier, F.; Juin, P.; Tasdemir, E.; Pierron, G.; Troulinaki, K.; Tavernarakis, N.; et al. Functional and physical interaction between Bcl-XL and a BH3-like domain in Beclin-1. EMBO J. 2007, 26, 2527–2539. [Google Scholar] [CrossRef]
- Hikisz, P.; Kiliańska, Z.M. Puma, a critical mediator of cell death—One decade on from its discovery. Cell. Mol. Biol. Lett. 2012, 17, 646–669. [Google Scholar] [CrossRef] [PubMed]
- Morsi, R.Z.; Hage-Sleiman, R.; Kobeissy, H.; Dbaibo, G. Noxa: Role in Cancer Pathogenesis and Treatment. Curr. Cancer Drug Targets 2018, 18, 914–928. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Kroemer, G. Cytoplasmic functions of the tumour suppressor p53. Nature 2009, 458. [Google Scholar] [CrossRef] [PubMed]
- Riedl, S.J.; Salvesen, G.S. The apoptosome: Signalling platform of cell death. Nat. Rev. Mol. Cell Biol. 2007, 8. [Google Scholar] [CrossRef] [PubMed]
- Cargill, M.; Altshuler, D.; Ireland, J.; Sklar, P.; Ardlie, K.; Patil, N.; Lane, C.R.; Lim, E.P.; Kalyanaraman, N.; Nemesh, J.; et al. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat. Genet. 1999, 22, 231–238. [Google Scholar] [CrossRef]
- Wang, D.G.; Fan, J.-B.; Siao, C.-J.; Berno, A.; Young, P.; Sapolsky, R.; Ghandour, G.; Perkins, N.; Winchester, E.; Spencer, J.; et al. Large-Scale Identification, Mapping, and Genotyping of Single-Nucleotide Polymorphisms in the. Science 1998, 280, 1077–1082. [Google Scholar] [CrossRef] [PubMed]
- Bouaoun, L.; Sonkin, D.; Ardin, M.; Hollstein, M.; Byrnes, G.; Zavadil, J.; Olivier, M. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum. Mutat. 2016, 37, 865–876. [Google Scholar] [CrossRef]
- Pavletich, N.P.; Chambers, K.A.; Pabo, C.O. The DNA-binding domain of 53 contains the four conserved regions the major mutation hot spots. Genes Dev. 1993, 7, 2556–2564. [Google Scholar] [CrossRef]
- Parkin, D.M.; Pisani, P.; Ferlay, J. Estimates of the worldwide incidence of 25 major cancers in 1990. Int. J. Cancer 1999, 80, 827–841. [Google Scholar] [CrossRef]
- Hieken, T.J.; Farolan, M.; Velasco, J.M.; D’Alessandro, S. Predicting the biologic behavior of ductal carcinoma in situ: An analysis of molecular markers. Surgery 2001, 130, 593–601. [Google Scholar] [CrossRef]
- Offutt, T.L.; Ieong, P.U.; Demir, Ö.; Amaro, R.E. Dynamics and Molecular Mechanisms of p53 Transcriptional Activation. Biochemistry 2018, 57, 6537. [Google Scholar] [CrossRef] [PubMed]
- Bullock, A.N.; Henckel, J.; Fersht, A.R. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: Definition of mutant states for rescue in cancer therapy. Oncogene 2000, 19, 1245–1256. [Google Scholar] [CrossRef] [PubMed]
- Rowan, S.; Ludwig, R.L.; Haupt, Y.; Bates, S.; Lu, X.; Oren, M.; Vousden, K.H. Specific loss of apoptotic but not cell-cycle arrest function in a human tumor derived p53 mutant. EMBO J. 1996, 15, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, R.A.; Chan, C.S.; Abstract, G. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2018, 25, 154–160. [Google Scholar] [CrossRef]
- Nigro, J.M.; Baker, S.J.; Preisinger, A.C.; Jessup, J.M.; Hostetter, R.; Cleary, K.; Bigner, S.H.; Davidson, N.; Baylin, S.; Devilee, P.; et al. Mutations in the p53 gene occur in diverse human tumour types. Nature 1989, 342, 705–708. [Google Scholar] [CrossRef]
- Xu, J.; Reumers, J.; Couceiro, J.R.; De Smet, F.; Gallardo, R.; Rudyak, S.; Cornelis, A.; Rozenski, J.; Zwolinska, A.; Marine, J.C.; et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat. Chem. Biol. 2011, 7, 285–295. [Google Scholar] [CrossRef]
- Dittmer, D.; Pati, S.; Zambetti, G.; Chu, S.; Teresky, A.K.; Moore, M.; Finlay, C.; Levine, A.J. Gain of function mutations in p53. Nat. Genet. 1993, 4, 42–46. [Google Scholar] [CrossRef]
- Gualberto, A.; Aldape, K.; Kozakiewicz, K.; Tlsty, T.D. An oncogenic form of p53 confers a dominant, gain-of-function phenotype that disrupts spindle checkpoint control. Proc. Natl. Acad. Sci. USA 1998, 95, 5166–5171. [Google Scholar] [CrossRef]
- Hsiao, M.; Low, J.; Dorn, E.; Ku, D.; Pattengale, P.; Yeargin, J.; Haas, M. Gain-of-function mutations of the p53 gene induce lymphohematopoietic metastatic potential and tissue invasiveness. Am. J. Pathol. 1994, 145, 702–714. [Google Scholar]
- Shaulian, E.; Zauberman, A.; Ginsberg, D.; Oren, M. Identification of a Minimal Transforming Domain of p53: Negative Dominance through Abrogation of Sequence-Specific DNA Binding. Mol. Cell. Biol. 1992, 12, 5581–5592. [Google Scholar] [CrossRef]
- Lee, J.-K.; Wang, J.; Sa, J.K.; Ladewig, E.; Lee, H.-O.; Lee, I.-H.; Ju Kang, H.; Rosenbloom, D.S.; Camara, P.G.; Liu, Z.; et al. Spatiotemporal genomic architecture informs precision oncology in glioblastoma. Nat. Genet. 2017, 49, 594–599. [Google Scholar] [CrossRef]
- Alexandrova, E.M.; Mirza, S.A.; Xu, S.; Schulz-Heddergott, R.; Marchenko, N.D.; Moll, U.M. p53 loss-of-heterozygosity is a necessary prerequisite for mutant p53 stabilization and gain-of-function in vivo. Cell Death Dis. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Xu, D.; Zhang, T.; Hu, W.; Feng, Z. Gain-of-function mutant p53 in cancer progression and therapy. J. Mol. Cell Biol. 2020, 12, 674–687. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Feng, Z. Tumor suppressor p53 and its gain-of-function mutants in cancer. Acta Biochim. Biophys. Sin. 2014, 46, 170. [Google Scholar] [CrossRef]
- Beyer, U.; Moll-Rocek, J.; Moll, U.M.; Dobbelstein, M. Endogenous retrovirus drives hitherto unknown proapoptotic p63 isoforms in the male germ line of humans and great apes. Proc. Natl. Acad. Sci. USA 2011, 108, 3624–3629. [Google Scholar] [CrossRef] [PubMed]
- Grande, L.; Bretones, G.; Rosa-Garrido, M.; Garrido-Martin, E.M.; Hernandez, T.; Fraile, S.; Botella, L.; De Alava, E.; Vidal, A.; Garcia Del Muro, X.; et al. Transcription Factors Sp1 and p73 Control the Expression of the Proapoptotic Protein NOXA in the Response of Testicular Embryonal Carcinoma Cells to Cisplatin. J. Biol. Chem. 2012, 287, 26495–26505. [Google Scholar] [CrossRef] [PubMed]
- Lobo, J.; Jerónimo, C.; Henrique, R. Cisplatin Resistance in Testicular Germ Cell Tumors: Current Challenges from Various Perspectives. Cancers 2020, 12, 1601. [Google Scholar] [CrossRef] [PubMed]
- Jost, C.A.; Marin, M.C.; Kaelin, W.G. p73 is a human p53-related protein that can induce apoptosis. Nature 1997, 389, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Kaghad, M.; Bonnet, H.; Yang, A.; Creancier, L.; Biscan, J.C.; Valent, A.; Minty, A.; Chalon, P.; Lelias, J.M.; Dumont, X.; et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell 1997, 90, 809–819. [Google Scholar] [CrossRef]
- Schmale, H.; Bamberger, C. A novel protein with strong homology to the tumor suppressor p53. Oncogene 1997, 15, 1363–1367. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Kaghad, M.; Wang, Y.; Gillett, E.; Fleming, M.D.; Dötsch, V.; Andrews, N.C.; Caput, D.; McKeon, F. p63, a p53 Homolog at 3q27-29, Encodes Multiple Products with Transactivating, Death-Inducing, and Dominant-Negative Activities. Mol. Cell 1998, 2, 305–316. [Google Scholar] [CrossRef]
- Dötsch, V.; Bernassola, F.; Coutandin, D.; Candi, E.; Melino, G. p63 and p73, the Ancestors of p53. Cold Spring Harb. Perspect. Biol. 2010, 2. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Zaika, E.; Zaika, A. P53 family: Role of protein isoforms in human cancer. J. Nucleic Acids 2012, 2012, 687359. [Google Scholar] [CrossRef] [PubMed]
- Pflaum, J.; Schlosser, S.; Müller, M. P53 family and cellular stress responses in cancer. Front. Oncol. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- DeYoung, M.P.; Ellisen, L.W. p63 and p73 in human cancer: Defining the network. Oncogene 2007, 26. [Google Scholar] [CrossRef]
- Yang, A.; Kaghad, M.; Caput, D.; Mckeon, F. On the shoulders of giants: p63, p73 and the rise of p53. TRENDS Genet. 2002, 18, 90–95. [Google Scholar] [CrossRef]
- Rutkowski, R.; Hofmann, K.; Gartner, A. Phylogeny and Function of the Invertebrate p53 Superfamily. Cold Spring Harb. Perspect. Biol. 2010, 2. [Google Scholar] [CrossRef]
- Duijf, P.H.G.; Vanmolkot, K.R.J.; Propping, P.; Friedl, W.; Krieger, E.; Mckeon, F.; Dötsch, V.; Brunner, H.G.; Van Bokhoven, H. Gain-of-function mutation in ADULT syndrome reveals the presence of a second transactivation domain in p63. Hum. Mol. Genet. 2002, 11, 799–804. [Google Scholar] [CrossRef]
- Ghioni, P.; Bolognese, F.; Duijf, P.H.G.; Van Bokhoven, H.; Mantovani, R.; Guerrini, L. Complex Transcriptional Effects of p63 Isoforms: Identification of Novel Activation and Repression Domains. Mol. Cell. Biol. 2002, 22, 8659–8668. [Google Scholar] [CrossRef]
- Helton, E.S.; Zhu, J.; Chen, X. The Unique NH 2-terminally Deleted (N) Residues, the PXXP Motif, and the PPXY Motif Are Required for the Transcriptional Activity of the N Variant of p63. J. Biol. Chem. 2006, 281. [Google Scholar] [CrossRef] [PubMed]
- Pietsch, E.C.; Sykes, S.M.; Mcmahon, S.B.; Murphy, M.E. The p53 family and programmed cell death. Oncogene 2008, 27, 6507–6521. [Google Scholar] [CrossRef] [PubMed]
- Flores, E.R.; Tsai, K.Y.; Crowley, D.; Sengupta, S.; Yang, A.; McKeon, F.; Jacks, T. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature 2002, 416. [Google Scholar] [CrossRef] [PubMed]
- Harms, K.; Nozell, S.; Chen, X. The common and distinct target genes of the p53 family transcription factors. Cell. Mol. Life Sci. 2004, 61, 822–842. [Google Scholar] [CrossRef] [PubMed]
- Melino, G.; Bernassola, F.; Ranalli, M.; Yee, K.; Xing Zong, W.; Corazzari, M.; Knight, R.A.; Green, D.R.; Thompson, C.; Vousden, K.H. p73 Induces Apoptosis via PUMA Transactivation and Bax Mitochondrial Translocation. J. Biol. Chem. 2004, 279. [Google Scholar] [CrossRef]
- Suh, E.-K.; Yang, A.; Kettenbach, A.; Bamberger, C.; Michaelis, A.H.; Zhu, Z.; Elvin, J.A.; Bronson, R.T.; Crum, C.P.; Mckeon, F. p63 protects the female germ line during meiotic arrest. Nature 2006, 444. [Google Scholar] [CrossRef]
- Amelio, I.; Grespi, F.; Annicchiarico-Petruzzelli, M.; Melino, G. P63 the Guardian of Human Reproduction. Cell Cycle 2012, 11, 4545–4551. [Google Scholar] [CrossRef]
- Gebel, J.; Tuppi, M.; Sänger, N.; Schumacher, B.; Dötsch, V. DNA Damaged Induced Cell Death in Oocytes. Molecules 2020, 25, 5714. [Google Scholar] [CrossRef]
- Talos, F.; Abraham, A.; Vaseva, A.; Holembowski, L.; Tsirka, S.; Scheel, A.; Bode, D.; Dobbelstein, M.; Brü ck, W.; Moll, U. p73 is an essential regulator of neural stem cell maintenance in embryonal and adult CNS neurogenesis. Cell Death Differ. 2010, 17, 1816–1829. [Google Scholar] [CrossRef]
- Vanbokhoven, H.; Melino, G.; Candi, E.; Declercq, W. P63, a story of mice and men. J. Investig. Dermatol. 2011, 131, 1196–1207. [Google Scholar] [CrossRef]
- Soares, E.; Zhou, H. Master regulatory role of p63 in epidermal development and disease. Cell. Mol. Life Sci. 2018, 75, 1179–1190. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Schweitzer, R.; Sun, D.; Kaghad, M.; Walker, N.; Bronsonk, R.T.; Tabin, C.; Sharpe, A.; Caput, D.; Crum, C.; et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 1999, 398, 714–718. [Google Scholar] [CrossRef] [PubMed]
- Mills, A.A.; Zheng, B.; Wang, X.-J.; Vogel, H.; Roop, D.R.; Bradley, A. P63 Is a P53 Homologue Required for Limb and Epidermal Morphogenesis. Nature 1999, 398, 708–713. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Walker, N.; Bronson, R.; Kaghad, M.; Oosterwegel, M.; Bonnin, J.; Vagner, C.; Bonnet, H.; Dikkesk, P.; Sharpe, A. P73-Deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Development 2000, 117, 1321–1331. [Google Scholar] [CrossRef]
- Ramos, H.; Raimundo, L.; Saraiva, L. p73: From the p53 shadow to a major pharmacological target in anticancer therapy. Pharmacol. Res. 2020, 162, 1043–6618. [Google Scholar] [CrossRef]
- Melino, G.; Lu, X.; Gasco, M.; Crook, T.; Knight, R.A. Functional regulation of p73 and p63: Development and cancer. Trends Biochem. Sci. 2003, 28, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Lamb, P.; Crawford, L. Characterization of the Human p53 Gene. Mol. Cell. Biol. 1986, 6, 1379–1385. [Google Scholar] [CrossRef]
- Marcel, V.; Perrier, S.; Aoubala, M.; Ageorges, S.; Groves, M.J.; Diot, A.; Fernandes, K.; Tauro, S.; Bourdon, J.-C. Δ160p53 is a novel N-terminal p53 isoform encoded by Δ133p53 transcript. FEBS Lett. 2010, 584, 4463–4468. [Google Scholar] [CrossRef]
- Bourdon, J.-C.; Fernandes, K.; Murray-Zmijewski, F.; Liu, G.; Diot, A.; Xirodimas, D.P.; Saville, M.K.; Lane, D.P. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005, 19, 2122–2137. [Google Scholar] [CrossRef]
- Billant, O.; Léon, A.; Le Guellec, S.; Friocourt, G.; Blondel, M.; Voisset, C. The dominant-negative interplay between p53, p63 and p73: A family affair. Oncotarget 2016, 7, 69549–69564. [Google Scholar] [CrossRef]
- Knezovi Florijan, M.; Ozreti, P.; Bujak, M.; Pezz, L.; Ciribilli, Y.; Ka stelan, Z.; Slade, N.; Hudolin, T. The role of p53 isoforms’ expression and p53 mutation status in renal cell cancer prognosis. Urol. Oncol. 2019, 37. [Google Scholar] [CrossRef] [PubMed]
- Hafsi, H.; Santos-Silva, D.; Courtois-Cox, S.; Hainaut, P. Effects of Δ40p53, an isoform of p53 lacking the N-terminus, on transactivation capacity of the tumor suppressor protein p53. BMC Cancer 2013, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Bernard, H.; Garmy-Susini, B.; Ainaoui, N.; Van Den Berghe, L.; Peurichard, A.; Javerzat, S.; Bikfalvi, A.; Lane, D.P.; Bourdon, J.C.; Prats, A.-C. The p53 isoform, Δ133p53a, stimulates angiogenesis and tumour progression. Oncogene 2013, 32, 2150–2160. [Google Scholar] [CrossRef] [PubMed]
- Hofstetter, G.; Berger, A.; Schuster, E.; Wolf, A.; Hager, G.; Vergote, I.; Cadron, I.; Sehouli, J.; Braicu, E.I.; Mahner, S.; et al. Δ133p53 is an independent prognostic marker in p53 mutant advanced serous ovarian cancer. Br. J. Cancer 2011, 105, 1593–1599. [Google Scholar] [CrossRef]
- Candeias, M.M.; Hagiwara, M.; Matsuda, M. Cancer-specific mutations in P53 induce the translation of Δ160P53 promoting tumorigenesis. EMBO Rep. 2016, 17, 1542–1551. [Google Scholar] [CrossRef]
- Zhang, Y.; Ma, W.-Y.; Kaji, A.; Bode, A.M.; Dong, Z. Requirement of ATM in UVA-induced Signaling and Apoptosis. J. Biol. Chem. 2001, 277, 3124–3131. [Google Scholar] [CrossRef]
- Midgley, C.A.; Owens, B.; Briscoe, C.V.; Thomas, D.B.; Lane, D.P.; Hall, P.A. Coupling between gamma irradiation, p53 induction and the apoptotic response depends upon cell type in vivo. J. Cell Sci. 1995, 108, 1843–1848. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, T.; Kato, S.; Otsuka, K.; Watanabe, G.; Kumabe, T.; Tominaga, T.; Yoshimoto, T.; Ishioka, C. The relationship among p53 oligomer formation, structure and transcriptional activity using a comprehensive missense mutation library. Oncogene 2005, 24, 6976–6981. [Google Scholar] [CrossRef]
- Liao, P.; Zeng, S.X.; Zhou, X.; Chen, T.; Zhou, F.; Cao, B.; Jung, J.H.; Del Sal, G.; Luo, S.; Lu, H. Mutant p53 Gains Its Function via c-Myc Activation upon CDK4 Phosphorylation at Serine 249 and Consequent PIN1 Binding. Mol. Cell 2017, 68, 1134–1146. [Google Scholar] [CrossRef]
- Nakamura, S.; Roth, J.A.; Mukhopadhyay, T. Multiple Lysine Mutations in the C-Terminal Domain of p53 Interfere with MDM2-Dependent Protein Degradation and Ubiquitination. Mol. Cell. Biol. 2000, 20, 9391–9398. [Google Scholar] [CrossRef]
- Liu, Y.; Tavana, O.; Gu, W. p53 modifications: Exquisite decorations of the powerful guardian. J. Mol. Cell Biol. 2019, 11, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Kachirskaia, I.; Yamaguchi, H.; West, L.E.; Wen, H.; Wang, E.W.; Dutta, S.; Appella, E.; Gozani, O. Modulation of p53 function by SET8-mediated methylation at lysine 382. Mol. Cell 2007, 27, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Chuikov, S.; Kurash, J.K.; Wilson, J.R.; Xiao, B.; Justin, N.; Ivanov, G.S.; Mckinney, K.; Tempst, P.; Prives, C.; Gamblin, S.J.; et al. Regulation of p53 activity through lysine methylation. Nature 2004, 432, 353–360. [Google Scholar] [CrossRef]
- Huang, J.; Perez-Burgos, L.; Placek, B.J.; Sengupta, R.; Richter, M.; Dorsey, J.A.; Kubicek, S.; Opravil, S.; Jenuwein, T.; Berger, S.L. Repression of p53 activity by Smyd2-mediated methylation. Nature 2006, 444. [Google Scholar] [CrossRef]
- Jansson, M.; Durant, S.T.; Cho, E.-C.; Sheahan, S.; Edelmann, M.; Kessler, B.; Thangue, N.B. La Arginine methylation regulates the p53 response. Nat. Cell Biol. 2008, 10. [Google Scholar] [CrossRef] [PubMed]
- Minamoto, T.; Buschmann, T.; Habelhah, H.; Matusevich, E.; Tahara, H.; Boerresen-Dale, A.-L.; Harris, C.; Sidransky, D.; Ronai, Z. Distinct pattern of p53 phosphorylation in human tumors. Oncogene 2001, 20, 3341–3347. [Google Scholar] [CrossRef] [PubMed]
- Gatti, A.; Li, H.-H.; Traugh, J.A.; Liu, X. Phosphorylation of Human p53 on Thr-55. Biochemistry 2000, 39, 9837–9842. [Google Scholar] [CrossRef]
- Waterman, M.J.F.; Stavridi, E.S.; Waterman, J.L.F.; Halazonetis, T.D. ATM-dependent activation of p53 involves dephosphorylation and association with 14-3-3 proteins. Nat. Genet. 1998, 19, 175–178. [Google Scholar] [CrossRef]
- Shieh, S.-Y.; Ahn, J.; Tamai, K.; Taya, Y.; Prives, C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev. 2000, 14, 289–300. [Google Scholar]
- Canman, C.E.; Lim, D.-S.; Cimprich, K.A.; Taya, Y.; Tamai, K.; Sakaguchi, K.; Appella, E.; Kastan, M.B.; Siliciano, J.D. Activation of the ATM Kinase by Ionizing Radiation and Phosphorylation of p53. Science 1998, 281, 1677–1679. [Google Scholar] [CrossRef] [PubMed]
- Tibbetts, R.S.; Brumbaugh, K.M.; Williams, J.M.; Sarkaria, J.N.; Cliby, W.A.; Shieh, S.-Y.; Taya, Y.; Prives, C.; Abraham, R.T. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 1999, 13, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Heller, H.; Elias, S.; Ciechanover, A. Components of Ubiquitin-Protein Ligase System R. J. Biol. Chem. 1983, 258, 8206–8214. [Google Scholar] [CrossRef]
- Rodriguez, M.S.; Desterro, J.M.P.; Lain, S.; Lane, D.P.; Hay, R.T. Multiple C-Terminal Lysine Residues Target p53 for Ubiquitin-Proteasome-Mediated Degradation. Mol. Cell. Biol. 2000, 20, 8458–8467. [Google Scholar] [CrossRef]
- Tasdemir, E.; Chiara Maiuri, M.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Stommel, J.M.; Marchenko, N.D.; Jimenez, G.S.; Moll, U.M.; Hope, T.J.; Wahl, G.M. A leucine-rich nuclear export signal in the p53 tetramerization domain: Regulation of subcellular localization and p53 activity by NES masking. EMBO J. 1999, 18, 1660–1672. [Google Scholar] [CrossRef]
- Momand, J.; Zambetti, G.P.; Olson, D.C.; George, D.; Levine, A.J. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992, 69, 1237–1245. [Google Scholar] [CrossRef]
- Oliner, J.D.; Pietenpol, J.A.; Thiagalingam, S.; Gyuris, J.; Kinzler, K.W.; Vogelstein, B. Oncoprotein Mdm2 conceals the activation domain of tumour suppressor p53. Nature 1993, 362, 857–860. [Google Scholar] [CrossRef]
- Leach, F.S.; Tokino, T.; Meltzer, P.; Burrell, M.; Oliner, J.D.; Smith, S.; Hill, D.E.; Sidransky, D.; Kinzler, K.W.; Vogelstein, B. p53 Mutation and MDM2 Amplification in Human Soft Tissue Sarcomas. Cancer Res. 1993, 53, 2231–2234. [Google Scholar]
- Jones, S.N.; Hancock, A.R.; Vogel, H.; Donehower, L.A.; Bradley, A. Overexpression of Mdm2 in mice reveals a p53-independent role for Mdm2 in tumorigenesis. Proc. Natl. Acad. Sci. USA 1998, 95, 15608–15612. [Google Scholar] [CrossRef]
- Momand, J.; Jung, D.; Wilczynski, S.; Niland, J. The MDM2 gene amplification database. Nucleic Acids Res. 1998, 26, 3453–3459. [Google Scholar] [CrossRef]
- Gostissa, M.; Hengstermann, A.; Fogal, V.; Sandy, P.; Schwarz, S.E.; Scheffner, M.; Sal, G. Del Activation of p53 by conjugation to the ubiquitin-like protein SUMO-1. EMBO J. 1999, 18, 6462–6471. [Google Scholar] [CrossRef]
- Bischof, O.; Schwamborn, K.; Martin, N.; Werner, A.; Sustmann, C.; Grosschedl, R.; Dejean, A. The E3 SUMO Ligase PIASy Is a Regulator of Cellular Senescence and Apoptosis. Mol. Cell 2006, 22, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Roeder, R.G. Activation of p53 Sequence-Specific DNA Binding by Acetylation of the p53 C-Terminal Domain. Cell 1997, 90, 595–606. [Google Scholar] [CrossRef]
- Tang, Y.; Zhao, W.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation Is Indispensable for p53 Activation. Cell 2008, 16, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Luo, J.; Brooks, C.L.; Gu, W. Acetylation of p53 Inhibits Its Ubiquitination by Mdm2. J. Biol. Chem. 2002, 277, 50607–50611. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Su, F.; Chen, D.; Shiloh, A.; Gu, W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature 2000, 408, 377–381. [Google Scholar] [CrossRef]
- Zhao, W.; Kruse, J.-P.; Tang, Y.; Jung, S.Y.; Qin, J.; Gu, W. Negative regulation of the deacetylase SIRT1 by DBC1. Nature 2008, 451, 587–590. [Google Scholar] [CrossRef]
- Huffman, D.M.; Grizzle, W.E.; Bamman, M.M.; Kim, J.-S.; Eltoum, I.A.; Elgavish, A.; Nagy, T.R. SIRT1 Is Significantly Elevated in Mouse and Human Prostate Cancer. Cancer Res. 2007, 67. [Google Scholar] [CrossRef]
- Parikh, N.; Hilsenbeck, S.; Creighton, C.J.; Dayaram, T.; Shuck, R.; Shinbrot, E.; Xi, L.; Gibbs, R.A.; Wheeler, D.A.; Donehower, L.A. Effects of TP53 Mutational Status on Gene Expression Patterns Across Ten Human Cancer Types. J. Pathol. 2014, 232, 522–533. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, C.; Xu, Z.; Scuoppo, C.; Rillahan, C.D.; Gao, J.; Spitzer, B.; Bosbach, B.; Kastenhuber, E.R.; Baslan, T.; et al. Deletions linked to TP53 loss drive cancer through p53-independent mechanisms. Nature 2016, 531, 471–475. [Google Scholar] [CrossRef]
- Petitjean, A.; Achatz, M.; Borresen-Dale, A.L.; Hainaut, P.; Olivier, M. TP53 mutations in human cancers: Functional selection and impact on cancer prognosis and outcomes. Oncogene 2007, 26, 2157–2165. [Google Scholar] [CrossRef]
- Nakayama, M.; Hong, C.P.; Oshima, H.; Sakai, E.; Kim, S.J.; Oshima, M. Loss of wild-type p53 promotes mutant p53-driven metastasis through acquisition of survival and tumor-initiating properties. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.N.; Roe, A.E.; Donehower, L.A.; Bradley, A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 1995, 378, 206–208. [Google Scholar] [CrossRef] [PubMed]
- Kuerbitz, S.J.; Plunkett, B.S.; Walsh, W.V.; Kastan, M.B. Wild-type p53 is a cell cycle checkpoint determinant following irradiation. Cell Biol. 1992, 89, 7491–7495. [Google Scholar] [CrossRef]
- Montes de Oca Luna, R.; Wagner, D.S.; Lozanot, G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 1995, 378, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Moyer, S.M.; Larsson, C.A.; Lozano, G. Mdm proteins: Critical regulators of embryogenesis and homoeostasis. J. Mol. Cell Biol. 2017, 9, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Shvarts, A.; TSteegenga, W.; Riteco, N.; van Laar, T.; Dekker, P.; Bazuine, M.; CAvan Ham, R.; van der Houven van Oordt, W.; Hateboer, G.; Jvan der Eb, A.; et al. MDMX: A novel p53-binding protein with some functional properties of MDM2. EMBO J. 1996, 15, 5349–5357. [Google Scholar] [CrossRef]
- Oliner, J.D.; Kinzler, K.W.; Meltzer, P.S.; George, D.L.; Vogelstein, B. Amplification of a gene encoding a pS3-associated protein in human sarcomas. Nature 1992, 358, 80–83. [Google Scholar] [CrossRef]
- Shadfan, M.; Lopez-Pajares, V.; Yuan, Z.-M. MDM2 and MDMX: Alone and together in regulation of p53. Transl. Cancer Res. 2012, 1, 88–89. [Google Scholar]
- Roth, J.; Dobbelstein, M.; Freedman, D.A.; Shenk, T.; Levine, A.J. Nucleo-cytoplasmic shuttling of the hdm2 oncoprotein regulates the levels of the p53 protein via a pathway used by the human immunodeficiency virus rev protein. EMBO J. 1998, 17, 554–564. [Google Scholar] [CrossRef]
- Fang, S.; Jensen, J.P.; Ludwig, R.L.; Vousden, K.H.; Weissman, A.M. Mdm2 Is a RING Finger-dependent Ubiquitin Protein Ligase for Itself and p53. J. Biochem. Chem. 2000, 275, 8945–8951. [Google Scholar] [CrossRef]
- Li, M.; Brooks, C.L.; Wu-Baer, F.; Chen, D.; Baer, R.; Gu, W. Mono-versus Polyubiquitination: Differential Control of p53 Fate. Science 2003, 302, 1972–1975. [Google Scholar] [CrossRef] [PubMed]
- Geyer, R.K.; Yu, Z.K.; Maki, C.G. The MDM2 RING-finger domain is required to promote p53 nuclear export. Nat. Cell Biol. 2000, 2, 569–573. [Google Scholar] [CrossRef]
- Marine, J.-C.; Lozano, G. Mdm2-mediated ubiquitylation: p53 and beyond. Cell Death Differ. 2010, 17, 93–102. [Google Scholar] [CrossRef]
- Haupt, Y.; Mayat, R.; Kazazt, A.; Orent, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef]
- Honda, R.; Tanaka, H.; Yasuda, H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. Fed. Eur. Biochem. Soc. 1997, 420, 25–27. [Google Scholar] [CrossRef]
- Kulikov, R.; Letienne, J.; Kaur, M.; Grossman, S.R.; Arts, J.; Blattner, C. Mdm2 facilitates the association of p53 with the proteasome. Proc. Natl. Acad. Sci. USA 2010, 107, 10038–10043. [Google Scholar] [CrossRef] [PubMed]
- Zauberman, A.; Barak, Y.; Ragimov, N.; Levy, N.; Oren, M. Sequence-specific DNA binding by p53: Identification of target sites and lack of binding to p53 MDM2 complexes. EMBO J. 1993, 12, 2799–2808. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-Y.; Zong, C.S.; Xia, W.; Wei, Y.; Ali-Seyed, M.; Li, Z.; Broglio, K.; Berry, D.A.; Hung, M.-C. MDM2 Promotes Cell Motility and Invasiveness by Regulating E-Cadherin Degradation. Mol. Cell. Biol. 2006, 26, 7269–7282. [Google Scholar] [CrossRef]
- Oliner, J.D.; Saiki, A.Y.; Caenepeel, S. The role of MDM2 amplification and overexpression in tumorigenesis. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo Activation of the p53 Pathway by Small-Molecule Antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Tovar, C.; Rosinski, J.; Filipovic, Z.; Higgins, B.; Kolinsky, K.; Hilton, H.; Zhao, X.; Vu, B.T.; Qing, W.; Packman, K.; et al. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: Implications for therapy. Proc. Natl. Acad. Sci. USA 2006, 103, 1888–1893. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Mühlenberg, T.; Leahy, M.; Hoiczyk, M.; Gauler, T.; Schuler, M.; Looijenga, L. Therapeutic Potential of Mdm2 Inhibition in Malignant Germ Cell Tumours. Eur. Urol. 2010, 57, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, D.-D.; Wu, Y.-P.; Su, D.; Zhou, T.-Y.; Gai, R.-H.; Fu, Y.-Y.; Zheng, L.; He, Q.-J.; Zhu, H.; et al. MDM2 promotes epithelial-mesenchymal transition and metastasis of ovarian cancre SKOV3 cells. Br. J. Cancer 2017, 117, 1192–1201. [Google Scholar] [CrossRef]
- Sdek, P.; Ying, H.; Chang, D.L.F.; Qiu, W.; Zheng, H.; Touitou, R.; Allday, M.J.; Xiao, Z.-X.J. MDM2 Promotes Proteasome-Dependent Ubiquitin-Independent Degradation of Retinoblastoma Protein. Mol. Cell 2005, 20, 699–708. [Google Scholar] [CrossRef]
- Bond, G.L.; Hu, W.; Bond, E.E.; Robins, H.; Lutzker, S.G.; Arva, N.C.; Bargonetti, J.; Bartel, F.; Taubert, H.; Wuerl, P.; et al. A Single Nucleotide Polymorphism in the MDM2 Promoter Attenuates the p53 Tumor Suppressor Pathway and Accelerates Tumor Formation in Humans. Cell 2004, 119, 591–602. [Google Scholar] [CrossRef]
- Bond, G.L.; Menin, C.; Bertorelle, R.; Alhopuro, P.; Aaltonen, L.A.; Levine, A.J. MDM2 SNP309 accelerates colorectal tumour formation in women. J. Med. Genet. 2006, 43, 950–952. [Google Scholar] [CrossRef]
- Sun, T.; Lee, G.-S.M.; Oh, W.K.; Pomerantz, M.; Yang, M.; Xie, W.; Freedman, M.L.; Kantoff, P.W. Single-nucleotide Polymorphisms in p53 Pathway and Aggressiveness of Prostate Cancer in a Caucasian Population. Clin. Cancer Res. 2010, 16, 5244–5251. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Du, M.; Gu, D.; Zhu, L.; Chu, H.; Tong, N.; Zhang, Z.; Xu, Z.; Wang, M. MDM2 SNP309 polymorphism is associated with colorectal cancer risk. Sci. Rep. 2014. [Google Scholar] [CrossRef]
- Knappskog, S.; Bjørnslett, M.; Myklebust, L.M.; Huijts, P.E.A.; Vreeswijk, M.P.; Edvardsen, H.; Guo, Y.; Zhang, X.; Yang, M.; Ylisaukko-Oja, S.K.; et al. Cancer Cell the MDM2 Promoter SNP285C/309G Haplotype Diminishes Sp1 Transcription Factor Binding and Reduces Risk for Breast and Ovarian Cancer in Caucasians. Cancer Cell 2011, 19, 273–282. [Google Scholar] [CrossRef]
- Knappskog, S.; Lønning, P.E. MDM2 promoter SNP285 and SNP309; phylogeny and impact on cancer risk. Oncotarget 2011, 2, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Bartel, F.; Taubert, H.; Harris, L.C. Alternative and aberrant splicing of MDM2 mRNA in human cancer. Cancer Cell 2002, 2, 9–15. [Google Scholar] [CrossRef]
- Sigalas, I.; Calvert, A.H.; Akdfrso, J.J.; Neal, D.E.; Lunec, J. Alternatively spliced mdm2 transcripts with loss of p53 binding domain sequences: Transforming ability and frequent detection in human cancer. Nat. Med. 1996, 2, 912–917. [Google Scholar] [CrossRef]
- Zheng, T.; Wang, J.; Zhao, Y.; Zhang, C.; Lin, M.; Wang, H.Y.; Liu, L.; Feng, Z.; Hu, W. Spliced MDM2 isoforms promote mutant p53 accumulation and gain-0f-function in tumorigenesis. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef]
- Evans, S.C.; Viswanathan, M.; Grier, J.D.; Narayana, M.; El-Naggar, A.K.; Lozano, G. An alternatively spliced HDM2 product increases p53 activity by inhibiting HDM2. Oncogene 2001, 20, 4041–4049. [Google Scholar] [CrossRef] [PubMed]
- Stommel, J.M.; Wahl, G.M. Accelerated MDM2 auto-degradation induced by DNA-damage kinases is required for p53 activation. EMBO J. 2004, 23, 1547–1556. [Google Scholar] [CrossRef] [PubMed]
- Itahana, K.; Mao, H.; Jin, A.; Itahana, Y.; Clegg, H.V.; Lindström, M.S.; Bhat, K.P.; Godfrey, V.L.; Evan, G.I.; Zhang, Y. Targeted inactivation of Mdm2 RING finger E3 Ubiquitin Ligase Activity in the Mouse Reveals Mechnaistic Insights into p53 regulation. Cancer Cell 2007, 12, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Ranaweera, R.S.; Yang, X. Auto-ubiquitination of Mdm2 Enhances Its Substrate Ubiquitin Ligase Activity. J. Biol. Chem. 2013, 288, 18939–18946. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Brooks, C.L.; Kon, N.; Gu, W. A dynamic role of HAUSP in the p53-Mdm2 pathway. Mol. Cell 2004, 13, 879–886. [Google Scholar] [CrossRef]
- Wang, X.; Taplick, J.; Geva, N.; Oren, M. Inhibition of p53 degradation by Mdm2 acetylation. FEBS Lett. 2004, 561, 195–201. [Google Scholar] [CrossRef]
- Mayo, L.D.; Donner, D.B. A phosphatidylinositol 3-kinaseAkt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc. Natl. Acad. Sci. USA 2001, 98, 11598–11603. [Google Scholar] [CrossRef]
- Gannon, H.S.; Woda, B.A.; Jones, S.N. ATM Phosphorylation of Mdm2 Ser394 Regulates the Amplitude and Duration of the DNA Damage Response in Mice. Cancer Cell 2012, 21, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Pajares, V.; Kim, M.M.; Yuan, Z.-M. Phosphorylation of MDMX Mediated by Akt Leads to Stabilization and Induces 14-3-3 Binding. J. Biol. Chem. 2008, 283, 13707–13713. [Google Scholar] [CrossRef]
- Anderson, J.S.; Lyon, C.E.; Fox, A.H.; Leung, A.K.L.; Lam, Y.W.; Steen, H.; Mann, M.; Lamond, A.I. Directed proteomic analysis of the human nucleolus. Curr. Biol. 2002, 12, 1–11. [Google Scholar] [CrossRef]
- Lindstrom, M.S.; Deisenroth, C.; Zhang, Y.; Lindström, M.S. Putting a Finger on Growth Surveillance: Insight into MDM2 Zinc Finger-Ribosomal Protein Interactions. Cell Cycle 2007, 6, 434–437. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, H. Signaling to p53: Ribosomal Proteins Find Their Way. Cancer Cell 2009, 16, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Gazda, H.T.; Sheen, M.R.; Vlachos, A.; Choesmel, V.; O’donohue, M.-F.; Schneider, H.; Darras, N.; Hasman, C.; Sieff, C.A.; Newburger, P.E.; et al. Ribosomal Protein L5 and L11 Mutations Are Associated with Cleft Palate and Abnormal Thumbs in Diamond-Blackfan Anemia Patients. Am. J. Hum. Genet. 2008, 83, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Derenzini, M.; Trere, D.; Pession, A.; Montanaro, L.; Sirri, V.; Ochst, R.L. Nucleolar Function and Size in Cancer Cells. Am. J. Pathol. 1998, 152, 1291–1297. [Google Scholar]
- Bee, A.; Ke, Y.; Forootan, S.; Lin, K.; Beesley, C.; Forrest, S.E.; Foster, C.S. Ribosomal Protein L19 Is a Prognostic Marker for Human Prostate Cancer. Clin. Cancer Res. 2006, 12. [Google Scholar] [CrossRef]
- Ishiguro, T.; Nakajima, M.; Naito, M.; Muto, T.; Tsuruo, T. Identification of genes differentially expressed in B16 murine melanoma sublines with different metastatic potentials. Cancer Res. 1996, 56, 875–879. [Google Scholar]
- Ebert, B.L.; Pretz, J.; Bosco, J.; Chang, C.Y.; Tamayo, P.; Galili, N.; Raza, A.; Root, D.E.; Attar, E.; Ellis, S.R.; et al. Identification of RPS14 as a 5q-syndrome gene by RNA interference screen HHS Public Access. Nature 2008, 451, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Martoriati, A.; Doumont, G.; Alcalay, M.; Bellefroid, E.; Pelicci, P.G.; Marine, J.-C. dapk1, encoding an activator of a p19 ARF-p53-mediated apoptotic checkpoint, is a transcription target of p53. Oncogene 2005, 24, 1461–1466. [Google Scholar] [CrossRef] [PubMed]
- Danovi, D.; Meulmeester, E.; Pasini, D.; Migliorini, D.; Capra, M.; Frenk, R.; de Graaf, P.; Francoz, S.; Gasparini, P.; Gobbi, A.; et al. Amplification of Mdmx (or Mdm4) Directly Contributes to Tumor Formation by Inhibiting p53 Tumor Suppressor Activity. Mol. Cell. Biol. 2004, 24, 5835–5843. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Reyes, A.; Parant, J.M.; Amelse, L.L.; Montes, R.; Luna, O.; Korsmeyer, S.J.; Lozano, G. Switching Mechanisms of Cell Death in mdm2-and mdm4-null Mice by Deletion of p53 Downstream Targets. Cancer Res. 2003, 63, 8664–8669. [Google Scholar]
- Parant, J.; Chavez-Reyes, A.; Little, N.A.; Yan, W.; Reinke, V.; Jochemsen, A.G.; Lozano, G. Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat. Genet. 2001, 29. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, S.; Gembarska, A.; Denecker, G.; Maetens, M.; Naessens, M.; Haigh, K.; Haigh, J.J.; Marine, J.-C. Widespread Overexpression of Epitope-Tagged Mdm4 Does Not Accelerate Tumor Formation In Vivo. Mol. Cell. Biol. 2010, 30, 5394–5405. [Google Scholar] [CrossRef] [PubMed]
- Francoz, S.; Froment, P.; Bogaerts, S.; De Clercq, S.; Maetens, M.; Doumont, G.; Bellefroid, E.; Marine, J.-C. Mdm4 and Mdm2 cooperate to inhibit p53 activity in proliferating and quiescent cells in vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 3232–3237. [Google Scholar] [CrossRef] [PubMed]
- Migliorini, D.; Danovi, D.; Colombo, E.; Carbone, R.; Pelicci, P.G.; Marine, J.-C. Hdmx Recruitment into the Nucleus by Hdm2 Is Essential for Its Ability to Regulate p53 Stability and Transactivation. J. Biol. Chem. 2001, 277, 7318–7323. [Google Scholar] [CrossRef]
- Tanimura, S.; Ohtsuka, S.; Mitsui, K.; Shirouzu, K.; Yoshimura, A.; Ohtsubo, M. MDM2 interacts with MDMX through their RING finger domains. FEBS Lett. 1999, 447, 5–9. [Google Scholar] [CrossRef]
- Pant, V.; Xiong, S.; Iwakuma, T.; Quintás-Cardama, A.; Lozano, G. Heterodimerization of Mdm2 and Mdm4 is critical for regulating p53 activity during embryogenesis but dispensable for p53 and Mdm2 stability. Proc. Natl. Acad. Sci. USA 2011, 108, 119995–120000. [Google Scholar] [CrossRef]
- Carrillo, A.M.; Bouska, A.; Arrate, M.P.; Eischen, C.M. Mdmx promotes genomic instability independent of p53 and Mdm2. Oncogene 2015, 34, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Arman Aksoy, B.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Onur Sumer, S.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortalS. Sci. Signal. 2013, 6. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Ramos, Y.F.M.; Stad, R.; Attema, J.; Peltenburg, L.T.C.; Van Der Eb, A.J.; Jochemsen, A.G. Aberrant Expression of HDMX Proteins in Tumor Cells Correlates with Wild-Type p53. Cancer Res. 2001, 61, 1839–1842. [Google Scholar] [PubMed]
- Dewaele, M.; Marine, J.; Invest, J.C.; Dewaele, M.; Tabaglio, T.; Willekens, K.; Bezzi, M.; Teo, S.X.; Low, D.H.P.; Koh, C.M. Antisense oligonucleotide—Mediated MDM4 exon 6 skipping impairs tumor growth. J. Clin. Investig. 2016, 126, 68–84. [Google Scholar] [CrossRef] [PubMed]
- Miranda, P.J.; Buckley, D.; Raghu, D.; Pang, J.-M.B.; Takano, E.A.; Vijayakumaran, R.; Teunisse, A.F.; Posner, A.; Procter, T.; Herold, M.J.; et al. MDM4 is a rational target for treating breast cancers with mutant p53. J. Pathol. 2017, 241, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Swetzig, W.M.; Wang, J.; Das, G.M. Estrogen receptor alpha (ERα/ESR1) mediates the p53-independent overexpression of MDM4/MDMX and MDM2 in human breast cancer. Oncotarget 2016, 7, 16049–16069. [Google Scholar] [CrossRef]
- Gilkes, D.M.; Pan, Y.; Coppola, D.; Yeatman, T.; Reuther, G.W.; Chen, J. Regulation of MDMX Expression by Mitogenic Signaling. Mol. Cell. Biol. 2008, 28, 1999–2010. [Google Scholar] [CrossRef] [PubMed]
- Haupt, S.; Mejía-Hernández, J.O.; Vijayakumaran, R.; Keam, S.P.; Haupt, Y. The long and the short of it: The MDM4 tail so far. J. Mol. Cell Biol. 2019, 11, 231–244. [Google Scholar] [CrossRef]
- Wynendaele, J.; Böhnke, A.; Leucci, E.; Nielsen, S.J.; Lambertz, I.; Hammer, S.; Sbrzesny, N.; Kubitza, D.; Wolf, A.; Gradhand, E.; et al. An illegitimate microRNA target site within the 3′ UTR of MDM4 affects ovarian cancer progression and chemosensitivity. Cancer Res. 2010, 70, 9641–9649. [Google Scholar] [CrossRef]
- Stegeman, S.; Moya, L.; Selth, L.A.; Spurdle, A.B.; Clements, J.A.; Batra, J. A genetic variant of MDM4 influences regulation by multiple microRNAs in prostate cancer. Endocr. Relat. Cancer 2015, 22, 265–276. [Google Scholar] [CrossRef]
- Lagos-Quintana, M.; Rauhut, R.; Lendeckel, W. Identification of Novel Genes Coding for Small Expressed RNAs. Science 2001, 294, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Elbashir, S.M.; Lendeckel, W.; Tuschl, T. RNA interference is mediated by 21-and 22-nucleotide RNAs. Genes Dev. 2001, 15, 188–200. [Google Scholar] [CrossRef]
- Zeng, Y.; Yi, R.; Cullen, B.R. MicroRNAs and small interfering RNAs can inhibit mRNA expression by similar mechanisms. Proc. Natl. Acad. Sci. USA 2003, 100, 9779–9784. [Google Scholar] [CrossRef] [PubMed]
- Prodosmo, A.; Giglio, S.; Moretti, S.; Mancini, F.; Barbi, F.; Avenia, N.; Di Conza, G.; Schünemann, H.J.; Pistola, L.; Ludovini, V.; et al. Analysis of human MDM4 variants in papillary thyroid carcinomas reveals new potential markers of cancer properties. J. Mol. Med. 2008, 86, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Bartel, F.; Schulz, J.; Böhnke, A.; Blümke, K.; Kappler, M.; Bache, M.; Schmidt, H.; Würl, P.; Taubert, H.; Hauptmann, S. Significance of HDMX-S (or MDM4) mRNA splice variant overexpression and HDMX gene amplification on primary soft tissue sarcoma prognosis. Int. J. Cancer 2005, 117, 469–475. [Google Scholar] [CrossRef]
- Pant, V.; Larsson, C.A.; Aryal, N.; Xiong, S.; James You, M.; Quintas-Cardama, A.; Lozano, G. Tumorigenesis promotes Mdm4-S overexpression. Oncotarget 2017, 8, 25837–25847. [Google Scholar] [CrossRef]
- Vin Chan, J.; Xin Ping Koh, D.; Liu, Y.; Joseph, T.L.; Lane, D.P.; Verma, C.S.; Sing Tan, Y. Role of the N-terminal lid in regulating the interaction of phosphorylated MDMX with p53. Oncotarget 2017, 8, 112825–112840. [Google Scholar] [CrossRef]
- Chen, L.; Gilkes, D.M.; Pan, Y.; Lane, W.S.; Chen, J. ATM and Chk2-dependent phosphorylation of MDMX contribute to p53 activation after DNA damage. EMBO J. 2005, 24, 3411–3422. [Google Scholar] [CrossRef]
- Lebron, C.; Chen, L.; Gilkes, D.M.; Chen, J. Regulation of MDMX nuclear import and degradation by Chk2 and 14-3-3. EMBO J. 2006, 25, 1196–1206. [Google Scholar] [CrossRef]
- Li, X.; Gilkes, D.; Li, B.; Cheng, Q.; Pernazza, D.; Lawrence, N.; Chen, J. Abnormal MDMX degradation in tumor cells due to ARF deficiency. Oncogene 2012, 9, 3721–3732. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, M.; Gu, W. A critical role for the non-coding 5S rRNA in regulating Mdmx stability. Mol. Cell 2011, 43, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef]
- Galanos, P.; Vougas, K.; Walter, D.; Polyzos, A.; Maya-Mendoza, A.; Haagensen, E.J.; Kokkalis, A.; Roumelioti, F.-M.; Gagos, S.; Tzetis, M.; et al. Protracted p53-independent stimulation of p21 WAF1/Cip1 fuels genomic instability by deregulating the replication licensing machinery Europe PMC Funders Group. Nat. Cell Biol. 2016, 18, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Galanos, P.; Pappas, G.; Polyzos, A.; Kotsinas, A.; Svolaki, I.; Giakoumakis, N.N.; Glytsou, C.; Pateras, I.S.; Swain, U.; Souliotis, V.L.; et al. Mutational signatures reveal the role of RAD52 in p53-independent p21-driven genomic instability. Genome Biol. 2018, 19. [Google Scholar] [CrossRef]
- Brugarolas, J.; Chandrasekaran, C.; Gordon, J.I.; Beach, D.; Jacks, T.; Hannon, G.J. Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature 1995, 377, 552–557. [Google Scholar] [CrossRef]
- Quelle, D.E.; Ashmun, R.A.; Shurtleff, S.A.; Kato, J.-Y.; Bar-Sagi, D.; Roussel, M.F.; Sherr, C.J. Overexpression of mouse D-type cyclins accelerates phase in rodent fibroblasts. Genes Dev. 1993, 7, 1559–1571. [Google Scholar] [CrossRef]
- Dou, Q.-P.; Levin, A.H.; Zhao, S.; Pardee, A.B. Cyclin E and Cyclin A as Candidates for the Restriction Point Protein1. Cancer Res. 1993, 53, 1493–1497. [Google Scholar]
- Waga, S.; Hannon, G.J.; Beach, D.; Stillman, B. The p21 inhibitor of cyclin-dependent kinases controls DNA replication by interaction with PCNA. Nature 1994, 369, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.M.; Kim, J.; Park, J.K.; Hwang, S.-G.; Kim, W.-J.; Lee, W.-J.; Kang, S.W.; Um, H.-D. Bcl-w promotes cell invasion by blocking the invasion-suppressing action of Bax. Cell. Signal. 2012, 24, 1163–1172. [Google Scholar] [CrossRef]
- Dotto, G.P. p21 WAF1aCip1: More than a break to the cell cycle? Biochim. Biophyis. Acta 2000, 1471, 43–56. [Google Scholar]
- Roninson, I.B. Oncogenic functions of tumour suppressor p21 Waf1/Cip1/Sdi1: Association with cell senescence and tumour-promoting activities of stromal fibroblasts. Cancer Lett. 2002, 179, 1–14. [Google Scholar] [CrossRef]
- Gartel, A.L.; Tyner, A.L. The Role of the Cyclin-dependent Kinase Inhibitor p21 in Apoptosis 1. Mol. Cancer Ther. 2002, 1, 349–639. [Google Scholar]
- Georgakilas, A.G.; Martin, O.A.; Bonner, W.M. p21: A Two-Faced Genome Guardian. Trends Mol. Med. 2017, 23. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.M.; Jung, C.H.; Kim, J.; Hwang, S.G.; Park, J.K.; Um, H.D. The p53/p21 complex regulates cancer cell invasion and apoptosis by targeting Bcl-2 family proteins. Cancer Res. 2017, 77, 3092–3100. [Google Scholar] [CrossRef]
- Chang, B.-D.; Watanabe, K.; Broude, E.V.; Fang, J.; Poole, J.C.; Kalinichenko, T.V.; Roninson, I.B. Effects of p21 Waf1/Cip1/Sdi1 on cellular gene expression: Implications for carcinogenesis, senescence, and age-related diseases. Proc. Natl. Acad. Sci. USA 2000, 97, 4291–4296. [Google Scholar] [CrossRef] [PubMed]
- Lukas, J.; Bártková, J.; Rohde, M.; Strauss, M.; Bartek, J. Cyclin D1 Is Dispensable for G 1 Control in Retinoblastoma Gene-Deficient Cells Independently of cdk4 Activity. Mol. Cell. Biol. 1995, 15, 2600–2611. [Google Scholar] [CrossRef]
- Ren, B.; Cam, H.; Takahashi, Y.; Volkert, T.; Terragni, J.; Young, R.A.; Dynlacht, B.D. E2F integrates cell cycle progression with DNA repair, replication, and G 2/M checkpoints. Genes Dev. 2002, 16, 245–256. [Google Scholar] [CrossRef]
- Shiohara, M.; El-Deiry, W.S.; Wada, M.; Nakamaki, T.; Takeuchi, S.; Yang, R.; Chen, D.-L.; Vogelstein, B.; Koeffler, H.P. Absence of WAF1 Mutations in a Variety of Human Malignancies. Blood 1994, 84, 3782–3784. [Google Scholar] [CrossRef]
- Deng, C.; Zhang, P.; Harper, J.W.; Elledge, S.J.; Leder, P. Mice Lacking p21 CIP1/WAF1 Undergo Normal Development, but Are Defective in Gl Checkpoint Control. Cell 1995, 82, 675–684. [Google Scholar] [CrossRef]
- Martín-Caballero, J.; Flores, J.M.; García-Palencia, P.; Serrano, M. Tumor Susceptibility of p21 Waf1/Cip1-deficient Mice 1. Cancer Res. 2001, 61, 6234–6238. [Google Scholar]
- Quereda, V.; Martinalbo, J.; Dubus, P.; Carnero, A.; Malumbres, M. Genetic cooperation between p21 Cip1 and INK4 inhibitors in cellular senescence and tumor suppression. Oncogene 2007, 26, 7665–7674. [Google Scholar] [CrossRef][Green Version]
- Adnane, J.; Jackson, R.J.; Nicosia, S.V.; Cantor, A.B.; Jack Pledger, W.; Sebti, S.M.M. Loss of p21 WAF1/CIP1 accelerates Ras oncogenesis in a transgenic/knockout mammary cancer model. Oncogene 2000, 19, 5338–5347. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Duan, Z.; Zarebski, A.; Montoya-Durango, D.; Leighton Grimes, H.; Horwitz, M. Gfi1 Coordinates Epigenetic Repression of p21 Cip/WAF1 by Recruitment of Histone Lysine Methyltransferase G9a and Histone Deacetylase 1. Mol. Cell. Biol. 2005, 25, 10338–10351. [Google Scholar] [CrossRef]
- Schmidt, T.; Karsunky, H.; Gau, E.; Zevnik, B.; Elsässer, H.-P.; Möröy, Y.T. Zinc finger protein GFI-1 has low oncogenic potential but cooperates strongly with pim and myc genes in T-cell lymphomagenesis. Oncogene 1998, 17, 2661–2667. [Google Scholar] [CrossRef]
- Kazanjian, A.; Wallis, D.; Au, N.; Nigam, R.; Venken, K.J.T.; Cagle, P.T.; Dickey, B.F.; Bellen, H.J.; Gilks, C.B.; Grimes, H.L. Growth Factor Independence-1 Is Expressed in Primary Human Neuroendocrine Lung Carcinomas and Mediates the Differentiation of Murine Pulmonary Neuroendocrine Cells. Cancer Res. 2004, 64, 6874–6882. [Google Scholar] [CrossRef]
- Wang, Y.A.; Elson, A.; Leder, P. Loss of p21 increases sensitivity to ionizing radiation and delays the onset of lymphoma in atm-deficient mice. Proc. Natl. Acad. Sci. USA 1997, 94, 14590–14595. [Google Scholar] [CrossRef]
- Gartel, A.L. Is p21 an oncogene? Mol. Cancer Ther. 2006, 5. [Google Scholar] [CrossRef]
- Missero, C.; Di Cunto, F.; Kiyokawa, H.; Koff, A.; Dotto, G.P. The absence of p21Cipl/wAF1 alters keratinocyte growth and differentiation and promotes ras-tumor progression. Genes Dev. 1996, 10, 3065–3075. [Google Scholar] [CrossRef]
- Evangelou, K.; Galanos, P.; Gorgoulis, V.G. The Janus face of p21. Mol. Cell. Oncol. 2016, 3. [Google Scholar] [CrossRef] [PubMed]
- Pomerantz, J.; Schreiber-Agus, N.; Liégeois, N.J.; Silverman, A.; Alland, L.; Chin, L.; Potes, J.; Chen, K.; Orlow, I.; Lee, H.W.; et al. The Ink4a tumor suppressor gene product, p19(Arf), interacts with MDM2 and neutralizes MDM2′s inhibition of p53. Cell 1998, 92, 713–723. [Google Scholar] [CrossRef]
- Quelle, D.E.; Zindy, F.; Ashmun, R.A.; Sherr, C.J. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell 1995, 83, 993–1000. [Google Scholar] [CrossRef]
- Mao, L.; Merlo, A.; Bedi, G.; Shapiro, G.I.; Edwards, C.D.; Rollins, B.J.; Sidransky, D. A Novel p16INK4A Transcript. Cancer Res. 1995, 55, 2995–2997. [Google Scholar]
- Zhang, Y.; Xiong, Y.; Yarbrough, W.G. ARF Promotes MDM2 Degradation and Stabilizes p53: ARF-INK4a Locus Deletion Impairs Both the Rb and p53 Tumor Suppression Pathways. Cell 1998, 92, 725–734. [Google Scholar] [CrossRef]
- Kamijo, T.; Zindy, F.; Roussel, M.F.; Quelle, D.E.; Downing, J.R.; Ashmun, R.A.; Grosveld, G.; Sherr, C.J. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19(ARF). Cell 1997, 91, 649–659. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiong, Y. Mutations in Human ARF Exon 2 Disrupt Its Nucleolar Localization and Impair Its Ability to Block Nuclear Export of MDM2 and p53. Mol. Cell 1999, 3, 579–591. [Google Scholar] [CrossRef]
- Weber, H.O.; Samuel, T.; Rauch, P.; Funk, J.O. Human p14 ARF-mediated cell cycle arrest strictly depends on intact p53 signaling pathways. Oncogene 2002, 21, 3207–3212. [Google Scholar] [CrossRef][Green Version]
- Honda, R.; Yasuda, H. Association of p19 ARF with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. EMBO J. 1999, 18, 22–27. [Google Scholar] [CrossRef]
- Weber, J.D.; Jeffers, J.R.; Rehg, J.E.; Randle, D.H.; Lozano, G.; Roussel, M.F.; Sherr, C.J.; Zambetti, G.P. p53-independent functions of the p19 ARF tumor suppressor. Genes Dev. 2000, 14, 2358–2365. [Google Scholar] [CrossRef]
- Eischen, C.M.; Weber, J.D.; Roussel, M.F.; Sherr, C.J.; Cleveland, J.L. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 1999, 13, 2658–2669. [Google Scholar] [CrossRef]
- Tompkins, V.; Hagen, J.; Zediak, V.P.; Quelle, D.E. Identification of novel ARF binding proteins by two-hybrid screening. Cell Cycle 2006, 5, 641–646. [Google Scholar] [CrossRef] [PubMed]
- Britigan, E.M.C.; Wan, J.; Zasadil, L.M.; Ryan, S.D.; Weaver, B.A. The ARF tumor suppressor prevents chromosomal instability and ensures mitotic checkpoint fidelity through regulation of Aurora B. Mol. Biol. Cell 2014, 25. [Google Scholar] [CrossRef]
- Churchman, M.L.; Roig, I.; Jasin, M.; Keeney, S.; Sherr, C.J. Expression of Arf Tumor Suppressor in Spermatogonia Facilitates Meiotic Progression in Male Germ Cells. PLoS Genet. 2011, 7, 1002157. [Google Scholar] [CrossRef]
- Gromley, A.; Churchman, M.L.; Zindy, F.; Sherr, C.J. Transient expression of the Arf tumor suppressor during male germ cell and eye development in Arf-Cre reporter mice. Proc. Natl. Acad. Sci. USA 2009, 106, 6285–6290. [Google Scholar] [CrossRef]
- Fulci, G.; Labuhn, M.; Maier, D.; Lachat, Y.; Hausmann, O.; Hegi, M.E.; Janzer, R.C.; Merlo, A.; Van Meir, E.G. p53 gene mutation and ink4a-arf deletion appear to be two mutually exclusive events in human glioblastoma. Oncogene 2000, 19, 3816–3822. [Google Scholar] [CrossRef]
- Hegi, M.E.; Zur Hausen, A.; Rüedi, D.; Malin, G.; Kleihues, P. Hemizygous or homozygous deletion of the chromosomal region containing the p16INK4a gene is associated with amplification of the EGF receptor gene in gioblastomas. J. Cancer 1997, 73, 57–63. [Google Scholar]
- Esteller, M.; Tortola, S.; Toyota, M.; Capella, G.; Peinado, M.A.; Baylin, S.B.; Herman, J.G. Hypermethylation-associated inactivation of p14(ARF) is independent of p16(INK4a) methylation and p53 mutational status. Cancer Res. 2000, 60, 129–133. [Google Scholar]
- Ren, Y.; Xiao, L.; Weng, G.; Shi, B. Clinical significance of p16 INK4A and p14 ARF promoter methylation in renal cell carcinoma: A meta-analysis. Oncotarget 2017, 8, 64385–64394. [Google Scholar] [CrossRef]
- Burri, N.; Shaw, P.; Bouzourene, H.; Sordat, I.; Sordat, B.; Gillet, M.; Schorderet, D.; Bosman, F.T.; Chaubert, P. Methylation Silencing and Mutations of the p14 ARF and p16 INK4a Genes in Colon Cancer. Lab. Investig. 2001, 81, 271. [Google Scholar] [CrossRef]
- Fotouhi, O.; Fahmideh, M.A.; Kjellman, M.; Sulaiman, L.; Höög, A.; Zedenius, J.; Hashemi, J.; Larsson, C. Global hypomethylation and promotor methylation in small intenstinal neuroendocrine tumors. Epigenetics 2014, 9, 987–997. [Google Scholar] [CrossRef]
- Esteller, M.; Cordon-Cardo, C.; Corn, P.G.; Meltzer, S.J.; Pohar, K.S.; Watkins, D.N.; Capella, G.; Peinado, M.A.; Matias-Guiu, X.; Prat, J.; et al. p14 ARF Silencing by Promoter Hypermethylation Mediates Abnormal Intracellular Localization of MDM2. Cancer Res. 2001, 61, 2816–2821. [Google Scholar]
- Budina-Kolomets, A.; Hontz, R.D.; Pimkina, J.; Murphy, M.E. A conserved domain in exon 2 coding for the human and murine ARF tumor suppressor protein is required for autophagy induction. Autophagy 2013, 9, 1553–1565. [Google Scholar] [CrossRef]
- Xie, Y.; Liu, S.; Lu, W.; Yang, Q.; Williams, K.D.; Binhazim, A.A.; Carver, B.S.; Matusik, R.J.; Chen, Z. Slug regulates E-cadherin repression via p19Arf in prostate tumorigenesis. Mol. Oncol. 2014, 8, 1355–1364. [Google Scholar] [CrossRef]
- Xie, Y.; Lu, W.; Liu, S.; Yang, Q.; Shawn Goodwin, J.; Sathyanarayana, S.A.; Pratap, S.; Chen, Z. MMP7 interacts with ARF in nucleus to potentiate tumor microenvironments for prostate cancer progression in vivo. Oncotarget 2016, 7, 47609–47619. [Google Scholar] [CrossRef]
- Owczarek, T.B.; Kobayashi, T.; Ramirez, R.; Rong, L.; Puzio-Kuter, A.M.; Teo, M.Y.; Sánchez-Vega, F.; Wang, J.; Schultz, N.; Zheng, T.; et al. ARF confers a context-dependent response to chemotherapy in muscle invasive bladder cancer. Cancer Res. 2017, 77, 1035–1046. [Google Scholar] [CrossRef]
- Harland, M.; Taylor, C.F.; Chambers, P.A.; Kukalizch, K.; Randerson-Moor, J.A.; Gruis, N.A.; De Snoo, F.A.; Ac Ter Huurne, J.; Goldstein, A.M.; Tucker, M.A.; et al. A mutation hotspot at the p14ARF splice site. Oncogene 2005, 24, 4604–4608. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.A.; Etkin, L.D.; Freemont, P.S. A novel zinc finger coiled-coil domain in a family of nuclear proteins. Trends Biochem. Sci. 1992, 17, 443–453. [Google Scholar] [CrossRef]
- Reymond, A.; Meroni, G.; Fantozzi, A.; Merla, G.; Cairo, S.; Luzi, L.; Riganelli, D.; Zanaria, E.; Messali, S.; Cainarca, S.; et al. The tripartite motif family identifies cell compartments. EMBO J. 2001, 20, 2140–2151. [Google Scholar] [CrossRef]
- Li, Y.; Wu, H.; Wu, W.; Zhuo, W.; Liu, W.; Zhang, Y.; Cheng, M.; Chen, Y.-G.; Gao, N.; Yu, H.; et al. Structural insights into the TRIM family of ubiquitin E3 ligases. Cell Res. 2014, 24. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Ji, Z.; Wang, Y.; Li, J.; Cao, H.; Zhu, H.H.; Gao, W.-Q. TRIM59 Is Up-regulated in Gastric Tumors, Promoting Ubiquitination and Degradation of p53. Gastroenterology 2014, 147, 1043–1054. [Google Scholar] [CrossRef]
- Han, Y.; Tan, Y.; Zhao, Y.; Zhang, Y.; He, X.; Yu, L.; Jiang, H.; Lu, H.; Tian, H. TRIM23 overexpression is a poor prognostic factor and contributes to carcinogenesis in colorectal cancer. J. Cell. Mol. Med. 2020, 24, 5491–5500. [Google Scholar] [CrossRef]
- Zhang, Y.; Du, H.; Li, Y.; Yuan, Y.; Chen, B.; Sun, S. Elevated TRIM23 expression predicts cisplatin resistance in lung adenocarcinoma. Cancer Sci. 2020, 111, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Li, Z.; Chen, J.; Hu, X. Expression and the potential functions of TRIM32 in lung cancer tumorigenesis. J. Cell. Biochem. 2018, 120. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Wang, X.L.; Ly, P.; Belyi, V.; Xu-Monette, Z.Y.; Young, K.H.; Hu, W.; Feng, Z. E3 ubiquitin ligase TRIM32 negatively regulates tumor suppressor p53 to promote tumorigenesis. Cell Death Differ. 2014, 21, 1792–1804. [Google Scholar] [CrossRef]
- Zhan, W.; Han, T.; Zhang, C.; Xie, C.; Gan, M.; Deng, K.; Fu, M.; Wang, J.-B. TRIM59 Promotes the Proliferation and Migration of Non-Small Cell Lung Cancer Cells by Upregulating Cell Cycle Related Proteins. PLoS ONE 2015, 10, e0142596. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ji, B.; Feng, Y.; Zhang, Y.; Ji, D.; Zhu, C.; Wang, S.; Zhang, C.; Zhang, D.; Sun, Y. TRIM59 facilitates the proliferation of colorectal cancer and promotes metastasis via the PI3K/AKT pathway. Oncol. Rep. 2017, 38, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Xing, D.; Li, Z.; Shen, J.; Zhao, H.; Li, S. TRIM59 is upregulated and promotes cell proliferation and migration in human osteosarcoma. Mol. Med. Rep. 2016, 13, 5200–5206. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Zhang, J.; Zhang, Y.; Feng, Q.; Wang, H.; Li, G.; Jiang, W.; Li, X. Knockdown of tripartite motif 59 (TRIM59) inhibits proliferation in cholangiocarcinoma via the PI3K/AKT/mTOR signalling pathway. Gene 2019, 698, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Frankish, A.; Diekhans, M.; Ferreira, A.-M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019, 47, 767. [Google Scholar] [CrossRef]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef]
- Congrains, A.; Kamide, K.; Ohishi, M.; Rakugi, H. ANRIL: Molecular Mechanisms and Implications in Human Health. Int. J. Mol. Sci. 2013, 14, 1278–1292. [Google Scholar] [CrossRef] [PubMed]
- Gordon, F.E.; Nutt, C.L.; Cheunsuchon, P.; Nakayama, Y.; Provencher, K.A.; Rice, K.A.; Zhou, Y.; Zhang, X.; Klibanski, A. Increased Expression of Angiogenic Genes in the Brains of Mouse Meg3-Null Embryos. Endocrinology 2010, 151, 2443–2452. [Google Scholar] [CrossRef]
- Brown, C.J.; Balabio, A.; Rupert, J.L.; Lafreniere, R.G.; Grompe, M.; Tonlorenzi, R.; Willard, H.F. A gene from the region of the human X inactivation centre is expressed exclusively form the inactive X chromosome. Nature 1991, 349, 38–44. [Google Scholar] [CrossRef]
- Guttman, M.; Donaghey, J.; Carey, B.W.; Garber, M.; Grenier, J.K.; Munson, G.; Young, G.; Lucas, A.B.; Ach, R.; Bruhn, L.; et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 2012, 477, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Yap, K.L.; Li, S.; Muñoz-Cabello, A.M.; Raguz, S.; Zeng, L.; Mujtaba, S.; Gil, J.; Walsh, M.J.; Zhou, M.-M. Molecular Interplay of the Non-coding RNA ANRIL and Methylated Histone H3 Lysine 27 by Polycomb CBX7 in Transcriptional Silencing of INK4a. Mol. Cell 2010, 38, 662–674. [Google Scholar] [CrossRef] [PubMed]
- Pasmant, E.; Laurendeau, I.; Héron, D.; Vidaud, M.; Vidaud, D.; Bièche, I. Characterization of a Germ-Line Deletion, Including the Entire INK4/ARF Locus, in a Melanoma-Neural System Tumor Family: Identification of ANRIL, an Antisense Noncoding RNA Whose Expression Coclusters with ARF. Cancer Res. 2007, 67. [Google Scholar] [CrossRef] [PubMed]
- Cunnington, M.S.; Koref, M.S.; Mayosi, B.M.; Burn, J.; Keavney, B. Chromosome 9p21 SNPs Associated with Multiple Disease Phenotypes Correlate with ANRIL Expression. PLoS Genet. 2010, 6. [Google Scholar] [CrossRef]
- Pal, S.; Datta, K.; Mukhopadhyay, D. Central Role of p53 on Regulation of Vascular Permeability Factor/Vascular Endothelial Growth Factor (VPF/VEGF) Expression in Mammary Carcinoma 1. Cancer Res. 2001, 61, 6952–6957. [Google Scholar]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Rådmark, O.; Kim, S.; et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef]
- Hutvágner, G.; Mclachlan, J.; Pasquinelli, A.E.; Bálint, É.; Tuschl, T.; Zamore, P.D. A Cellular Function for the RNA-Interference Enzyme Dicer in the Maturation of the let-7 Small Temporal RNA. Science 2001, 293, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, E.; Kim, S.Y.; Carmell, M.A.; Murchison, E.P.; Alcorn, H.; Li, M.Z.; Mills, A.A.; Elledge, S.J.; Anderson, K.V.; Hannon, G.J. Dicer is essential for mouse development. Nat. Genet. 2003, 35. [Google Scholar] [CrossRef]
- Doench, J.G.; Sharp, P.A. Specificity of microRNA target selection in translational repression. Genes Dev. 2004, 18, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Hammond, S.M.; Bernstein, E.; Beach, D.; Hannon, G.J. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature 2000, 404, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Le, M.T.N.; Teh, C.; Shyh-Chang, N.; Xie, H.; Zhou, B.; Korzh, V.; Lodish, H.F.; Lim, B. MicroRNA-125b is a novel negative regulator of p53. Genes Dev. 2009, 23, 862–876. [Google Scholar] [CrossRef]
- Shi, X.-B.; Xue, L.; Yang, J.; Ma, A.-H.; Zhao, J.; Xu, M.; Tepper, C.G.; Evans, C.P.; Kung, H.-J.; deVere White, R.W. An androgen-regulated miRNA suppresses Bak1 expression and induces androgen-independent growth of prostate cancer cells. Proc. Natl. Acad. Sci. USA 2007, 104, 19983–19988. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, M.; Quelen, C.; Rosati, R.; De Mas, V.M.; La Starza, R.; Bastard, C.; Lippert, E.; Talmant, P.; Lafage-Pochitaloff, M.; Leroux, D.; et al. Myeloid cell differentiation arrest by miR-125b-1 in myelodysplasic syndrome and acute myeloid leukemia with the t(2;11)(p21;q23) translocation. J. Exp. Med. 2008, 205, 2499–2506. [Google Scholar] [CrossRef]
- Chen, L.; Luo, L.; Chen, W.; Xu, H.-X.; Chen, F.; Chen, L.-Z.; Zeng, W.-T.; Chen, J.-S.; Huang, X.-H. MicroRNA-24 increases hepatocellular carcinoma cell metastasis and invasion by targeting p53: miR-24 targeted p53. Biomed. Pharmacother. 2016, 84, 1113–1118. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Wang, X.; Wu, R.; Lin, M.; Laddha, S.V.; Yang, Q.; Chan, C.S.; Feng, Z. MicroRNA-339-5p inhibits colorectal tumorigenesis through regulation of the MDM2/p53 signaling. Oncotarget 2014, 15, 9106–9117. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Tan, C.; Yue, X.; Zhao, Y.; Peng, J.; Wang, X.; Laddha, S.V.; Chan, C.S.; Zheng, S.; et al. microRNA-1827 represses MDM2 to positively regulate tumor suppressor p53 and suppress tumorigenesis. Oncotarget 2016, 7, 8783–8796. [Google Scholar] [CrossRef]
- Zhou, C.; Liu, G.; Wang, L.; Lu, Y.; Yuan, L.; Zheng, L.; Chen, F.; Peng, F.; Li, X. MiR-339-5p Regulates the Growth, Colony Formation and Metastasis of Colorectal Cancer Cells by Targeting PRL-1. PLoS ONE 2013, 8, e0063142. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.-S.; Wu, Q.; Wang, C.-Q.; Wang, X.-N.; Wang, Y.; Zhao, J.-J.; Mao, S.-S.; Zhang, G.-H.; Zhang, N.; Xu, X.-C. MiR-339-5p inhibits breast cancer cell migration and invasion in vitro and may be a potential biomarker for breast cancer prognosis. BMC Cancer 2010, 10. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.U.; Son, Y.; Kim, C.H.; Kim, K.S.; Hyun, S.H.; Woo, H.G.; Jee, B.A.; Choi, J.H.; Sung, H.K.; Choi, H.C.; et al. Embryonic Stem Cell-Derived mmu-miR-291a-3p Inhibits Cellular Senescence in Human Dermal Fibroblasts Through the TGF-β Receptor 2 Pathway. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 1359–1367. [Google Scholar] [CrossRef]
- Rasko, J.E.J.; Klenova, E.M.; Leon, J.; Filippova, G.N.; Loukinov, D.I.; Vatolin, S.; Robinson, A.F.; Hu, Y.J.; Ulmer, J.; Ward, M.D.; et al. Cell Growth Inhibition by the Multifunctional Multivalent Zinc-Finger Factor CTCF. Cancer Res. 2001, 61, 6002–6007. [Google Scholar] [PubMed]
- Lobanenkov, V.V.; Nicolas, R.H.; Adler, V.V.; Paterson, H.; Klenova, E.M.; Polotskaja, A.V.; Goodwin, G.H. A novel sequence-specific DNA binding protein which interacts with three regularly spaced direct repeats of the CCCTC-motif in the 5′-flanking sequence of the chicken c-myc gene. Oncogene 1990, 5, 1743–1753. [Google Scholar] [PubMed]
- Filippova, G.N.; Fagerlie, S.; Klenova, E.M.; Myers, C.; Dehner, Y.; Goodwin, G.; Neiman, P.E.; Collins, S.J.; Lobanenkov, V.V. An Exceptionally Conserved Transcriptional Repressor, CTCF, Employs Different Combinations of Zinc Fingers to Bind Diverged Promoter Sequences of Avian and Mammalian c-myc Oncogenes. Mol. Cell. Biol. 1996, 16, 2802–2813. [Google Scholar] [CrossRef]
- Klenova, E.M.; Nicolas, R.H.; Paterson, H.F.; Carne, A.F.; Heath, C.M.; Goodwin, G.H.; Neiman, P.E.; Lobanenkov3, V.V.; Lobanenkov, V.V.; Nicolas, R.H.; et al. CTCF, a Conserved Nuclear Factor Required for Optimal Transcriptional Activity of the Chicken c-myc Gene, Is an 11-Zn-Finger Protein Differentially Expressed in Multiple Forms. Mol. Cell. Biol. 1993, 13, 7612–7624. [Google Scholar] [CrossRef]
- Kanduri, C.; Pant, V.; Loukinov, D.; Pugacheva, E.; Qi, C.-F.; Wolffe, A.; Ohlsson, R.; Lobanenkov, V.V. Functional association of CTCF with the insulator upstream of the H19 gene is parent of origin-specific and methylation-sensitive. Curr. Biol. 2000, 10, 853–856. [Google Scholar] [CrossRef]
- Quitschke, W.W.; Taheny, M.J.; Fochtmann, L.J.; Vostrov, A.A. Differential effect of zinc finger deletions on the binding of CTCF to the promoter of the amyloid precursor protein gene. Nucleic Acids Res. 2000, 28, 3370–3378. [Google Scholar] [CrossRef][Green Version]
- Chen, H.; Tian, Y.; Shu, W.; Bo, X.; Wang, S. Comprehensive Identification and Annotation of Cell Type-Specific and Ubiquitous CTCF-Binding Sites in the Human Genome. PLoS ONE 2012, 7, e41374. [Google Scholar] [CrossRef]
- Moore, J.M.; Rabaia, N.A.; Smith, L.E.; Fagerlie, S.; Gurley, K. Loss of Maternal CTCF Is Associated with Peri-Implantation Lethality of Ctcf Null Embryos. PLoS ONE 2012, 7, e34915. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, M.J.; Beatty, L.G.; Zhou, W.; Du, M.; Sadowski, P.D. The CTCF Insulator Protein Is Posttranslationally Modified by SUMO. Mol. Cell. Biol. 2009, 29, 714–725. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Ginjala, V.; Pant, V.; Chernukhin, I.; Whitehead, J.; Docquier, F.; Farrar, D.; Tavoosidana, G.; Mukhopadhyay, R.; Kanduri, C.; et al. Poly(ADP-ribosyl)ation regulates CTCF-dependent chromatin insulation. Nat. Genet. 2004, 36. [Google Scholar] [CrossRef]
- Kemp, C.J.; Moore, J.M.; Moser, R.; Bernard, B.; Teater, M.; Smith, L.E.; Rabaia, N.A.; Gurley, K.E.; Guinney, J.; Busch, S.E.; et al. CTCF haploinsufficiency destabilizes DNA methylation and predisposes to cancer. Cell Rep. 2014, 7, 1020–1029. [Google Scholar] [CrossRef]
- Filippova, G.N.; Lindblom, A.; Meincke, L.J.; Klenova, E.M.; Neiman, P.E.; Collins, S.J.; Doggett, N.A.; Lobanenkov, V.V. A Widely Expressed Transcription Factor With Multiple DNA Sequence Specificity, CTCF, Is Localized at Chromosome Segment 16q22.1 Within One of the Smallest Regions of Overlap for Common Deletions in Breast and Prostate Cancers. Genes Chromosomes Cancer 1998, 22, 26–36. [Google Scholar] [CrossRef]
- Soto-Reyes, E.; Recillas-Targa, F. Epigenetic regulation of the human p53 gene promoter by the CTCF transcription factor in transformed cell lines. Oncogene 2010, 29, 2217–2227. [Google Scholar] [CrossRef] [PubMed]
- Witcher, M.; Emerson, B.M. Epigenetic Silencing of the p16 INK4a Tumor Suppressor is Associated with Loss of CTCF Binding and a Chromatin Boundary. Mol. Cell 2009, 34, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Docquier, F.; Farrar, D.; Arcy, V.D.; Chernukhin, I.; Robinson, A.F.; Loukinov, D.; Vatolin, S.; Pack, S.; Mackay, A.; Harris, R.A.; et al. Heightened Expression of CTCF in Breast Cancer Cells Is Associated with Resistance to Apoptosis. Cancer Res. 2005, 65, 5112–5122. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Catalá, C.F.; Gretton, S.; Vostrov, A.; Pugacheva, E.; Farrar, D.; Ito, Y.; Docquier, F.; Kita, G.-X.; Murrell, A.; Lobanenkov, V.; et al. A novel mechanism for CTCF in the epigenetic regulation of Bax in breast cancer cells. Neoplasia 2013, 15, 912. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Mustafa, M.; Yuri Kim, C.; Hee Kim, M. Depletion of CTCF in Breast Cancer Cells Selectively Induces Cancer Cell Death via p53. J. Cancer 2017, 8, 2124–2131. [Google Scholar] [CrossRef]
- Basu, S.; Murphy, M.E. Genetic modifiers of the p53 pathway. Cold Spring Harb. Perspect. Med. 2016, 6, a026302. [Google Scholar] [CrossRef] [PubMed]
- Ruggero, D.; Grisendi, S.; Piazza, F.; Rego, E.; Mari, F.; Rao, P.H.; Cordon-Cardo, C.; Pandolfi, P.P. Dyskeratosis Congenita and Cancer in Mice Deficient in Ribosomal RNA Modification. Science 2003, 299, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wengrod, J.; Gardner, L.B. Overexpression of the c-myc Oncogene Inhibits Nonsense-mediated RNA Decay in B Lymphocytes. J. Biol. Chem. 2011, 286, 40038–40043. [Google Scholar] [CrossRef] [PubMed]
- Kalkat, M.; De Melo, J.; Hickman, K.A.; Lourenco, C.; Redel, C.; Resetca, D.; Tamachi, A.; Tu, W.B.; Penn, L.Z. MYC Deregulation in Primary Human Cancers. Genes 2017, 8, 151. [Google Scholar] [CrossRef]
- He, X.; Zhang, P. Serine/arginine-rich splicing factor 3 (SRSF3) regulates homologous recombination-mediated DNA repair. Mol. Cancer 2015, 14. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Li, C.; Mccoy, J.P.; Deng, C.-X.; Zheng, Z.-M. SRp20 is a proto-oncogene critical for cell proliferation and tumor induc-tion and maintenance. Int. J. Biol. Sci. 2010, 6, 806–826. [Google Scholar] [CrossRef]
- Mitsuuchi, Y.; Johnson, S.W.; Selvakumaran, M.; Williams, S.J.; Hamilton, T.C.; Testa, J.R. The Phosphatidylinositol 3-Kinase/AKT Signal Transduction Pathway Plays a Critical Role in the Expression of p21 WAF1/CIP1/SDI1 Induced by Cisplatin and Paclitaxel. Cancer Res. 2000, 60, 5390–5394. [Google Scholar]
- Sun, L.; Li, Y.; Yang, B. Downregulated long non-coding RNA MEG3 in breast cancer regulates proliferation, migration and invasion by depending on p53′s transcriptional activity. Biochem. Biophys. Res. Commun. 2016, 478, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Chernukhin, I.; Shamsuddin, S.; Kang, S.Y.; Bergström, R.; Kwon, Y.-W.; Yu, W.; Whitehead, J.; Mukhopadhyay, R.; Docquier, F.; Farrar, D.; et al. CTCF Interacts with and Recruits the Largest Subunit of RNA Polymerase II to CTCF Target Sites Genome-Wide. Mol. Cell. Biol. 2007, 27, 1631–1648. [Google Scholar] [CrossRef]
- Zhang, C.; Yu, M.; Li, X.; Zhang, Z.; Han, C.; Yan, B. Overexpression of long non-coding RNA MEG3 suppresses breast cancer cell proliferation, invasion, and angiogenesis through AKT pathway. Tumor Biol. 2017, 1–12. [Google Scholar] [CrossRef]
- Lee, J.; Go, Y.; Kang, I.; Han, Y.-M.; Kim, J. Oct-4 controls cell-cycle progression of embryonic stem cells. Biochem. J. 2010, 426, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Gutekunst, M.; Mueller, T.; Weilbacher, A.; Dengler, M.A.; Bedke, J.; Kruck, S.; Oren, M.; Aulitzky, W.E.; Van Der Kuip, H. Cisplatin Hypersensitivity of Testicular Germ Cell Tumors Is Determined by High Constitutive Noxa Levels Mediated by Oct-4. Cancer Res. 2013, 73. [Google Scholar] [CrossRef]
- Oing, C.; Alsdorf, W.H.; Von Amsberg, G.; Oechsle, K.; Bokemeyer, C. Platinum-refractory germ cell tumors: An update on current treatment options and developments. World J. Urol. 2017, 35, 1167–1175. [Google Scholar] [CrossRef] [PubMed]
- Tisato, V.; Voltan, R.; Gonelli, A.; Secchiero, P.; Zauli, G. MDM2/X inhibitors under clinical evaluation: Perspectives for the management of hematological malignancies and pediatric cancer. J. Hematol. Oncol. 2017, 10. [Google Scholar] [CrossRef] [PubMed]








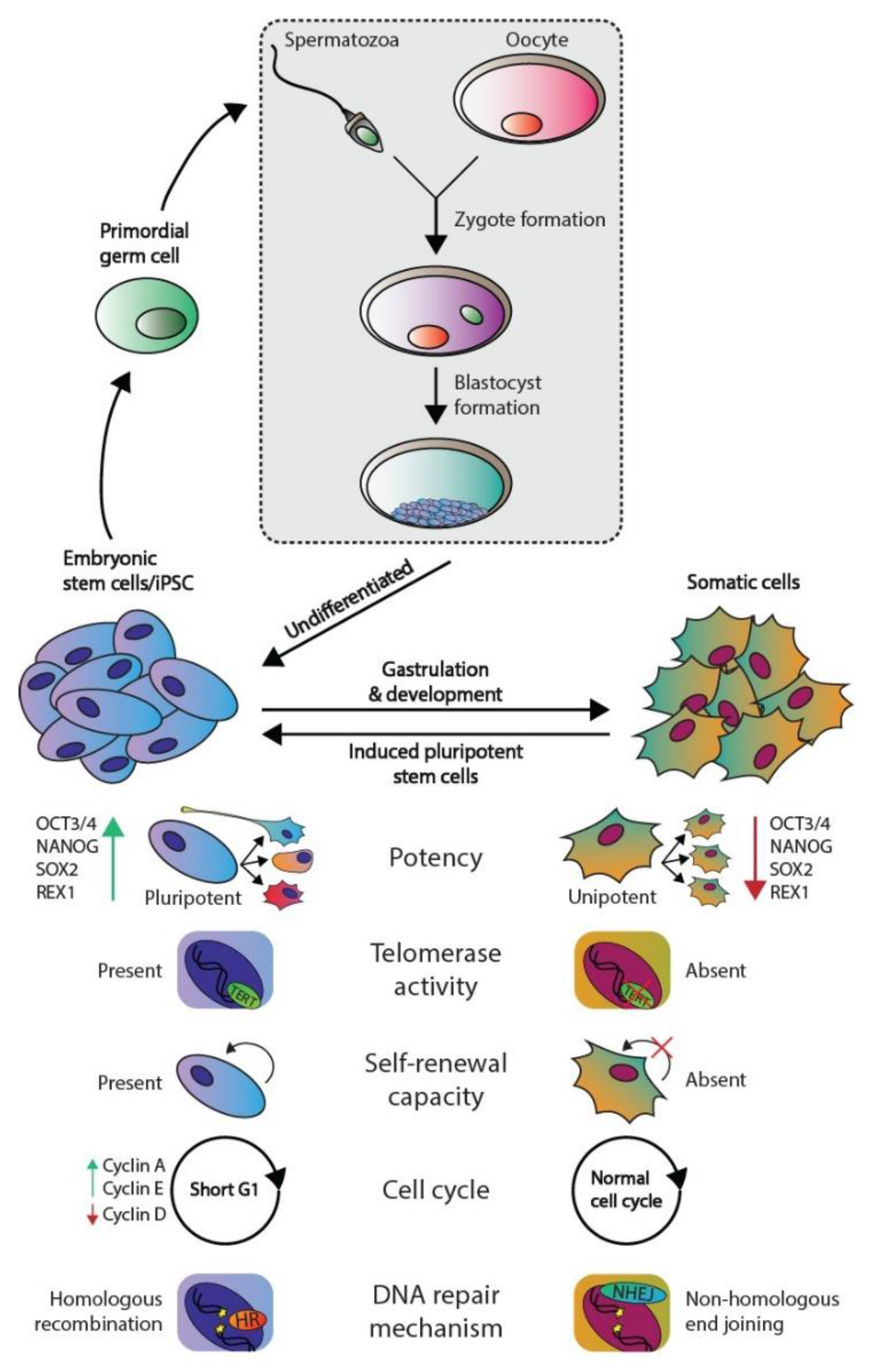{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
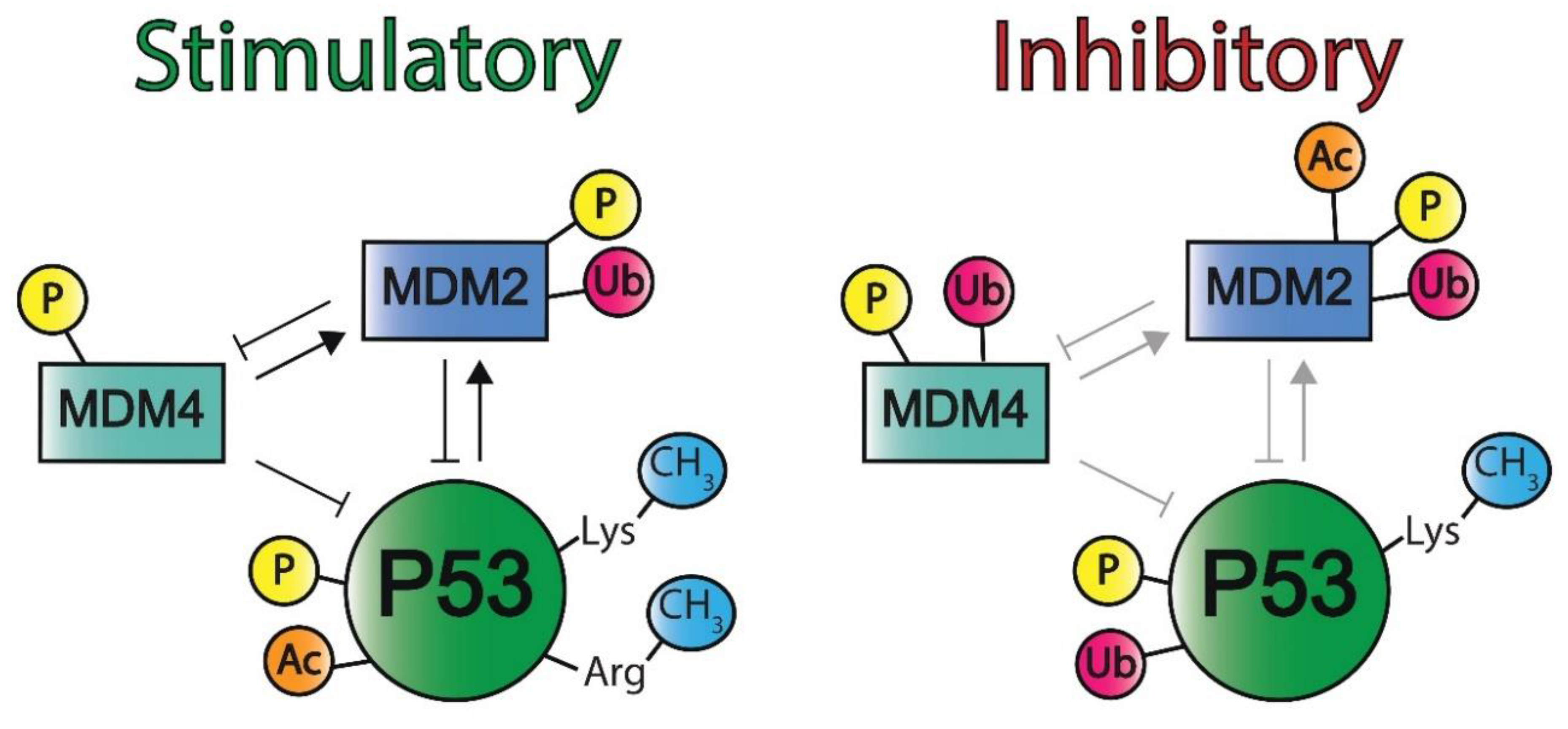{kind=link}
{kind=link}
| Targeting Agents | Mechanism(s) | NCT Number |
|---|---|---|
| Phase I | ||
| Vaccine therapy (i.e., gene therapy) | Virus-based adaption of P53 expression in cancerous cells, mostly combined with chemotherapeutics | P53MVA: NCT02275039 Ad5CMVP53: NCT00004225 |
| APR-246 | A small molecule that binds mutant P53 and aids in folding to restore its wild-type conformation | NCT02098343 |
| AMG-232 (KRT-232) | A small molecule that inhibits MDM2 and thereby activates P53 | NCT03217266 |
| ALRN-6924 | A peptide that binds MDM2 and MDM4 to activate WT P53 in noncancerous cells, thus preventing side effects while enhancing chemotherapy effects in cancer cells. | NCT02264613 |
| Phase I/II | ||
| APG-115 | MDM2 inhibitor that activates WT P53 with and without platinum-based chemotherapeutics | NCT03781986 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Timmerman, D.M.; Remmers, T.L.; Hillenius, S.; Looijenga, L.H.J. Mechanisms of TP53 Pathway Inactivation in Embryonic and Somatic Cells—Relevance for Understanding (Germ Cell) Tumorigenesis. Int. J. Mol. Sci. 2021, 22, 5377. https://doi.org/10.3390/ijms22105377
Timmerman DM, Remmers TL, Hillenius S, Looijenga LHJ. Mechanisms of TP53 Pathway Inactivation in Embryonic and Somatic Cells—Relevance for Understanding (Germ Cell) Tumorigenesis. International Journal of Molecular Sciences. 2021; 22(10):5377. https://doi.org/10.3390/ijms22105377
Chicago/Turabian StyleTimmerman, Dennis M., Tessa L. Remmers, Sanne Hillenius, and Leendert H. J. Looijenga. 2021. "Mechanisms of TP53 Pathway Inactivation in Embryonic and Somatic Cells—Relevance for Understanding (Germ Cell) Tumorigenesis" International Journal of Molecular Sciences 22, no. 10: 5377. https://doi.org/10.3390/ijms22105377
APA StyleTimmerman, D. M., Remmers, T. L., Hillenius, S., & Looijenga, L. H. J. (2021). Mechanisms of TP53 Pathway Inactivation in Embryonic and Somatic Cells—Relevance for Understanding (Germ Cell) Tumorigenesis. International Journal of Molecular Sciences, 22(10), 5377. https://doi.org/10.3390/ijms22105377