
Nonalcoholic Fatty Liver Disease (NAFLD). Mitochondria as Players and Targets of Therapies?

 ,
, 
 and
and 
Abstract
1. Introduction
- -
- Adiponutrin patatin-like phospholipase domain-containing protein 3 (PNPLA3) is expressed on the surface of intrahepatocyte lipid droplets and has lipase or lysophosphatidic acyltransferase activity. Carriers of the variant p.I148M have an increased risk of developing NAFLD [7], liver fibrosis and cirrhosis [8], and hepatocellular carcinoma (HCC) [9,10,11];
- -
- Membrane-bound O-acyltransferase domain-containing 7 gene (MBOAT7) has lysophosphatidylinositol acyltransferase activity with the regulation of arachidonic acid levels and shows anti-inflammatory activity. Carriers of the variant rs641738 C > T display deranged MBOAT7 activity [12];
- -
- Transmembrane 6 superfamily member 2 gene (TM6SF2) is involved in hepatic VLDL secretion. Carriers of the variant p.E167K show decreased circulating VLDL and increased liver steatosis [13];
- -
- Glucokinase regulatory protein gene (GCKR) variant p. P446L [14];
- -
- Missense variant in the mitochondrial amidoxime reducing component 1 (MARC1) might have protective effects in NAFLD [15];
- -
- Hydroxysteroid 17-beta dehydrogenase 13 (HSD17B13). The genetic variant is associated with serum levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST). The rs72613567:TA variant confers a reduced risk of nonalcoholic steatohepatitis (not steatosis) in human liver samples [16].
2. Physiological Homeostasis of Free Fatty Acids (FFA) in the Hepatocyte
2.1. Uptake of Circulating FFA
2.2. De Novo Lipogenesis
2.3. Uptake of Dietary FFA
3. Mitochondrial Function in the Hepatocyte
4. Epidemiology and Manifestations of NAFLD
5. General Features of Diagnosis of NAFLD
6. Lipotoxicity during Insulin Resistance and the Onset of Liver Steatosis
7. FFA and Toxic Lipids in NAFLD
7.1. Free Fatty Acids (FFA)
7.2. Triglycerides (TG)
7.3. Lysophosphatidylcholine (LPC)
7.4. Ceramides
7.5. Free Cholesterol
8. Mitochondrial Dysfunction in NAFLD and NASH
8.1. FFA Import in Mitochondria, Electron Transfer Chain Efficiency
8.2. Diet and Mitochondrial Disfunction with ROS Production
8.3. Metabolism Alterations, Lipotoxicity and Apoptosis
8.4. Nitrosative Stress and Cell Death
9. Therapy of NAFLD
9.1. Modification of Lifestyles and General Measures
9.2. Drugs
10. Therapies Targeting Mitochondria in NAFLD
10.1. Physical Exercise
10.2. Antidiabetic Drugs
10.3. Bile Acids (BA)
10.4. Antioxidant Agents
10.5. Mitotherapy
10.6. Novel Agents
11. Combination Therapy
12. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wong, V.W.-S.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Baj, J.; Garruti, G.; Celano, G.; De Angelis, M.; Wang, H.H.; Di Palo, D.M.; Bonfrate, L.; Wang, D.Q.-H.; Portincasa, P. Liver Steatosis, Gut-Liver Axis, Microbiome and Environmental Factors. A Never-Ending Bidirectional Cross-Talk. J. Clin. Med. 2020, 9, 2648. [Google Scholar] [CrossRef] [PubMed]
- Di Palo, D.M.; Garruti, G.; Di Ciaula, A.; Molina-Molina, E.; Shanmugam, H.; De Angelis, M.; Portincasa, P. Increased Colonic Permeability and Lifestyles as Contributing Factors to Obesity and Liver Steatosis. Nutrients 2020, 12, 564. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Carbone, F.; Shanmugham, H.; Molina-Molina, E.; Bonfrate, L.; Ministrini, S.; Montecucco, F.; Portincasa, P. Adiponectin involved in portal flow hepatic extraction of 13C-metacethin in obesity and non-alcoholic fatty liver. Eur. J. Intern. Med. 2021. [Google Scholar] [CrossRef] [PubMed]
- Caussy, C.; Soni, M.; Cui, J.; Bettencourt, R.; Schork, N.; Chen, C.H.; Ikhwan, M.A.; Bassirian, S.; Cepin, S.; Gonzalez, M.P.; et al. Nonalcoholic fatty liver disease with cirrhosis increases familial risk for advanced fibrosis. J. Clin. Investig. 2017, 127, 2697–2704. [Google Scholar] [CrossRef]
- Krawczyk, M.; Liebe, R.; Lammert, F. Toward Genetic Prediction of Nonalcoholic Fatty Liver Disease Trajectories: PNPLA3 and Beyond. Gastroenterology 2020, 158, 1865–1880.e1. [Google Scholar] [CrossRef] [PubMed]
- Dai, G.; Liu, P.; Li, X.; Zhou, X.; He, S. Association between PNPLA3 rs738409 polymorphism and nonalcoholic fatty liver disease (NAFLD) susceptibility and severity: A meta-analysis. Medicine 2019, 98. [Google Scholar] [CrossRef] [PubMed]
- Lauridsen, B.K.; Stender, S.; Kristensen, T.S.; Kofoed, K.F.; Kober, L.; Nordestgaard, B.G.; Tybjaerg-Hansen, A. Liver fat content, non-alcoholic fatty liver disease, and ischaemic heart disease: Mendelian randomization and meta-analysis of 279 013 individuals. Eur. Heart J. 2018, 39, 385–393. [Google Scholar] [CrossRef]
- Huang, Z.; Guo, X.; Zhang, G.; Liang, L.; Nong, B. Correlation between PNPLA3 rs738409 polymorphism and hepatocellular carcinoma: A meta-analysis of 10,330 subjects. Int. J. Biol. Markers 2019, 34, 117–122. [Google Scholar] [CrossRef]
- Krawczyk, M.; Bonfrate, L.; Portincasa, P. Nonalcoholic fatty liver disease. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 695–708. [Google Scholar] [CrossRef]
- Krawczyk, M.; Portincasa, P.; Lammert, F. PNPLA3-associated steatohepatitis: Toward a gene-based classification of fatty liver disease. Semin. Liver Dis. 2013, 33, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Mancina, R.M.; Dongiovanni, P.; Petta, S.; Pingitore, P.; Meroni, M.; Rametta, R.; Boren, J.; Montalcini, T.; Pujia, A.; Wiklund, O.; et al. The MBOAT7-TMC4 Variant rs641738 Increases Risk of Nonalcoholic Fatty Liver Disease in Individuals of European Descent. Gastroenterology 2016, 150, 1219–1230.e6. [Google Scholar] [CrossRef] [PubMed]
- Kahali, B.; Liu, Y.L.; Daly, A.K.; Day, C.P.; Anstee, Q.M.; Speliotes, E.K. TM6SF2: Catch-22 in the fight against nonalcoholic fatty liver disease and cardiovascular disease? Gastroenterology 2015, 148, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, B.; Linden, D.; Brolen, G.; Liljeblad, M.; Bjursell, M.; Romeo, S.; Loomba, R. Review article: The emerging role of genetics in precision medicine for patients with non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2020, 51, 1305–1320. [Google Scholar] [CrossRef]
- Emdin, C.A.; Haas, M.E.; Khera, A.V.; Aragam, K.; Chaffin, M.; Klarin, D.; Hindy, G.; Jiang, L.; Wei, W.Q.; Feng, Q.; et al. A missense variant in Mitochondrial Amidoxime Reducing Component 1 gene and protection against liver disease. PLoS Genet. 2020, 16, e1008629. [Google Scholar] [CrossRef] [PubMed]
- Abul-Husn, N.S.; Cheng, X.; Li, A.H.; Xin, Y.; Schurmann, C.; Stevis, P.; Liu, Y.; Kozlitina, J.; Stender, S.; Wood, G.C.; et al. A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease. N. Engl. J. Med. 2018, 378, 1096–1106. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Ono, M.; Okamoto, N.; Saibara, T. The latest idea in NAFLD/NASH pathogenesis. Clin. J. Gastroenterol. 2010, 3, 263–270. [Google Scholar] [CrossRef]
- Bai, L.; Li, H. Innate immune regulatory networks in hepatic lipid metabolism. J. Mol. Med. 2019, 97, 593–604. [Google Scholar] [CrossRef]
- Lafontan, M.; Langin, D. Lipolysis and lipid mobilization in human adipose tissue. Prog. Lipid Res. 2009, 48, 275–297. [Google Scholar] [CrossRef]
- Nassir, F.; Ibdah, J.A. Role of mitochondria in nonalcoholic fatty liver disease. Int. J. Mol. Sci. 2014, 15, 8713–8742. [Google Scholar] [CrossRef]
- Kazantzis, M.; Stahl, A. Fatty acid transport proteins, implications in physiology and disease. Biochim. Biophys. Acta 2012, 1821, 852–857. [Google Scholar] [CrossRef]
- Horton, J.; Goldstein, J.; Brown, M. SREBPs: Transcriptional mediators of lipid homeostasis. Cold Spring Harb. Symp. Quant. Biol. 2002, 67, 491–498. [Google Scholar] [CrossRef]
- Arab, J.P.; Arrese, M.; Trauner, M. Recent Insights into the Pathogenesis of Nonalcoholic Fatty Liver Disease. Annu. Rev. Pathol. 2018, 13, 321–350. [Google Scholar] [CrossRef]
- Arab, J.P.; Karpen, S.J.; Dawson, P.A.; Arrese, M.; Trauner, M. Bile acids and nonalcoholic fatty liver disease: Molecular insights and therapeutic perspectives. Hepatology 2017, 65, 350–362. [Google Scholar] [CrossRef] [PubMed]
- Di Ciaula, A.; Garruti, G.; Lunardi Baccetto, R.; Molina-Molina, E.; Bonfrate, L.; Wang, D.Q.; Portincasa, P. Bile Acid Physiology. Ann. Hepatol. 2017, 16, s4–s14. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef]
- Portincasa, P.; Di Ciaula, A.; Garruti, G.; Vacca, M.; De Angelis, M.; Wang, D.Q. Bile Acids and GPBAR-1: Dynamic Interaction Involving Genes, Environment and Gut Microbiome. Nutrients 2020, 12, 3709. [Google Scholar] [CrossRef] [PubMed]
- Thiam, A.R.; Farese, R.V., Jr.; Walther, T.C. The biophysics and cell biology of lipid droplets. Nat. Rev. Mol. Cell Biol. 2013, 14, 775–786. [Google Scholar] [CrossRef]
- Greenberg, A.S.; Coleman, R.A. Expanding roles for lipid droplets. Trends Endocrinol. Metab. TEM 2011, 22, 195–196. [Google Scholar] [CrossRef] [PubMed]
- Degli Esposti, D.; Hamelin, J.; Bosselut, N.; Saffroy, R.; Sebagh, M.; Pommier, A.; Martel, C.; Lemoine, A. Mitochondrial roles and cytoprotection in chronic liver injury. Biochem. Res. Int. 2012, 2012, 387626. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Rector, R.S.; Thyfault, J.P.; Ibdah, J.A. Nonalcoholic fatty liver disease and mitochondrial dysfunction. World J. Gastroenterol. WJG 2008, 14, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Guerrieri, F.; Nicoletti, C.; Adorisio, E.; Caraccio, G.; Leonetti, P.; Zanotti, F.; Cantatore, P. Correlation between decreased expression of mitochondrial F0F1-ATP synthase and low regenerating capability of the liver after partial hepatectomy in hypothyroid rats. J. Bioenerg. Biomembr. 2000, 32, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Pizzuto, R.; Paventi, G.; Atlante, A.; Passarella, S. Pyruvate kinase in pig liver mitochondria. Arch. Biochem. Biophys. 2010, 495, 42–48. [Google Scholar] [CrossRef]
- Paventi, G.; Pizzuto, R.; Passarella, S. The occurrence of l-lactate dehydrogenase in the inner mitochondrial compartment of pig liver. Biochem. Biophys. Res. Commun. 2017, 489, 255–261. [Google Scholar] [CrossRef]
- Passarella, S.; de Bari, L.; Valenti, D.; Pizzuto, R.; Paventi, G.; Atlante, A. Mitochondria and L-lactate metabolism. FEBS Lett. 2008, 582, 3569–3576. [Google Scholar] [CrossRef]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2020, 152, 116–141. [Google Scholar] [CrossRef]
- Samuel, V.T.; Shulman, G.I. Nonalcoholic fatty liver disease as a nexus of metabolic and hepatic diseases. Cell Metab. 2018, 27, 22–41. [Google Scholar] [CrossRef]
- Passarella, S. Phosphoenolpyruvate (PEP) and Mitochondria in Perspectives on Mitochondrial Transport and Energy Metabolism, 1st ed.; Aracne Editrice: Rome, Italy, 2019; pp. 233–269. [Google Scholar]
- Chen, Z.; Yu, Y.; Cai, J.; Li, H. Emerging Molecular Targets for Treatment of Nonalcoholic Fatty Liver Disease. Trends Endocrinol. Metab. TEM 2019, 30, 903–914. [Google Scholar] [CrossRef]
- Grattagliano, I.; Montezinho, L.P.; Oliveira, P.J.; Fruhbeck, G.; Gomez-Ambrosi, J.; Montecucco, F.; Carbone, F.; Wieckowski, M.R.; Wang, D.Q.; Portincasa, P. Targeting mitochondria to oppose the progression of nonalcoholic fatty liver disease. Biochem. Pharmacol. 2019, 160, 34–45. [Google Scholar] [CrossRef]
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: A prospective study. Gastroenterology 2011, 140, 124–131. [Google Scholar] [CrossRef]
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef]
- Lazo, M.; Hernaez, R.; Eberhardt, M.S.; Bonekamp, S.; Kamel, I.; Guallar, E.; Koteish, A.; Brancati, F.L.; Clark, J.M. Prevalence of nonalcoholic fatty liver disease in the United States: The Third National Health and Nutrition Examination Survey, 1988–1994. Am. J. Epidemiol. 2013, 178, 38–45. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Stepanova, M.; Afendy, M.; Fang, Y.; Younossi, Y.; Mir, H.; Srishord, M. Changes in the prevalence of the most common causes of chronic liver diseases in the United States from 1988 to 2008. Clin. Gastroenterol. Hepatol. 2011, 9, 524–530.e1. [Google Scholar] [CrossRef] [PubMed]
- Molina-Molina, E.; Lunardi Baccetto, R.; Wang, D.Q.; de Bari, O.; Krawczyk, M.; Portincasa, P. Exercising the hepatobiliary-gut axis. The impact of physical activity performance. Eur. J. Clin. Investig. 2018, 48, e12958. [Google Scholar] [CrossRef]
- Molina-Molina, E.; Krawczyk, M.; Stachowska, E.; Lammert, F.; Portincasa, P. Non-Alcoholic Fatty Liver Disease in Non-Obese Individuals: Prevalence, Pathogenesis and Treatment. Clin. Res. Hepatol. Gastroenterol. 2019, 43, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhou, F.; Wang, W.; Zhang, X.J.; Ji, Y.X.; Zhang, P.; She, Z.G.; Zhu, L.; Cai, J.; Li, H. Epidemiological Features of NAFLD From 1999 to 2018 in China. Hepatology 2020, 71, 1851–1864. [Google Scholar] [CrossRef] [PubMed]
- Schwenger, K.J.; Fischer, S.E.; Jackson, T.D.; Okrainec, A.; Allard, J.P. Non-alcoholic fatty liver disease in morbidly obese individuals undergoing bariatric surgery: Prevalence and effect of the pre-bariatric very low calorie diet. Obes. Surg. 2018, 28, 1109–1116. [Google Scholar] [CrossRef] [PubMed]
- Silverman, J.F.; O’Brien, K.F.; Long, S.; Leggett, N.; Khazanie, P.G.; Pories, W.J.; Norris, H.T.; Caro, J.F. Liver pathology in morbidly obese patients with and without diabetes. Am. J. Gastroenterol. Springer Nat. 1990, 85, 1349–1355. [Google Scholar]
- Meex, R.C.R.; Blaak, E.E. Mitochondrial Dysfunction is a Key Pathway that Links Saturated Fat Intake to the Development and Progression of NAFLD. Mol. Nutr. Food Res. 2021, 65, e1900942. [Google Scholar] [CrossRef]
- Loomba, R.; Sanyal, A.J. The global NAFLD epidemic. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.J.; Choi, K.M. Hepatokines as a link between obesity and cardiovascular diseases. Diabetes Metab. J. 2015, 39, 10. [Google Scholar] [CrossRef] [PubMed]
- Bril, F.; Cusi, K. Nonalcoholic Fatty Liver Disease: The New Complication of Type 2 Diabetes Mellitus. Endocrinol. Metab. Clin. N. Am. 2016, 45, 765–781. [Google Scholar] [CrossRef] [PubMed]
- Meex, R.C.; Hoy, A.J.; Morris, A.; Brown, R.D.; Lo, J.C.; Burke, M.; Goode, R.J.; Kingwell, B.A.; Kraakman, M.J.; Febbraio, M.A. Fetuin B is a secreted hepatocyte factor linking steatosis to impaired glucose metabolism. Cell Metab. 2015, 22, 1078–1089. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef]
- Ludwig, J.; Viggiano, T.R.; McGill, D.B.; Oh, B.J. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnaMed. disease. Mayo Clin. Proc. Mayo Clin. 1980, 55, 434–438. [Google Scholar] [PubMed]
- Caldwell, S.H.; Oelsner, D.H.; Iezzoni, J.C.; Hespenheide, E.E.; Battle, E.H.; Driscoll, C.J. Cryptogenic cirrhosis: Clinical characterization and risk factors for underlying disease. Hepatology 1999, 29, 664–669. [Google Scholar] [CrossRef]
- Browning, J.D.; Kumar, K.S.; Saboorian, M.H.; Thiele, D.L. Ethnic differences in the prevalence of cryptogenic cirrhosis. Am. J. Gastroenterol. 2004, 99, 292–298. [Google Scholar] [CrossRef]
- Nasr, P.; Ignatova, S.; Kechagias, S.; Ekstedt, M. Natural history of nonalcoholic fatty liver disease: A prospective follow-up study with serial biopsies. Hepatol. Commun. 2018, 2, 199–210. [Google Scholar] [CrossRef]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Mittal, S.; El-Serag, H.B.; Sada, Y.H.; Kanwal, F.; Duan, Z.; Temple, S.; May, S.B.; Kramer, J.R.; Richardson, P.A.; Davila, J.A. Hepatocellular Carcinoma in the Absence of Cirrhosis in United States Veterans is Associated With Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2016, 14, 124–131.e1. [Google Scholar] [CrossRef] [PubMed]
- National Institute of Alcohol Abuse and Alcoholism (NIH). Available online: https://pubs.niaaa.nih.gov/publications/practitioner/pocketguide/pocket_guide2.htm (accessed on 16 March 2021).
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Lindenmeyer, C.C.; McCullough, A.J. The Natural History of Nonalcoholic Fatty Liver Disease—An Evolving View. Clin. Liver Dis. 2018, 22, 11–21. [Google Scholar] [CrossRef]
- Rinella, M.E.; Sanyal, A.J. Management of NAFLD: A stage-based approach. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Vecchie, A.; Dallegri, F.; Carbone, F.; Bonaventura, A.; Liberale, L.; Portincasa, P.; Fruhbeck, G.; Montecucco, F. Obesity phenotypes and their paradoxical association with cardiovascular diseases. Eur. J. Intern. Med. 2018, 48, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Portincasa, P.; Wang, D.Q.H. Nonalcoholic fatty liver and gallstone disease. In Gallstones. Recent Advances in Epidemiology, Pathogenesis, Diagnosis and Management; Wang, D.Q.H., Portincasa, P., Eds.; Nova Science Publisher Inc.: New York, NY, USA, 2017; pp. 387–414. [Google Scholar]
- Grattagliano, I.; De Bari, O.; Di Palo, D.; Montecucco, F.; Carbone, F.; Oliveira, P.; Wang, D.Q.H.; Portincasa, P. Mitochondria in liver diseases. In Mitochondrial Biology and Experimental Therapeutics; Oliveira, P., Ed.; Springer Nature: Cham, Switzerland, 2018; pp. 91–126. [Google Scholar]
- Wilson, C.G.; Tran, J.L.; Erion, D.M.; Vera, N.B.; Febbraio, M.; Weiss, E.J. Hepatocyte-Specific Disruption of CD36 Attenuates Fatty Liver and Improves Insulin Sensitivity in HFD-Fed Mice. Endocrinology 2016, 157, 570–585. [Google Scholar] [CrossRef]
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef]
- Sunny, N.E.; Parks, E.J.; Browning, J.D.; Burgess, S.C. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 2011, 14, 804–810. [Google Scholar] [CrossRef]
- Marra, F.; Svegliati-Baroni, G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J. Hepatol. 2018, 68, 280–295. [Google Scholar] [CrossRef]
- Meex, R.C.R.; Watt, M.J. Hepatokines: Linking nonalcoholic fatty liver disease and insulin resistance. Nat. Rev. Endocrinol. 2017, 13, 509–520. [Google Scholar] [CrossRef]
- Li, Z.Z.; Berk, M.; McIntyre, T.M.; Feldstein, A.E. Hepatic Lipid Partitioning and Liver Damage in Nonalcoholic Fatty Liver Disease: Role of stearoyl-CoA desaturase. J. Biol. Chem. 2009, 284, 5637–5644. [Google Scholar] [CrossRef]
- Akazawa, Y.; Cazanave, S.; Mott, J.L.; Elmi, N.; Bronk, S.F.; Kohno, S.; Charlton, M.R.; Gores, G.J. Palmitoleate attenuates palmitate-induced Bim and PUMA up-regulation and hepatocyte lipoapoptosis. J. Hepatol. 2010, 52, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.Y.; Watt, M.J.; Rensen, S.; Greve, J.W.; Huynh, K.; Jayawardana, K.S.; Meikle, P.J.; Meex, R.C.R. Mitochondrial dysfunction-related lipid changes occur in nonalcoholic fatty liver disease progression. J. Lipid Res. 2018, 59, 1977–1986. [Google Scholar] [CrossRef] [PubMed]
- Parry, S.A.; Rosqvist, F.; Mozes, F.E.; Cornfield, T.; Hutchinson, M.; Piche, M.E.; Hulsmeier, A.J.; Hornemann, T.; Dyson, P.; Hodson, L. Intrahepatic Fat and Postprandial Glycemia Increase After Consumption of a Diet Enriched in Saturated Fat Compared With Free Sugars. Diabetes Care 2020, 43, 1134–1141. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.X.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374. [Google Scholar] [CrossRef] [PubMed]
- Papazyan, R.; Sun, Z.; Kim, Y.H.; Titchenell, P.M.; Hill, D.A.; Lu, W.; Damle, M.; Wan, M.; Zhang, Y.; Briggs, E.R.; et al. Physiological Suppression of Lipotoxic Liver Damage by Complementary Actions of HDAC3 and SCAP/SREBP. Cell Metab. 2016, 24, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, A.R.; Brasaemle, D.L.; McAndrews-Hill, M.; Sztalryd, C.; Londos, C. Adoption of PERILIPIN as a unifying nomenclature for the mammalian PAT-family of intracellular lipid storage droplet proteins. J. Lipid Res. 2010, 51, 468–471. [Google Scholar] [CrossRef] [PubMed]
- Brasaemle, D.L.; Rubin, B.; Harten, I.A.; Gruia-Gray, J.; Kimmel, A.R.; Londos, C. Perilipin A increases triacylglycerol storage by decreasing the rate of triacylglycerol hydrolysis. J. Biol. Chem. 2000, 275, 38486–38493. [Google Scholar] [CrossRef]
- Wang, C.; Zhao, Y.; Gao, X.; Li, L.; Yuan, Y.; Liu, F.; Zhang, L.; Wu, J.; Hu, P.; Zhang, X.; et al. Perilipin 5 improves hepatic lipotoxicity by inhibiting lipolysis. Hepatology 2015, 61, 870–882. [Google Scholar] [CrossRef]
- Han, M.S.; Park, S.Y.; Shinzawa, K.; Kim, S.; Chung, K.W.; Lee, J.H.; Kwon, C.H.; Lee, K.W.; Lee, J.H.; Park, C.K.; et al. Lysophosphatidylcholine as a death effector in the lipoapoptosis of hepatocytes. J. Lipid Res. 2008, 49, 84–97. [Google Scholar] [CrossRef]
- Mota, M.; Banini, B.A.; Cazanave, S.C.; Sanyal, A.J. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metab. Clin. Exp. 2016, 65, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Pagadala, M.; Kasumov, T.; McCullough, A.J.; Zein, N.N.; Kirwan, J.P. Role of ceramides in nonalcoholic fatty liver disease. Trends Endocrinol. Metab. TEM 2012, 23, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Raichur, S.; Wang, S.T.; Chan, P.W.; Li, Y.; Ching, J.; Chaurasia, B.; Dogra, S.; Ohman, M.K.; Takeda, K.; Sugii, S.; et al. CerS2 haploinsufficiency inhibits beta-oxidation and confers susceptibility to diet-induced steatohepatitis and insulin resistance. Cell Metab. 2014, 20, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, G.N. The Role of Cholesterol in the Pathogenesis of NASH. Trends Endocrinol. Metab. TEM 2016, 27, 84–95. [Google Scholar] [CrossRef]
- Bellanti, F.; Mitarotonda, D.; Tamborra, R.; Blonda, M.; Iannelli, G.; Petrella, A.; Sanginario, V.; Iuliano, L.; Vendemiale, G.; Serviddio, G. Oxysterols induce mitochondrial impairment and hepatocellular toxicity in non-alcoholic fatty liver disease. Free Radic. Biol. Med. 2014, 75 (Suppl. 1), S16–S17. [Google Scholar] [CrossRef]
- Müller, F.A.; Sturla, S.J. Human in vitro models of nonalcoholic fatty liver disease. Curr. Opin. Toxicol. 2019, 16, 9–16. [Google Scholar] [CrossRef]
- Fu, S.; Watkins, S.M.; Hotamisligil, G.S. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab. 2012, 15, 623–634. [Google Scholar] [CrossRef]
- Perry, R.J.; Samuel, V.T.; Petersen, K.F.; Shulman, G.I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 2014, 510, 84–91. [Google Scholar] [CrossRef]
- Ertunc, M.E.; Hotamisligil, G.S. Lipid signaling and lipotoxicity in metaflammation: Indications for metabolic disease pathogenesis and treatment. J. Lipid Res. 2016, 57, 2099–2114. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Xu, M.; Zhang, X.; Li, H. Innate Immune Signaling in Nonalcoholic Fatty Liver Disease and Cardiovascular Diseases. Annu. Rev. Pathol. 2019, 14, 153–184. [Google Scholar] [CrossRef]
- Cai, J.; Zhang, X.J.; Li, H. The Role of Innate Immune Cells in Nonalcoholic Steatohepatitis. Hepatology 2019, 70, 1026–1037. [Google Scholar] [CrossRef]
- Wang, X.A.; Zhang, R.; She, Z.G.; Zhang, X.F.; Jiang, D.S.; Wang, T.; Gao, L.; Deng, W.; Zhang, S.M.; Zhu, L.H.; et al. Interferon regulatory factor 3 constrains IKKbeta/NF-kappaB signaling to alleviate hepatic steatosis and insulin resistance. Hepatology 2014, 59, 870–885. [Google Scholar] [CrossRef] [PubMed]
- Passarella, S.; Atlante, A.; Valenti, D.; de Bari, L. The role of mitochondrial transport in energy metabolism. Mitochondrion 2003, 2, 319–343. [Google Scholar] [CrossRef]
- Serviddio, G.; Giudetti, A.M.; Bellanti, F.; Priore, P.; Rollo, T.; Tamborra, R.; Siculella, L.; Vendemiale, G.; Altomare, E.; Gnoni, G.V. Oxidation of hepatic carnitine palmitoyl transferase-I (CPT-I) impairs fatty acid beta-oxidation in rats fed a methionine-choline deficient diet. PLoS ONE 2011, 6, e24084. [Google Scholar] [CrossRef]
- Perez-Carreras, M.; Del Hoyo, P.; Martin, M.A.; Rubio, J.C.; Martin, A.; Castellano, G.; Colina, F.; Arenas, J.; Solis-Herruzo, J.A. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 2003, 38, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Einer, C.; Hohenester, S.; Wimmer, R.; Wottke, L.; Artmann, R.; Schulz, S.; Gosmann, C.; Simmons, A.; Leitzinger, C.; Eberhagen, C.; et al. Mitochondrial adaptation in steatotic mice. Mitochondrion 2018, 40, 1–12. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Campbell-Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J.N. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Zheng, F.; Pan, Q.; Zhang, S.; Yu, D.; Xu, Z.; Li, H. Glucose fluctuation increased hepatocyte apoptosis under lipotoxicity and the involvement of mitochondrial permeability transition opening. J. Mol. Endocrinol. 2015, 55, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Navarro, C.D.C.; Figueira, T.R.; Francisco, A.; Dal’Bo, G.A.; Ronchi, J.A.; Rovani, J.C.; Escanhoela, C.A.F.; Oliveira, H.C.F.; Castilho, R.F.; Vercesi, A.E. Redox imbalance due to the loss of mitochondrial NAD(P)-transhydrogenase markedly aggravates high fat diet-induced fatty liver disease in mice. Free Radic. Biol. Med. 2017, 113, 190–202. [Google Scholar] [CrossRef] [PubMed]
- King, A.L.; Swain, T.M.; Mao, Z.; Udoh, U.S.; Oliva, C.R.; Betancourt, A.M.; Griguer, C.E.; Crowe, D.R.; Lesort, M.; Bailey, S.M. Involvement of the mitochondrial permeability transition pore in chronic ethanol-mediated liver injury in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G265–G277. [Google Scholar] [CrossRef] [PubMed]
- Begriche, K.; Massart, J.; Robin, M.A.; Borgne-Sanchez, A.; Fromenty, B. Drug-induced toxicity on mitochondria and lipid metabolism: Mechanistic diversity and deleterious consequences for the liver. J. Hepatol. 2011, 54, 773–794. [Google Scholar] [CrossRef]
- Caldwell, S.H.; Crespo, D.M. The spectrum expanded: Cryptogenic cirrhosis and the natural history of non-alcoholic fatty liver disease. J. Hepatol. 2004, 40, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Petrosillo, G.; Portincasa, P.; Grattagliano, I.; Casanova, G.; Matera, M.; Ruggiero, F.M.; Ferri, D.; Paradies, G. Mitochondrial dysfunction in rat with nonalcoholic fatty liver Involvement of complex I, reactive oxygen species and cardiolipin. Biochim. Biophys. Acta 2007, 1767, 1260–1267. [Google Scholar] [CrossRef] [PubMed]
- Vecchione, G.; Grasselli, E.; Cioffi, F.; Baldini, F.; Oliveira, P.J.; Sardao, V.A.; Cortese, K.; Lanni, A.; Voci, A.; Portincasa, P.; et al. The Nutraceutic Silybin Counteracts Excess Lipid Accumulation and Ongoing Oxidative Stress in an In Vitro Model of Non-Alcoholic Fatty Liver Disease Progression. Front. Nutr. 2017, 4, 42. [Google Scholar] [CrossRef] [PubMed]
- Sunny, N.E.; Bril, F.; Cusi, K. Mitochondrial Adaptation in Nonalcoholic Fatty Liver Disease: Novel Mechanisms and Treatment Strategies. Trends Endocrinol. Metab.TEM 2017, 28, 250–260. [Google Scholar] [CrossRef]
- Portincasa, P.; Grattagliano, I.; Lauterburg, B.H.; Palmieri, V.O.; Palasciano, G.; Stellaard, F. Liver breath tests non-invasively predict higher stages of non-alcoholic steatohepatitis. Clin. Sci. 2006, 111, 135–143. [Google Scholar] [CrossRef]
- Miele, L.; Grieco, A.; Armuzzi, A.; Candelli, M.; Forgione, A.; Gasbarrini, A.; Gasbarrini, G. Hepatic mitochondrial beta-oxidation in patients with nonalcoholic steatohepatitis assessed by 13 C-octanoate breath test. Am. J. Gastroenterol. 2003, 98, 2335. [Google Scholar] [CrossRef]
- Grattagliano, I.; Bonfrate, L.; Oliveira, P.J.; Castorani, L.; Ruggiero, V.; Valenzano, A.T.; Ascensao, A.; Buzoianu, A.; Portincasa, P. Breath tests with novel 13C-substrates for clinical studies of liver mitochondrial function in health and disease. Eur. Rev. Med. Pharmacol. Sci. 2013, 17 (Suppl. 2), 72–81. [Google Scholar]
- Grattagliano, I.; Bonfrate, L.; Lorusso, M.; Castorani, L.; de Bari, O.; Portincasa, P. Exploring liver mitochondrial function by (1)(3)C-stable isotope breath tests: Implications in clinical biochemistry. Methods Mol. Biol. 2015, 1241, 137–152. [Google Scholar] [CrossRef]
- Bonfrate, L.; Grattagliano, I.; Palasciano, G.; Portincasa, P. Dynamic carbon 13 breath tests for the study of liver function and gastric emptying. Gastroenterol. Rep. 2015, 3, 12–21. [Google Scholar] [CrossRef]
- Schmid, A.I.; Szendroedi, J.; Chmelik, M.; Krssak, M.; Moser, E.; Roden, M. Liver ATP synthesis is lower and relates to insulin sensitivity in patients with type 2 diabetes. Diabetes Care 2011, 34, 448–453. [Google Scholar] [CrossRef]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.A.; Deja, S.; Satapati, S.; Fu, X.; Burgess, S.C.; Browning, J.D. Impaired ketogenesis and increased acetyl-CoA oxidation promote hyperglycemia in human fatty liver. JCI Insight 2019, 5. [Google Scholar] [CrossRef]
- Satapati, S.; Kucejova, B.; Duarte, J.A.; Fletcher, J.A.; Reynolds, L.; Sunny, N.E.; He, T.; Nair, L.A.; Livingston, K.A.; Fu, X.; et al. Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J. Clin. Investig. 2015, 125, 4447–4462. [Google Scholar] [CrossRef]
- Festi, D.; Capodicasa, S.; Sandri, L.; Colaiocco-Ferrante, L.; Staniscia, T.; Vitacolonna, E.; Vestito, A.; Simoni, P.; Mazzella, G.; Portincasa, P.; et al. Measurement of hepatic functional mass by means of 13C-methacetin and 13C-phenylalanine breath tests in chronic liver disease: Comparison with Child-Pugh score and serum bile acid levels. World J. Gastroenterol. WJG 2005, 11, 142–148. [Google Scholar] [CrossRef]
- Grattagliano, I.; Lauterburg, B.H.; Palasciano, G.; Portincasa, P. 13C-breath tests for clinical investigation of liver mitochondrial function. Eur. J. Clin. Investig. 2010, 40, 843–850. [Google Scholar] [CrossRef]
- Palmieri, V.O.; Grattagliano, I.; Minerva, F.; Pollice, S.; Palasciano, G.; Portincasa, P. Liver function as assessed by breath tests in patients with hepatocellular carcinoma. J. Surg. Res. 2009, 157, 199–207. [Google Scholar] [CrossRef]
- Perri, F.; Bellini, M.; Portincasa, P.; Parodi, A.; Bonazzi, P.; Marzio, L.; Galeazzi, F.; Usai, P.; Citrino, A.; Usai-Satta, P. (13)C-octanoic acid breath test (OBT) with a new test meal (EXPIROGer): Toward standardization for testing gastric emptying of solids. Dig. Liver Dis. 2010, 42, 549–553. [Google Scholar] [CrossRef]
- Win, S.; Than, T.A.; Le, B.H.; Garcia-Ruiz, C.; Fernandez-Checa, J.C.; Kaplowitz, N. Sab (Sh3bp5) dependence of JNK mediated inhibition of mitochondrial respiration in palmitic acid induced hepatocyte lipotoxicity. J. Hepatol. 2015, 62, 1367–1374. [Google Scholar] [CrossRef]
- Gariani, K.; Menzies, K.J.; Ryu, D.; Wegner, C.J.; Wang, X.; Ropelle, E.R.; Moullan, N.; Zhang, H.; Perino, A.; Lemos, V.; et al. Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice. Hepatology 2016, 63, 1190–1204. [Google Scholar] [CrossRef]
- Sunny, N.E.; Kalavalapalli, S.; Bril, F.; Garrett, T.J.; Nautiyal, M.; Mathew, J.T.; Williams, C.M.; Cusi, K. Cross-talk between branched-chain amino acids and hepatic mitochondria is compromised in nonalcoholic fatty liver disease. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E311–E319. [Google Scholar] [CrossRef]
- Mehta, R.; Jeiran, K.; Koenig, A.B.; Otgonsuren, M.; Goodman, Z.; Baranova, A.; Younossi, Z. The role of mitochondrial genomics in patients with non-alcoholic steatohepatitis (NASH). BMC Med. Genet. 2016, 17, 63. [Google Scholar] [CrossRef]
- Musso, G.; Gambino, R.; De Michieli, F.; Biroli, G.; Premoli, A.; Pagano, G.; Bo, S.; Durazzo, M.; Cassader, M. Nitrosative stress predicts the presence and severity of nonalcoholic fatty liver at different stages of the development of insulin resistance and metabolic syndrome: Possible role of vitamin A intake. Am. J. Clin. Nutr. 2007, 86, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Mantena, S.K.; King, A.L.; Andringa, K.K.; Landar, A.; Darley-Usmar, V.; Bailey, S.M. Novel interactions of mitochondria and reactive oxygen/nitrogen species in alcohol mediated liver disease. World J. Gastroenterol. WJG 2007, 13, 4967–4973. [Google Scholar] [CrossRef] [PubMed]
- Vanni, E.; Marengo, A.; Mezzabotta, L.; Bugianesi, E. Systemic complications of nonalcoholic fatty liver disease: When the liver is not an innocent bystander. Semin. Liver Dis. 2015, 35, 236–249. [Google Scholar] [CrossRef] [PubMed]
- Martel, C.; Allouche, M.; Esposti, D.D.; Fanelli, E.; Boursier, C.; Henry, C.; Chopineau, J.; Calamita, G.; Kroemer, G.; Lemoine, A.; et al. GSK3-mediated VDAC phosphorylation controls outer mitochondrial membrane permeability during lipid accumulation. Hepatology 2013, 57, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E.; Bayir, H.A.; Belikova, N.A.; Kapralov, O.; Tyurina, Y.Y.; Tyurin, V.A.; Jiang, J.; Stoyanovsky, D.A.; Wipf, P.; Kochanek, P.M.; et al. Cytochrome c/cardiolipin relations in mitochondria: A kiss of death. Free Radic. Biol. Med. 2009, 46, 1439–1453. [Google Scholar] [CrossRef] [PubMed]
- Afonso, M.B.; Rodrigues, P.M.; Simao, A.L.; Ofengeim, D.; Carvalho, T.; Amaral, J.D.; Gaspar, M.M.; Cortez-Pinto, H.; Castro, R.E.; Yuan, J.; et al. Activation of necroptosis in human and experimental cholestasis. Cell Death Dis. 2016, 7, e2390. [Google Scholar] [CrossRef]
- Haouzi, D.; Lekehal, M.; Moreau, A.; Moulis, C.; Feldmann, G.; Robin, M.A.; Letteron, P.; Fau, D.; Pessayre, D. Cytochrome P450-generated reactive metabolites cause mitochondrial permeability transition, caspase activation, and apoptosis in rat hepatocytes. Hepatology 2000, 32, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, Y.; Shimizu, S. VDAC regulation by the Bcl-2 family of proteins. Cell Death Differ. 2000, 7, 1174–1181. [Google Scholar] [CrossRef] [PubMed]
- Atlante, A.; Bobba, A.; De Bari, L.; Fontana, F.; Calissano, P.; Marra, E.; Passarella, S. Caspase-dependent alteration of the ADP/ATP translocator triggers the mitochondrial permeability transition which is not required for the low-potassium-dependent apoptosis of cerebellar granule cells. J. Neurochem. 2006, 97, 1166–1181. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Morganti, C.; Morciano, G.; Pedriali, G.; Lebiedzinska-Arciszewska, M.; Aquila, G.; Giorgi, C.; Rizzo, P.; Campo, G.; Ferrari, R.; et al. Mitochondrial permeability transition involves dissociation of F1FO ATP synthase dimers and C-ring conformation. Embo Rep. 2017, 18, 1077–1089. [Google Scholar] [CrossRef]
- Elustondo, P.A.; Nichols, M.; Negoda, A.; Thirumaran, A.; Zakharian, E.; Robertson, G.S.; Pavlov, E.V. Mitochondrial permeability transition pore induction is linked to formation of the complex of ATPase C-subunit, polyhydroxybutyrate and inorganic polyphosphate. Cell Death Discov. 2016, 2, 16070. [Google Scholar] [CrossRef]
- He, J.; Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Permeability transition in human mitochondria persists in the absence of peripheral stalk subunits of ATP synthase. Proc. Natl. Acad. Sci. USA 2017, 114, 9086–9091. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Ford, H.C.; Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Persistence of the mitochondrial permeability transition in the absence of subunit c of human ATP synthase. Proc. Natl. Acad. Sci. USA 2017, 114, 3409–3414. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Papucci, L.; Formigli, L.; Schiavone, N.; Tani, A.; Donnini, M.; Lapucci, A.; Perna, F.; Tempestini, A.; Witort, E.; Morganti, M.; et al. Apoptosis shifts to necrosis via intermediate types of cell death by a mechanism depending on c-myc and bcl-2 expression. Cell Tissue Res. 2004, 316, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Fouret, G.; Gaillet, S.; Lecomte, J.; Bonafos, B.; Djohan, F.; Barea, B.; Badia, E.; Coudray, C.; Feillet-Coudray, C. 20-Week follow-up of hepatic steatosis installation and liver mitochondrial structure and activity and their interrelation in rats fed a high-fat–high-fructose diet. Br. J. Nutr. 2018, 119, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Seki, S.; Kitada, T.; Yamada, T.; Sakaguchi, H.; Nakatani, K.; Wakasa, K. In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver diseases. J. Hepatol. 2002, 37, 56–62. [Google Scholar] [CrossRef]
- Wang, J.; He, W.; Tsai, P.-J.; Chen, P.-H.; Ye, M.; Guo, J.; Su, Z. Mutual interaction between endoplasmic reticulum and mitochondria in nonalcoholic fatty liver disease. Lipids Health Dis. 2020, 19, 1–19. [Google Scholar] [CrossRef]
- Sheka, A.C.; Adeyi, O.; Thompson, J.; Hameed, B.; Crawford, P.A.; Ikramuddin, S. Nonalcoholic Steatohepatitis: A Review. JAMA 2020, 323, 1175–1183. [Google Scholar] [CrossRef]
- Promrat, K.; Kleiner, D.E.; Niemeier, H.M.; Jackvony, E.; Kearns, M.; Wands, J.R.; Fava, J.L.; Wing, R.R. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology 2010, 51, 121–129. [Google Scholar] [CrossRef]
- Keating, S.E.; Hackett, D.A.; George, J.; Johnson, N.A. Exercise and non-alcoholic fatty liver disease: A systematic review and meta-analysis. J. Hepatol. 2012, 57, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Keating, S.E.; Hackett, D.A.; Parker, H.M.; O’Connor, H.T.; Gerofi, J.A.; Sainsbury, A.; Baker, M.K.; Chuter, V.H.; Caterson, I.D.; George, J.; et al. Effect of aerobic exercise training dose on liver fat and visceral adiposity. J. Hepatol. 2015, 63, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Vilar-Gomez, E.; Martinez-Perez, Y.; Calzadilla-Bertot, L.; Torres-Gonzalez, A.; Gra-Oramas, B.; Gonzalez-Fabian, L.; Friedman, S.L.; Diago, M.; Romero-Gomez, M. Weight Loss Through Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroenterology 2015, 149, 367–378.e5. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.F.; Dufour, S.; Befroy, D.; Lehrke, M.; Hendler, R.E.; Shulman, G.I. Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes 2005, 54, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Cassader, M.; Rosina, F.; Gambino, R. Impact of current treatments on liver disease, glucose metabolism and cardiovascular risk in non-alcoholic fatty liver disease (NAFLD): A systematic review and meta-analysis of randomised trials. Diabetologia 2012, 55, 885–904. [Google Scholar] [CrossRef] [PubMed]
- Molina-Molina, E.; Shanmugam, H.; Di Ciaula, A.; Grattagliano, I.; Di Palo, D.M.; Palmieri, V.O.; Portincasa, P. ((13)C)-Methacetin breath test provides evidence of subclinical liver dysfunction linked to fat storage but not lifestyle. JHEP Rep. 2021, 3, 100203. [Google Scholar] [CrossRef]
- Cerqueira, F.M.; Cunha, F.M.d.; Silva, C.C.; Chausse, B.; Romano, R.L.; Garcia, C.; Colepicolo, P.; Medeiros, M.H.G.d.; Kowaltowski, A.J. Redox state, insulin sensitivity and aging. In Resumos; Reunião Anual da Federação de Sociedades de Biologia Experimental: São Paulo, Brazil, 2011. [Google Scholar]
- Kowaltowski, A.J. Caloric restriction and redox state: Does this diet increase or decrease oxidant production? Redox Rep. 2011, 16, 237–241. [Google Scholar] [CrossRef]
- Walsh, M.E.; Shi, Y.; Van Remmen, H. The effects of dietary restriction on oxidative stress in rodents. Free Radic. Biol. Med. 2014, 66, 88–99. [Google Scholar] [CrossRef]
- Tetri, L.H.; Basaranoglu, M.; Brunt, E.M.; Yerian, L.M.; Neuschwander-Tetri, B.A. Severe NAFLD with hepatic necroinflammatory changes in mice fed trans fats and a high-fructose corn syrup equivalent. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G987–G995. [Google Scholar] [CrossRef] [PubMed]
- Romero-Gomez, M.; Zelber-Sagi, S.; Trenell, M. Treatment of NAFLD with diet, physical activity and exercise. J. Hepatol. 2017, 67, 829–846. [Google Scholar] [CrossRef]
- Bower, G.; Toma, T.; Harling, L.; Jiao, L.R.; Efthimiou, E.; Darzi, A.; Athanasiou, T.; Ashrafian, H. Bariatric Surgery and Non-Alcoholic Fatty Liver Disease: A Systematic Review of Liver Biochemistry and Histology. Obes. Surg. 2015, 25, 2280–2289. [Google Scholar] [CrossRef]
- Mathurin, P.; Hollebecque, A.; Arnalsteen, L.; Buob, D.; Leteurtre, E.; Caiazzo, R.; Pigeyre, M.; Verkindt, H.; Dharancy, S.; Louvet, A. Prospective study of the long-term effects of bariatric surgery on liver injury in patients without advanced disease. Gastroenterology 2009, 137, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.B.; Bhathal, P.S.; Hughes, N.R.; O’Brien, P.E. Nonalcoholic fatty liver disease: Improvement in liver histological analysis with weight loss. Hepatology 2004, 39, 1647–1654. [Google Scholar] [CrossRef]
- Clark, J.M.; Alkhuraishi, A.R.; Solga, S.F.; Alli, P.; Diehl, A.M.; Magnuson, T.H. Roux-en-Y gastric bypass improves liver histology in patients with non-alcoholic fatty liver disease. Obes. Res. 2005, 13, 1180–1186. [Google Scholar] [CrossRef]
- Tai, C.M.; Huang, C.K.; Hwang, J.C.; Chiang, H.; Chang, C.Y.; Lee, C.T.; Yu, M.L.; Lin, J.T. Improvement of nonalcoholic fatty liver disease after bariatric surgery in morbidly obese Chinese patients. Obes. Surg. 2012, 22, 1016–1021. [Google Scholar] [CrossRef]
- Lee, Y.; Doumouras, A.G.; Yu, J.; Brar, K.; Banfield, L.; Gmora, S.; Anvari, M.; Hong, D. Complete Resolution of Nonalcoholic Fatty Liver Disease After Bariatric Surgery: A Systematic Review and Meta-analysis. Clin. Gastroenterol. Hepatol. 2019, 17, 1040–1060.e11. [Google Scholar] [CrossRef]
- De Almeida, S.R.; Rocha, P.R.; Sanches, M.D.; Leite, V.H.; da Silva, R.A.; Diniz, M.T.; Diniz Mde, F.; Rocha, A.L. Roux-en-Y gastric bypass improves the nonalcoholic steatohepatitis (NASH) of morbid obesity. Obes. Surg. 2006, 16, 270–278. [Google Scholar] [CrossRef]
- Lassailly, G.; Caiazzo, R.; Buob, D.; Pigeyre, M.; Verkindt, H.; Labreuche, J.; Raverdy, V.; Leteurtre, E.; Dharancy, S.; Louvet, A.; et al. Bariatric Surgery Reduces Features of Nonalcoholic Steatohepatitis in Morbidly Obese Patients. Gastroenterology 2015, 149, 379–388. [Google Scholar] [CrossRef]
- Chavez-Tapia, N.C.; Tellez-Avila, F.I.; Barrientos-Gutierrez, T.; Mendez-Sanchez, N.; Lizardi-Cervera, J.; Uribe, M. Bariatric surgery for non-alcoholic steatohepatitis in obese patients. Cochrane Database Syst. Rev. 2010, CD007340. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, I.O.; Passos, E.; Rocha-Rodrigues, S.; Torrella, J.R.; Rizo, D.; Santos-Alves, E.; Portincasa, P.; Martins, M.J.; Ascensao, A.; Magalhaes, J. Physical exercise antagonizes clinical and anatomical features characterizing Lieber-DeCarli diet-induced obesity and related metabolic disorders. Clin. Nutr. 2015, 34, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Ekstedt, M.; Franzen, L.E.; Holmqvist, M.; Bendtsen, P.; Mathiesen, U.L.; Bodemar, G.; Kechagias, S. Alcohol consumption is associated with progression of hepatic fibrosis in non-alcoholic fatty liver disease. Scand. J. Gastroenterol. 2009, 44, 366–374. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, Z.; Li, H.; Zhao, J.; Wei, X.; Lin, W.; Zhao, X.; Jiang, A.; Yuan, J. Endogenous ethanol produced by intestinal bacteria induces mitochondrial dysfunction in non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2020, 35, 2009–2019. [Google Scholar] [CrossRef] [PubMed]
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Masciana, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Rotman, Y.; Sanyal, A.J. Current and upcoming pharmacotherapy for non-alcoholic fatty liver disease. Gut 2017, 66, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, R.; Onali, S.; Thorburn, D.; Davidson, B.R.; Gurusamy, K.S.; Tsochatzis, E. Pharmacological interventions for non-alcohol related fatty liver disease (NAFLD): An attempted network meta-analysis. Cochrane Database Syst. Rev. 2017, 3, CD011640. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef]
- Barreyro, F.J.; Holod, S.; Finocchietto, P.V.; Camino, A.M.; Aquino, J.B.; Avagnina, A.; Carreras, M.C.; Poderoso, J.J.; Gores, G.J. The pan-caspase inhibitor Emricasan (IDN-6556) decreases liver injury and fibrosis in a murine model of non-alcoholic steatohepatitis. Liver Int. 2015, 35, 953–966. [Google Scholar] [CrossRef]
- Alkhouri, N.; Carter-Kent, C.; Feldstein, A.E. Apoptosis in nonalcoholic fatty liver disease: Diagnostic and therapeutic implications. Expert Rev. Gastroenterol. Hepatol. 2011, 5, 201–212. [Google Scholar] [CrossRef]
- Rakoski, M.O.; Singal, A.G.; Rogers, M.A.; Conjeevaram, H. Meta-analysis: Insulin sensitizers for the treatment of non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2010, 32, 1211–1221. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, L.; Wang, B.; Wang, J.; Chen, D. Metformin in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Biomed. Rep. 2013, 1, 57–64. [Google Scholar] [CrossRef]
- Cusi, K.; Orsak, B.; Bril, F.; Lomonaco, R.; Hecht, J.; Ortiz-Lopez, C.; Tio, F.; Hardies, J.; Darland, C.; Musi, N.; et al. Long-Term Pioglitazone Treatment for Patients With Nonalcoholic Steatohepatitis and Prediabetes or Type 2 Diabetes Mellitus: A Randomized Trial. Ann. Intern. Med. 2016, 165, 305–315. [Google Scholar] [CrossRef]
- Aithal, G.P.; Thomas, J.A.; Kaye, P.V.; Lawson, A.; Ryder, S.D.; Spendlove, I.; Austin, A.S.; Freeman, J.G.; Morgan, L.; Webber, J. Randomized, placebo-controlled trial of pioglitazone in nondiabetic subjects with nonalcoholic steatohepatitis. Gastroenterology 2008, 135, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Belfort, R.; Harrison, S.A.; Brown, K.; Darland, C.; Finch, J.; Hardies, J.; Balas, B.; Gastaldelli, A.; Tio, F.; Pulcini, J.; et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N. Engl. J. Med. 2006, 355, 2297–2307. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Mofrad, P.S.; Contos, M.J.; Sargeant, C.; Luketic, V.A.; Sterling, R.K.; Stravitz, R.T.; Shiffman, M.L.; Clore, J.; Mills, A.S. A pilot study of vitamin E versus vitamin E and pioglitazone for the treatment of nonalcoholic steatohepatitis. Clin. Gastroenterol. Hepatol. 2004, 2, 1107–1115. [Google Scholar] [CrossRef]
- Mahady, S.E.; Wong, G.; Craig, J.C.; George, J. Pioglitazone and vitamin E for nonalcoholic steatohepatitis: A cost utility analysis. Hepatology 2012, 56, 2172–2179. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Cassader, M.; Paschetta, E.; Gambino, R. Thiazolidinediones and Advanced Liver Fibrosis in Nonalcoholic Steatohepatitis: A Meta-analysis. JAMA Intern. Med. 2017, 177, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Khera, R.; Allen, A.M.; Murad, M.H.; Loomba, R. Comparative effectiveness of pharmacological interventions for nonalcoholic steatohepatitis: A systematic review and network meta-analysis. Hepatology 2015, 62, 1417–1432. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A.; Brunt, E.M.; Wehmeier, K.R.; Oliver, D.; Bacon, B.R. Improved nonalcoholic steatohepatitis after 48 weeks of treatment with the PPAR-gamma ligand rosiglitazone. Hepatology 2003, 38, 1008–1017. [Google Scholar] [CrossRef]
- Ratziu, V.; Charlotte, F.; Bernhardt, C.; Giral, P.; Halbron, M.; Lenaour, G.; Hartmann-Heurtier, A.; Bruckert, E.; Poynard, T.; Group, L.S. Long-term efficacy of rosiglitazone in nonalcoholic steatohepatitis: Results of the fatty liver improvement by rosiglitazone therapy (FLIRT 2) extension trial. Hepatology 2010, 51, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Giral, P.; Jacqueminet, S.; Charlotte, F.; Hartemann-Heurtier, A.; Serfaty, L.; Podevin, P.; Lacorte, J.M.; Bernhardt, C.; Bruckert, E.; et al. Rosiglitazone for nonalcoholic steatohepatitis: One-year results of the randomized placebo-controlled Fatty Liver Improvement with Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology 2008, 135, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E.; Lominadze, Z.; Loomba, R.; Charlton, M.; Neuschwander-Tetri, B.A.; Caldwell, S.H.; Kowdley, K.; Harrison, S.A. Practice patterns in NAFLD and NASH: Real life differs from published guidelines. Ther. Adv. Gastroenterol. 2016, 9, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Alkhouri, N.; Davison, B.A.; Sanyal, A.; Edwards, C.; Colca, J.R.; Lee, B.H.; Loomba, R.; Cusi, K.; Kolterman, O.; et al. Insulin sensitizer MSDC-0602K in non-alcoholic steatohepatitis: A randomized, double-blind, placebo-controlled phase IIb study. J. Hepatol. 2020, 72, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Staels, B.; Rubenstrunk, A.; Noel, B.; Rigou, G.; Delataille, P.; Millatt, L.J.; Baron, M.; Lucas, A.; Tailleux, A.; Hum, D.W.; et al. Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2013, 58, 1941–1952. [Google Scholar] [CrossRef]
- Hanf, R.; Millatt, L.J.; Cariou, B.; Noel, B.; Rigou, G.; Delataille, P.; Daix, V.; Hum, D.W.; Staels, B. The dual peroxisome proliferator-activated receptor alpha/delta agonist GFT505 exerts anti-diabetic effects in db/db mice without peroxisome proliferator-activated receptor gamma–associated adverse cardiac effects. Diabetes Vasc. Dis. Res. 2014, 11, 440–447. [Google Scholar] [CrossRef]
- Cariou, B.; Hanf, R.; Lambert-Porcheron, S.; Zair, Y.; Sauvinet, V.; Noel, B.; Flet, L.; Vidal, H.; Staels, B.; Laville, M. Dual peroxisome proliferator-activated receptor alpha/delta agonist GFT505 improves hepatic and peripheral insulin sensitivity in abdominally obese subjects. Diabetes Care 2013, 36, 2923–2930. [Google Scholar] [CrossRef]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-alpha and -delta, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening. Gastroenterology 2016, 150, 1147–1159.e5. [Google Scholar] [CrossRef]
- Gawrieh, S.; Noureddin, M.; Loo, N.M.; Mohseni, R.; Awasty, V.R.; Cusi, K.; Kowdley, K.V.; Lai, M.; Schiff, E.R.; Parmar, D.V. A phase 2, prospective, multicenter, double-blind, randomized study of saroglitazar magnesium 1 mg, 2 mg or 4 mg versus placebo in patients with nonalcoholic fatty liver disease and/or nonalcoholic steatohepatitis (EVIDENCES IV). Hepatology 2019, 70, 1484A–1485A. [Google Scholar]
- Jinnouchi, H.; Sugiyama, S.; Yoshida, A.; Hieshima, K.; Kurinami, N.; Suzuki, T.; Miyamoto, F.; Kajiwara, K.; Matsui, K.; Jinnouchi, T. Liraglutide, a glucagon-like peptide-1 analog, increased insulin sensitivity assessed by hyperinsulinemic-euglycemic clamp examination in patients with uncontrolled type 2 diabetes mellitus. J. Diabetes Res. 2015, 2015, 706416. [Google Scholar] [CrossRef]
- Petit, J.M.; Cercueil, J.P.; Loffroy, R.; Denimal, D.; Bouillet, B.; Fourmont, C.; Chevallier, O.; Duvillard, L.; Verges, B. Effect of Liraglutide Therapy on Liver Fat Content in Patients With Inadequately Controlled Type 2 Diabetes: The Lira-NAFLD Study. J. Clin. Endocrinol. Metab. 2017, 102, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Houlihan, D.D.; Rowe, I.A.; Clausen, W.H.; Elbrond, B.; Gough, S.C.; Tomlinson, J.W.; Newsome, P.N. Safety and efficacy of liraglutide in patients with type 2 diabetes and elevated liver enzymes: Individual patient data meta-analysis of the LEAD program. Aliment. Pharmacol. Ther. 2013, 37, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; LEAN Trial Team; Abouda, G.; et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef]
- Nahra, R.; Wang, T.; Gadde, K.; Stumvoll, M.; Jermutus, L.; Hirshberg, B.; Ambery, P. Holistic effects of cotadutide (medi0382) on metabolic, cardiovascular, and hepatic parameters in overweight or obese subjects with type 2 diabetes mellitus (T2DM): A 26-week analysis of a randomized phase 2b study. Hepatology 2019, 70, 1354A–1355A. [Google Scholar]
- Loomba, R.; Kayali, Z.; Noureddin, M.; Ruane, P.; Lawitz, E.J.; Bennett, M.; Wang, L.; Harting, E.; Tarrant, J.M.; McColgan, B.J.; et al. GS-0976 Reduces Hepatic Steatosis and Fibrosis Markers in Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2018, 155, 1463–1473.e6. [Google Scholar] [CrossRef]
- Kim, C.W.; Addy, C.; Kusunoki, J.; Anderson, N.N.; Deja, S.; Fu, X.; Burgess, S.C.; Li, C.; Ruddy, M.; Chakravarthy, M.; et al. Acetyl CoA Carboxylase Inhibition Reduces Hepatic Steatosis but Elevates Plasma Triglycerides in Mice and Humans: A Bedside to Bench Investigation. Cell Metab. 2017, 26, 576. [Google Scholar] [CrossRef]
- Lambrecht, J.; Tacke, F. Acetyl-CoA Carboxylase Inhibition as a Therapeutic Tool in the Battle Against NASH: Hitting More Than Just One Mechanism? Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 859–861. [Google Scholar] [CrossRef]
- Amin, N.; Carvajal-Gonzalez, S.; Aggarwal, N.; Tuthill, T.; Inglot, M.; Bergman, A.; Esler, W. PF-05221304 (PF’1304), A liver-targeted Acetyl-CoA Carboxylase Inhibitor (ACCI), in adults with nonalcoholic fatty liver disease (NAFLD) demonstrates robust reductions in liver fat and ALT-phase 2a, dose-ranging study. Hepatology 2019, 70, 21A–22A. [Google Scholar]
- Dufour, J.F.; Caussy, C.; Loomba, R. Combination therapy for non-alcoholic steatohepatitis: Rationale, opportunities and challenges. Gut 2020, 69, 1877–1884. [Google Scholar] [CrossRef]
- Calle, R.; Bergman, A.; Somayaji, V.; Chidsey, K.; Kazierad, D. PS-110-Ketohexokinase inhibitor PF-06835919 administered for 6 weeks reduces whole liver fat as measured by magnetic resonance imaging-proton density fat fraction in subjects with non-alcoholic fatty liver disease. J. Hepatol. 2019, 70, e69–e70. [Google Scholar] [CrossRef]
- Verschueren, K.H.G.; Blanchet, C.; Felix, J.; Dansercoer, A.; De Vos, D.; Bloch, Y.; Van Beeumen, J.; Svergun, D.; Gutsche, I.; Savvides, S.N.; et al. Structure of ATP citrate lyase and the origin of citrate synthase in the Krebs cycle. Nature 2019, 568, 571–575. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef]
- Mudaliar, S.; Henry, R.R.; Sanyal, A.J.; Morrow, L.; Marschall, H.U.; Kipnes, M.; Adorini, L.; Sciacca, C.I.; Clopton, P.; Castelloe, E.; et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 2013, 145, 574–582.e1. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Sanyal, A.J.; Loomba, R.; Rinella, M.; Harrison, S.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; MacConell, L.; Shringarpure, R.; et al. REGENERATE: Design of a pivotal, randomised, phase 3 study evaluating the safety and efficacy of obeticholic acid in patients with fibrosis due to nonalcoholic steatohepatitis. Contemp. Clin. Trials 2019, 84, 105803. [Google Scholar] [CrossRef]
- Min, H.K.; Kapoor, A.; Fuchs, M.; Mirshahi, F.; Zhou, H.; Maher, J.; Kellum, J.; Warnick, R.; Contos, M.J.; Sanyal, A.J. Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell Metab. 2012, 15, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; Geier, A.; Beckebaum, S.; Newsome, P.N.; et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: Interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2019, 394, 2184–2196. [Google Scholar] [CrossRef]
- Sanyal, A.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.; Goodman, Z.; Bedossa, P.; Khalili, M.; Boursier, J.; Stinton, L. Obeticholic acid treatment in patients with non-alcoholic steatohepatitis: A secondary analysis in the regenerate study across fibrosis stages. In Proceedings of the AISF Annual Meeting, Rome, Italy, 27 February 2020; pp. 46–47. [Google Scholar]
- Lucas, K.; Lopez, P.; Lawitz, E.; Sheikh, A.; Aizenberg, D.; Hsia, S.; Bee, G.B.; Vierling, J.; Frias, J.; White, J. Tropifexor, a highly potent FXR agonist, produces robust and dose-dependent reductions in hepatic fat and serum alanine aminotransferase in patients with fibrotic NASH after 12 weeks of therapy: FLIGHT-FXR Part C interim results. Dig. Liver Dis. 2020, 52, e38. [Google Scholar] [CrossRef]
- An, P.; Wei, G.; Huang, P.; Li, W.; Qi, X.; Lin, Y.; Vaid, K.A.; Wang, J.; Zhang, S.; Li, Y.; et al. A novel non-bile acid FXR agonist EDP-305 potently suppresses liver injury and fibrosis without worsening of ductular reaction. Liver Int. 2020, 40, 1655–1669. [Google Scholar] [CrossRef]
- Ajmera, V.H.; Cachay, E.; Ramers, C.; Vodkin, I.; Bassirian, S.; Singh, S.; Mangla, N.; Bettencourt, R.; Aldous, J.L.; Park, D.; et al. MRI Assessment of Treatment Response in HIV-associated NAFLD: A Randomized Trial of a Stearoyl-Coenzyme-A-Desaturase-1 Inhibitor (ARRIVE Trial). Hepatology 2019, 70, 1531–1545. [Google Scholar] [CrossRef] [PubMed]
- Iruarrizaga-Lejarreta, M.; Varela-Rey, M.; Fernandez-Ramos, D.; Martinez-Arranz, I.; Delgado, T.C.; Simon, J.; Juan, V.G.; delaCruz-Villar, L.; Azkargorta, M.; Lavin, J.L.; et al. Role of Aramchol in steatohepatitis and fibrosis in mice. Hepatol. Commun. 2017, 1, 911–927. [Google Scholar] [CrossRef]
- Safadi, R.; Konikoff, F.M.; Mahamid, M.; Zelber-Sagi, S.; Halpern, M.; Gilat, T.; Oren, R.; Group, F. The fatty acid-bile acid conjugate Aramchol reduces liver fat content in patients with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2014, 12, 2085–2091.e1. [Google Scholar] [CrossRef]
- Ratziu, V.; Ladron-De-Guevara, L.; Safadi, R.; Poordad, F.; Fuster, F.; Flores-Figueroa, J.; Harrison, S.A.; Arrese, M.; Fargion, S.; Ben-Bashat, D. One-year results of the global phase 2b randomized placebo-controlled arrest trial of aramchol, a stearoyl CoA desaturase inhibitor, in patients with NASH. Hepatology 2018, 68, 1448A–1449A. [Google Scholar] [CrossRef]
- Traussnigg, S.; Schattenberg, J.M.; Demir, M.; Wiegand, J.; Geier, A.; Teuber, G.; Hofmann, W.P.; Kremer, A.E.; Spreda, F.; Kluwe, J.; et al. Norursodeoxycholic acid versus placebo in the treatment of non-alcoholic fatty liver disease: A double-blind, randomised, placebo-controlled, phase 2 dose-finding trial. Lancet Gastroenterol. Hepatol. 2019, 4, 781–793. [Google Scholar] [CrossRef]
- Nies, V.J.; Sancar, G.; Liu, W.; van Zutphen, T.; Struik, D.; Yu, R.T.; Atkins, A.R.; Evans, R.M.; Jonker, J.W.; Downes, M.R. Fibroblast Growth Factor Signaling in Metabolic Regulation. Front. Endocrinol. 2015, 6, 193. [Google Scholar] [CrossRef]
- Fu, L.; John, L.M.; Adams, S.H.; Yu, X.X.; Tomlinson, E.; Renz, M.; Williams, P.M.; Soriano, R.; Corpuz, R.; Moffat, B.; et al. Fibroblast growth factor 19 increases metabolic rate and reverses dietary and leptin-deficient diabetes. Endocrinology 2004, 145, 2594–2603. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Rinella, M.E.; Abdelmalek, M.F.; Trotter, J.F.; Paredes, A.H.; Arnold, H.L.; Kugelmas, M.; Bashir, M.R.; Jaros, M.J.; Ling, L.; et al. NGM282 for treatment of non-alcoholic steatohepatitis: A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2018, 391, 1174–1185. [Google Scholar] [CrossRef]
- Kharitonenkov, A.; Shiyanova, T.L.; Koester, A.; Ford, A.M.; Micanovic, R.; Galbreath, E.J.; Sandusky, G.E.; Hammond, L.J.; Moyers, J.S.; Owens, R.A.; et al. FGF-21 as a novel metabolic regulator. J. Clin. Investig. 2005, 115, 1627–1635. [Google Scholar] [CrossRef]
- Staiger, H.; Keuper, M.; Berti, L.; Hrabe de Angelis, M.; Haring, H.U. Fibroblast Growth Factor 21-Metabolic Role in Mice and Men. Endocr. Rev. 2017, 38, 468–488. [Google Scholar] [CrossRef]
- Sanyal, A.; Charles, E.D.; Neuschwander-Tetri, B.A.; Loomba, R.; Harrison, S.A.; Abdelmalek, M.F.; Lawitz, E.J.; Halegoua-DeMarzio, D.; Kundu, S.; Noviello, S.; et al. Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: A randomised, double-blind, placebo-controlled, phase 2a trial. Lancet 2019, 392, 2705–2717. [Google Scholar] [CrossRef]
- Alvarado, T.F.; Puliga, E.; Preziosi, M.; Poddar, M.; Singh, S.; Columbano, A.; Nejak-Bowen, K.; Monga, S.P. Thyroid Hormone Receptor beta Agonist Induces beta-Catenin-Dependent Hepatocyte Proliferation in Mice: Implications in Hepatic Regeneration. Gene Expr. 2016, 17, 19–34. [Google Scholar] [CrossRef]
- Ogawa, Y.; Yoneda, M.; Kobayashi, T.; Honda, Y.; Kessoku, T.; Imajo, K.; Saito, S.; Nakajima, A. Present and emerging pharmacotherapies for non-alcoholic steatohepatitis in adults. Expert Opin. Pharmacother. 2019, 20, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.C.; Cherney, D.Z.I. The actions of SGLT2 inhibitors on metabolism, renal function and blood pressure. Diabetologia 2018, 61, 2098–2107. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, J.W.; Lundkvist, P.; Jansson, P.A.; Johansson, L.; Kvarnstrom, M.; Moris, L.; Miliotis, T.; Forsberg, G.B.; Riserus, U.; Lind, L.; et al. Effects of dapagliflozin and n-3 carboxylic acids on non-alcoholic fatty liver disease in people with type 2 diabetes: A double-blind randomised placebo-controlled study. Diabetologia 2018, 61, 1923–1934. [Google Scholar] [CrossRef] [PubMed]
- Latva-Rasku, A.; Honka, M.-J.; Kullberg, J.; Mononen, N.; Lehtimäki, T.; Saltevo, J.; Kirjavainen, A.K.; Saunavaara, V.; Iozzo, P.; Johansson, L. The SGLT2 inhibitor dapagliflozin reduces liver fat but does not affect tissue insulin sensitivity: A randomized, double-blind, placebo-controlled study with 8-week treatment in type 2 diabetes patients. Diabetes Care 2019, 42, 931–937. [Google Scholar] [CrossRef]
- Sattar, N.; Fitchett, D.; Hantel, S.; George, J.T.; Zinman, B. Empagliflozin is associated with improvements in liver enzymes potentially consistent with reductions in liver fat: Results from randomised trials including the EMPA-REG OUTCOME® trial. Diabetologia 2018, 61, 2155–2163. [Google Scholar] [CrossRef]
- Seko, Y.; Sumida, Y.; Sasaki, K.; Itoh, Y.; Iijima, H.; Hashimoto, T.; Ishii, S.; Inagaki, N. Effects of canagliflozin, an SGLT2 inhibitor, on hepatic function in Japanese patients with type 2 diabetes mellitus: Pooled and subgroup analyses of clinical trials. J. Gastroenterol. 2018, 53, 140–151. [Google Scholar] [CrossRef]
- Harrison, S.A.; Manghi, F.P.; Smith, W.B.; Alpenidze, D.; Aizenberg, D.; Burggraaf, K.; Chen, C.-Y.; Zuckerman, E.; Ravussin, E.; Charatcharoenwitthaya, P. LIK066 (Licogliflozin), AN SGLT1/2 inhibitor, robustly decreases alt and improves markers of hepatic and metabolic health in patients with non-alcoholic fatty liver disease: Interim analysis of a 12-week, randomized, placebo-controlled, phase 2a study. Hepatology 2019, 70, 1482A–1483A. [Google Scholar]
- Kuchay, M.S.; Krishan, S.; Mishra, S.K.; Farooqui, K.J.; Singh, M.K.; Wasir, J.S.; Bansal, B.; Kaur, P.; Jevalikar, G.; Gill, H.K.; et al. Effect of Empagliflozin on Liver Fat in Patients With Type 2 Diabetes and Nonalcoholic Fatty Liver Disease: A Randomized Controlled Trial (E-LIFT Trial). Diabetes Care 2018, 41, 1801–1808. [Google Scholar] [CrossRef]
- Bai, L.; Chen, M.M.; Chen, Z.D.; Zhang, P.; Tian, S.; Zhang, Y.; Zhu, X.Y.; Liu, Y.; She, Z.G.; Ji, Y.X.; et al. F-box/WD Repeat-Containing Protein 5 Mediates the Ubiquitination of Apoptosis Signal-Regulating Kinase 1 and Exacerbates Nonalcoholic Steatohepatitis in Mice. Hepatology 2019, 70, 1942–1957. [Google Scholar] [CrossRef]
- Loomba, R.; Lawitz, E.; Mantry, P.S.; Jayakumar, S.; Caldwell, S.H.; Arnold, H.; Diehl, A.M.; Djedjos, C.S.; Han, L.; Myers, R.P.; et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: A randomized, phase 2 trial. Hepatology 2018, 67, 549–559. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, P.X.; Zhao, L.P.; Zhang, X.; Ji, Y.X.; Zhang, X.J.; Fang, C.; Lu, Y.X.; Yang, X.; Gao, M.M.; et al. The deubiquitinating enzyme TNFAIP3 mediates inactivation of hepatic ASK1 and ameliorates nonalcoholic steatohepatitis. Nat. Med. 2018, 24, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.-X.; Ji, Y.-X.; Zhang, X.-J.; Zhao, L.-P.; Yan, Z.-Z.; Zhang, P.; Shen, L.-J.; Yang, X.; Fang, J.; Tian, S. Targeting CASP8 and FADD-like apoptosis regulator ameliorates nonalcoholic steatohepatitis in mice and nonhuman primates. Nat. Med. 2017, 23, 439. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Ratziu, V.; Harrison, S.A.; Abdelmalek, M.F.; Aithal, G.P.; Caballeria, J.; Francque, S.; Farrell, G.; Kowdley, K.V.; Craxi, A.; et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology 2018, 67, 1754–1767. [Google Scholar] [CrossRef]
- Ratziu, V.; Sanyal, A.; Francque, S.; Sun, W.; Wong, V.; Loomba, R.; Goodman, Z.; Lefebvre, E.; Aithal, G.; Harrison, S. Cenicriviroc treatment for adults with non-alcoholic steatohepatitis: Year 2 analysis of the phase 2B CENTAUR study. J. Hepatol. 2018, 68, S1–S2. [Google Scholar] [CrossRef]
- Wang, P.X.; Zhang, X.J.; Luo, P.; Jiang, X.; Zhang, P.; Guo, J.; Zhao, G.N.; Zhu, X.; Zhang, Y.; Yang, S.; et al. Hepatocyte TRAF3 promotes liver steatosis and systemic insulin resistance through targeting TAK1-dependent signalling. Nat. Commun. 2016, 7, 10592. [Google Scholar] [CrossRef]
- Ji, Y.X.; Huang, Z.; Yang, X.; Wang, X.; Zhao, L.P.; Wang, P.X.; Zhang, X.J.; Alves-Bezerra, M.; Cai, L.; Zhang, P.; et al. The deubiquitinating enzyme cylindromatosis mitigates nonalcoholic steatohepatitis. Nat. Med. 2018, 24, 213–223. [Google Scholar] [CrossRef]
- Chalasani, N.; Abdelmalek, M.F.; Garcia-Tsao, G.; Vuppalanchi, R.; Alkhouri, N.; Rinella, M.; Noureddin, M.; Pyko, M.; Shiffman, M.; Sanyal, A.; et al. Effects of Belapectin, an Inhibitor of Galectin-3, in Patients With Nonalcoholic Steatohepatitis With Cirrhosis and Portal Hypertension. Gastroenterology 2020, 158, 1334–1345.e5. [Google Scholar] [CrossRef]
- Carino, A.; Cipriani, S.; Marchiano, S.; Biagioli, M.; Santorelli, C.; Donini, A.; Zampella, A.; Monti, M.C.; Fiorucci, S. BAR502, a dual FXR and GPBAR1 agonist, promotes browning of white adipose tissue and reverses liver steatosis and fibrosis. Sci. Rep. 2017, 7, 42801. [Google Scholar] [CrossRef]
- Puri, P.; Sanyal, A.J. The Intestinal Microbiome in Nonalcoholic Fatty Liver Disease. Clin. Liver Dis. 2018, 22, 121–132. [Google Scholar] [CrossRef]
- Georgescu, E.F.; Georgescu, M. Therapeutic Options in Non-Alcoholic Steatohepatitis (NASH). Are all Agents Alike? Results of a Preliminary Study. J. Gastrointestin. Liver Dis. 2007, 16, 39–46. [Google Scholar]
- Hyogo, H.; Tazuma, S.; Arihiro, K.; Iwamoto, K.; Nabeshima, Y.; Inoue, M.; Ishitobi, T.; Nonaka, M.; Chayama, K. Efficacy of atorvastatin for the treatment of nonalcoholic steatohepatitis with dyslipidemia. Metab. Clin. Exp. 2008, 57, 1711–1718. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Petta, S.; Mannisto, V.; Mancina, R.M.; Pipitone, R.; Karja, V.; Maggioni, M.; Kakela, P.; Wiklund, O.; Mozzi, E. Statin use and non-alcoholic steatohepatitis in at risk individuals. J. Hepatol. 2015, 63, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Echeverria, F.; Valenzuela, R.; Bustamante, A.; Alvarez, D.; Ortiz, M.; Espinosa, A.; Illesca, P.; Gonzalez-Manan, D.; Videla, L.A. High-fat diet induces mouse liver steatosis with a concomitant decline in energy metabolism: Attenuation by eicosapentaenoic acid (EPA) or hydroxytyrosol (HT) supplementation and the additive effects upon EPA and HT co-administration. Food Funct. 2019, 10, 6170–6183. [Google Scholar] [CrossRef]
- Parker, H.M.; Johnson, N.A.; Burdon, C.A.; Cohn, J.S.; O’Connor, H.T.; George, J. Omega-3 supplementation and non-alcoholic fatty liver disease: A systematic review and meta-analysis. J. Hepatol. 2012, 56, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Sofi, F.; Giangrandi, I.; Cesari, F.; Corsani, I.; Abbate, R.; Gensini, G.F.; Casini, A. Effects of a 1-year dietary intervention with n-3 polyunsaturated fatty acid-enriched olive oil on non-alcoholic fatty liver disease patients: A preliminary study. Int. J. Food Sci. Nutr. 2010, 61, 792–802. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.; Baillie, R.A.; Wiest, M.M.; Mirshahi, F.; Choudhury, J.; Cheung, O.; Sargeant, C.; Contos, M.J.; Sanyal, A.J. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 2007, 46, 1081–1090. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Abdelmalek, M.F.; Suzuki, A.; Cummings, O.W.; Chojkier, M. No Significant Effects of Ethyl-Eicosapentanoic Acid on Histologic Features of Nonalcoholic Steatohepatitis in a Phase 2 Trial. Gastroenterology 2014, 147, 377–384.e1. [Google Scholar] [CrossRef] [PubMed]
- Simon, T.G.; Henson, J.; Osganian, S.; Masia, R.; Chan, A.T.; Chung, R.T.; Corey, K.E. Daily Aspirin Use Associated With Reduced Risk For Fibrosis Progression In Patients With Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2019, 17, 2776–2784.e2774. [Google Scholar] [CrossRef]
- Teodoro, J.S.; Duarte, F.V.; Gomes, A.P.; Varela, A.T.; Peixoto, F.M.; Rolo, A.P.; Palmeira, C.M. Berberine reverts hepatic mitochondrial dysfunction in high-fat fed rats: A possible role for SirT3 activation. Mitochondrion 2013, 13, 637–646. [Google Scholar] [CrossRef]
- Yan, H.M.; Xia, M.F.; Wang, Y.; Chang, X.X.; Yao, X.Z.; Rao, S.X.; Zeng, M.S.; Tu, Y.F.; Feng, R.; Jia, W.P.; et al. Efficacy of Berberine in Patients with Non-Alcoholic Fatty Liver Disease. PLoS ONE 2015, 10, e0134172. [Google Scholar] [CrossRef]
- Wei, Y.; Clark, S.E.; Thyfault, J.P.; Uptergrove, G.M.; Li, W.; Whaley-Connell, A.T.; Ferrario, C.M.; Sowers, J.R.; Ibdah, J.A. Oxidative stress-mediated mitochondrial dysfunction contributes to angiotensin II-induced nonalcoholic fatty liver disease in transgenic Ren2 rats. Am. J. Pathol. 2009, 174, 1329–1337. [Google Scholar] [CrossRef]
- Yan, J.; Jiang, J.; He, L.; Chen, L. Mitochondrial superoxide/hydrogen peroxide: An emerging therapeutic target for metabolic diseases. Free Radic. Biol. Med. 2020, 152, 33–42. [Google Scholar] [CrossRef]
- Pessayre, D.; Berson, A.; Fromenty, B.; Mansouri, A. Mitochondria in steatohepatitis. Semin. Liver Dis. 2001, 21, 57–69. [Google Scholar] [CrossRef]
- Thoma, C.; Day, C.P.; Trenell, M.I. Lifestyle interventions for the treatment of non-alcoholic fatty liver disease in adults: A systematic review. J. Hepatol. 2012, 56, 255–266. [Google Scholar] [CrossRef]
- Li, Z.; Li, Y.; Zhang, H.X.; Guo, J.R.; Lam, C.W.K.; Wang, C.Y.; Zhang, W. Mitochondria-Mediated Pathogenesis and Therapeutics for Non-Alcoholic Fatty Liver Disease. Mol. Nutr. Food Res. 2019, 63, e1900043. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.; Ju, L.; Qiu, M.; Xie, Q.; Chen, Y.; Shen, W.; Sun, W.; Wang, W.; Tian, J. Liraglutide ameliorates non-alcoholic fatty liver disease by enhancing mitochondrial architecture and promoting autophagy through the SIRT1/SIRT3-FOXO3a pathway. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2016, 46, 933–943. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Sabet, A.; Djedjos, S.; Miller, R.; Sun, X.; Hussain, M.A.; Radovick, S.; Wondisford, F.E. Metformin and insulin suppress hepatic gluconeogenesis through phosphorylation of CREB binding protein. Cell 2009, 137, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Yuan, Q.; Xu, T.; Yao, L.; Feng, J.; Ma, J.; Wang, L.; Lu, C.; Wang, D. Pioglitazone Improves Mitochondrial Function in the Remnant Kidney and Protects against Renal Fibrosis in 5/6 Nephrectomized Rats. Front. Pharmacol. 2017, 8, 545. [Google Scholar] [CrossRef]
- Chen, Y.S.; Liu, H.M.; Lee, T.Y. Ursodeoxycholic Acid Regulates Hepatic Energy Homeostasis and White Adipose Tissue Macrophages Polarization in Leptin-Deficiency Obese Mice. Cells 2019, 8, 253. [Google Scholar] [CrossRef]
- Xie, C.; Jiang, C.; Shi, J.; Gao, X.; Sun, D.; Sun, L.; Wang, T.; Takahashi, S.; Anitha, M.; Krausz, K.W.; et al. An Intestinal Farnesoid X Receptor-Ceramide Signaling Axis Modulates Hepatic Gluconeogenesis in Mice. Diabetes 2017, 66, 613–626. [Google Scholar] [CrossRef]
- Ferramosca, A.; Di Giacomo, M.; Zara, V. Antioxidant dietary approach in treatment of fatty liver: New insights and updates. World J. Gastroenterol. WJG 2017, 23, 4146–4157. [Google Scholar] [CrossRef]
- Ding, S.; Jiang, J.; Zhang, G.; Bu, Y.; Zhang, G.; Zhao, X. Resveratrol and caloric restriction prevent hepatic steatosis by regulating SIRT1-autophagy pathway and alleviating endoplasmic reticulum stress in high-fat diet-fed rats. PLoS ONE 2017, 12, e0183541. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Ma, J.; Wang, W.; Zhang, L.; Xu, J.; Wang, K.; Li, D. Resveratrol supplement inhibited the NF-κB inflammation pathway through activating AMPKα-SIRT1 pathway in mice with fatty liver. Mol. Cell. Biochem. 2016, 422, 75–84. [Google Scholar] [CrossRef]
- Shang, J.; Chen, L.L.; Xiao, F.X.; Sun, H.; Ding, H.C.; Xiao, H. Resveratrol improves non-alcoholic fatty liver disease by activating AMP-activated protein kinase. Acta Pharmacol. Sin. 2008, 29, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Asin-Cayuela, J.; Manas, A.R.; James, A.M.; Smith, R.A.; Murphy, M.P. Fine-tuning the hydrophobicity of a mitochondria-targeted antioxidant. FEBS Lett. 2004, 571, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Rokitskaya, T.I.; Klishin, S.S.; Severina, I.I.; Skulachev, V.P.; Antonenko, Y.N. Kinetic analysis of permeation of mitochondria-targeted antioxidants across bilayer lipid membranes. J. Membr. Biol. 2008, 224, 9–19. [Google Scholar] [CrossRef]
- Smith, R.A.; Porteous, C.M.; Gane, A.M.; Murphy, M.P. Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 5407–5412. [Google Scholar] [CrossRef]
- Grattagliano, I.; Diogo, C.V.; Mastrodonato, M.; de Bari, O.; Persichella, M.; Wang, D.Q.; Liquori, A.; Ferri, D.; Carratu, M.R.; Oliveira, P.J.; et al. A silybin-phospholipids complex counteracts rat fatty liver degeneration and mitochondrial oxidative changes. World J. Gastroenterol. WJG 2013, 19, 3007–3017. [Google Scholar] [CrossRef]
- Vecchione, G.; Grasselli, E.; Voci, A.; Baldini, F.; Grattagliano, I.; Wang, D.Q.; Portincasa, P.; Vergani, L. Silybin counteracts lipid excess and oxidative stress in cultured steatotic hepatic cells. World J. Gastroenterol. WJG 2016, 22, 6016–6026. [Google Scholar] [CrossRef]
- Wu, N.; Zu, Y.; Fu, Y.; Kong, Y.; Zhao, J.; Li, X.; Li, J.; Wink, M.; Efferth, T. Antioxidant activities and xanthine oxidase inhibitory effects of extracts and main polyphenolic compounds obtained from Geranium sibiricum L. J. Agric. Food Chem. 2010, 58, 4737–4743. [Google Scholar] [CrossRef]
- Ling, W.H.; Shen, T.R.; Tang, X.L.; Jiang, X.W. Anthocyanins Improved Mitochondrial Dysfunction in Mice of Non-alcoholic Fatty Liver Disease Induced by High Fat Diet. FASEB J. 2016, 30, 915–929. [Google Scholar]
- Tang, X.; Shen, T.; Jiang, X.; Xia, M.; Sun, X.; Guo, H.; Ling, W. Purified anthocyanins from bilberry and black currant attenuate hepatic mitochondrial dysfunction and steatohepatitis in mice with methionine and choline deficiency. J. Agric. Food Chem. 2015, 63, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Yang, J.; Hu, O.; Huang, J.; Ran, L.; Chen, M.; Zhang, Y.; Zhou, X.; Zhu, J.; Zhang, Q. Dihydromyricetin ameliorates nonalcoholic fatty liver disease by improving mitochondrial respiratory capacity and redox homeostasis through modulation of SIRT3 signaling. Antioxid. Redox Signal. 2019, 30, 163–183. [Google Scholar] [CrossRef]
- Schwimmer, J.B.; Lavine, J.E.; Wilson, L.A.; Neuschwander-Tetri, B.A.; Xanthakos, S.A.; Kohli, R.; Barlow, S.E.; Vos, M.B.; Karpen, S.J.; Molleston, J.P. In children with nonalcoholic fatty liver disease, cysteamine bitartrate delayed release improves liver enzymes but does not reduce disease activity scores. Gastroenterology 2016, 151, 1141–1154.e9. [Google Scholar] [CrossRef] [PubMed]
- Dohil, R.; Schmeltzer, S.; Cabrera, B.L.; Wang, T.; Durelle, J.; Duke, K.B.; Schwimmer, J.B.; Lavine, J.E. Enteric-coated cysteamine for the treatment of paediatric non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2011, 33, 1036–1044. [Google Scholar] [CrossRef]
- Ye, J.H.; Chao, J.; Chang, M.L.; Peng, W.H.; Cheng, H.Y.; Liao, J.W.; Pao, L.H. Pentoxifylline ameliorates non-alcoholic fatty liver disease in hyperglycaemic and dyslipidaemic mice by upregulating fatty acid beta-oxidation. Sci. Rep. 2016, 6, 33102. [Google Scholar] [CrossRef]
- Zein, C.O.; Lopez, R.; Yerian, L.; Anderson, K.A.; McCullough, A.J.; Rinella, M.E. 932 Pentoxifylline Improves Non-Invasive Serum Markers of Fibrosis: Combined Results From 2 Randomized, Placebo-Controlled Trials. Gastroenterology 2012, 142, S-936. [Google Scholar] [CrossRef]
- Zein, C.O.; Yerian, L.M.; Gogate, P.; Lopez, R.; Kirwan, J.P.; Feldstein, A.E.; McCullough, A.J. Pentoxifylline improves nonalcoholic steatohepatitis: A randomized placebo-controlled trial. Hepatology 2011, 54, 1610–1619. [Google Scholar] [CrossRef]
- Rendon, D.A. Letter to the Editor: The bioenergetics of hepatic mitochondria isolated from avocado oil-treated rats: Typical experimental errors in the study of the bioenergetics of isolated mitochondria. J. Bioenerg. Biomembr. 2015, 47, 451–453. [Google Scholar] [CrossRef][Green Version]
- Ortiz-Avila, O.; Gallegos-Corona, M.A.; Sanchez-Briones, L.A.; Calderon-Cortes, E.; Montoya-Perez, R.; Rodriguez-Orozco, A.R.; Campos-Garcia, J.; Saavedra-Molina, A.; Mejia-Zepeda, R.; Cortes-Rojo, C. Protective effects of dietary avocado oil on impaired electron transport chain function and exacerbated oxidative stress in liver mitochondria from diabetic rats. J. Bioenerg. Biomembr. 2015, 47, 337–353. [Google Scholar] [CrossRef]
- Garcia-Berumen, C.I.; Olmos-Orizaba, B.E.; Marquez-Ramirez, C.A.; Orozco, A.R.R.; Gonzalez-Cortez, A.; Saavedra-Molina, A.; Montoya-Perez, R.; Cortes-Rojo, C. Avocado Oil Ameliorates Non-Alcoholic Fatty Liver Disease by Down-Regulating Inflammatory Cytokines and Improving Mitochondrial Dynamics. FASEB J. 2019, 33, 660–666. [Google Scholar]
- Fu, A.; Shi, X.; Zhang, H.; Fu, B. Mitotherapy for Fatty Liver by Intravenous Administration of Exogenous Mitochondria in Male Mice. Front. Pharmacol. 2017, 8, 241. [Google Scholar] [CrossRef] [PubMed]
- Ajith, T.A. Role of mitochondria and mitochondria-targeted agents in non-alcoholic fatty liver disease. Clin. Exp. Pharmacol. Physiol. 2018, 45, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Pessayre, D.; Fromenty, B. NASH: A mitochondrial disease. J. Hepatol. 2005, 42, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Liang, K.; Zhao, S.; Jia, W.; Liu, Y.; Wu, H.; Lv, J.; Cao, C.; Chen, T.; Zhuang, S.; et al. Chemoproteomics reveals baicalin activates hepatic CPT1 to ameliorate diet-induced obesity and hepatic steatosis. Proc. Natl. Acad. Sci. USA 2018, 115, E5896–E5905. [Google Scholar] [CrossRef] [PubMed]
- Fazzari, M.; Chartoumpekis, D.; Li, L.; Guimaraes, D.A.; Shiva, S.; Freeman, B.A.; Khoo, N. Nitro-oleic Acid Protects Mice from Diet-Induced Hepatic Steatosis and Insulin Resistance without the Adverse Side Effects of Thiazolidinediones. Free Radic. Biol. Med. 2017, 112, 152. [Google Scholar] [CrossRef]
- Cho, J.; Zhang, Y.; Park, S.-Y.; Joseph, A.-M.; Han, C.; Park, H.-J.; Kalavalapalli, S.; Chun, S.-K.; Morgan, D.; Kim, J.-S. Mitochondrial ATP transporter depletion protects mice against liver steatosis and insulin resistance. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Amanat, S.; Eftekhari, M.H.; Fararouei, M.; Bagheri Lankarani, K.; Massoumi, S.J. Genistein supplementation improves insulin resistance and inflammatory state in non-alcoholic fatty liver patients: A randomized, controlled trial. Clin. Nutr. 2018, 37, 1210–1215. [Google Scholar] [CrossRef]
- Lawitz, E.J.; Neff, G.; Ruane, P.J.; Younes, Z.; Zhang, J.; Jia, C.; Chuang, J.; Huss, R.; Chung, C.; Subramanian, M. Fenofibrate mitigates increases in serum triglycerides due to the ACC inhibitor firsocostat in patients with advanced fibrosis due to NASH: A phase 2 randomized trial. Hepatology 2019, 70, 1489A–1490A. [Google Scholar]
- Stevanovic, J.; Beleza, J.; Coxito, P.; Ascensao, A.; Magalhaes, J. Physical exercise and liver “fitness”: Role of mitochondrial function and epigenetics-related mechanisms in non-alcoholic fatty liver disease. Mol. Metab. 2020, 32, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Little, J.P.; Safdar, A.; Benton, C.R.; Wright, D.C. Skeletal muscle and beyond: The role of exercise as a mediator of systemic mitochondrial biogenesis. Appl. Physiol. Nutr. Metab. Physiol. Appl. Nutr. Metab. 2011, 36, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Venditti, P.; Di Meo, S. Antioxidants, tissue damage, and endurance in trained and untrained young male rats. Arch. Biochem. Biophys. 1996, 331, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Ascensao, A.; Martins, M.J.; Santos-Alves, E.; Goncalves, I.O.; Portincasa, P.; Oliveira, P.J.; Magalhaes, J. Modulation of hepatic redox status and mitochondrial metabolism by exercise: Therapeutic strategy for liver diseases. Mitochondrion 2013, 13, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Rector, R.S.; Thyfault, J.P.; Laye, M.J.; Morris, R.T.; Borengasser, S.J.; Uptergrove, G.M.; Chakravarthy, M.V.; Booth, F.W.; Ibdah, J.A. Cessation of daily exercise dramatically alters precursors of hepatic steatosis in Otsuka Long-Evans Tokushima Fatty (OLETF) rats. J. Physiol. 2008, 586, 4241–4249. [Google Scholar] [CrossRef] [PubMed]
- Rector, R.S.; Thyfault, J.P.; Morris, R.T.; Laye, M.J.; Borengasser, S.J.; Booth, F.W.; Ibdah, J.A. Daily exercise increases hepatic fatty acid oxidation and prevents steatosis in Otsuka Long-Evans Tokushima Fatty rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G619–G626. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Shen, W.; Liu, Z.; Guan, S.; Liu, J.; Ding, S. Endurance exercise causes mitochondrial and oxidative stress in rat liver: Effects of a combination of mitochondrial targeting nutrients. Life Sci. 2010, 86, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Shulman, G.I. Mechanisms for insulin resistance: Common threads and missing links. Cell 2012, 148, 852–871. [Google Scholar] [CrossRef]
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human fatty liver disease: Old questions and new insights. Science 2011, 332, 1519–1523. [Google Scholar] [CrossRef]
- Chastin, S.F.; Buck, C.; Freiberger, E.; Murphy, M.; Brug, J.; Cardon, G.; O’Donoghue, G.; Pigeot, I.; Oppert, J.M.; DEDIPAC Consortium. Systematic literature review of determinants of sedentary behaviour in older adults: A DEDIPAC study. Int. J. Behav. Nutr. Phys. Act. 2015, 12, 127. [Google Scholar] [CrossRef]
- Gross, B.; Pawlak, M.; Lefebvre, P.; Staels, B. PPARs in obesity-induced T2DM, dyslipidaemia and NAFLD. Nat. Rev. Endocrinol. 2017, 13, 36–49. [Google Scholar] [CrossRef]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef]
- Musso, G.; Gambino, R.; Cassader, M.; Pagano, G. A meta-analysis of randomized trials for the treatment of nonalcoholic fatty liver disease. Hepatology 2010, 52, 79–104. [Google Scholar] [CrossRef]
- Bojic, L.A.; Huff, M.W. Peroxisome proliferator-activated receptor delta: A multifaceted metabolic player. Curr. Opin. Lipidol. 2013, 24, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Red Eagle, A.; Vats, D.; Morel, C.R.; Goforth, M.H.; Subramanian, V.; Mukundan, L.; Ferrante, A.W.; Chawla, A. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab. 2008, 7, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Riserus, U.; Sprecher, D.; Johnson, T.; Olson, E.; Hirschberg, S.; Liu, A.; Fang, Z.; Hegde, P.; Richards, D.; Sarov-Blat, L.; et al. Activation of peroxisome proliferator-activated receptor (PPAR)delta promotes reversal of multiple metabolic abnormalities, reduces oxidative stress, and increases fatty acid oxidation in moderately obese men. Diabetes 2008, 57, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Promrat, K.; Lutchman, G.; Uwaifo, G.I.; Freedman, R.J.; Soza, A.; Heller, T.; Doo, E.; Ghany, M.; Premkumar, A.; Park, Y.; et al. A pilot study of pioglitazone treatment for nonalcoholic steatohepatitis. Hepatology 2004, 39, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Colca, J.R.; McDonald, W.G.; Cavey, G.S.; Cole, S.L.; Holewa, D.D.; Brightwell-Conrad, A.S.; Wolfe, C.L.; Wheeler, J.S.; Coulter, K.R.; Kilkuskie, P.M.; et al. Identification of a mitochondrial target of thiazolidinedione insulin sensitizers (mTOT)--relationship to newly identified mitochondrial pyruvate carrier proteins. PLoS ONE 2013, 8, e61551. [Google Scholar] [CrossRef] [PubMed]
- Kalavalapalli, S.; Bril, F.; Koelmel, J.P.; Abdo, K.; Guingab, J.; Andrews, P.; Li, W.Y.; Jose, D.; Yost, R.A.; Frye, R.F.; et al. Pioglitazone improves hepatic mitochondrial function in a mouse model of nonalcoholic steatohepatitis. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E163–E173. [Google Scholar] [CrossRef] [PubMed]
- McCommis, K.S.; Chen, Z.; Fu, X.; McDonald, W.G.; Colca, J.R.; Kletzien, R.F.; Burgess, S.C.; Finck, B.N. Loss of Mitochondrial Pyruvate Carrier 2 in the Liver Leads to Defects in Gluconeogenesis and Compensation via Pyruvate-Alanine Cycling. Cell Metab. 2015, 22, 682–694. [Google Scholar] [CrossRef]
- Shannon, C.E.; Daniele, G.; Galindo, C.; Abdul-Ghani, M.A.; DeFronzo, R.A.; Norton, L. Pioglitazone inhibits mitochondrial pyruvate metabolism and glucose production in hepatocytes. FEBS J. 2017, 284, 451–465. [Google Scholar] [CrossRef]
- Cui, J.; Philo, L.; Nguyen, P.; Hofflich, H.; Hernandez, C.; Bettencourt, R.; Richards, L.; Salotti, J.; Bhatt, A.; Hooker, J.; et al. Sitagliptin vs. placebo for non-alcoholic fatty liver disease: A randomized controlled trial. J. Hepatol. 2016, 65, 369–376. [Google Scholar] [CrossRef]
- Joy, T.R.; McKenzie, C.A.; Tirona, R.G.; Summers, K.; Seney, S.; Chakrabarti, S.; Malhotra, N.; Beaton, M.D. Sitagliptin in patients with non-alcoholic steatohepatitis: A randomized, placebo-controlled trial. World J. Gastroenterol. WJG 2017, 23, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.B.; Song, Q.; Yu, Y.J.; Yu, H.G.; Luo, H.S.; Tan, S.Y. Effect of metformin on mitochondrial pathway of apoptosis and oxidative stress in cell model of nonalcoholic fatty liver disease. Zhonghua Gan Zang Bing Za Zhi 2020, 28, 64–68. [Google Scholar] [CrossRef]
- Yu, X.; Hao, M.; Liu, Y.; Ma, X.; Lin, W.; Xu, Q.; Zhou, H.; Shao, N.; Kuang, H. Liraglutide ameliorates non-alcoholic steatohepatitis by inhibiting NLRP3 inflammasome and pyroptosis activation via mitophagy. Eur. J. Pharmacol. 2019, 864, 172715. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Houten, S.M.; Wang, L.; Moschetta, A.; Mangelsdorf, D.J.; Heyman, R.A.; Moore, D.D.; Auwerx, J. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J. Clin. Investig. 2004, 113, 1408–1418. [Google Scholar] [CrossRef] [PubMed]
- Porez, G.; Prawitt, J.; Gross, B.; Staels, B. Bile acid receptors as targets for the treatment of dyslipidemia and cardiovascular disease Thematic Review Series: New Lipid and Lipoprotein Targets for the Treatment of Cardiometabolic Diseases. J. Lipid Res. 2012, 53, 1723–1737. [Google Scholar] [CrossRef] [PubMed]
- Calkin, A.C.; Tontonoz, P. Transcriptional integration of metabolism by the nuclear sterol-activated receptors LXR and FXR. Nat. Rev. Mol. Cell Biol. 2012, 13, 213–224. [Google Scholar] [CrossRef] [PubMed]
- De Aguiar Vallim, T.Q.; Tarling, E.J.; Edwards, P.A. Pleiotropic roles of bile acids in metabolism. Cell Metab. 2013, 17, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Jahn, D.; Rau, M.; Hermanns, H.M.; Geier, A. Mechanisms of enterohepatic fibroblast growth factor 15/19 signaling in health and disease. Cytokine Growth Factor Rev. 2015, 26, 625–635. [Google Scholar] [CrossRef]
- Lazaridis, K.N.; Gores, G.J.; Lindor, K.D. Ursodeoxycholic acid ‘mechanisms of action and clinical use in hepatobiliary disorders’. J. Hepatol. 2001, 35, 134–146. [Google Scholar] [CrossRef]
- Laurin, J.; Lindor, K.D.; Crippin, J.S.; Gossard, A.; Gores, G.J.; Ludwig, J.; Rakela, J.; McGill, D.B. Ursodeoxycholic acid or clofibrate in the treatment of non-alcohol-induced steatohepatitis: A pilot study. Hepatology 1996, 23, 1464–1467. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.; Thorell, A.; Claudel, T.; Jha, P.; Koefeler, H.; Lackner, C.; Hoesel, B.; Fauler, G.; Stojakovic, T.; Einarsson, C.; et al. Ursodeoxycholic acid exerts farnesoid X receptor-antagonistic effects on bile acid and lipid metabolism in morbid obesity. J. Hepatol. 2015, 62, 1398–1404. [Google Scholar] [CrossRef]
- Krahenbuhl, S.; Talos, C.; Fischer, S.; Reichen, J. Toxicity of bile acids on the electron transport chain of isolated rat liver mitochondria. Hepatology 1994, 19, 471–479. [Google Scholar] [CrossRef]
- Krahenbuhl, S.; Talos, C.; Lauterburg, B.H.; Reichen, J. Reduced antioxidative capacity in liver mitochondria from bile duct ligated rats. Hepatology 1995, 22, 607–612. [Google Scholar] [CrossRef]
- Rolo, A.P.; Oliveira, P.J.; Moreno, A.J.; Palmeira, C.M. Bile acids affect liver mitochondrial bioenergetics: Possible relevance for cholestasis therapy. Toxicol. Sci. An. Off. J. Soc. Toxicol. 2000, 57, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Krahenbuhl, S.; Fischer, S.; Talos, C.; Reichen, J. Ursodeoxycholate protects oxidative mitochondrial metabolism from bile acid toxicity: Dose-response study in isolated rat liver mitochondria. Hepatology 1994, 20, 1595–1601. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Luangmonkong, T.; Suriguga, S.; Mutsaers, H.A.M.; Groothuis, G.M.M.; Olinga, P.; Boersema, M. Targeting Oxidative Stress for the Treatment of Liver Fibrosis. Rev. Physiol. Biochem. Pharmacol. 2018, 175, 71–102. [Google Scholar] [PubMed]
- Jiang, C.; Xie, C.; Li, F.; Zhang, L.; Nichols, R.G.; Krausz, K.W.; Cai, J.; Qi, Y.; Fang, Z.Z.; Takahashi, S.; et al. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J. Clin. Investig. 2015, 125, 386–402. [Google Scholar] [CrossRef] [PubMed]
- Chachay, V.S.; Macdonald, G.A.; Martin, J.H.; Whitehead, J.P.; O’Moore–Sullivan, T.M.; Lee, P.; Franklin, M.; Klein, K.; Taylor, P.J.; Ferguson, M. Resveratrol does not benefit patients with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2014, 12, 2092–2103.e6. [Google Scholar] [CrossRef]
- Feillet-Coudray, C.; Fouret, G.; Ebabe Elle, R.; Rieusset, J.; Bonafos, B.; Chabi, B.; Crouzier, D.; Zarkovic, K.; Zarkovic, N.; Ramos, J.; et al. The mitochondrial-targeted antioxidant MitoQ ameliorates metabolic syndrome features in obesogenic diet-fed rats better than Apocynin or Allopurinol. Free Radic. Res. 2014, 48, 1232–1246. [Google Scholar] [CrossRef] [PubMed]
- Fouret, G.; Tolika, E.; Lecomte, J.; Bonafos, B.; Aoun, M.; Murphy, M.P.; Ferreri, C.; Chatgilialoglu, C.; Dubreucq, E.; Coudray, C.; et al. The mitochondrial-targeted antioxidant, MitoQ, increases liver mitochondrial cardiolipin content in obesogenic diet-fed rats. Biochim. Biophys. Acta 2015, 1847, 1025–1035. [Google Scholar] [CrossRef]
- Mercer, J.R.; Yu, E.; Figg, N.; Cheng, K.K.; Prime, T.A.; Griffin, J.L.; Masoodi, M.; Vidal-Puig, A.; Murphy, M.P.; Bennett, M.R. The mitochondria-targeted antioxidant MitoQ decreases features of the metabolic syndrome in ATM+/-/ApoE-/- mice. Free Radic. Biol. Med. 2012, 52, 841–849. [Google Scholar] [CrossRef]
- Dhanasekaran, A.; Kotamraju, S.; Kalivendi, S.V.; Matsunaga, T.; Shang, T.; Keszler, A.; Joseph, J.; Kalyanaraman, B. Supplementation of endothelial cells with mitochondria-targeted antioxidants inhibit peroxide-induced mitochondrial iron uptake, oxidative damage, and apoptosis. J. Biol. Chem. 2004, 279, 37575–37587. [Google Scholar] [CrossRef] [PubMed]
- Gane, E.J.; Weilert, F.; Orr, D.W.; Keogh, G.F.; Gibson, M.; Lockhart, M.M.; Frampton, C.M.; Taylor, K.M.; Smith, R.A.; Murphy, M.P. The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase II study of hepatitis C patients. Liver Int. 2010, 30, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Baselga-Escudero, L.; Souza-Mello, V.; Pascual-Serrano, A.; Rachid, T.; Voci, A.; Demori, I.; Grasselli, E. Beneficial effects of the Mediterranean spices and aromas on non-alcoholic fatty liver disease. Trends Food Sci. Technol. 2017, 61, 141–159. [Google Scholar] [CrossRef]
- Saller, R.; Meier, R.; Brignoli, R. The use of silymarin in the treatment of liver diseases. Drugs 2001, 61, 2035–2063. [Google Scholar] [CrossRef]
- Trappoliere, M.; Caligiuri, A.; Schmid, M.; Bertolani, C.; Failli, P.; Vizzutti, F.; Novo, E.; di Manzano, C.; Marra, F.; Loguercio, C.; et al. Silybin, a component of sylimarin, exerts anti-inflammatory and anti-fibrogenic effects on human hepatic stellate cells. J. Hepatol. 2009, 50, 1102–1111. [Google Scholar] [CrossRef]
- Solhi, H.; Ghahremani, R.; Kazemifar, A.M.; Hoseini Yazdi, Z. Silymarin in treatment of non-alcoholic steatohepatitis: A randomized clinical trial. Casp. J. Intern. Med. 2014, 5, 9–12. [Google Scholar]
- Loguercio, C.; Andreone, P.; Brisc, C.; Brisc, M.C.; Bugianesi, E.; Chiaramonte, M.; Cursaro, C.; Danila, M.; de Sio, I.; Floreani, A.; et al. Silybin combined with phosphatidylcholine and vitamin E in patients with nonalcoholic fatty liver disease: A randomized controlled trial. Free Radic. Biol. Med. 2012, 52, 1658–1665. [Google Scholar] [CrossRef]
- Colman, E. Dinitrophenol and obesity: An early twentieth-century regulatory dilemma. Regul. Toxicol. Pharmacol. 2007, 48, 115–117. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Chu, K.; Zhao, N.; Wu, J.; Ma, L.; Zhu, C.; Chen, X.; Wei, G.; Liao, M. Corilagin Alleviates Nonalcoholic Fatty Liver Disease in High-Fat Diet-Induced C57BL/6 Mice by Ameliorating Oxidative Stress and Restoring Autophagic Flux. Front. Pharmacol. 2019, 10, 1693. [Google Scholar] [CrossRef]
- Perry, R.J.; Zhang, D.; Zhang, X.M.; Boyer, J.L.; Shulman, G.I. Controlled-release mitochondrial protonophore reverses diabetes and steatohepatitis in rats. Science 2015, 347, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Fu, A. Mitotherapy as a Novel Therapeutic Strategy for Mitochondrial Diseases. Curr. Mol. Pharmacol. 2020, 13, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Dobrzyn, P.; Dobrzyn, A.; Miyazaki, M.; Cohen, P.; Asilmaz, E.; Hardie, D.G.; Friedman, J.M.; Ntambi, J.M. Stearoyl-CoA desaturase 1 deficiency increases fatty acid oxidation by activating AMP-activated protein kinase in liver. Proc. Natl. Acad. Sci. USA 2004, 101, 6409–6414. [Google Scholar] [CrossRef]
- Dobrzyn, A.; Ntambi, J.M. Stearoyl-CoA desaturase as a new drug target for obesity treatment. Obes. Rev. 2005, 6, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Atlante, A.; Bobba, A.; Paventi, G.; Pizzuto, R.; Passarella, S. Genistein and daidzein prevent low potassium-dependent apoptosis of cerebellar granule cells. Biochem. Pharmacol. 2010, 79, 758–767. [Google Scholar] [CrossRef][Green Version]
- Pockros, P.J.; Fuchs, M.; Freilich, B.; Schiff, E.; Kohli, A.; Lawitz, E.J.; Hellstern, P.A.; Owens-Grillo, J.; Van Biene, C.; Shringarpure, R.; et al. CONTROL: A randomized phase 2 study of obeticholic acid and atorvastatin on lipoproteins in nonalcoholic steatohepatitis patients. Liver Int. 2019, 39, 2082–2093. [Google Scholar] [CrossRef] [PubMed]










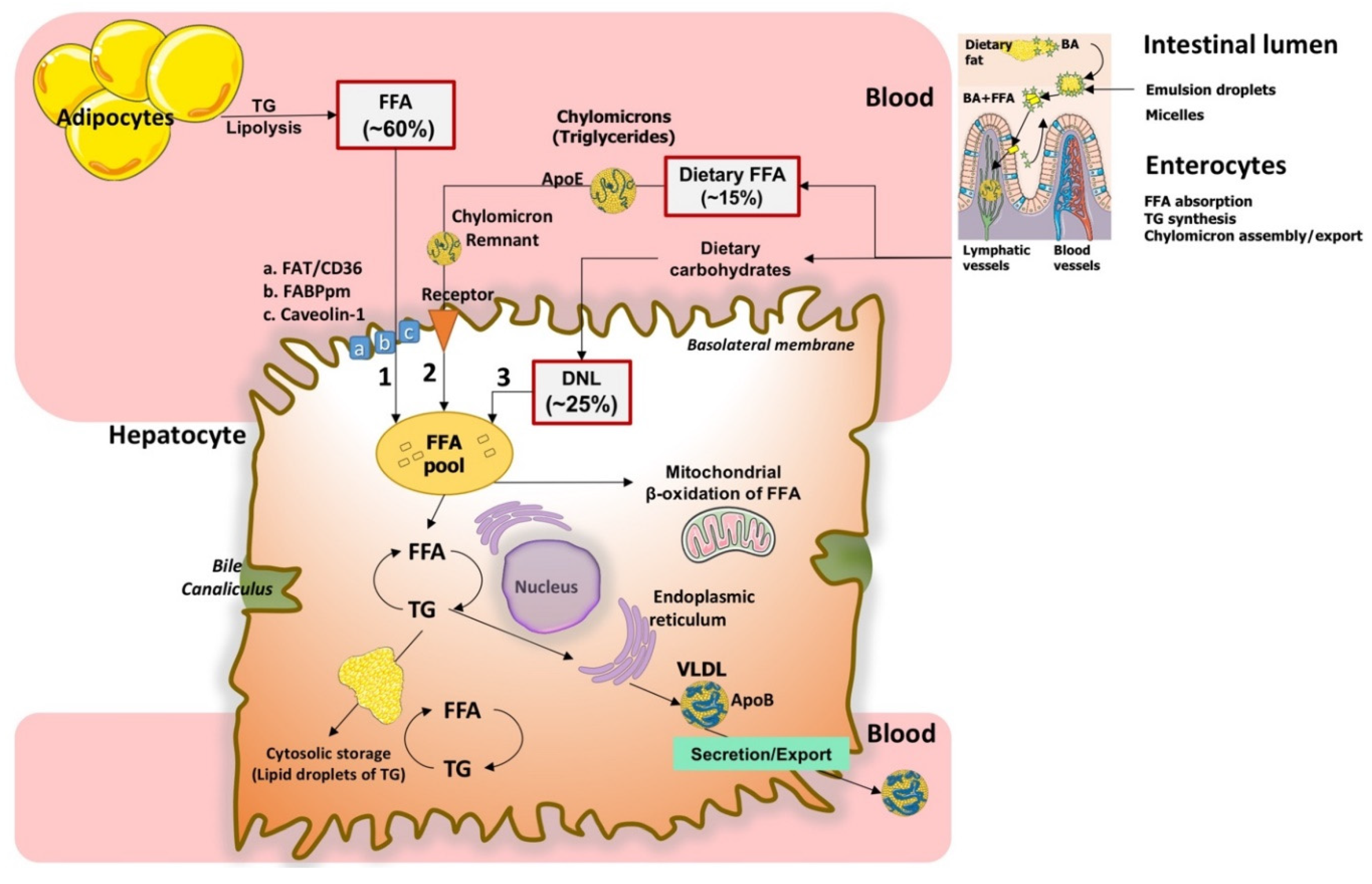{kind=link}
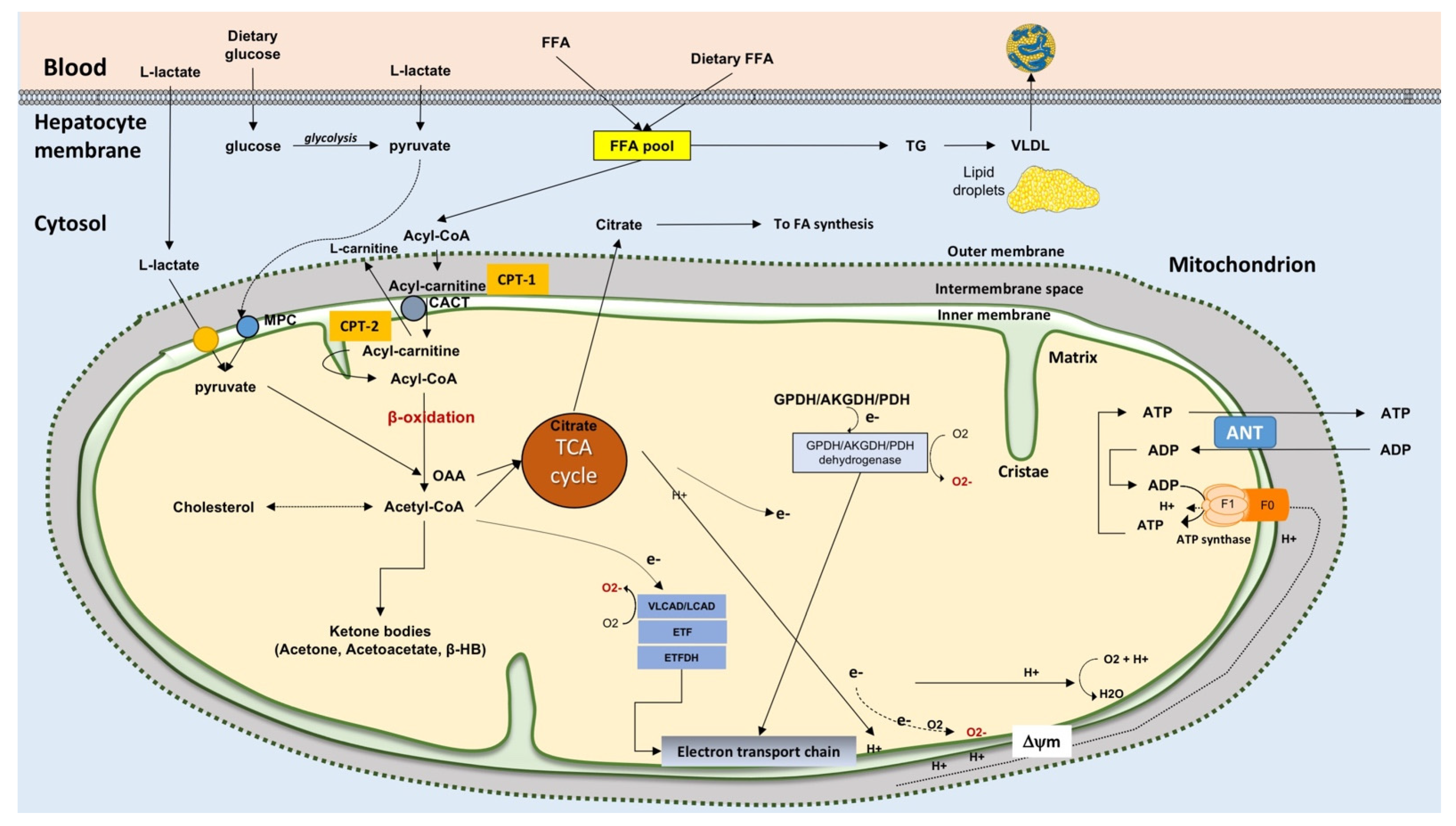{kind=link}
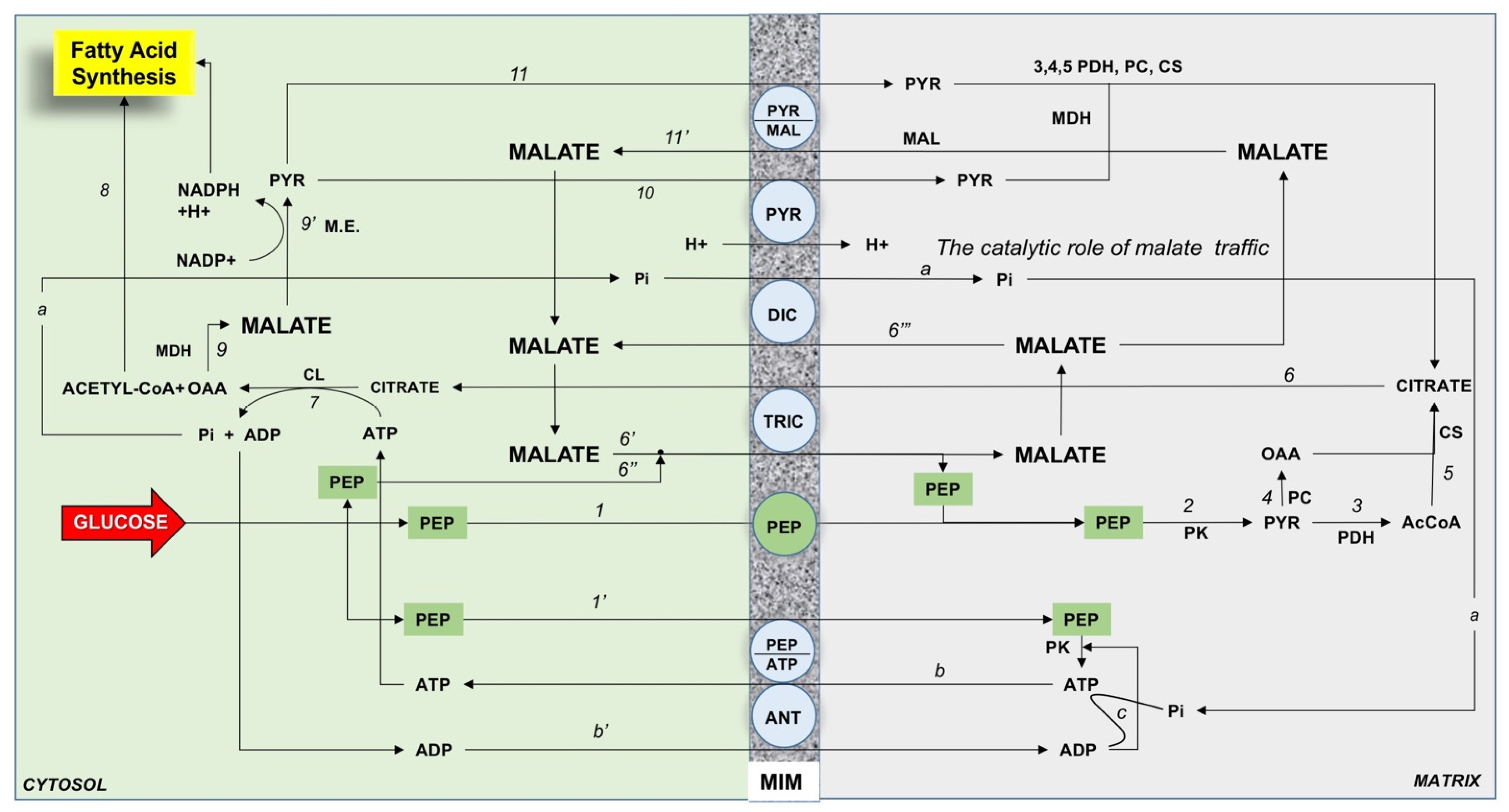{kind=link}
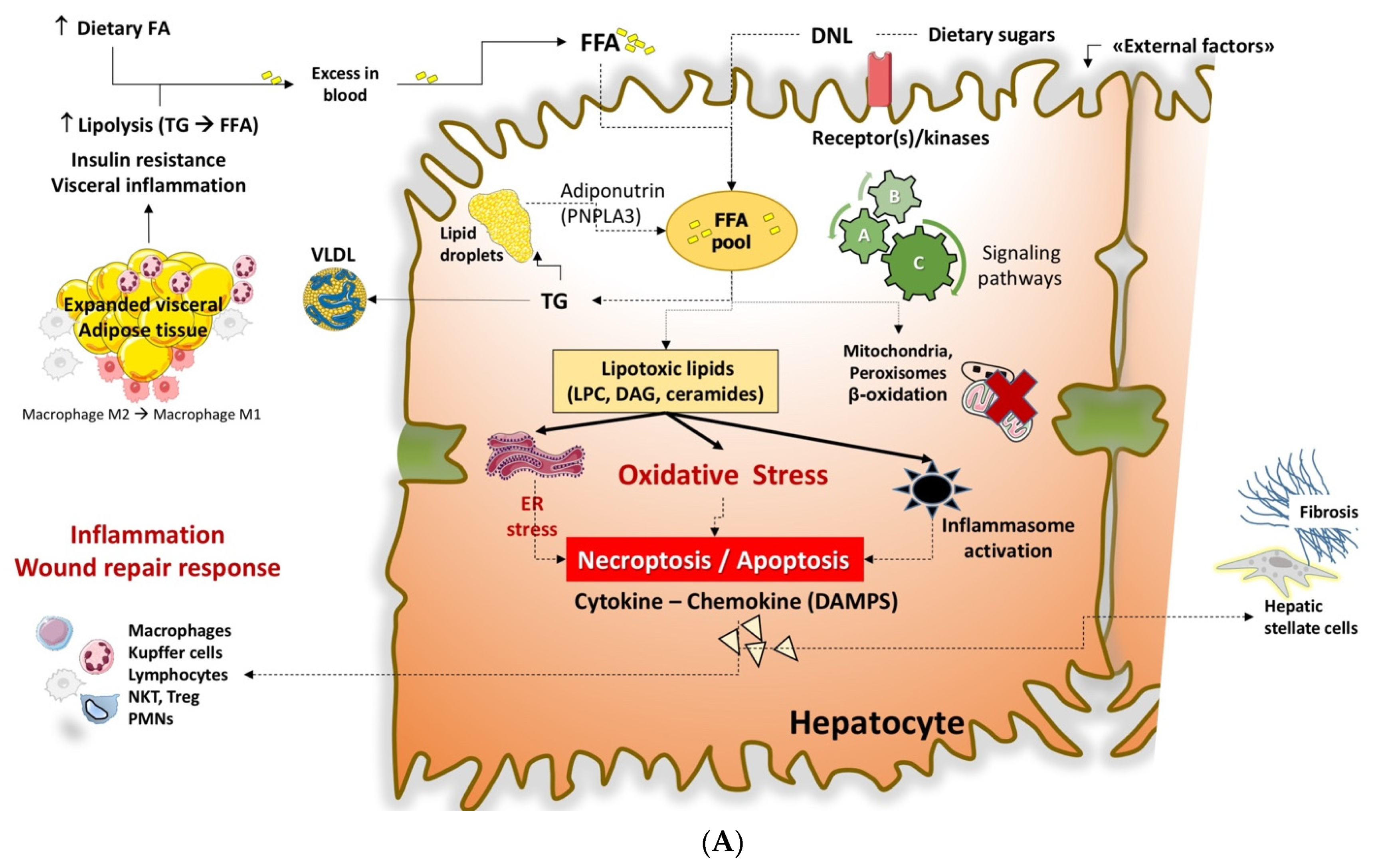{kind=link}
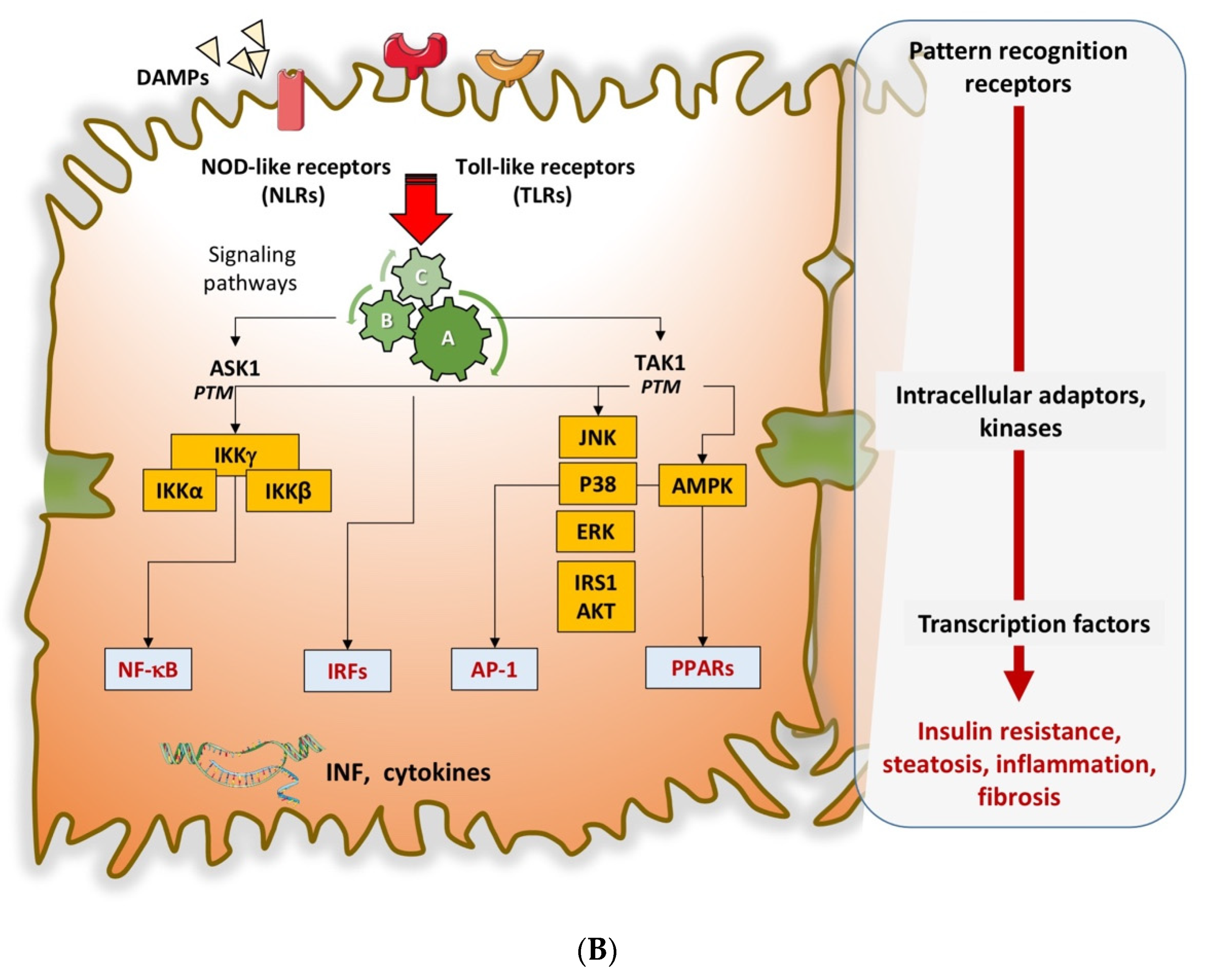{kind=link}
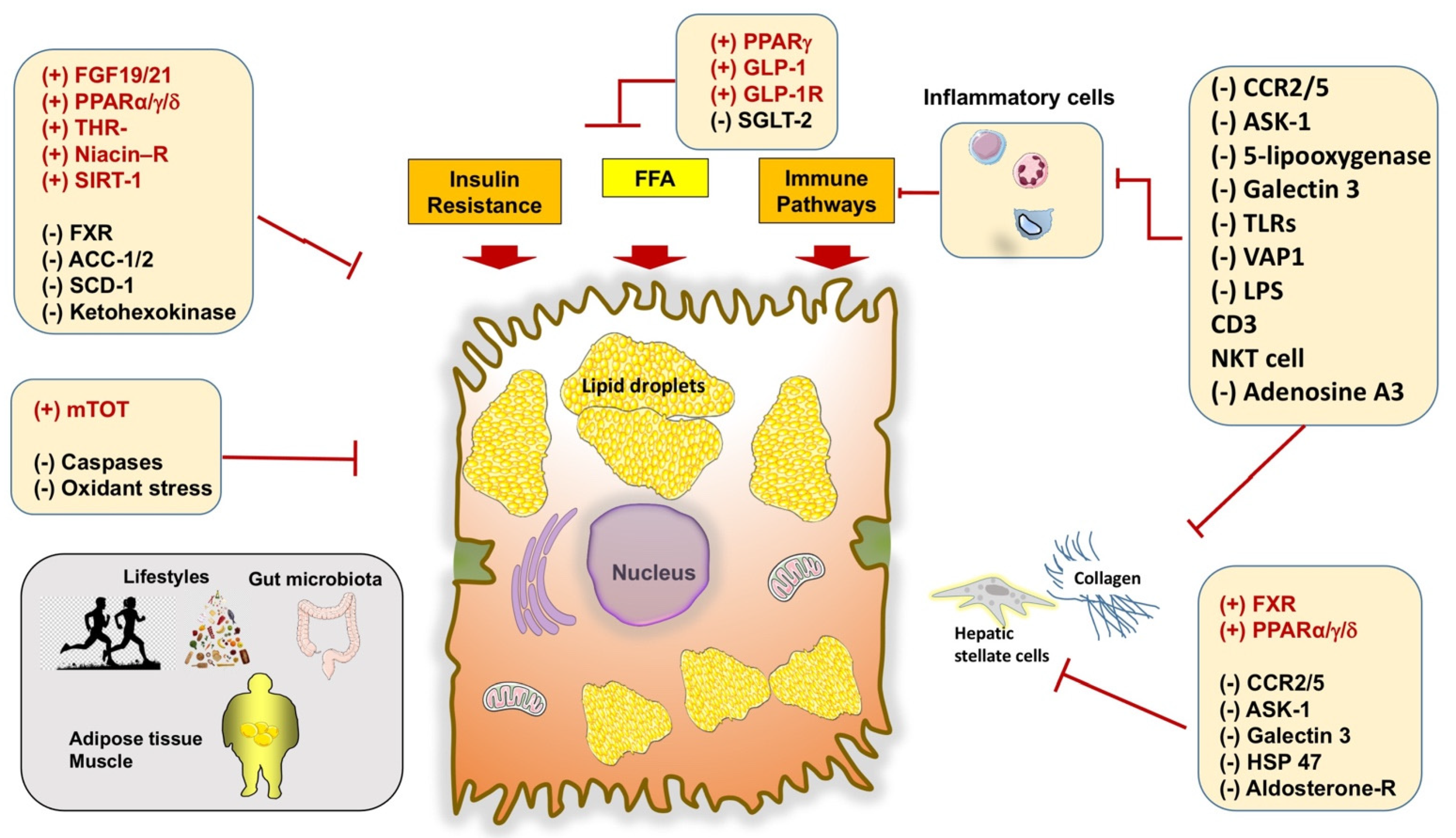{kind=link}
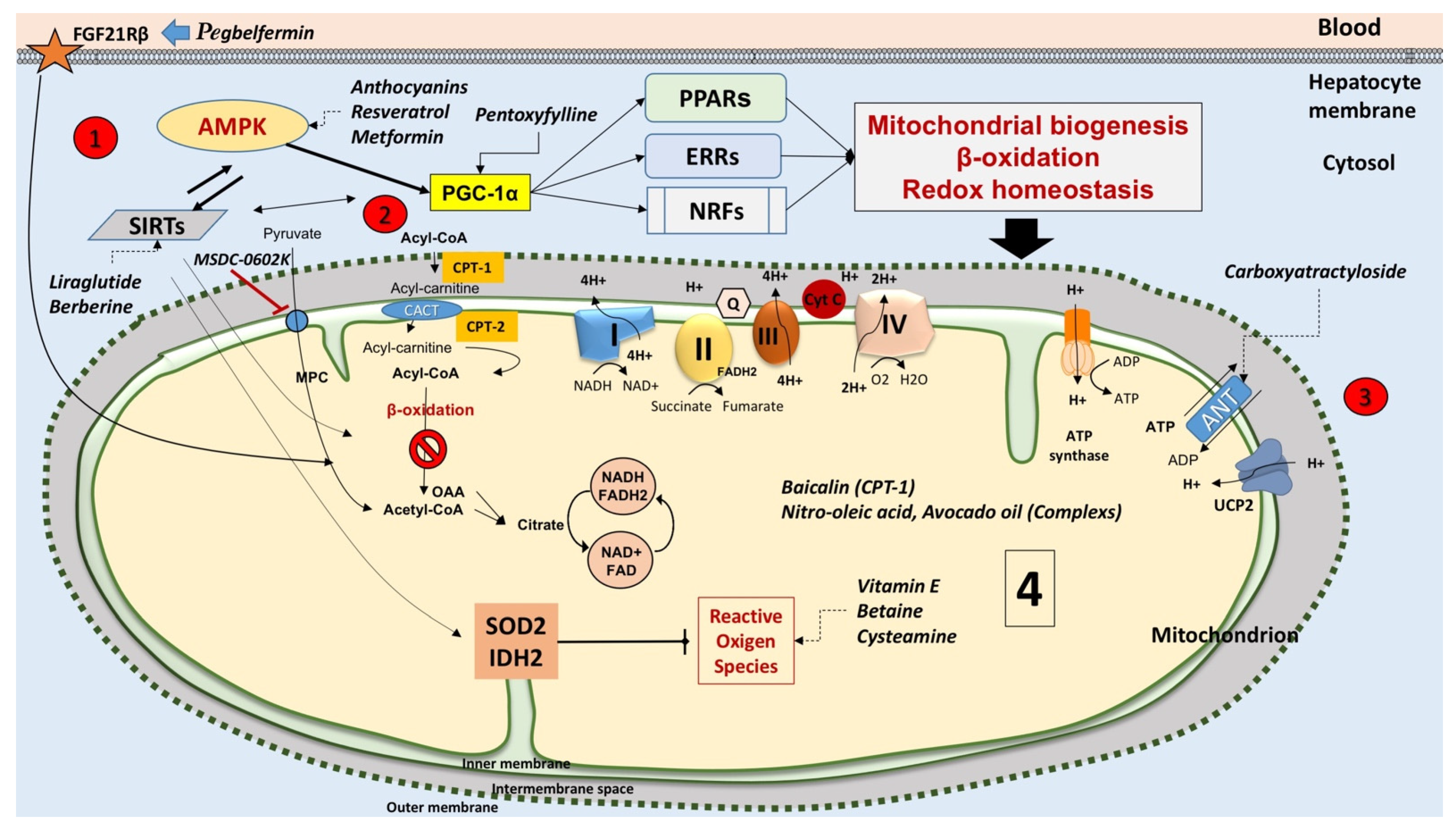{kind=link}
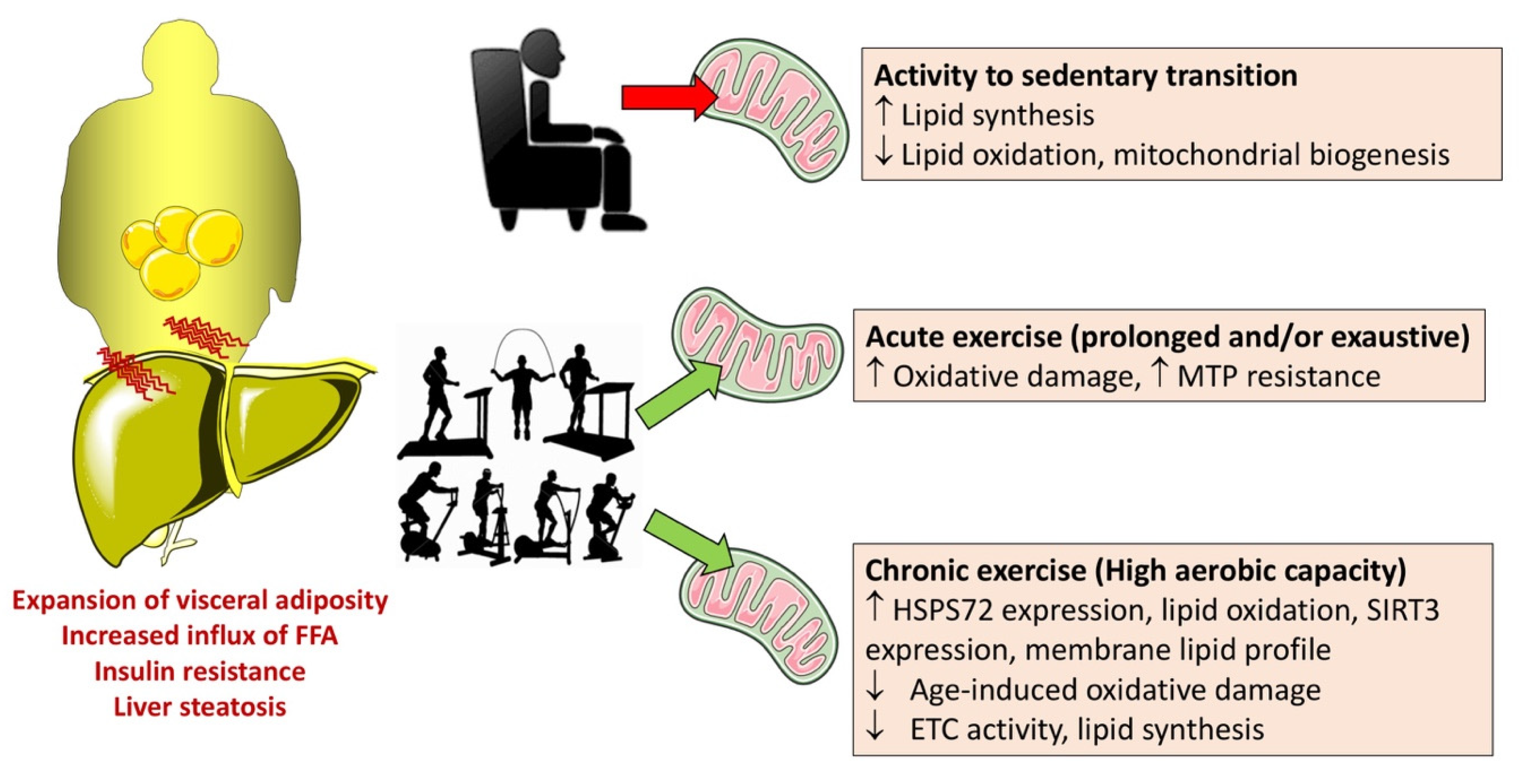{kind=link}
{kind=link}
| Condition | Features |
|---|---|
| Nonalcoholic fatty liver (NAFL) | Simple steatosis No evidence of inflammation, necrosis, and fibrosis |
| Nonalcoholic steatohepatitis (NASH) | Steatosis associated with pericellular fibrosis, lobular inflammation, and apoptosis. Histological findings are indistinguishable from alcoholic steatohepatitis [57]. NASH affects 3% to 6% of the U.S. population with a risk of progression to cirrhosis in about 20% of the cases [58,59,60,61] Possible progression to hepatocellular carcinoma (HCC) [62] |
| Cryptogenic cirrhosis | Late stage of progressive hepatic fibrosis and steatotic chronic liver disease Distortion of the architecture of the liver and growth of regenerative nodules May progress to HCC |
| Hepatocellular carcinoma (HCC) | Primary tumor of the liver that usually develops in the setting of chronic liver disease |
| Outcome | Causes |
|---|---|
| Increased influx of the circulating free fatty acids (FFA) | Obesity |
| Overfeeding (also saturated fatty acids) | |
| Rapid weight loss | |
| Total parenteral nutrition (transformation of carbohydrates/proteins to TG) | |
| Decreased mitochondrial β-oxidation of FFA | Vitamin B5 (pantothenic acid) deficiency |
| Excessive alcohol consumption | |
| Drugs: valproic acid or chronic aspirin → coenzyme A deficiency | |
| Decreased activity acyl-CoA:diacylglycerol acyltransferase 1 (DGAT1) | |
| Possible decreased activity of miRNAs | |
| Increased accumulation of ceramides | |
| Decreased secretion/export of very low-density lipoproteins (VLDL) | Abetalipoproteinemia, protein malnutrition, or choline deficiency |
| Defective secretion of postprandial Apo B | |
| Drugs: amiodarone and tetracycline → defective lipidation of Apo B (inhibition of microsomal triglyceride transfer protein (MTP)) | |
| Ongoing NAFLD/NASH |
| Class (Type of Compounds) | Observed Clinical Effects |
|---|---|
| Vitamin (Vitamin E) |
|
| Anti-apoptotic agents (Emricasan) |
|
| Insulin sensitizer (Metformin) |
|
| PPARγ-agonists (Thiazolidinediones: pioglitazone, rosiglitazone, MSDC-0602K) |
|
| Dual PPAR activators (Elafibranor, Saroglitazar) |
|
| Pan-PPAR activator (Lanifibranor) |
|
| Glucagon-like peptide (GLP)-1 and GLP-1 agonists (Liraglutide, Semaglutide, Tirzepatide, CotadutideDulaglutide, Exenatide, Albiglutide) |
|
| Inhibitors of metabolic enzymes (Acetyl-CoA carboxylase [ACC] inhibitor; Firsocostat [GS-0976], PF-05221304, PF-06865571, PF-06835919) |
|
| Cleavage of citrate to generate oxaloacetate and acetyl-CoA (ATP-Citrate Lyase [ACLY]) |
|
| Liver farnesoid X receptor (FXR) agonist—bile acid (Obeticolic acid [OCA]) |
|
| FXR agonist—non-bile acids (Tropifexor, Cilofexor, EYP001Nidufexor, EDP-305) |
|
| Enzyme inhibitors—Inhibition of stearoyl-CoA desaturase 1 (SCD1) (Arachidyl-amido cholanoic acid [aramchol]) |
|
| Bile acids derivative (Norursodeoxycholic acid) |
|
| Intestinal hormones (Fibroblast growth factor-19 [FGF-19]; Fibroblast growth factor-21 [FGF-21] and its analog Pegbelfermin) |
|
| Hepatic thyroid hormone receptor (THR)-β-selective agonists (Resmetirom; VK2809) |
|
| Sodium/glucose transport protein 2 (SGLT2) inhibitors (Empagliflozin, Canagliflozin, Dapagliflozin, Lipogliflozin) | |
| Immune response (Selonsertib [GS-4997]) |
|
| Chemokine inhibitors (CCR2/CCR5 receptor inhibitor Cenicriviroc) |
|
| Deubiquitinase function (Cylindromatosis[CYLD]) |
|
| Antifibrotic agents (ND-L02-s0201 anti-heat shock protein 47 [HSP47]) |
|
| Inhibitor of galectin (Belapectin) |
|
| Agent acting at extrahepatic levels (BAR502) |
|
| Agents acting at extrahepatic levels (Probiotics) |
|
| Statin (Atorvastatin) |
|
| Fatty acids (Omega-3 fatty acids, Polyunsaturated fatty acids [PUFA]) | |
| Antinflammatory agent (Aspirin) |
|
| Natural pentacyclic isoquinoline alkaloid (Berberine) |
|
| Inhibitor of mitochondrial pyruvate carrier (MSDC-0602K) |
|
| General Measures | Notes |
|---|---|
| Lifestyles | Moderately hypocaloric diet plus physical exercise might improve mitochondrial function and alleviate inflammation [259,260,261] |
| Antidiabetic drugs | Elafibranor [190,193] |
| Liraglutide [262] | |
| Metformin [263] | |
| Thiazolidinediones (pioglitazone) [264], MSDC-0602K [189] | |
| Bile acids | Obeticholic acid [27,208,209] |
| Ursodeoxycholic acid [265] | |
| Agents acting as antioxidants, on nuclear receptors or mitochondrial metabolism | Vitamin E (α-Tocopherol) [64] |
| Tempol [266] 1 | |
| Resveratrol [267,268,269,270] 1 | |
| Mitoquinone (Mito-Q) and Mitovitamin E (MitoVit-E) [271,272,273] 1,2 | |
| Silymarin (major component is Silybin) [108,274,275] | |
| Corilagin [276] 2 | |
| Anthocyanins (i.e., Cyanidin) [277,278] 1 | |
| Dihydromyricetin [279] 1 | |
| Berberine [255] 1 | |
| Hydroxytyrosol [249] 1 | |
| Cysteamine [280,281] | |
| Pentoxifilline [282,283,284] | |
| Avocado oil [285,286,287] 1 | |
| Pegbelfermin (via FGF21R beta) [225] | |
| Mitotherapy | Exogenous mitochondria tagged with green-fluorescence protein (GFP) and retrieved in mouse liver, lungs, brain, muscle, and kidneys [288,289] 1. Improved energy production may restore hepatocyte function [290] 1 |
| Miscellanea | Aramchol [216,217] |
| Baicalin [291] 1,2 | |
| Nitro-oleic acid [292] 1 | |
| Carboxyatractyloside [293] 1 | |
| Genistein [294] | |
| Firsocostat (acetyl-CoA carboxylase (ACC) inhibitor) [295] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Ciaula, A.; Passarella, S.; Shanmugam, H.; Noviello, M.; Bonfrate, L.; Wang, D.Q.-H.; Portincasa, P. Nonalcoholic Fatty Liver Disease (NAFLD). Mitochondria as Players and Targets of Therapies? Int. J. Mol. Sci. 2021, 22, 5375. https://doi.org/10.3390/ijms22105375
Di Ciaula A, Passarella S, Shanmugam H, Noviello M, Bonfrate L, Wang DQ-H, Portincasa P. Nonalcoholic Fatty Liver Disease (NAFLD). Mitochondria as Players and Targets of Therapies? International Journal of Molecular Sciences. 2021; 22(10):5375. https://doi.org/10.3390/ijms22105375
Chicago/Turabian StyleDi Ciaula, Agostino, Salvatore Passarella, Harshitha Shanmugam, Marica Noviello, Leonilde Bonfrate, David Q.-H. Wang, and Piero Portincasa. 2021. "Nonalcoholic Fatty Liver Disease (NAFLD). Mitochondria as Players and Targets of Therapies?" International Journal of Molecular Sciences 22, no. 10: 5375. https://doi.org/10.3390/ijms22105375
APA StyleDi Ciaula, A., Passarella, S., Shanmugam, H., Noviello, M., Bonfrate, L., Wang, D. Q.-H., & Portincasa, P. (2021). Nonalcoholic Fatty Liver Disease (NAFLD). Mitochondria as Players and Targets of Therapies? International Journal of Molecular Sciences, 22(10), 5375. https://doi.org/10.3390/ijms22105375