
Tandem Repeats in Bacillus: Unique Features and Taxonomic Distribution

Abstract
1. Introduction
2. Materials and Methods
2.1. Detection of Tandem Repeats
2.2. Identification of Tandem Repeat Families
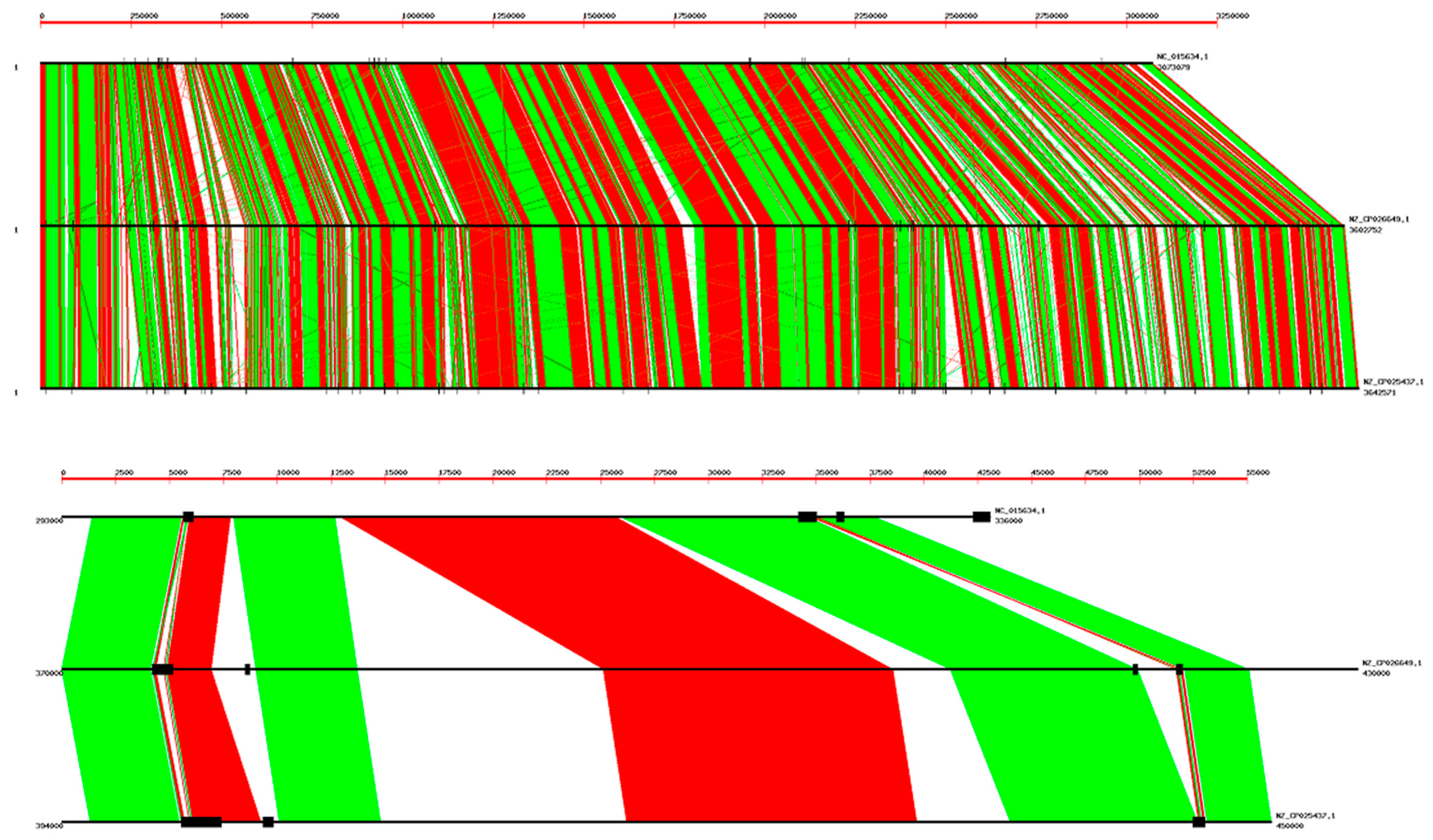
2.3. Genome Alignments
3. Results
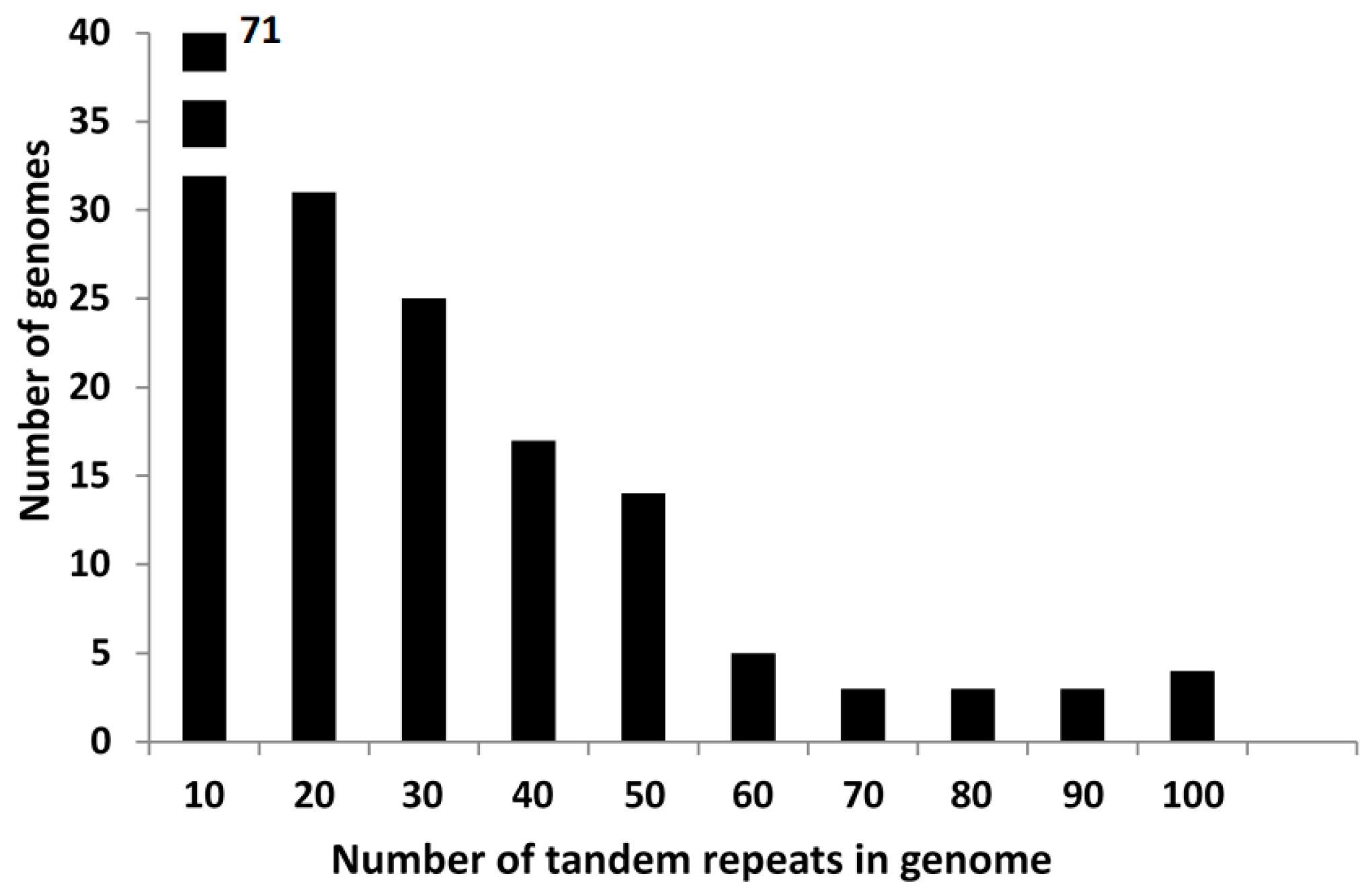
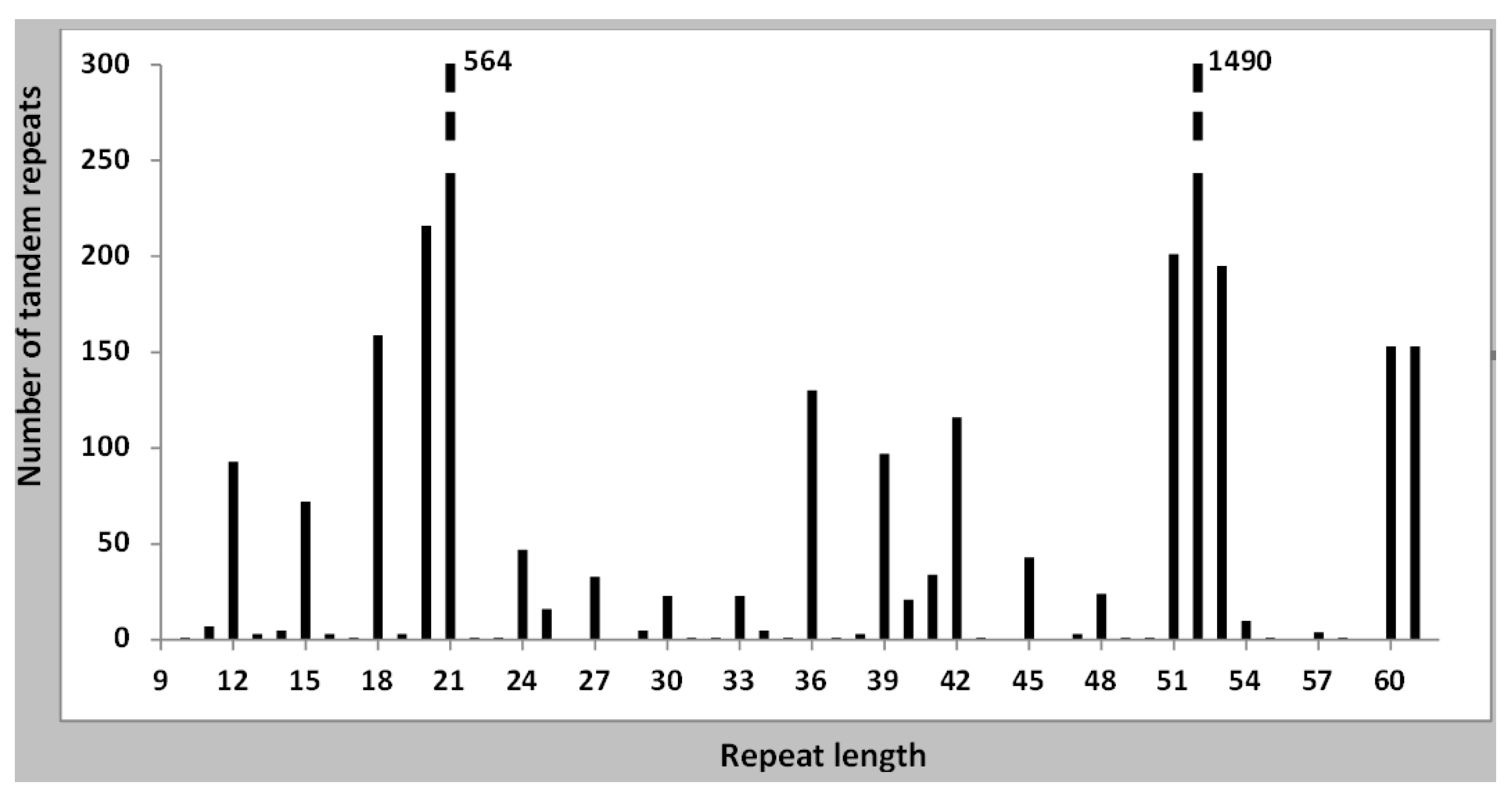
3.1. General Features of Tandem Repeats
3.2. Tandem Repeats with a 52 nt Repeat
3.3. Tandem Repeats with a 20–21 nt Repeat
3.4. Tandem Repeats with Repeat Length Multiple of Three
3.5. Taxonomic Distribution of Tandem Repeats
4. Discussion
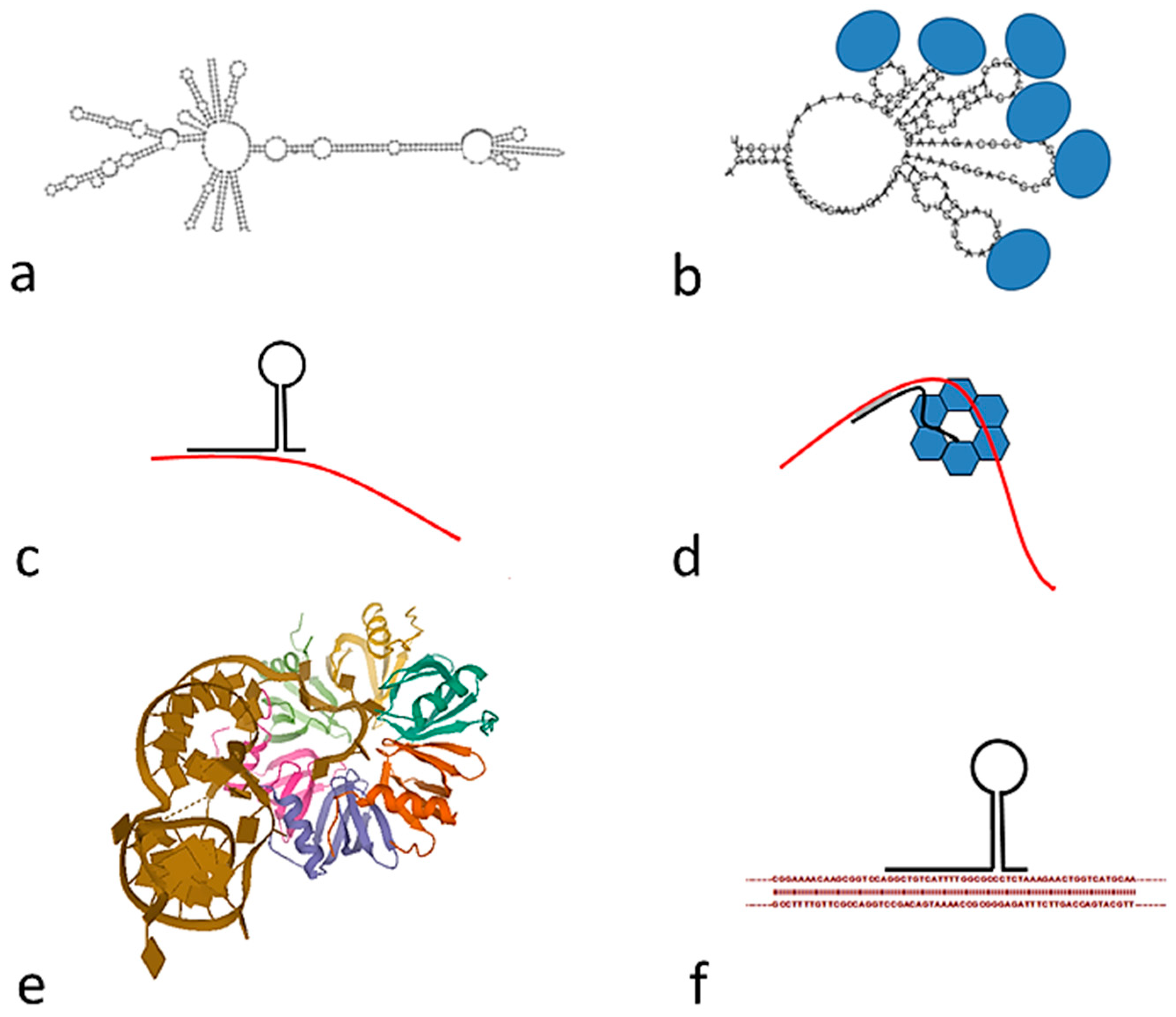
4.1. Unique Features of Tandem Repeats
4.2. Origin of Tandem Repeats and Comparison with Eukaryotes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Treangen, T.J.; Abraham, A.; Touchon, M.; Rocha, E.P.C. Genesis, effects and fates of repeats in prokaryotic genomes. FEMS Microbiol. Rev. 2009, 33, 539–571. [Google Scholar] [CrossRef] [PubMed]
- Denoeud, F.; Vernaud, G. Identification of polymorphic tandem repeats by direct comparison of genome sequence from different bacterial strains: A web-based resource. BMC Bioinform. 2004, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Subirana, J.A.; Messeguer, X. Unique features of tandem repeats in bacteria. J. Bacteriol. 2020, 202, e00229-20. [Google Scholar] [CrossRef] [PubMed]
- Khurana, H.; Sharma, M.; Verma, H.; Lopes, B.S.; Lal, R.; Negi, R.N. Genomic insights into the phylogeny of Bacillus strains and elucidation of their secondary metabolic potential. Genomics 2020, 112, 3191–3200. [Google Scholar] [CrossRef]
- Maughan, H.; Van der Auwera, G. Bacillus taxonomy in the genomic era finds phenotypes to be essential though often misleading. Infect. Gen. Evol. 2011, 11, 789–797. [Google Scholar] [CrossRef]
- Hernández-González, I.L.; Moreno-Hagelsieb, G.; Olmedo-Álvarez, G. Environmentally-driven gene content convergence and the Bacillus phylogeny. BMC Evol. Biol. 2018, 18, 148. [Google Scholar] [CrossRef]
- Secaira-Morocho, H.; Castillo, J.A.; Driks, A. Diversity and evolutionary dynamics of spore-coat proteins in spore-forming species of Bacillales. Microb. Genom. 2020, 6, 000451. [Google Scholar] [CrossRef]
- NCBI. Available online: https://www.ncbi.nlm.nih.gov/genome/browse#!/prokaryotes/ (accessed on April 2021).
- ALGGEN Algorísmica I Genètica. Available online: http://alggen.lsi.upc.edu (accessed on April 2021).
- Subirana, J.A.; Albà, M.M.; Messeguer, X. High evolutionary turnover of tandem repeat families in Caenorhabditis. BMC Evol. Biol. 2015, 15, 218. [Google Scholar] [CrossRef]
- Subirana, J.A.; Messeguer, X. Evolution of Tandem Repeat Satellite Sequences in Two Closely Related Caenorhabditis Species. Diminution of Satellites in Hermaphrodites. Genes 2017, 8, 351. [Google Scholar] [CrossRef]
- Subirana, J.A.; Messeguer, X. How Long Are Long Tandem Repeats? A Challenge for Current Methods of Whole-Genome Sequence Assembly: The Case of Satellites in Caenorhabditis elegans. Genes 2018, 9, 500. [Google Scholar] [CrossRef]
- Tørresen, O.K.; Star, B.; Mier, B.; Andrade-Navarro, M.A.; Bateman, A.; Jarnot, P.; Gruca, A.; Grynberg, M.; Kajava, A.V.; Promponas, V.J.; et al. Tandem repeats lead to sequence assembly errors and impose multi-level challenges for genome and protein databases. Nucleic Acids Res. 2019, 47, 10994–11006. [Google Scholar] [CrossRef] [PubMed]
- Treangen, T.J.; Messeguer, X. M-GCAT: Interactively and efficiently constructing large-scale multiple genome comparison frameworks in closely related species. BMC Bioinform. 2006, 7, 433. [Google Scholar] [CrossRef] [PubMed]
- Ishino, Y.; Krupovic, M.; Forterre, P. History of CRISPR-Cas from Encounter with a Mysterious Repeated Sequence to Genome Editing Technology. J. Bacteriol. 2018, 200, e00580-17. [Google Scholar] [CrossRef]
- Patel, S.; Gupta, R.S. A phylogenomic and comparative genomic framework for resolving the polyphyly of the genus Bacillus: Proposal for six new genera of Bacillus species, Peribacillus gen.nov., Cytobacillus gen. nov., Mesobacillus gen. nov., Neobacillus gen. nov., Metabacillus gen. nov. and Alkalihalobacillus gen. nov. Int. J. Syst. Evol. Microbiol. 2020, 70, 406–438. [Google Scholar] [PubMed]
- Gupta, R.S.; Patel, S.; Saini, N.; Chen, S. Robust demarcation of 17 distinct Bacillus species clades, proposed as novel Bacillaceae genera, by phylogenomics and comparative genomic analyses: Description of Robertmurraya kyonggiensis sp. nov. and proposal for an emended genus Bacillus limiting it only to the members of the Subtilis and Cereus clades of species. Int. J. Syst. Evol. Microbiol. 2020, 70, 5753–5798, 6531–6533. [Google Scholar]
- Barco, R.A.; Garrity, G.M.; Scott, J.J.; Amend, J.P.; Nealson, K.H.; Emerson, D. A Genus Definition for Bacteria and Archaea Based on a Standard Genome Relatedness Index. mBio 2020, 11, e02475-19. [Google Scholar] [CrossRef]
- Ash, C.; Priest, F.G.; Collins, M.D. Molecular identification of rRNA group 3 bacilli (Ash, Farrow, Wallbanks and Collins) using a PCR probe test. Antonie Van Leeuwenhoek 1993, 64, 253–260. [Google Scholar] [CrossRef]
- Wagner, G.H.; Romby, P. Small RNAs in Bacteria and Archaea: Who They Are, What They Do, and How They Do It. In Advances in Genetics; Friedmann, T., Dunlap, J., Goodwin, S.F., Eds.; Elsevier Science Direct: Amsterdam, The Netherlands, 2015; Volume 90, pp. 133–208. ISBN 978-0-12-803694-5. [Google Scholar]
- Qin, J.; Wang, X.; Wang, L.; Zhu, B.; Zhang, X.; Yao, Q.; Xu, P. Comparative transcriptome analysis reveals different molecular mechanisms of Bacillus coagulans 2-6 response to sodium lactate and calcium lactate during lactic acid production. PLoS ONE 2015, 10, e0124316. [Google Scholar] [CrossRef]
- Bechhofer, D.H.; Deutscher, M.P. Bacterial ribonucleases and their roles in RNA metabolism. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 242–300. [Google Scholar] [CrossRef]
- RNAfold web server. Available online: http://rna.tbi.univie.ac.at//cgi-bin/RNAWebSuite/RNAfold.cgi (accessed on April 2021).
- Peer, A.; Margalit, H. Accessibility and Evolutionary Conservation Mark Bacterial Small-RNA Target-Binding Regions. J. Bacteriol. 2011, 193, 1690–1701. [Google Scholar] [CrossRef]
- Quendera, A.P.; Seixas, A.F.; dos Santos, R.F.; Santos, I.; Silva, J.P.N.; Arraiano, C.M.; Andrade, J.M. RNA-Binding Proteins Driving the Regulatory Activity of Small Non-coding RNAs in Bacteria. Front. Mol. Biosci. 2020, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Updegrove, T.B.; Zhang, A.; Storz, G. Hfq: The flexible RNA matchmaker. Curr. Opin. Microbiol. 2016, 30, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Dimastrogiovanni, D.; Fröhlich, K.S.; Bandyra, K.J.; Bruce, H.A.; Hohensee, S.; Vogel, J.; Luisi, B.F. Recognition of the small regulatory RNA RydC by the bacterial Hfq protein. eLife 2014, 3, e05375. [Google Scholar] [CrossRef] [PubMed]
- Orans, J.; Kovach, A.R.; Hoff, K.E.; Horstmann, N.M.; Brennan, R.G. Crystal structure of an Escherichia coli Hfq Core(residues 2–69)–DNA complex reveals multifunctional nucleic acid binding sites. Nucleic Acids Res. 2020, 38, 3987–3997. [Google Scholar] [CrossRef]
- PDB, Protein Data Bank. Available online: https://www.rcsb.org/3d-view/4V2S/1 (accessed on April 2021).
- Castresana, J.; Guigó, R.; Albà, M.M. Clustering of genes coding for DNA binding proteins in a region of atypical evolution of the human genome. J. Mol. Evol. 2004, 59, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Kirchberger, P.C.; Schmidt, M.L.; Ochman, H. The Ingenuity of Bacterial Genomes. Annu. Rev. Microbiol. 2020, 74, 815–834. [Google Scholar] [CrossRef]
- Slomka, S.; Françoise, I.; Hornung, G.; Asraf, O.; Biniashvili, T.; Pilpel, Y.; Dahan, O. Experimental Evolution of Bacillus subtilis Reveals the Evolutionary Dynamics of Horizontal Gene Transfer and Suggests Adaptive and Neutral Effects. Genetics 2020, 216, 543–558. [Google Scholar] [CrossRef]
- Dubnau, D.; Blokesch, M. Mechanisms of DNA Uptake by Naturally Competent Bacteria. Annu. Rev. Genet. 2019, 53, 217–237. [Google Scholar] [CrossRef]
- Liehr, T. Repetitive Elements in Humans. Int. J. Mol. Sci. 2021, 22, 2072. [Google Scholar] [CrossRef]
- Miga, K.H. Centromeric satellite DNAs: Hidden sequence variation in the human population. Genes 2019, 10, 352. [Google Scholar] [CrossRef]
- Phillips, C.M.; Meng, X.; Zhang, L.; Chretien, J.H.; Urnov, F.D.; Dernburg, A.F. Identification of chromosome sequence motifs that mediate meiotic pairing and synapsis in C. elegans. Nat. Cell Biol. 2009, 11, 934–942. [Google Scholar] [CrossRef] [PubMed]
- Shatskikh, A.S.; Kotov, A.A.; Adashev, V.E.; Bazylev, S.S.; Olenina, L.V. Functional Significance of Satellite DNAs: Insights from Drosophila. Front. Cell Dev. Biol. 2020, 8, 312. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacillus Species | Number of Tandem Repeats | % 52 nt Repeat | Family Code | Consensus Repeat |
|---|---|---|---|---|
| cellulosilyticus | 64 | 73.4 | 37_52_10 | gTGTaTCATACgaaggCAATGACACgtGAgAAAGtaGaaGaaacgnAATAAa |
| 61_52_6 | CAcTCAACGAAGGTcATCATAAGcAAGCAATGCTaCCCCAAAACCAAAcCcn | |||
| coagulans | 53av | 90.8 | 1_52_139 | GTgAAGgAAGgcCnTCnTTTTTcCncGCTTcCTTAACGTAGACGcgCTCTAT |
| 2_52_35 | TTTTGTCCTTTTGaCaGcTTCAAAAnGACATTTCGgGCCCgGATgCAgCntG | |||
| 8_52_18 | TGTCCTTCATaagggtGATGAAaGACAAAACaCnGGcCgggaAAcGgCgAAt | |||
| horikoshii | 64 | 71.9 | 22_52_12 | ATGAAGACaTcAGTGAcGAGaAAtcAGGAGgAGAGAaGTCcTCATcGncGTt |
| kochii | 59 | 88.1 | 52_51_7 | TTCnTTCTGAGTGACtTGCtAatCCcTTTTGCGAAGCgCTCAnCtCtgGtt |
| 92_54_4 | CTTTTaTTAGTcGCGaTTcTcacTTTTgCcCTACTtATCttnTcacTnatTCTT | |||
| litoralis | 80 | 73.7 | 42_52_9 | TGTCGTTCATaAGggtgATGAACGACAAAAGtGnTnaGAAAAGagngnGtag |
| 77_52_5 | AATCGGGACAGAAAAaaGAgcgaGCAGtgaAAnTgaGTCTCGATAgTgggng | |||
| oceanisediminis | 38 | 84.2 | 79_52_5 | nCgCcaAcTTCGGACTCatTCtCtCggTTTTccgcttCTtCTGTCCGAAgtn |
| 96_54_4 | cGATTAACTACCATTTTnCctTncaCCnnccTcaTTTTCGtCGTtAatcCaTcc | |||
| thermoamylovorans | 70 | 78.6 | 4_52_21 | AAAAtGacGACGAGAAnnGGTCTCGTCGCCAAAAAatGGAGTTTTcCGgctc |
| 5_52_21 | TTGgCGACGAGaCcnatTCTCGTCaCCaTTTTgaGGtGAAAAAnGCtCnaTT | |||
| 30_53_11 | TGTCCAATAGAACGGcTCTCgTGGACAAAATnGAGgnnTCAATCAGgAAAAAc | |||
| New species | ||||
| pseudalcaliphilus | 103 | 92.2 | 3_50_31 | gAATCnCGGGGTTGCGaGCnGAAAAAGaGGAGAAAgCCCGAGCAAAcGcn |
| 5_52_25 | GTTAATGTGaAGATACgGAgGCcAAACcttGgAGTAtCTGCACAAAGAGggG | |||
| 26_53_11 | nGtTGGTCGACATGATCAtGgGnAAAAAAGGncaAGAACcTGTCGATGAaGGn | |||
| alkalitelluris | 91 | 74.7 | 4_52_27 | AAgGGAATCAaACAAcgCtTTTCaTTCCCTTTgttggGCTTTtGGCATgAGA |
| 71_52_7 | TGTCTGAAGTaGCCTnGnGTTCGGACAGcTTTgaTtgtTTnnaaGcnAAAGC | |||
| 72_52_7 | CATAGgCctTCTATGATtcAGTTGCcgaaGCgAAAACAAGGAGnAAGTgAAT | |||
| indicus | 90av | 96.6 | 7_52_18 | ATCGTACCCTcgnAAACcGaaAAaCGATnTgggAGGGTAAGCAAanGcnnGA |
| alcalophilus | 103 | 86.4 | 9_52_16 | CCTtTGATTCCCTTTncGGCTtTtATTcaatgGCTTTtGgcaTCATTGcnGn |
| 11_51_15 | TTTTCATTACcTATtcCnntTTaTTCGCACCcTAATtcnncaCAgCnCgGc | |||
| 41_51_9 | TTTTTCATTACcTATcCncnnTTTTTCGCACcnTAATTTggcCtgCtcggC | |||
| sp.m3-13 | 72 | 73.6 | 20_52_13 | ATGAAGACnTcAGTGACgAggAAaaAGGAGgAGAGAaGTCCTCATcGccGTt |
| sp.SG-1 | 77 | 81.8 | 42_53_9 | CanaCCAACAtCcCTcncAtAATcCatTCTCaTTGGnctGaTTActCCcTTTT |
| selenatarsenatis | 82 | 59.8 | Many | |
| mesonae | 95 | 88.4 | Many |
| Bacillus Species | NCBI Code | Repeat (nt) | Characteristic Signals |
|---|---|---|---|
| oagulans | NZ_CP026649.1 | 52 | TCTAYG AARGACA |
| cellulosilyticus | NC_014829.1 | 52 | GGTCATCAT CAATGCT ACGAAGG ATGACAC |
| alkalitelluris | NZ_KV917374.1 | 52 | AAAgGGAAT AAAGCTGTC AATCATAG |
| mesonae | NZ_KV440949.1 | 52 | TTTTC TTCAT |
| weihaiensis | NZ_CP016020.1 | 21 | TCGCGG |
| NCBI Code | Bacillus Species | Tandem Repeat | Protein Gene | |||
|---|---|---|---|---|---|---|
| Start | Length | Start | Length | NCBI Code | ||
| NC_015634.1 | coagulans | 2718394 | 301 | 2718351 | 447 | WP_013860576 |
| NZ_CP026649.1 | coagulans | Heavily mutated | 3215973 | 387 | WP_035183339 | |
| NZ_LT603683.1 | glycinifermentans | 580499 | 301 | 580438 | 438 | WP_065894177 |
| NC_006582.1 | clausii | 3644328 | 241 | 3644228 | 402 | WP_011248345 |
| NC_017190.1 | amyloliquefaciens | 438961 | 301 | 438923 | 369 | WP_014471456 |
| NC_014551.1 | amyloliquefaciens | 456884 | 241 | 456846 | 309 | WP_013351072 |
| NC_006322.1 | licheniformis | 540638 | 301 | 540531 | 441 | WP_011197566 |
| NZ_CP007640.1 | atrophaeus | 4062283 | 301 | 4062244 | 375 | WP_010789649 |
| NC_000964 | subtilis | 494545 | 301 | 494506 | 372 | WP_003246542 |
| NCBI Code | Bacillus Species | Genome Size (Mb) | GC% | Number of Tandem Repeats with a Given Repeat Size | |||||
|---|---|---|---|---|---|---|---|---|---|
| Total | 10–19 | 20–21 | 22–50 | 51–53 | >53 | ||||
| Rich in 52 nt repeat | |||||||||
| NZ_KV917374.1 | alkalitelluris | 5.43 | 36.4 | 91 | 7 | 7 | 3 | 69 | 5 |
| NC_014829.1 | cellulosilyticus | 4.68 | 36.5 | 64 | 7 | 0 | 2 | 47 | 8 |
| NC_015634.1 | coagulans | 3.07 | 47.3 | 26 | 0 | 0 | 1 | 21 | 4 |
| NC_016023.1 | coagulans | 3.55 | 46.5 | 63 | 0 | 1 | 1 | 57 | 4 |
| NZ_CP023704.1 | thermoamylovorans | 4.02 | 37.5 | 70 | 2 | 4 | 7 | 55 | 2 |
| Intermediate | |||||||||
| NC_022524.1 | infantis | 4.88 | 46 | 50 | 2 | 12 | 8 | 27 | 1 |
| NZ_BASE01000145 | selenatarsenitis | 4.76 | 42.1 | 82 | 1 | 30 | 1 | 49 | 1 |
| Rich in 21 nt repeat | |||||||||
| NZ_CP016020.1 | weihaiensis | 4.36 | 36.5 | 32 | 2 | 29 | 0 | 0 | 1 |
| NZ_CP011008.1 | simplex | 5.52 | 39.8 | 40 | 3 | 27 | 9 | 0 | 1 |
| NZ_CP017080.1 | muralis | 5.01 | 42.3 | 38 | 1 | 22 | 13 | 0 | 2 |
| Cereus group | |||||||||
| NC_004722.1 | cereus | 5.51 | 35.3 | 25 | 9 | 1 | 15 | 0 | 0 |
| NZ_CP018931.1 | cereus | 5.24 | 35.4 | 31 | 5 | 3 | 21 | 0 | 2 |
| NZ_CP007512.1 | bombysepticus | 5.88 | 35.0 | 31 | 9 | 4 | 15 | 0 | 3 |
| NC_003997.3 | anthracis | 5.23 | 35.4 | 19 | 3 | 5 | 11 | 0 | 0 |
| NZ_CP009692.1 | mycoides | 5.64 | 35.4 | 26 | 7 | 2 | 15 | 0 | 2 |
| Tandem repeat poor | |||||||||
| NC_014103.1 | megaterium | 5.1 | 38.1 | 8 | 7 | 1 | 0 | 0 | 0 |
| NC_017138.1 | megaterium | 5.08 | 38.1 | 5 | 2 | 2 | 0 | 0 | 1 |
| NZ_CP011007.1 | pumilus | 3.88 | 41.5 | 4 | 0 | 0 | 4 | 0 | 0 |
| NC_014551.1 | amyloliquefaciens | 3.98 | 46.1 | 3 | 1 | 0 | 0 | 0 | 2 |
| NC_000964.3 | subtilis | 4.22 | 43.5 | 1 | 0 | 0 | 0 | 0 | 1 |
| NZ_CP012024.1 | smithii | 3.38 | 40.8 | 0 | |||||
| NZ_CP012502.1 | beveridgei | 3.58 | 46.1 | 0 | |||||
| NZ_CP017786.1 | xiamenensis | 3.64 | 41.5 | 0 | |||||
| Group | Genome (Mb) | CG% | Number of Tandem Repeats | 52 nt Tandem Repeats |
|---|---|---|---|---|
| CEREUS | 5.5 | 35.4 | 25.7 | NO |
| SUBTILIS | 4.2 | 44.9 | 3.8 | NO |
| PUMILUS | 3.8 | 41.3 | 3.2 | NO |
| METHANOLICUS | 3.3–6.4 | 36–42 | 12–54 | Variable |
| MEGATERIUM | 3.9–5.5 | 35–38 | 2–7 | NO |
| SIMPLEX | 4.6–5.5 | 39–42 | 12–40 | NO |
| HALODURANS-A | 4.6 | 38.7 | 80.3 | YES |
| HALODURANS-B | 4.1 | 42.3 | 4 | NO |
| COAGULANS | 3.6 | 37–46 | 56 | YES |
| MISCELLANEOUS | 3.2–5.3 | 33–45 | 0–49 | Variable |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subirana, J.A.; Messeguer, X. Tandem Repeats in Bacillus: Unique Features and Taxonomic Distribution. Int. J. Mol. Sci. 2021, 22, 5373. https://doi.org/10.3390/ijms22105373
Subirana JA, Messeguer X. Tandem Repeats in Bacillus: Unique Features and Taxonomic Distribution. International Journal of Molecular Sciences. 2021; 22(10):5373. https://doi.org/10.3390/ijms22105373
Chicago/Turabian StyleSubirana, Juan A., and Xavier Messeguer. 2021. "Tandem Repeats in Bacillus: Unique Features and Taxonomic Distribution" International Journal of Molecular Sciences 22, no. 10: 5373. https://doi.org/10.3390/ijms22105373
APA StyleSubirana, J. A., & Messeguer, X. (2021). Tandem Repeats in Bacillus: Unique Features and Taxonomic Distribution. International Journal of Molecular Sciences, 22(10), 5373. https://doi.org/10.3390/ijms22105373