
Dadaist2: A Toolkit to Automate and Simplify Statistical Analysis and Plotting of Metabarcoding Experiments



Abstract
1. Introduction
2. Results
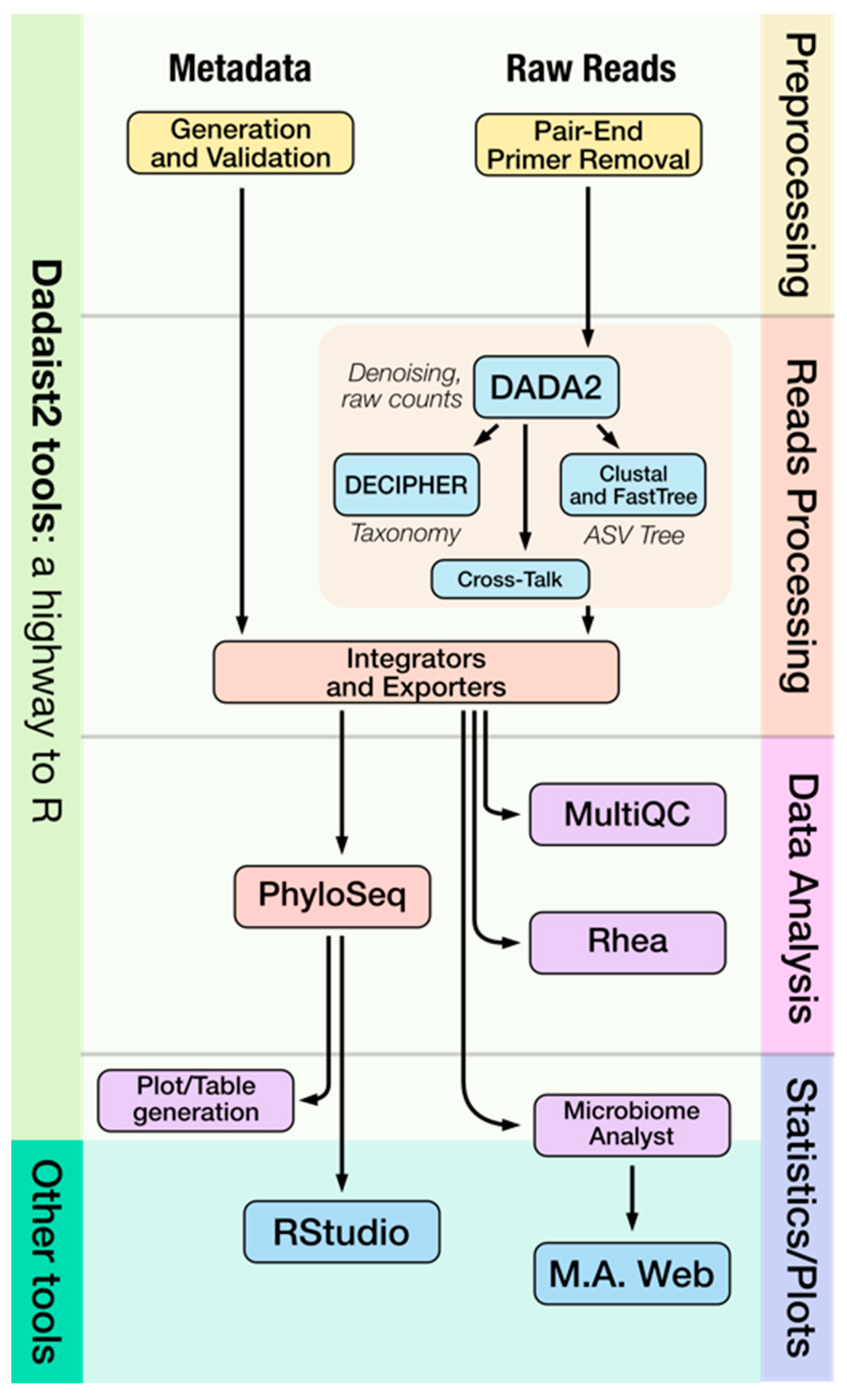
2.1. Dadaist2 Primary Analysis
2.1.1. Cross Talk Removal
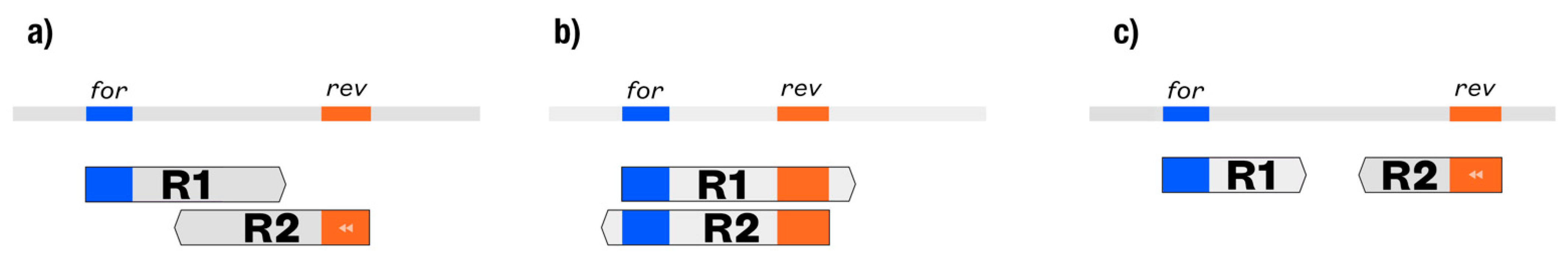
2.1.2. Dedicated Workflow for Amplicons of Variable Lengths
2.1.3. Modular Access to Individual Steps and User-Friendly Reports
2.2. Dadaist2 Secondary Analysis
2.2.1. Automatic Secondary Analyses from the Command Line Using Rhea
2.2.2. Interactive Secondary Analysis Using the MicrobiomeAnalyst Webserver
2.2.3. Custom Secondary Analyses Using PhyloSeq
2.3. Examples and Validation
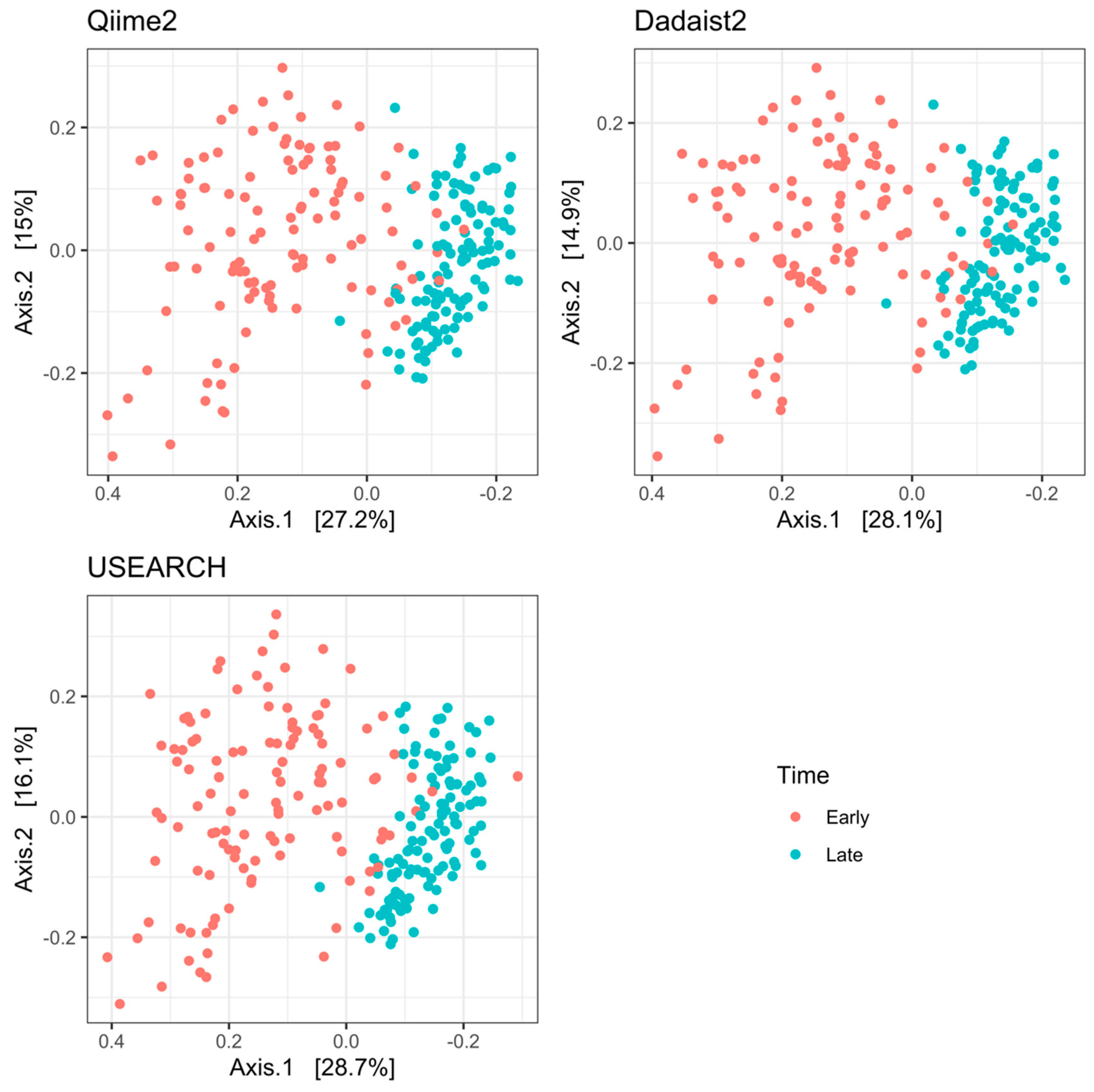
2.3.1. Validation and Comparison of Dadaist2 Components with Common Tools
2.3.2. Reproduction of Biological Results on Real Data Using Dadaist2
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- The Human Microbiome Project Consortium Structure, Function and Diversity of the Healthy Human Microbiome. Nature 2012, 486, 207–214. [CrossRef]
- Bharti, R.; Grimm, D.G. Current Challenges and Best-Practice Protocols for Microbiome Analysis. Brief. Bioinform. 2021, 22, 178–193. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, F.; Laiola, M.; Blaiotta, G.; Ercolini, D. Different Amplicon Targets for Sequencing-Based Studies of Fungal Diversity. Appl. Environ. Microbiol. 2017, 83, e00905-17. [Google Scholar] [CrossRef]
- Westcott, S.L.; Schloss, P.D. De Novo Clustering Methods Outperform Reference-Based Methods for Assigning 16S RRNA Gene Sequences to Operational Taxonomic Units. PeerJ 2015, 3, e1487. [Google Scholar] [CrossRef] [PubMed]
- Kunin, V.; Engelbrektson, A.; Ochman, H.; Hugenholtz, P. Wrinkles in the Rare Biosphere: Pyrosequencing Errors Can Lead to Artificial Inflation of Diversity Estimates. Environ. Microbiol. 2010, 12, 118–123. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Estaki, M.; Jiang, L.; Bokulich, N.A.; McDonald, D.; González, A.; Kosciolek, T.; Martino, C.; Zhu, Q.; Birmingham, A.; Vázquez-Baeza, Y.; et al. QIIME 2 Enables Comprehensive End-to-End Analysis of Diverse Microbiome Data and Comparative Studies with Publicly Available Data. Curr. Protoc. Bioinform. 2020, 70, e100. [Google Scholar] [CrossRef] [PubMed]
- Weißbecker, C.; Schnabel, B.; Heintz-Buschart, A. Dadasnake, a Snakemake Implementation of DADA2 to Process Amplicon Sequencing Data for Microbial Ecology. GigaScience 2020, 9, giaa135. [Google Scholar] [CrossRef]
- Hildebrand, F.; Tadeo, R.; Voigt, A.; Bork, P.; Raes, J. LotuS: An Efficient and User-Friendly OTU Processing Pipeline. Microbiome 2014, 2, 30. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Lagkouvardos, I.; Fischer, S.; Kumar, N.; Clavel, T. Rhea: A Transparent and Modular R Pipeline for Microbial Profiling Based on 16S RRNA Gene Amplicons. PeerJ 2017, 5, e2836. [Google Scholar] [CrossRef]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. MicrobiomeAnalyst: A Web-Based Tool for Comprehensive Statistical, Visual and Meta-Analysis of Microbiome Data. Nucleic Acids Res. 2017, 45, W180–W188. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and Clustering Orders of Magnitude Faster than BLAST. Bioinform. Oxf. Engl. 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME Improves Sensitivity and Speed of Chimera Detection. Bioinform. Oxf. Engl. 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed]
- Köster, J.; Rahmann, S. Snakemake-a Scalable Bioinformatics Workflow Engine. Bioinform. Oxf. Engl. 2018, 34, 3600. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Wright, E.S. Using DECIPHER v2.0 to Analyze Big Biological Sequence Data in R. R J. 2016, 8, 352. [Google Scholar] [CrossRef]
- Nelson, M.C.; Morrison, H.G.; Benjamino, J.; Grim, S.L.; Graf, J. Analysis, Optimization and Verification of Illumina-Generated 16S RRNA Gene Amplicon Surveys. PLoS ONE 2014, 9, e94249. [Google Scholar] [CrossRef]
- Edgar, R.C. UNCROSS2: Identification of Cross-Talk in 16S RRNA OTU Tables. bioRxiv 2018. [Google Scholar] [CrossRef]
- The Bioconda Team; Grüning, B.; Dale, R.; Sjödin, A.; Chapman, B.A.; Rowe, J.; Tomkins-Tinch, C.H.; Valieris, R.; Köster, J. Bioconda: Sustainable and Comprehensive Software Distribution for the Life Sciences. Nat. Methods 2018, 15, 475–476. [Google Scholar] [CrossRef]
- Di Tommaso, P.; Chatzou, M.; Floden, E.W.; Barja, P.P.; Palumbo, E.; Notredame, C. Nextflow Enables Reproducible Computational Workflows. Nat. Biotechnol. 2017, 35, 316–319. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinform. Oxf. Engl. 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Edgar, R.C.; Flyvbjerg, H. Octave Plots for Visualizing Diversity of Microbial OTUs. bioRxiv 2018. [Google Scholar] [CrossRef]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a Dual-Index Sequencing Strategy and Curation Pipeline for Analyzing Amplicon Sequence Data on the MiSeq Illumina Sequencing Platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing Mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Rideout, J.R.; Mercurio, W.G.; Shiffer, A.; Wolfe, B.; Maurice, C.F.; Dutton, R.J.; Turnbaugh, P.J.; Knight, R.; Caporaso, J.G. Mockrobiota: A Public Resource for Microbiome Bioinformatics Benchmarking. mSystems 2016, 1. [Google Scholar] [CrossRef]
- Tang, J.; Iliev, I.D.; Brown, J.; Underhill, D.M.; Funari, V.A. Mycobiome: Approaches to Analysis of Intestinal Fungi. J. Immunol. Methods 2015, 421, 112–121. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, Scalable Generation of High-quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing Large Minimum Evolution Trees with Profiles Instead of a Distance Matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef]
- Telatin, A.; Fariselli, P.; Birolo, G. SeqFu: A Suite of Utilities for the Robust and Reproducible Manipulation of Sequence Files. Bioengineering 2021, 8, 59. [Google Scholar] [CrossRef]
- Telatin, A. Qiime Artifact EXtractor (Qax): A Fast and Versatile Tool to Interact with Qiime2 Archives. BioTech 2021, 10, 5. [Google Scholar] [CrossRef]
- Müller, V.M.; Zietek, T.; Rohm, F.; Fiamoncini, J.; Lagkouvardos, I.; Haller, D.; Clavel, T.; Daniel, H. Gut Barrier Impairment by High-Fat Diet in Mice Depends on Housing Conditions. Mol. Nutr. Food Res. 2016, 60, 897–908. [Google Scholar] [CrossRef]
- James, S.A.; Phillips, S.; Telatin, A.; Baker, D.; Ansorge, R.; Clarke, P.J.; Hall, L.R.; Carding, S. Preterm Infants Harbour a Rapidly Changing Mycobiota That Includes Candida Pathobionts. J. Fungi 2020, 6, 273. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Dadaist2 | DadaSnake | Qiime2 |
|---|---|---|---|
| Command Line Interface | ✔ | ✔ | ✔ |
| Modular design | ✔ | — | ✔ |
| Framework with plugins | — | — | ✔ |
| Alternative denoisers to DADA2 | — | — | ✔ |
| Easy installation | ✔ | — | ✔ |
| Start to End pipeline | ✔ | ✔ | — |
| Crosstalk reduction | ✔ | — | — |
| MultiQC summary report | ✔ | — | — |
| Export to Rhea | ✔ | — | — |
| Export to Microbiome Analyst | ✔ | — | — |
| Export to PhyloSeq | ✔ | ✔ | — |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ansorge, R.; Birolo, G.; James, S.A.; Telatin, A. Dadaist2: A Toolkit to Automate and Simplify Statistical Analysis and Plotting of Metabarcoding Experiments. Int. J. Mol. Sci. 2021, 22, 5309. https://doi.org/10.3390/ijms22105309
Ansorge R, Birolo G, James SA, Telatin A. Dadaist2: A Toolkit to Automate and Simplify Statistical Analysis and Plotting of Metabarcoding Experiments. International Journal of Molecular Sciences. 2021; 22(10):5309. https://doi.org/10.3390/ijms22105309
Chicago/Turabian StyleAnsorge, Rebecca, Giovanni Birolo, Stephen A. James, and Andrea Telatin. 2021. "Dadaist2: A Toolkit to Automate and Simplify Statistical Analysis and Plotting of Metabarcoding Experiments" International Journal of Molecular Sciences 22, no. 10: 5309. https://doi.org/10.3390/ijms22105309
APA StyleAnsorge, R., Birolo, G., James, S. A., & Telatin, A. (2021). Dadaist2: A Toolkit to Automate and Simplify Statistical Analysis and Plotting of Metabarcoding Experiments. International Journal of Molecular Sciences, 22(10), 5309. https://doi.org/10.3390/ijms22105309