
NRF2 DLG Domain Mutations Identified in Japanese Liver Cancer Patients Affect the Transcriptional Activity in HCC Cell Lines

 , ,
, ,  ,
, 
Abstract
1. Introduction
2. Results
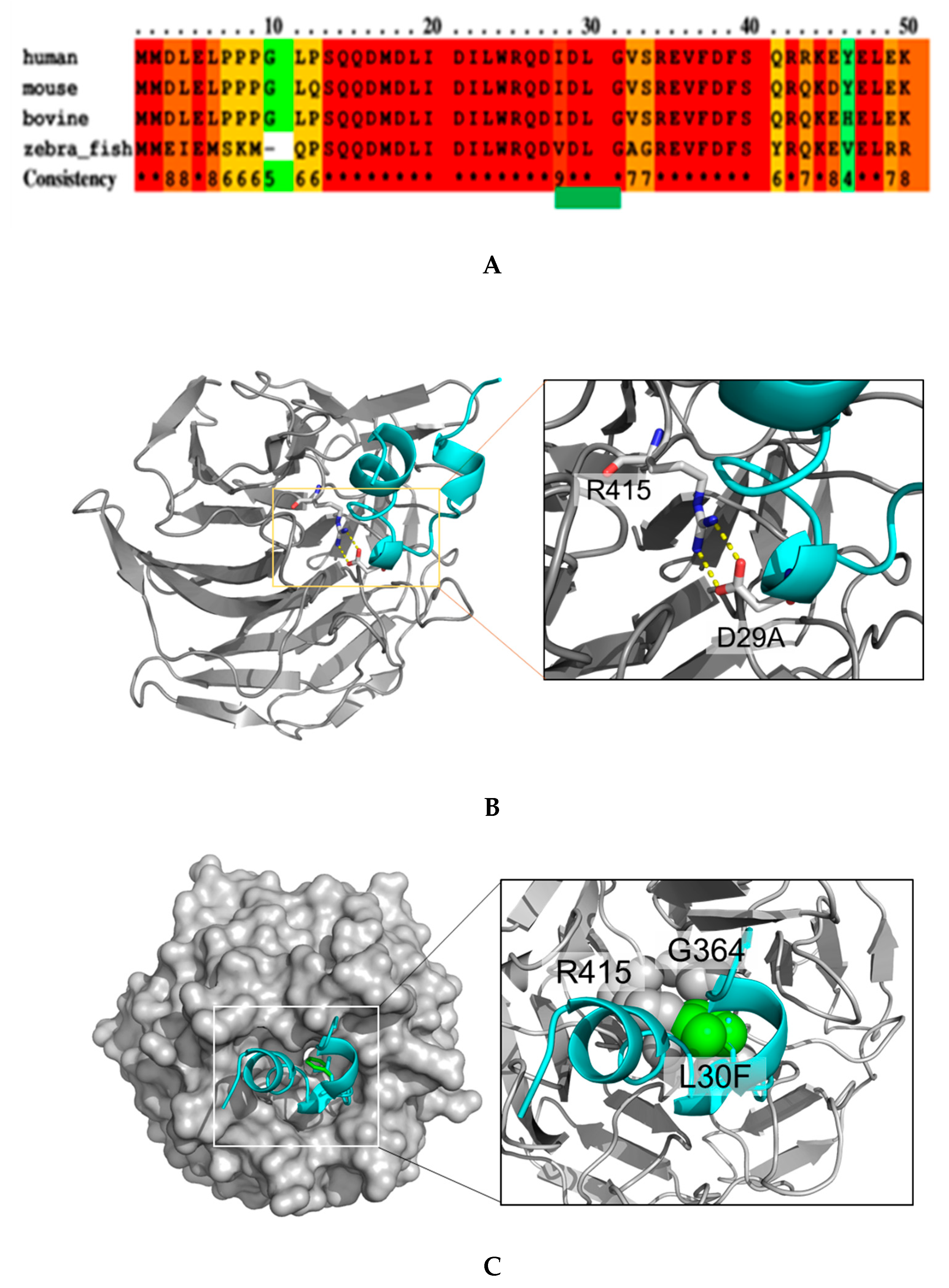
2.1. NRF2 Mutations Are Mostly Located in the DLG Motif of NRF2
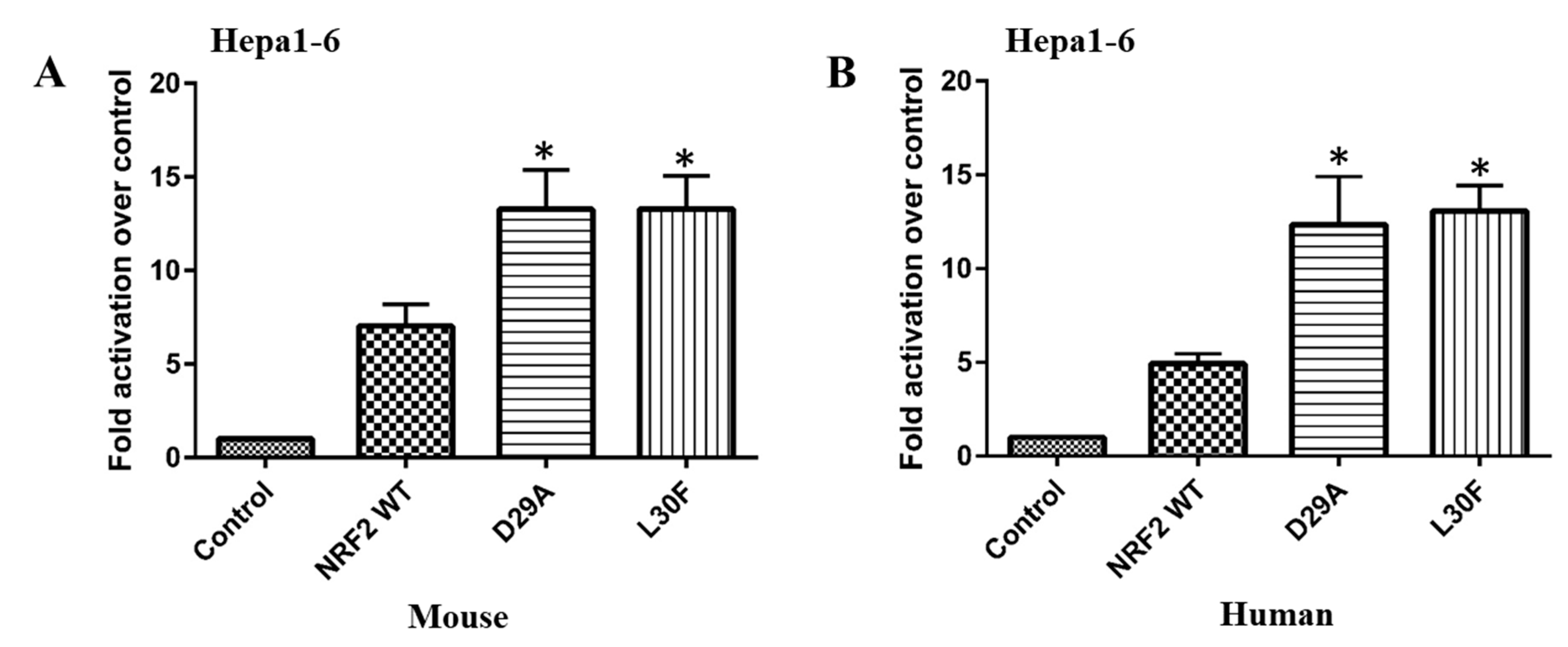
2.2. NRF2 DLG Mutations Have a Gain-of-Function Activity
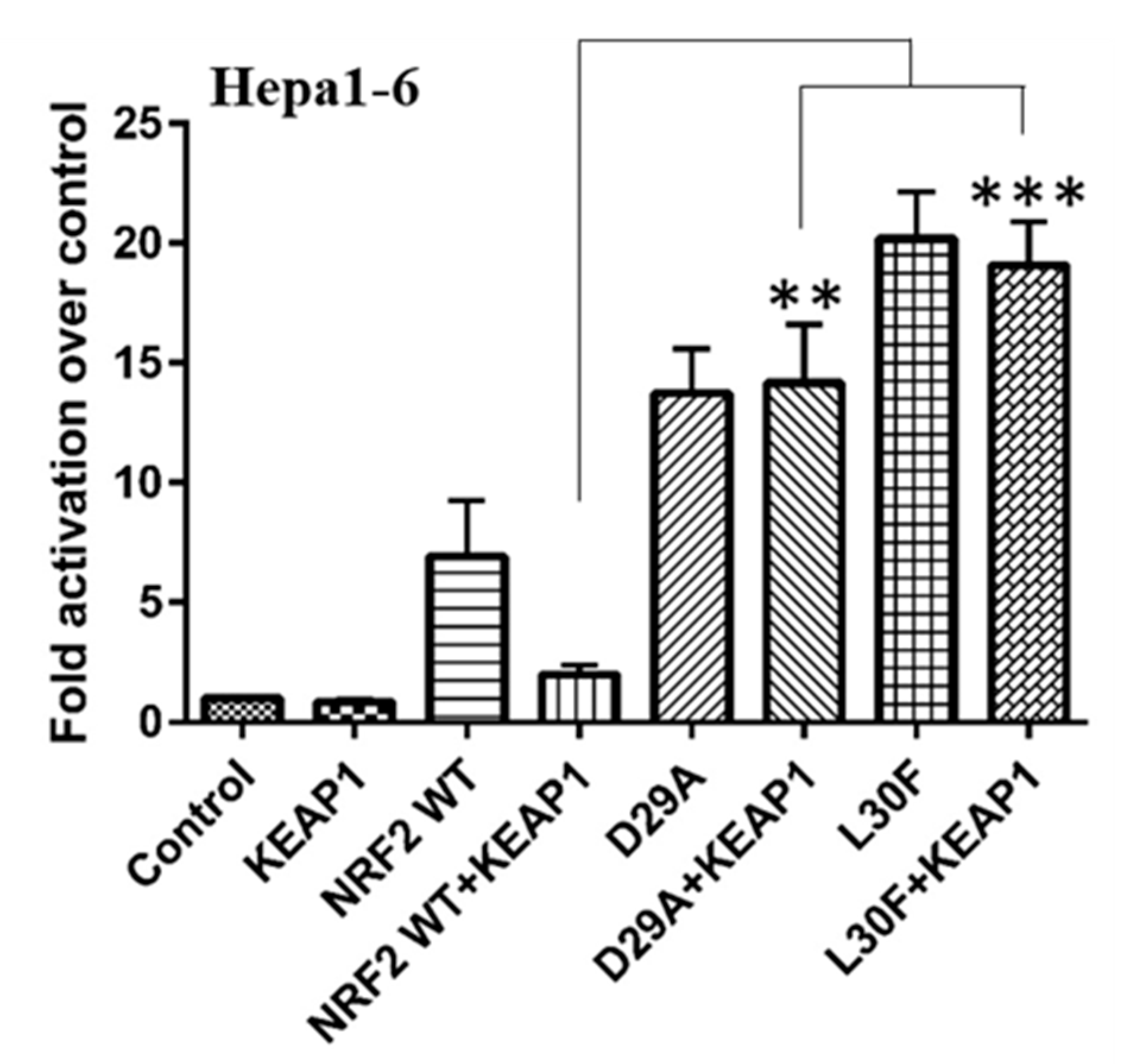
2.3. KEAP1 Expression Fails to Reduce the Transcriptional Activity of NRF2 MTs
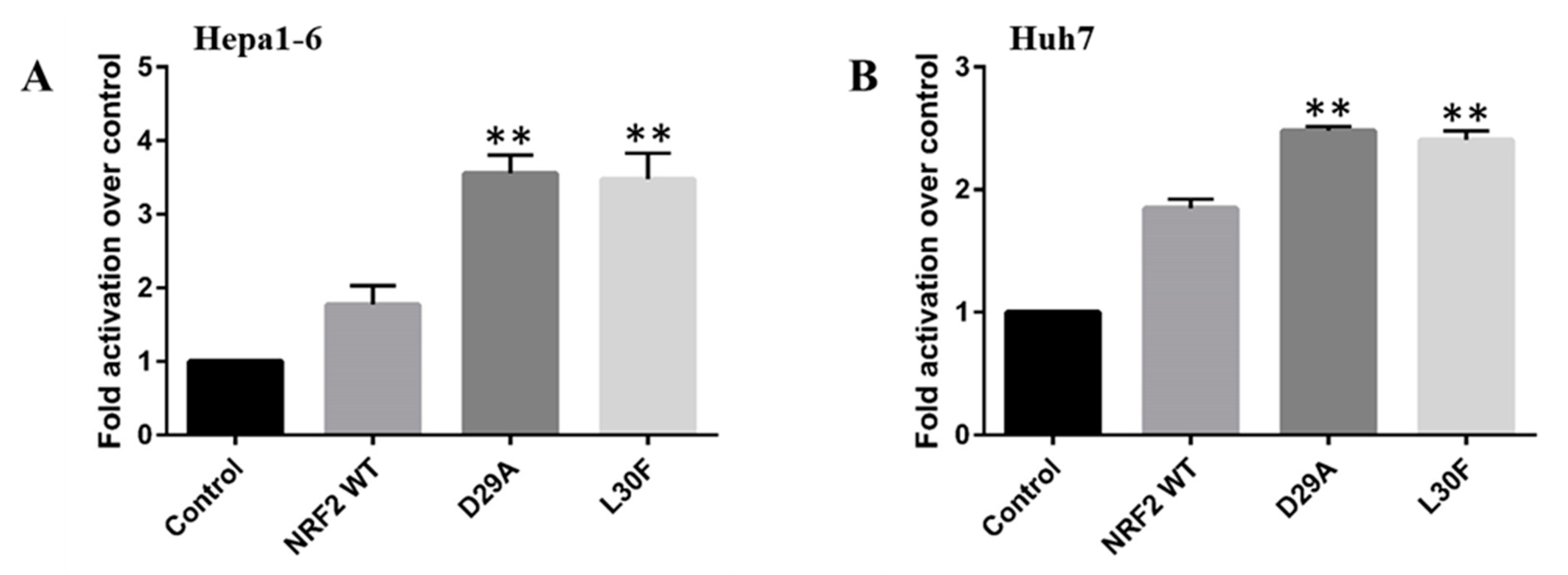
2.4. NRF2 Mutations Increase the Transcriptional Activity of the MMP9 Promoter
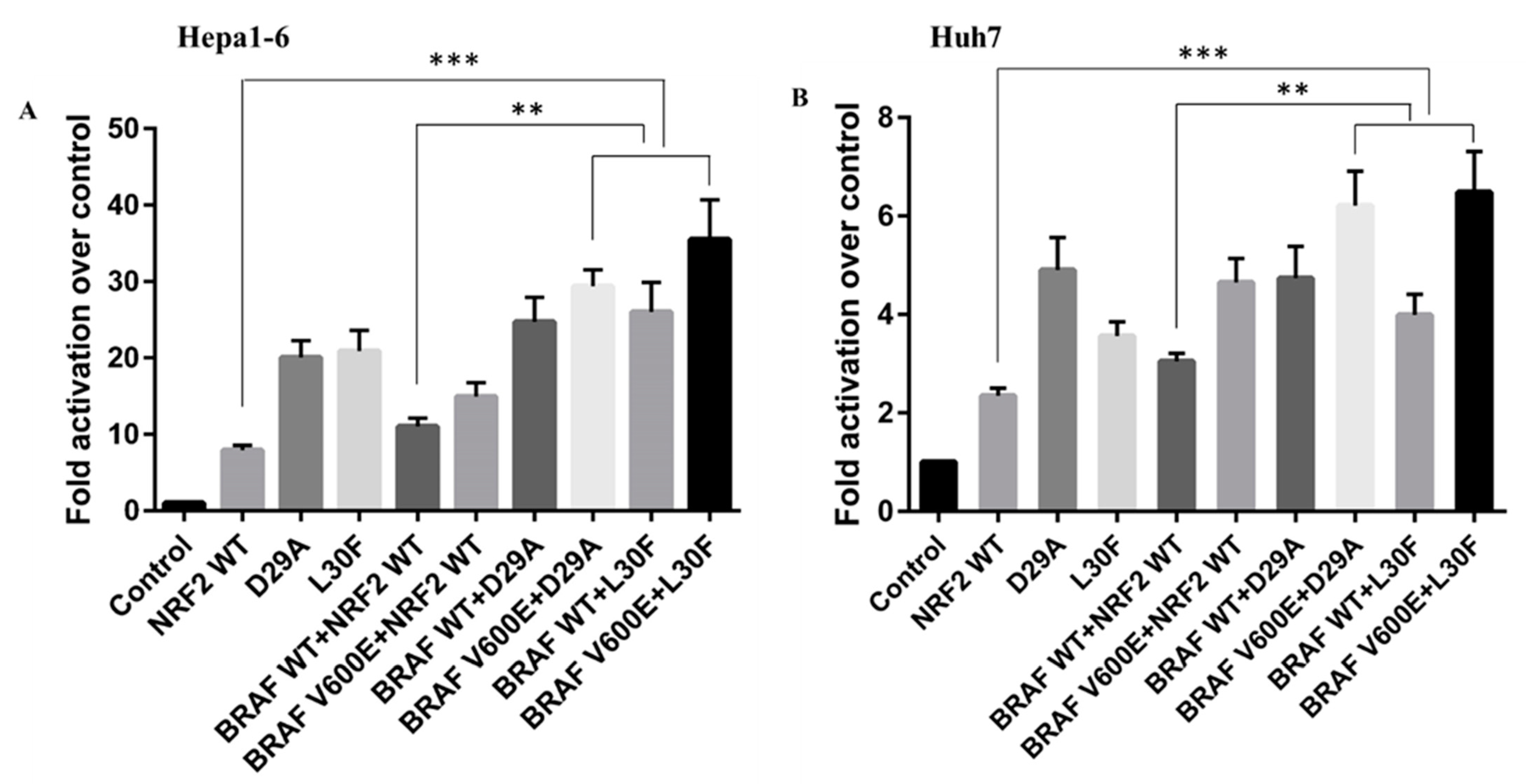
2.5. Both NRF2 and BRAF Mutations Increase the Transcriptional Activity of Target Promoters
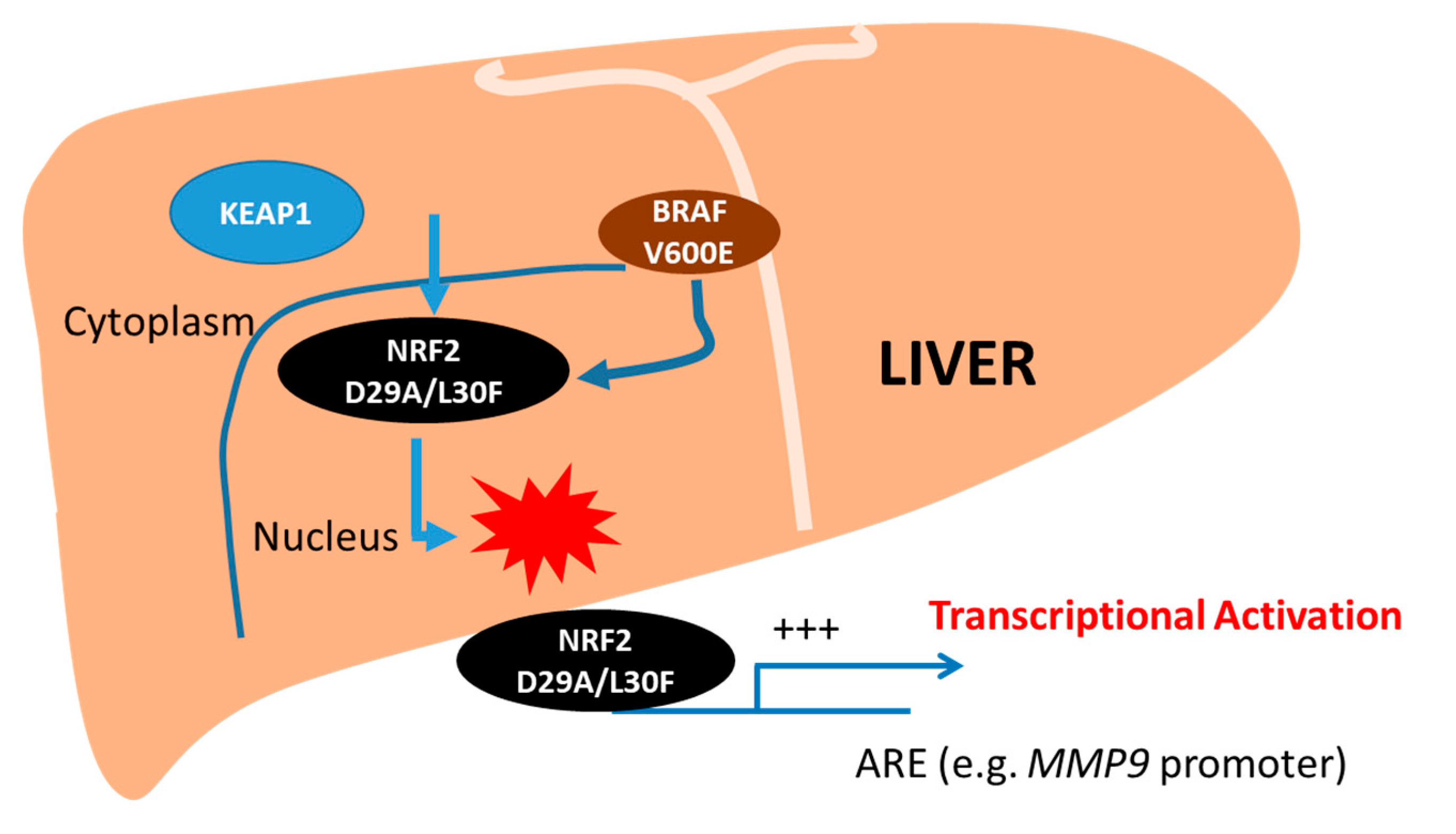
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Plasmids and Primers
4.3. Cell Transfections and Luciferase Assays
4.4. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ARE | Antioxidant response element |
| BRAF | V-raf murine sarcoma viral oncogene homolog B1 |
| CMD | Choline- devoid methionine-deficient |
| CUL3 E3 | Cullin 3-RING E3 |
| DEN | Diethylnitrosamine |
| DMEM | Dulbecco’s modified eagle medium |
| ERK | Extracellular signal-regulated kinases |
| ESCA | Esophageal carcinoma |
| FBS | Fetal bovine serum |
| Hepa 1-6 | Mouse hepatoma cell |
| HBV | Hepatitis B virus |
| HCC | Hepatocellular carcinoma |
| HCV | Hepatitis C virus |
| HepG2 | Human hepatoma cell line |
| Huh-7 | Human hepatoma cells-7 |
| ICGC | International Cancer Genome Consortium |
| JUN | Putative transforming gene of avian sarcoma virus 17 |
| KEAP1 | Kelch-like ECH-associated protein 1 |
| KO | Knockout |
| K-Ras | Kirsten rat sarcoma viral oncogene homolog |
| LUSC | Lung squamous cell carcinoma |
| MAPK | Mitogen-activated protein kinase |
| MEK | MAPK or ERK kinases |
| MMP-9 | Matrix metalloproteinase 9 |
| MYC | Cellular homolog of the retroviral v-Myc oncogene |
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| NSCLC | Non-small cell lung cancer |
| NRF2 | NF-E2-related factor 2 |
| UCEC | Uterine corpus endometrial carcinoma |
References
- Lin, L.; Yan, L.; Liu, Y.; Qu, C.; Ni, J.; Li, H. The Burden and Trends of Primary Liver Cancer Caused by Specific Etiologies from 1990 to 2017 at the Global, Regional, National, Age, and Sex Level Results from the Global Burden of Disease Study 2017. Liver Cancer 2020, 9, 563–582. [Google Scholar] [CrossRef] [PubMed]
- De Martel, C.; Maucort-Boulch, D.; Plummer, M.; Franceschi, S. World-wide relative contribution of hepatitis B and C viruses in hepatocellular carcinoma. Hepatology 2015, 62, 1190–1200. [Google Scholar] [CrossRef] [PubMed]
- Kanda, T.; Goto, T.; Hirotsu, Y.; Masuzaki, R.; Moriyama, M.; Omata, M. Molecular Mechanisms: Connections between Nonalcoholic Fatty Liver Disease, Steatohepatitis and Hepatocellular Carcinoma. Int. J. Mol. Sci. 2020, 21, 1525. [Google Scholar] [CrossRef] [PubMed]
- Michelotti, G.A.; Machado, M.V.; Diehl, A.M. NAFLD, NASH and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 656–665. [Google Scholar] [CrossRef]
- Sidharthan, S.; Kottilil, S. Mechanisms of alcohol-induced hepatocellular carcinoma. Hepatol. Int. 2014, 8, 452–457. [Google Scholar] [CrossRef] [PubMed]
- White, D.L.; Kanwal, F.; El-Serag, H.B. Association between nonalcoholic fatty liver disease and risk for hepatocellular cancer, based on systematic review. Clin. Gastroenterol. Hepatol. 2012, 10, 1342–1359.e1342. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.I.; Lu, S.N.; Liaw, Y.F.; You, S.L.; Sun, C.A.; Wang, L.Y.; Hsiao, C.K.; Chen, P.J.; Chen, D.S.; Chen, C.J. Hepatitis B e antigen and the risk of hepatocellular carcinoma. N. Engl. J. Med. 2002, 347, 168–174. [Google Scholar] [CrossRef]
- Yang, J.D.; Kim, W.R.; Coelho, R.; Mettler, T.A.; Benson, J.T.; Sanderson, S.O.; Therneau, T.M.; Kim, B.; Roberts, L.R. Cirrhosis is present in most patients with hepatitis B and hepatocellular carcinoma. Clin. Gastroenterol. Hepatol. 2011, 9, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Ramezani, A.; Nahad, M.P.; Faghihloo, E. The role of Nrf2 transcription factor in viral infection. J. Cell. Biochem. 2018, 119, 6366–6382. [Google Scholar] [CrossRef] [PubMed]
- Ngo, H.K.C.; Kim, D.H.; Cha, Y.N.; Na, H.K.; Surh, Y.J. Nrf2 Mutagenic Activation Drives Hepatocarcinogenesis. Cancer Res. 2017, 77, 4797–4808. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, C.; Zhang, L.; Yang, Q.; Zhou, S.; Wen, Q.; Wang, J. Nrf2 is a potential prognostic marker and promotes proliferation and invasion in human hepatocellular carcinoma. BMC Cancer 2015, 15, 531. [Google Scholar] [CrossRef] [PubMed]
- Shibata, T.; Ohta, T.; Tong, K.I.; Kokubu, A.; Odogawa, R.; Tsuta, K.; Asamura, H.; Yamamoto, M.; Hirohashi, S. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc. Natl. Acad. Sci. USA 2008, 105, 13568–13573. [Google Scholar] [CrossRef] [PubMed]
- Guichard, C.; Amaddeo, G.; Imbeaud, S.; Ladeiro, Y.; Pelletier, L.; Maad, I.B.; Calderaro, J.; Bioulac-Sage, P.; Letexier, M.; Degos, F.; et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 2012, 44, 694–698. [Google Scholar] [CrossRef] [PubMed]
- Fukutomi, T.; Takagi, K.; Mizushima, T.; Ohuchi, N.; Yamamoto, M. Kinetic, thermodynamic, and structural characterizations of the association between Nrf2-DLGex degron and Keap1. Mol. Cell. Biol. 2014, 34, 832–846. [Google Scholar] [CrossRef]
- Orrù, C.; Perra, A.; Kowalik, M.A.; Rizzolio, S.; Puliga, E.; Cabras, L.; Giordano, S.; Columbano, A. Distinct Mechanisms Are Responsible for Nrf2-Keap1 Pathway Activation at Different Stages of Rat Hepatocarcinogenesis. Cancers (Basel) 2020, 12, 2305. [Google Scholar] [CrossRef]
- Shibata, T.; Kokubu, A.; Saito, S.; Narisawa-Saito, M.; Sasaki, H.; Aoyagi, K.; Yoshimatsu, Y.; Tachimori, Y.; Kushima, R.; Kiyono, T.; et al. NRF2 mutation confers malignant potential and resistance to chemoradiation therapy in advanced esophageal squamous cancer. Neoplasia 2011, 13, 864–873. [Google Scholar] [CrossRef]
- Ohta, T.; Iijima, K.; Miyamoto, M.; Nakahara, I.; Tanaka, H.; Ohtsuji, M.; Suzuki, T.; Kobayashi, A.; Yokota, J.; Sakiyama, T.; et al. Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Res. 2008, 68, 1303–1309. [Google Scholar] [CrossRef]
- Endo, H.; Owada, S.; Inagaki, Y.; Shida, Y.; Tatemichi, M. Glucose starvation induces LKB1-AMPK-mediated MMP-9 expression in cancer cells. Sci. Rep. 2018, 8, 10122. [Google Scholar] [CrossRef]
- Chen, H.H.; Chang, H.H.; Chang, J.Y.; Tang, Y.C.; Cheng, Y.C.; Lin, L.M.; Cheng, S.Y.; Huang, C.H.; Sun, M.W.; Chen, C.T.; et al. Enhanced B-Raf-mediated NRF2 gene transcription and HATs-mediated NRF2 protein acetylation contributes to ABCC1-mediated chemoresistance and glutathione-mediated survival in acquired topoisomerase II poison-resistant cancer cells. Free Radic. Biol. Med. 2017, 113, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Maurer, G.; Tarkowski, B.; Baccarini, M. Raf kinases in cancer-roles and therapeutic opportunities. Oncogene 2011, 30, 3477–3488. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef]
- Pratilas, C.A.; Taylor, B.S.; Ye, Q.; Viale, A.; Sander, C.; Solit, D.B.; Rosen, N. (V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc. Natl. Acad. Sci. USA 2009, 106, 4519–4524. [Google Scholar] [CrossRef] [PubMed]
- Ritt, D.A.; Monson, D.M.; Specht, S.I.; Morrison, D.K. Impact of feedback phosphorylation and Raf heterodimerization on normal and mutant B-Raf signaling. Mol. Cell. Biol. 2010, 30, 806–819. [Google Scholar] [CrossRef] [PubMed]
- Mitsuishi, Y.; Motohashi, H.; Yamamoto, M. The Keap1-Nrf2 system in cancers: Stress response and anabolic metabolism. Front. Oncol. 2012, 2, 200. [Google Scholar] [CrossRef] [PubMed]
- Salemi, R.; Falzone, L.; Madonna, G.; Polesel, J.; Cinà, D.; Mallardo, D.; Ascierto, P.A.; Libra, M.; Candido, S. MMP-9 as a Candidate Marker of Response to BRAF Inhibitors in Melanoma Patients With BRAF(V600E) Mutation Detected in Circulating-Free DNA. Front. Pharmacol. 2018, 9, 856. [Google Scholar] [CrossRef] [PubMed]
- Khamari, R.; Trinh, A.; Gabert, P.E.; Corazao-Rozas, P.; Riveros-Cruz, S.; Balayssac, S.; Malet-Martino, M.; Dekiouk, S.; Joncquel Chevalier Curt, M.; Maboudou, P.; et al. Glucose metabolism and NRF2 coordinate the antioxidant response in melanoma resistant to MAPK inhibitors. Cell Death Dis. 2018, 9, 325. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Bajari, R.; Andric, D.; Gerthoffert, F.; Lepsa, A.; Nahal-Bose, H.; Stein, L.D.; Ferretti, V. The International Cancer Genome Consortium Data Portal. Nat. Biotechnol. 2019, 37, 367–369. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Wang, H.; Zhu, L.; Mao, L.; Qiao, L.; Su, X. The role of Nrf2 in migration and invasion of human glioma cell U251. World Neurosurg. 2013, 80, 363–370. [Google Scholar] [CrossRef]
- Frasca, F.; Nucera, C.; Pellegriti, G.; Gangemi, P.; Attard, M.; Stella, M.; Loda, M.; Vella, V.; Giordano, C.; Trimarchi, F.; et al. BRAF(V600E) mutation and the biology of papillary thyroid cancer. Endocr. Relat. Cancer 2008, 15, 191–205. [Google Scholar] [CrossRef]
- Guarneri, C.; Bevelacqua, V.; Polesel, J.; Falzone, L.; Cannavò, P.S.; Spandidos, D.A.; Malaponte, G.; Libra, M. NF-κB inhibition is associated with OPN/MMP-9 downregulation in cutaneous melanoma. Oncol. Rep. 2017, 37, 737–746. [Google Scholar] [CrossRef]
- Chu, X.Y.; Li, Z.J.; Zheng, Z.W.; Tao, Y.L.; Zou, F.X.; Yang, X.F. KEAP1/NRF2 signaling pathway mutations in cervical cancer. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 4458–4466. [Google Scholar] [CrossRef] [PubMed]
- Kerins, M.J.; Ooi, A. A catalogue of somatic NRF2 gain-of-function mutations in cancer. Sci. Rep. 2018, 8, 12846. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, L.; Li, Y.; Sun, F.; Fang, Y.; Zhang, R.; Wu, J.; Zhou, G.; Song, H.; Xue, L.; et al. Exome sequencing identifies somatic mutations in novel driver genes in non-small cell lung cancer. Aging 2020, 12, 13701–13715. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J. Biol. Chem. 2004, 279, 31556–31567. [Google Scholar] [CrossRef]
- Tong, K.I.; Katoh, Y.; Kusunoki, H.; Itoh, K.; Tanaka, T.; Yamamoto, M. Keap1 recruits Neh2 through binding to ETGE and DLG motifs: Characterization of the two-site molecular recognition model. Mol. Cell. Biol. 2006, 26, 2887–2900. [Google Scholar] [CrossRef] [PubMed]
- Tong, K.I.; Padmanabhan, B.; Kobayashi, A.; Shang, C.; Hirotsu, Y.; Yokoyama, S.; Yamamoto, M. Different electrostatic potentials define ETGE and DLG motifs as hinge and latch in oxidative stress response. Mol. Cell. Biol. 2007, 27, 7511–7521. [Google Scholar] [CrossRef] [PubMed]
- Orrù, C.; Szydlowska, M.; Taguchi, K.; Zavattari, P.; Perra, A.; Yamamoto, M.; Columbano, A. Genetic inactivation of Nrf2 prevents clonal expansion of initiated cells in a nutritional model of rat hepatocarcinogenesis. J. Hepatol. 2018, 69, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Ziv, E.; Zhang, Y.; Kelly, L.; Nikolovski, I.; Boas, F.E.; Erinjeri, J.P.; Cai, L.; Petre, E.N.; Brody, L.A.; Covey, A.M.; et al. NRF2 Dysregulation in Hepatocellular Carcinoma and Ischemia: A Cohort Study and Laboratory Investigation. Radiology 2020, 297, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, A.; Furuta, M.; Totoki, Y.; Tsunoda, T.; Kato, M.; Shiraishi, Y.; Tanaka, H.; Taniguchi, H.; Kawakami, Y.; Ueno, M.; et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat. Genet. 2016, 48, 500–509. [Google Scholar] [CrossRef]
- Inami, Y.; Waguri, S.; Sakamoto, A.; Kouno, T.; Nakada, K.; Hino, O.; Watanabe, S.; Ando, J.; Iwadate, M.; Yamamoto, M.; et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J. Cell Biol. 2011, 193, 275–284. [Google Scholar] [CrossRef]
- Zavattari, P.; Perra, A.; Menegon, S.; Kowalik, M.A.; Petrelli, A.; Angioni, M.M.; Follenzi, A.; Quagliata, L.; Ledda-Columbano, G.M.; Terracciano, L.; et al. Nrf2, but not β-catenin, mutation represents an early event in rat hepatocarcinogenesis. Hepatology 2015, 62, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [PubMed]
- Gong, M.; Li, Y.; Ye, X.; Zhang, L.; Wang, Z.; Xu, X.; Shen, Y.; Zheng, C. Loss-of-function mutations in KEAP1 drive lung cancer progression via KEAP1/NRF2 pathway activation. Cell Commun. Signal. 2020, 18, 98. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Brekken, R.; McMahon, G.; Vu, T.H.; Itoh, T.; Tamaki, K.; Tanzawa, K.; Thorpe, P.; Itohara, S.; Werb, Z.; et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2000, 2, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Zipper, L.M.; Mulcahy, R.T. Erk activation is required for Nrf2 nuclear localization during pyrrolidine dithiocarbamate induction of glutamate cysteine ligase modulatory gene expression in HepG2 cells. Toxicol. Sci. 2003, 73, 124–134. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MT ID | DNA Change | Type | Amino Acid Change | Project | Tumor Type | Tumor Subtype | Donors Affected |
|---|---|---|---|---|---|---|---|
| MU1324215 | chr2:g.178098960C>G | single base substitution | D29H | LUSC-US | Lung cancer | Squamous cell carcinoma | 5/485 (1.03%) |
| CESC-US | Cervical cancer | Cervical squamous cell carcinoma | 2/289 (0.69%) | ||||
| HNSC-US | Head and neck cancer | Squamous cell carcinoma | 3/508 (0.59%) | ||||
| LUSC-KR | Lung cancer | Adenocarcinoma, squamous cell carcinoma | 1/170 (0.59%) | ||||
| BLCA-US | Bladder cancer | Invasive urothelial bladder cancer | 2/411 (0.49%) | ||||
| LICA-FR | Liver cancer | Hepatocellular carcinoma (secondary to alcohol and adiposity) | 1/252 (0.40%) | ||||
| ESCA-CN | Esophageal cancer | Squamous carcinoma | 1/332 (0.30%) | ||||
| UCEC-US | Endometrial cancer | Uterine corpus endometrial carcinoma | 1/531 (0.19%) | ||||
| LUAD-US | Lung cancer | Adenocarcinoma | 1/516 (0.19%) | ||||
| MU1327674 | chr2:g.178098960C>T | single base substitution | D29N | LUSC-US | Lung cancer | Squamous cell carcinoma | 5/485 (1.03%) |
| LUSC-KR | Lung cancer | Adenocarcinoma, squamous cell carcinoma | 1/170 (0.59%) | ||||
| LICA-CN | Liver cancer | Hepatocellular carcinoma HBV-associated | 1/402 (0.25%) | ||||
| LINC-JP | Liver cancer | Hepatocellular carcinoma (virus associated) | 1/394 (0.25%) | ||||
| MU1316143 | chr2:g.178098960C>A | single base substitution | D29Y | LUSC-US | Lung cancer | Squamous cell carcinoma | 2/485 (0.41%) |
| CESC-US | Cervical cancer | Cervical squamous cell carcinoma | 1/289 (0.35%) | ||||
| BLCA-US | Bladder cancer | Invasive urothelial bladder cancer | 1/411 (0.24%) | ||||
| HNSC-US | Head and Neck cancer | Squamous cell carcinoma | 1/508 (0.20%) | ||||
| MU871836 | chr2:g.178098959T>C | single base substitution | D29G | LICA-FR | Liver cancer | Hepatocellular carcinoma (secondary to alcohol and adiposity) | 2/252 (0.79%) |
| LUSC-KR | Lung cancer | Adenocarcinoma, squamous cell carcinoma | 1/170 (0.59%) | ||||
| ORCA-IN | Oral cancer | Gingivobuccal | 1/178 (0.56%) | ||||
| LINC-JP | Liver cancer | Hepatocellular carcinoma (virus associated) | 2/394 (0.51%) | ||||
| LUSC-US | Lung cancer | Squamous cell carcinoma | 2/485 (0.41%) | ||||
| LICA-CN | Liver cancer | Hepatocellular carcinoma HBV-associated | 1/402 (0.25%) | ||||
| LUAD-US | Lung cancer | Adenocarcinoma | 1/516 (0.19%) | ||||
| MU1330977 | chr2:g.178098957G>A | single base substitution | L30F | LUSC-US | Lung cancer | Squamous cell carcinoma | 4/485 (0.82%) |
| LIRI-JP | Liver cancer | Hepatocellular carcinoma (virus associated) | 1/258 (0.39%) | ||||
| PACA-CA | Pancreatic cancer | Ductal adenocarcinoma | 1/268 (0.37%) | ||||
| HNSC-US | Head and neck cancer | Squamous cell carcinoma | 1/508 (0.20%) | ||||
| MU1292484 | chr2:g.178098953C>G | single base substitution | G31A | LUSC-US | Lung cancer | Squamous cell carcinoma | 5/485 (1.03%) |
| ESCA-CN | Esophageal cancer | Squamous carcinoma | 1/332 (0.30%) | ||||
| BLCA-US | Bladder cancer | Invasive urothelial bladder cancer | 1/411 (0.24%) | ||||
| MU866686 | chr2:g.178098953C>T | single base substitution | G31E | LUSC-KR | Lung cancer | Adenocarcinoma, squamous cell carcinoma | 2/170 (1.18%) |
| LINC-JP | Liver cancer | Hepatocellular carcinoma (Virus associated) | 2/394 (0.51%) | ||||
| MU83818151 | chr2:g.178098954C>T | single base substitution | G31R | LICA-FR | Liver cancer | Hepatocellular carcinoma (secondary to alcohol and adiposity) | 1/252 (0.40%) |
| BLCA-US | Bladder cancer | Invasive urothelial bladder cancer | 1/411 (0.24%) | ||||
| LUAD-US | Lung cancer | Adenocarcinoma | 1/516 (0.19%) | ||||
| MU623518 | chr2:g.178098956A>T | single base substitution | L30H | KIRC-US | Renal cancer | Clear cell carcinoma | 1/361 (0.28%) |
| HNSC-US | Head and neck cancer | Squamous cell carcinoma | 1/508 (0.20%) | ||||
| MU130685128 | chr2:g.178098953C>A | single base substitution | G31V | LUSC-US | Lung cancer | Squamous cell carcinoma | 1/485 (0.21%) |
| MU830878 | chr2:g.178098956A>C | single base substitution | L30R | LINC-JP | Liver cancer | Hepatocellular carcinoma (virus associated) | 3/394 (0.76%) |
| KIRP-US | Renal cancer | Papillary carcinoma | 1/278 (0.36%) | ||||
| MU131168581 | chr2:g.178098956A>G | single base substitution | L30P | HNSC-US | Head and neck cancer | Squamous cell carcinoma | 1/508 (0.20%) |
| MU29615597 | chr2:g.178098959T>G | single base substitution | D29A | LIRI-JP | Liver cancer | Hepatocellular carcinoma (virus associated) | 1/258 (0.39%) |
| Primer Name | Primer Sequence | Species |
|---|---|---|
| D29A | F: CTCGACTTACTCCAAGAGCTATATCTTGCCTCCAAAGTA R: TACTTTGGAGGCAAGATATAGCTCTTGGAGTAAGTCGAG | Human |
| L30F | F: CTCGACTTACTCCAAAATCTATATCTTGCCTCCAAAGTATGTCA R: TGACATACTTTGGAGGCAAGATATAGATTTTGGAGTAAGTCGAG | Human |
| D29A | F: CTCGACTTACTCCAAGAGCTATGTCTTGCCTCCAA R: TTGGAGGCAAGACATAGCTCTTGGAGTAAGTCGAG | Mouse |
| L30F | F: CGACTTACTCCAAAATCTATGTCTTGCCTCCAAAGGAT R: ATCCTTTGGAGGCAAGACATAGATTTTGGAGTAAGTCG | Mouse |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haque, E.; Śmiech, M.; Łuczyńska, K.; Bouchard, M.F.; Viger, R.; Kono, H.; Pierzchała, M.; Taniguchi, H. NRF2 DLG Domain Mutations Identified in Japanese Liver Cancer Patients Affect the Transcriptional Activity in HCC Cell Lines. Int. J. Mol. Sci. 2021, 22, 5296. https://doi.org/10.3390/ijms22105296
Haque E, Śmiech M, Łuczyńska K, Bouchard MF, Viger R, Kono H, Pierzchała M, Taniguchi H. NRF2 DLG Domain Mutations Identified in Japanese Liver Cancer Patients Affect the Transcriptional Activity in HCC Cell Lines. International Journal of Molecular Sciences. 2021; 22(10):5296. https://doi.org/10.3390/ijms22105296
Chicago/Turabian StyleHaque, Effi, Magdalena Śmiech, Kamila Łuczyńska, Marie France Bouchard, Robert Viger, Hidetoshi Kono, Mariusz Pierzchała, and Hiroaki Taniguchi. 2021. "NRF2 DLG Domain Mutations Identified in Japanese Liver Cancer Patients Affect the Transcriptional Activity in HCC Cell Lines" International Journal of Molecular Sciences 22, no. 10: 5296. https://doi.org/10.3390/ijms22105296
APA StyleHaque, E., Śmiech, M., Łuczyńska, K., Bouchard, M. F., Viger, R., Kono, H., Pierzchała, M., & Taniguchi, H. (2021). NRF2 DLG Domain Mutations Identified in Japanese Liver Cancer Patients Affect the Transcriptional Activity in HCC Cell Lines. International Journal of Molecular Sciences, 22(10), 5296. https://doi.org/10.3390/ijms22105296