
Understanding the Role of Perilipin 5 in Non-Alcoholic Fatty Liver Disease and Its Role in Hepatocellular Carcinoma: A Review of Novel Insights



Abstract
1. Introduction
2. An Overview of Plin5 Transcriptional Regulation and Interactions
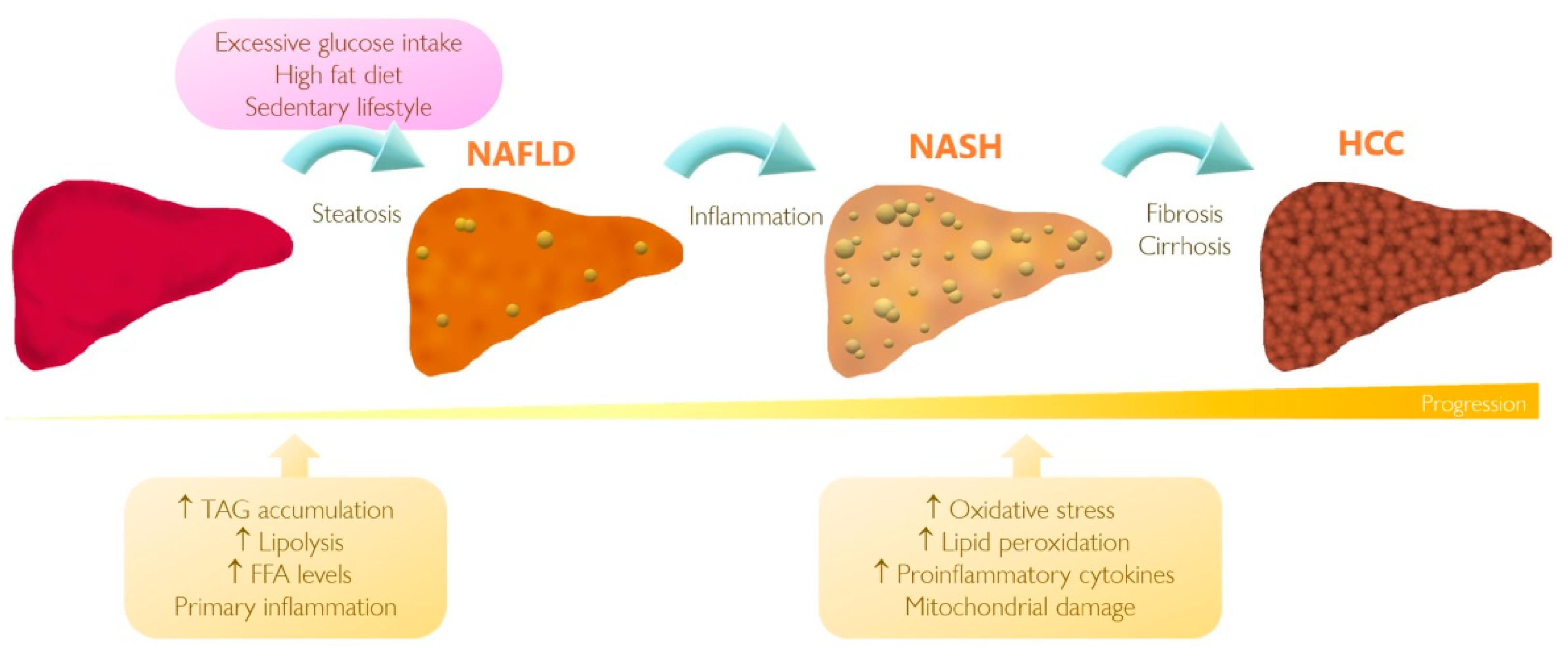
3. Understanding NAFLD and Its Progression to HCC
4. The Role of PLIN5 in Lipid Metabolism and NAFLD
5. PLIN5 Is Responsive to Cellular Processes Altered in NAFLD
6. Perilipins in Cancer: The Unrevealed Role of PLIN5
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABHD5 | α-β-hydrolase domain-containing 5 |
| ACAA2 | Acetyl-CoA acyltransferase 2 |
| ACAT1 | Acetyl-CoA acetyltransferase 1 |
| ACAT2 | Acetyl-CoA acetyltransferase 2 |
| AKT | Protein kinase B |
| ALP | Alkaline phosphatase |
| ALT | Alanine aminotransferase |
| AMPK | 5’-AMP-activated protein kinase |
| Arf-COPI | ADP-ribosylation factor1-coat protein complex I |
| AST | Aspartate aminotransferase |
| ATF | Activating transcription factor |
| ATGL | Adipose triglyceride lipase |
| BAT | Brown adipocyte tissue |
| C/EBPα | Factor CCAAT/enhancer-binding protein alpha |
| CAT | Catalase |
| ccRCC | Clear cell renal cell carcinoma |
| CD | Cluster of differentiation |
| CtrD | Control diet |
| CGI-58 | Comparative gene identification-58 |
| CHOP | C/EBP homologous protein |
| CM-Plin5 | Cardiac muscle-specific overexpression of Plin5 |
| COX2 | Cytochrome c oxidase subunit 2 |
| COX4 | Cytochrome c oxidase subunit 4 |
| CS | Citrate synthase |
| DBC1 | Deleted in breast cancer-1 |
| DGAT1 | Diacylglycerol acyltransferase1 |
| DGAT2 | Diacylglycerol acyltransferase 2 |
| DRP1 | Dynamin related protein 1 |
| ECM | Extracellualr matrix |
| EDL | Extensor digitorum longus (muscle) |
| EpCAM | Epithelial cell adhesion molecule |
| ER | Endoplasmic reticulum |
| ERK | Extracellular-regulated kinase |
| FA | Fatty acid |
| FASN | Fatty acid synthase |
| FFA | Free fatty acid |
| FGF21 | Fibroblast growth factor 21 |
| FSN-1 | Fission protein 1 |
| G6P | Gucose-6-phosphatase |
| GMC | Glycycoumarin |
| GNAI3 | G Protein subunit alpha I3 |
| GPX2 | Glutathione Peroxidase 2 |
| GSK-3β | Glycogen synthase kinase 3 beta |
| HBV | Hepatitis B virus |
| HCC | Hepatocellular carcinoma |
| HCV | Hepatitis C virus |
| HFD | High fat diet |
| HO-1 | Heme oxygenase-1 |
| HSC | Hepatic stellate cell(s) |
| HSL | Hormone-sensitive lipase |
| HSP70 | Heat shock protein 70 |
| IL-1β | Interleukin-1 beta |
| IL-6 | Interleukin-6 |
| JNK | c-Jun N-terminal kinase |
| JUN | Jun proto-oncogene |
| KO | Knockout |
| LCN2 | Lipocalin 2 |
| LD | Lipid droplets |
| LPS | Lipopolysaccharide |
| LSDP5 | Lipid storage droplet protein 5 |
| MCK-Plin5 | Skeletal muscle-specific overexpression of Plin5 |
| MCP1 | Monocyte chemoattractant protein-1 |
| MFN1 | Mitofusin 1 |
| MFN2 | Mitofusin 2 |
| MGL | Monoglyceride lipase |
| MKO | Muscle-specific Plin5 knockout |
| MLDP | Myocardial LD protein |
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| NF-κB | Nuclear factor-kappa B |
| NHLRC2 | NHL repeat containing 2 |
| NLRP3 | NLR family pyrin domain containing 3 |
| NOTCH1 | Notch homolog 1 |
| NQO1 | NAD(P)H quinone dehydrogenase 1 |
| NRF2 | Nuclear factor erythroid 2-related factor 2 |
| OA | Oleic acid |
| OPA1 | Mitochondrial dynamin-like GTPase |
| OXPAT | Oxidative tissues-enriched PAT protein |
| p38 | p38 mitogen-activated protein kinase |
| p53 | Tumor protein P53 |
| PGC-1α | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| PI3K | Phosphatidylinositol-3-kinase |
| PKA | Protein kinase A |
| Plin5−/− | Plin5 deficient |
| PLIN5 | Perilipin 5 |
| PPAR | Peroxisome proliferator-activated receptor |
| PPARα | Peroxisome proliferator-activated receptor alpha |
| PPRE | PPAR regulatory element |
| RAB | Ras superfamily of GTPases |
| ROS | Reactive oxygen species |
| SCD1 | Stearoyl-CoA desaturase-1 |
| SF | Splicing factor |
| SIG1R | Sigma 1 receptor |
| SIRT1 | Sirtuin-1 |
| SNPs | Single-nucleotide polymorphisms |
| SREBF1 | Sterol regulatory element binding factor 1 |
| SREBP1 | Sterol regulatory element-binding protein 1 |
| SREBP2 | Sterol regulatory element-binding protein 2 |
| TAG | Triacylglycerides |
| TCGA | The cancer genome atlas |
| TME | Tumor microenvironment |
| TNF-α | Tumor necorsis factor-alpha |
| VDAC1 | Voltage-dependent anion-selective channel 1 |
| WAT | White adipocyte tissue |
References
- Minehira, K.; Gual, P. Role of Lipid Droplet Proteins in the Development of NAFLD and Hepatic Insulin Resistance. Non-Alcoholic Fat Liver Disease; IntechOpen: London, UK, 2018; pp. 55–77. [Google Scholar] [CrossRef]
- Olzmann, J.A.; Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef]
- Sahini, N.; Borlak, J. Recent insights into the molecular pathophysiology of lipid droplet formation in hepatocytes. Prog. Lipid Res. 2014, 54, 86–112. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, A.S.; Egan, J.J.; Wek, S.A.; Garty, N.B.; Blanchette-Mackie, E.J.; Londos, C. Perilipin, a major hormonally regulated adipocyte-specific phosphoprotein associated with the periphery of lipid storage droplets. J. Biol. Chem. 1991, 266, 11341–11346. [Google Scholar] [CrossRef]
- Jiang, H.P.; Serrero, G. Isolation and characterization of a full-length cDNA coding for an adipose differentiation-related protein. Proc. Natl. Acad. Sci. USA 1992, 89, 7856–7860. [Google Scholar] [CrossRef]
- Díaz, E.; Pfeffer, S.R. Tip47: A cargo selection device for mannose 6-phosphate receptor trafficking. Cell 1998, 93, 433–443. [Google Scholar] [CrossRef]
- Kimmel, A.R.; Brasaemle, D.L.; McAndrews-Hill, M.; Sztalryd, C.; Londos, C. Adoption of PERILIPIN as a unifying nomenclature for the mammalian PAT-family of intracellular lipid storage droplet proteins. J. Lipid Res. 2010, 51, 468–471. [Google Scholar] [CrossRef] [PubMed]
- Brasaemle, D.L.; Subramanian, V.; Garcia, A.; Marcinkiewicz, A.; Rothenberg, A. Perilipin A and the control of triacylglycerol metabolism. Mol. Cell. Biochem. 2009, 326, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Itabe, H.; Yamaguchi, T.; Nimura, S.; Sasabe, N. Perilipins: A diversity of intracellular lipid droplet proteins. Lipids Health Dis. 2017, 16, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Listenberger, L.L.; Ostermeyer-Fay, A.G.; Goldberg, E.B.; Brown, W.J.; Brown, D.A. Adipocyte differentiation-related protein reduces the lipid droplet association of adipose triglyceride lipase and slows triacylglycerol turnover. J. Lipid Res. 2007, 48, 2751–2761. [Google Scholar] [CrossRef] [PubMed]
- Wolins, N.E.; Skinner, J.R.; Schoenfish, M.J.; Tzekov, A.; Bensch, K.G.; Bickel, P.E. Adipocyte protein S3-12 coats nascent lipid droplets. J. Biol. Chem. 2003, 278, 37713–37721. [Google Scholar] [CrossRef]
- Ma, S.Y.; Sun, K.S.; Zhang, M.; Zhou, X.; Zheng, X.H.; Tian, S.Y.; Liu, Y.S.; Chen, L.; Gao, X.; Ye, J.; et al. Disruption of Plin5 degradation by CMA causes lipid homeostasis imbalance in NAFLD. Liver Int. 2020, 40, 2427–2438. [Google Scholar] [CrossRef] [PubMed]
- Hayes, C.N.; Zhang, P.; Chayama, K. The role of lipids in hepatocellular carcinoma. In Hepatocellular Carcinoma; Tirnitz-Parker, J.E.E., Ed.; Codon Publications: Brisbane, Australia, 2019; Chapter 5. [Google Scholar] [CrossRef]
- Corn, K.C.; Windham, M.A.; Rafat, M. Lipids in the tumor microenvironment: From cancer progression to treatment. Prog. Lipid Res. 2020, 80. [Google Scholar] [CrossRef] [PubMed]
- Calvisi, D.F.; Wang, C.; Ho, C.; Ladu, S.; Lee, S.A.; Mattu, S.; Destefanis, G.; Delogu, S.; Zimmermann, A.; Ericsson, J.; et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 2011, 140, 1071–1083. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, N.U.; Sheikh, T.A. Endoplasmic reticulum stress and Oxidative stress in the pathogenesis of Non-alcoholic fatty liver disease. Free Radic. Res. 2015, 49, 1405–1418. [Google Scholar] [CrossRef]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2020, 20, 116–141. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef]
- Saha, S.K.; Lee, S.B.; Won, J.; Choi, H.Y.; Kim, K.; Yang, G.M.; Dayem, A.A.; Cho, S.G. Correlation between oxidative stress, nutrition, and cancer initiation. Int. J. Mol. Sci. 2017, 18, 1544. [Google Scholar] [CrossRef]
- Mason, R.R.; Mokhtar, R.; Matzaris, M.; Selathurai, A.; Kowalski, G.M.; Mokbel, N.; Meikle, P.J.; Bruce, C.R.; Watt, M.J. PLIN5 deletion remodels intracellular lipid composition and causes insulin resistance in muscle. Mol. Metab. 2014, 3, 652–663. [Google Scholar] [CrossRef]
- Keenan, S.N.; Meex, R.C.; Lo, J.C.Y.; Ryan, A.; Nie, S.; Montgomery, M.K.; Watt, M.J. Perilipin 5 Deletion in hepatocytes remodels lipid metabolism and causes hepatic insulin resistance in mice. Diabetes 2019, 68, 543–555. [Google Scholar] [CrossRef]
- Wang, H.; Sreenivasan, U.; Gong, D.W.; O’Connell, K.A.; Dabkowski, E.R.; Hecker, P.A.; Ionica, N.; Konig, M.; Mahurkar, A.; Sun, Y.; et al. Cardiomyocyte-specific perilipin 5 overexpression leads to myocardial steatosis and modest cardiac dysfunction. J. Lipid Res. 2013, 54, 953–965. [Google Scholar] [CrossRef]
- Wang, C.; Zhao, Y.; Gao, X.; Li, L.; Yuan, Y.; Liu, F.; Zhang, L.; Wu, J.; Hu, P.; Zhang, X.; et al. Perilipin 5 improves hepatic lipotoxicity by inhibiting lipolysis. Hepatology 2015, 61, 870–882. [Google Scholar] [CrossRef]
- Lin, J.; Chen, A. Perilipin 5 restores the formation of lipid droplets in activated hepatic stellate cells and inhibits their activation. Lab. Investig. 2016, 96, 791–806. [Google Scholar] [CrossRef]
- Montgomery, M.K.; Mokhtar, R.; Bayliss, J.; Parkington, H.C.; Suturin, V.M.; Bruce, C.R.; Watt, M.J. Perilipin 5 deletion unmasks an endoplasmic reticulum Stress-Fibroblast growth factor 21 axis in skeletal muscle. Diabetes 2018, 67, 594–606. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Jin, Y.; Wang, Q.; Huang, J.; Wu, X.; Ren, Z. Perilipin 5 protects against cellular oxidative stress by enhancing mitochondrial function in HepG2 cells. Cells 2019, 8, 1241. [Google Scholar] [CrossRef] [PubMed]
- Asimakopoulou, A.; Engel, K.M.; Gassler, N.; Bracht, T.; Sitek, B.; Buhl, E.M.; Kalampoka, S.; Pinoé-Schmidt, M.; van Helden, J.; Schiller, J.; et al. Deletion of Perilipin 5 protects against hepatic injury in nonalcoholic fatty liver disease via missing inflammasome activation. Cells 2020, 9, 1346. [Google Scholar] [CrossRef]
- Zhang, E.; Cui, W.; Lopresti, M.; Mashek, M.T.; Najt, C.P.; Hu, H.; Mashek, D.G. Hepatic PLIN5 signals via SIRT1 to promote autophagy and prevent inflammation during fasting. J. Lipid Res. 2020, 61, 338–350. [Google Scholar] [CrossRef]
- Zhu, Y.; Ren, C.; Zhang, M.; Zhong, Y. Perilipin 5 reduces oxidative damage associated with lipotoxicity by activating the PI3K/ERK-mediated Nrf2-ARE signaling pathway in INS-1 pancreatic β-cells. Front. Endocrinol. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Feng, J.; Xie, L.; Yu, X.; Liu, C.; Dong, H.; Lu, W.; Kong, R. Perilipin 5 ameliorates high-glucose-induced podocyte injury via Akt/GSK-3β/Nrf2-mediated suppression of apoptosis, oxidative stress, and inflammation. Biochem. Biophys. Res. Commun. 2021, 12, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Dalen, K.T.; Dahl, T.; Holter, E.; Arntsen, B.; Londos, C.; Sztalryd, C. LSDP5 is a PAT protein specifically expressed in fatty acid oxidizing tissues. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2007, 1771, 210–227. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Matsushita, S.; Motojima, K.; Hirose, F.; Osumi, T. MLDP, a novel PAT family protein localized to lipid droplets and enriched in the heart, is regulated by peroxisome proliferator-activated receptor α. J. Biol. Chem. 2006, 281, 14232–14240. [Google Scholar] [CrossRef]
- Wolins, N.E.; Quaynor, B.K.; Skinner, J.R.; Tzekov, A.; Croce, M.A.; Gropler, M.C. OXPAT/PAT-1 is a PPAR-induced lipid droplet protein that promotes fatty acid utilization. Diabetes 2006, 55, 3418–3428. [Google Scholar] [CrossRef]
- Kimmel, A.R.; Sztalryd, C. Perilipin 5, a lipid droplet protein adapted to mitochondrial energy utilization. Curr. Opin. Lipidol. 2014, 25, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhang, L.; Meng, Q.; Niu, C.; Jin, D.; Yu, A. C/EBPα promotes transcription of the porcine perilipin 5 gene. Mol. Cell. Endocrinol. 2012, 364, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Granneman, J.G.; Moore, H.P.H.; Mottillo, E.P.; Zhu, Z.; Zhou, L. Interactions of Perilipin-5 (Plin5) with adipose triglyceride lipase. J. Biol. Chem. 2011, 286, 5126–5135. [Google Scholar] [CrossRef]
- Wang, H.; Bell, M.; Sreenevasan, U.; Hu, H.; Liu, J.; Dalen, K.; Londos, C.; Yamaguchi, T.; Rizzo, M.A.; Coleman, R.; et al. Unique regulation of adipose triglyceride lipase (ATGL) by perilipin 5, a lipid droplet-associated protein. J. Biol. Chem. 2011, 286, 15707–15715. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Romeo, S.; Valenti, L. Hepatocellular carcinoma in nonalcoholic fatty liver: Role of environmental and genetic factors. World J. Gastroenterol. 2014, 20, 12945–12955. [Google Scholar] [CrossRef]
- Huang, D.Q.; El-Serag, H.B.; Loomba, R. Global epidemiology of NAFLD-related HCC: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2020. [Google Scholar] [CrossRef]
- Burt, A.D.; Tiniakos, D.G.; Lackner, C. Diagnosis and assessment of NAFLD: Definitions and histopathological classification. Semin. Liver Dis. 2015, 35, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Lian, C.Y.; Zhai, Z.Z.; Li, Z.F.; Wang, L. High fat diet-triggered non-alcoholic fatty liver disease: A review of proposed mechanisms. Chem. Biol. Interact. 2020, 330. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.-H.; et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075. [Google Scholar] [CrossRef]
- Del Campo, J.A.; Gallego-Durán, R.; Gallego, P.; Grande, L. Genetic and epigenetic regulation in nonalcoholic fatty liver disease (NAFLD). Int. J. Mol. Sci. 2018, 19, 911. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Deng, R.; Liu, S.; Wang, K.; Sun, J. Hepatitis B virus-related hepatocellular carcinoma in the era of antiviral therapy: The emerging role of non-viral risk factors. Liver Int. 2020, 40, 2316–2325. [Google Scholar] [CrossRef]
- Angulo, P. Nonalcoholic fatty liver disease. N. Engl. J. Med. 2002, 346, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P. Pathogenesis of steatohepatitis. Best Pract. Res. Clin. Gastroenterol. 2002, 16, 663–678. [Google Scholar] [CrossRef]
- Diehl, A.M.; Li, Z.P.; Lin, H.Z.; Yang, S.Q. Cytokines and the pathogenesis of non-alcoholic steatohepatitis. Gut 2005, 54, 303–306. [Google Scholar] [CrossRef]
- Tilg, H. The role of cytokines in non-alcoholic fatty liver disease. Dig. Dis. 2010, 28, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “Hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Arab, J.P.; Arrese, M.; Trauner, M. Recent insights into the pathogenesis of nonalcoholic fatty liver disease. Annu. Rev. Pathol. Mech. Dis. 2018, 13, 321–350. [Google Scholar] [CrossRef]
- Kutlu, O.; Kaleli, H.N.; Ozer, E. Molecular pathogenesis of nonalcoholic steatohepatitis-(NASH-) related hepatocellular carcinoma. Can. J. Gastroenterol. Hepatol. 2018, 2018. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef]
- Jiang, C.M.; Pu, C.W.; Hou, Y.H.; Chen, Z.; Alanazy, M.; Hebbard, L. Non alcoholic steatohepatitis a precursor for hepatocellular carcinoma development. World J. Gastroenterol. 2014, 20, 16464–16473. [Google Scholar] [CrossRef]
- Takaki, A.; Kawai, D.; Yamamoto, K. Multiple hits, including oxidative stress, as pathogenesis and treatment target in non-alcoholic steatohepatitis (NASH). Int. J. Mol. Sci. 2013, 14, 20704–20728. [Google Scholar] [CrossRef] [PubMed]
- Van Der Windt, D.J.; Sud, V.; Zhang, H.; Tsung, A.; Huang, H. The effects of physical exercise on fatty liver disease. Gene Expr. 2018, 18, 89–101. [Google Scholar] [CrossRef]
- Choudhary, N.S.; Saraf, N.; Saigal, S.; Gautam, D.; Lipi, L.; Rastogi, A.; Goja, S.; Menon, P.B.; Bhangui, P.; Ramchandra, S.K.; et al. Rapid reversal of liver steatosis with life style modification in highly motivated liver donors. J. Clin. Exp. Hepatol. 2015, 5, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Grasselli, E.; Voci, A.; Pesce, C.; Canesi, L.; Fugassa, E.; Gallo, G.; Vergani, L. PAT protein mRNA expression in primary rat hepatocytes: Effects of exposure to fatty acids. Int. J. Mol. Med. 2010, 25, 505–512. [Google Scholar] [CrossRef]
- Li, H.; Song, Y.; Zhang, L.J.; Gu, Y.; Li, F.F.; Pan, S.Y. LSDP5 enhances triglyceride storage in hepatocytes by influencing lipolysis and fatty acid β-oxidation of lipid droplets. PLoS ONE 2012, 7, e36712. [Google Scholar] [CrossRef]
- Kuramoto, K.; Okamura, T.; Yamaguchi, T.; Nakamura, T.Y.; Wakabayashi, S.; Morinaga, H.; Nomura, M.; Yanase, T.; Otsu, K.; Usuda, N.; et al. Perilipin 5, a lipid droplet-binding protein, protects heart from oxidative burden by sequestering fatty acid from excessive oxidation. J. Biol. Chem. 2012, 287, 23852–23863. [Google Scholar] [CrossRef] [PubMed]
- Pollak, N.M.; Schweiger, M.; Jaeger, D.; Kolb, D.; Kumari, M.; Schreiber, R.; Kolleritsch, S.; Markolin, P.; Grabner, G.F.; Heier, C.; et al. Cardiac-Specific overexpression of perilipin 5 provokes severe cardiac steatosis via the formation of a lipolytic barrier. J. Lipid Res. 2013, 54, 1092–1102. [Google Scholar] [CrossRef]
- Harris, L.A.; Skinner, J.R.; Shew, T.M.; Pietka, T.A.; Abumrad, N.A.; Wolins, N.E. Perilipin 5-driven lipid droplet accumulation in skeletal muscle stimulates the expression of fibroblast growth factor 21. Diabetes 2015, 64, 2757–2768. [Google Scholar] [CrossRef]
- Bernsmeier, C.; Dill, M.T.; Provenzano, A.; Makowska, Z.; Krol, I.; Muscogiuri, G.; Semela, D.; Tornillo, L.; Marra, F.; Heim, M.H. Hepatic Notch1 deletion predisposes to diabetes and steatosis via glucose-6-phosphatase and perilipin-5 upregulation. Lab. Investig. 2016, 96, 972–980. [Google Scholar] [CrossRef] [PubMed]
- Langhi, C.; Marquart, T.J.; Allen, R.M.; Baldán, A. Perilipin-5 is regulated by statins and controls triglyceride contents in the hepatocyte. J. Hepatol. 2014, 61, 358–365. [Google Scholar] [CrossRef]
- Zhong, W.; Fan, B.; Cong, H.; Wang, T.; Gu, J. Oleic acid-induced perilipin 5 expression and lipid droplets formation are regulated by the PI3K/PPARα pathway in HepG2 cells. Appl. Physiol. Nutr. Metab. 2019, 44, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Ning, H.; Sun, Z.; Liu, Y.; Liu, L.; Hao, L.; Ye, Y.; Feng, R.; Li, J.; Li, Y.; Chu, X.; et al. Insulin protects hepatic lipotoxicity by regulating ER stress through the PI3K/Akt/p53 involved pathway independently of autophagy inhibition. Nutrients 2016, 8, 227. [Google Scholar] [CrossRef] [PubMed]
- Gallardo-Montejano, V.I.; Saxena, G.; Kusminski, C.M.; Yang, C.; McAfee, J.L.; Hahner, L.; Hoch, K.; Dubinsky, W.; Narkar, V.A.; Bickel, P.E. Nuclear Perilipin 5 integrates lipid droplet lipolysis with PGC-1α/SIRT1-dependent transcriptional regulation of mitochondrial function. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Gao, X.; Nan, Y.; Zhao, Y.; Yuan, Y.; Ren, B.; Sun, C.; Cao, K.; Yu, M.; Feng, X.; Ye, J. Atorvastatin reduces lipid accumulation in the liver by activating protein kinase A-mediated phosphorylation of perilipin 5. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1512–1519. [Google Scholar] [CrossRef]
- Najt, C.P.; Khan, S.A.; Heden, T.D.; Witthuhn, B.A.; Perez, M.; Heier, J.L.; Mead, L.E.; Franklin, M.P.; Karanja, K.K.; Graham, M.J.; et al. Lipid droplet-derived monounsaturated fatty acids traffic via PLIN5 to allosterically activate SIRT1. Mol. Cell. 2020, 77, 810–824. [Google Scholar] [CrossRef] [PubMed]
- London, E.; Nesterova, M.; Stratakis, C.A. Acute vs chronic exposure to high fat diet leads to distinct regulation of PKA. J. Mol. Endocrinol. 2017, 59, 1–12. [Google Scholar] [CrossRef]
- Keenan, S.N.; DeNardo, W.; Lou, J.; Schittenhelm, R.B.; Montgomery, M.K.; Granneman, J.G.; Hinde, E.; Watt, M.J. Perilipin 5 S155 phosphorylation by PKA is required for the control of hepatic lipid metabolism and glycemic control. J. Lipid Res. 2021, 62. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.; Yin, S.; Zhao, C.; Fan, L.; Hu, H. Involvement of activation of PLIN5-Sirt1 axis in protective effect of glycycoumarin on hepatic lipotoxicity. Biochem. Biophys. Res. Commun. 2020, 528, 7–13. [Google Scholar] [CrossRef]
- Geng, L.; Lam, K.S.L.; Xu, A. The therapeutic potential of FGF21 in metabolic diseases: From bench to clinic. Nat. Rev. Endocrinol. 2020, 16, 654–667. [Google Scholar] [CrossRef]
- Wan, X.S.; Lu, X.H.; Xiao, Y.C.; Lin, Y.; Zhu, H.; Ding, T.; Yang, Y.; Huang, Y.; Zhang, Y.; Liu, Y.L.; et al. ATF4- and CHOP-dependent induction of FGF21 through endoplasmic reticulum stress. BioMed Res. Int. 2014, 2014. [Google Scholar] [CrossRef]
- Park, G.B.; Choi, Y.; Kim, Y.S.; Lee, H.K.; Kim, D.; Hur, D.Y. ROS-mediated JNK/p38-MAPK activation regulates Bax translocation in Sorafenib-induced apoptosis of EBV-transformed B cells. Int. J. Oncol. 2014, 44, 977–985. [Google Scholar] [CrossRef]
- Hocsak, E.; Szabo, V.; Kalman, N.; Antus, C.; Cseh, A.; Sumegi, K.; Eros, K.; Hegedus, Z.; Gallyas, F.; Sumegi, B.; et al. PARP inhibition protects mitochondria and reduces ROS production via PARP-1-ATF4-MKP-1-MAPK retrograde pathway. Free Radic. Biol. Med. 2017, 108, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Kolleritsch, S.; Kien, B.; Schoiswohl, G.; Diwoky, C.; Schreiber, R.; Heier, C.; Maresch, L.K.; Schweiger, M.; Eichmann, T.O.; Stryeck, S.; et al. Low cardiac lipolysis reduces mitochondrial fission and prevents lipotoxic heart dysfunction in Perilipin 5 mutant mice. Cardiovasc. Res. 2020, 116, 339–352. [Google Scholar] [CrossRef]
- Asimakopoulou, A.; Borkham-Kamphorst, E.; Henning, M.; Yagmur, E.; Gassler, N.; Liedtke, C.; Berger, T.; Mak, T.W.; Weiskirchen, R. Lipocalin-2 (LCN2) regulates PLIN5 expression and intracellular lipid droplet formation in the liver. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 1513–1524. [Google Scholar] [CrossRef] [PubMed]
- Andersson, L.; Drevinge, C.; Mardani, I.; Dalen, K.T.; Ståhlman, M.; Klevstig, M.; Lundqvist, A.; Haugen, F.; Adiels, M.; Fogelstrand, P.; et al. Deficiency in perilipin 5 reduces mitochondrial function and membrane depolarization in mouse hearts. Int. J. Biochem. Cell Biol. 2017, 91, 9–13. [Google Scholar] [CrossRef]
- Gemmink, A.; Daemen, S.; Kuijpers, H.J.H.; Schaart, G.; Duimel, H.; López-Iglesias, C.; van Zandvoort, M.A.M.J.; Knoops, K.; Hesselink, M.K.C. Super-Resolution microscopy localizes perilipin 5 at lipid droplet-mitochondria interaction sites and at lipid droplets juxtaposing to perilipin 2. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 1423–1432. [Google Scholar] [CrossRef] [PubMed]
- Varghese, M.; Kimler, V.A.; Ghazi, F.R.; Rathore, G.K.; Perkins, G.A.; Ellisman, M.H.; Granneman, J.G. Adipocyte lipolysis affects Perilipin 5 and cristae organization at the cardiac lipid droplet-mitochondrial interface. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Kanwal, F.; Kramer, J.R.; Mapakshi, S.; Natarajan, Y.; Chayanupatkul, M.; Richardson, P.A.; Li, L.; Desiderio, R.; Thrift, A.P.; Asch, S.M.; et al. Risk of hepatocellular cancer in patients with non-alcoholic fatty liver disease. Gastroenterology 2018, 155, 1828–1837. [Google Scholar] [CrossRef]
- Maan, M.; Peters, J.M.; Dutta, M.; Patterson, A.D. Lipid metabolism and lipophagy in cancer. Biochem. Biophys. Res. Commun. 2018, 504, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Honda, M.; Takatori, H.; Nishino, R.; Minato, H.; Takamura, H.; Ohta, T.; Kaneko, S. Activation of lipogenic pathway correlates with cell proliferation and poor prognosis in hepatocellular carcinoma. J. Hepatol. 2009, 50, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, I.; Shaban, M.D.R.; Shereen, F.; El-Goday, M.D.M.; Sweed, M.S.D. Progressive and prognosis role of lipogenic pathway in hepatocellular carcinoma: A systematic review and meta-analysis. Med. J. Cairo Univ. 2019, 87, 249–256. [Google Scholar] [CrossRef]
- Zhou, C.; Wang, M.; Zhou, L.; Zhang, Y.; Liu, W.; Qin, W.; He, R.; Lu, Y.; Wang, Y.; Chen, X.Z.; et al. Prognostic significance of PLIN1 expression in human breast cancer. Oncotarget 2016, 7, 54488–54502. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Ruan, H.; Wang, K.; Song, Z.; Bao, L.; Xu, T.; Xiao, H.; Wang, C.; Cheng, G.; Tong, J.; et al. Overexpression of PLIN2 is a prognostic marker and attenuates tumor progression in clear cell renal cell carcinoma. Int. J. Oncol. 2018, 53, 137–147. [Google Scholar] [CrossRef]
- Wang, K.; Ruan, H.; Song, Z.S.; Cao, Q.; Bao, L.; Liu, D.; Xu, T.; Xiao, H.; Wang, C.; Cheng, G.; et al. PLIN3 is up-regulated and correlates with poor prognosis in clear cell renal cell carcinoma. Urol. Oncol. Semin. Orig. Investig. 2018, 36. [Google Scholar] [CrossRef]
- Zhou, L.; Song, Z.; Hu, J.; Liu, L.; Hou, Y.; Zhang, X.; Yang, X.; Chen, K. ACSS3 represses prostate cancer progression through downregulating lipid droplet-associated protein PLIN3. Theranostics 2020, 841–860. [Google Scholar] [CrossRef]
- Straub, B.K.; Herpel, E.; Singer, S.; Zimbelmann, R.; Breuhahn, K.; MacHer-Goeppinger, S.; Warth, A.; Lehmann-Koch, J.; Longerich, T.; Heid, H.; et al. Lipid droplet-associated PAT-proteins show frequent and differential expression in neoplastic steatogenesis. Mod. Pathol. 2010, 23, 480–492. [Google Scholar] [CrossRef]
- Ye, J.; Liu, H.; Xu, Z.L.; Zheng, L.; Liu, R.Y. Identification of a multidimensional transcriptome prognostic signature for lung adenocarcinoma. J. Clin. Lab. Anal. 2019, 33. [Google Scholar] [CrossRef]
- Hashani, M.; Witzel, H.R.; Pawella, L.M.; Lehmann-Koch, J.; Schumacher, J.; Mechtersheimer, G.; Schnölzer, M.; Schirmacher, P.; Roth, W.; Straub, B.K. Widespread expression of perilipin 5 in normal human tissues and in diseases is restricted to distinct lipid droplet subpopulations. Cell Tissue Res. 2018, 374, 121–136. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, P.; Li, B.; Dang, H.; Jiang, J.; Meng, L.; Zhang, H.; Zhang, Y.; Wang, X.; Li, Q.; et al. The expression of perilipin family proteins can be used as diagnostic markers of liposarcoma and to differentiate subtypes. J. Cancer 2020, 11, 4081–4090. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xu, D.; Wan, Y.; Xi, Q. Methylation of PLIN5 is a crucial biomarker and is involved in ovarian cancer development. Transl. Cancer Res. 2020, 9, 2919–2930. [Google Scholar] [CrossRef]
- Asimakopoulou, A.; Vucur, M.; Luedde, T.; Schneiders, S.; Kalampoka, S.; Weiss, T.S.; Weiskirchen, R. Perilipin 5 and Lipocalin 2 expression in hepatocellular carcinoma. Cancers 2019, 11, 385. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Yan, G.; Sun, H.; Zhang, J.; Song, D.; Kong, R.; Yan, P.; Hu, P.; Xie, A.; Wang, S.; et al. Identification of prognostic and metastasis-related alternative splicing signatures in hepatocellular carcinoma. Biosci. Rep. 2020, 40, 1–14. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Model | Tissue | Findings | References |
|---|---|---|---|
| Plin5−/− mice | Heart | No LD formation in fed and fasting state ↓ TAG content in fed and fasting state | [59] |
| Soleus muscle | ↓ TAG content | ||
| Liver | ↓ TAG content in fed state ↑ TAG content in fasting state | ||
| BAT | ↓ TAG content in fed and fasting state | ||
| WAT | ↓ TAG content in fasting mice | ||
| Plin5−/− mice | Whole body | ↑ Carbohydrate oxidation | [20] |
| Muscle | ↑ Skeletal muscle insulin resistance | ||
| Liver | Improvement of insulin sensitivity | ||
| Plin5−/− mice | Heart | ↓ Cardiac LD formation ↑ Cardiac FA oxidation | [34] |
| Plin5−/− mice | Liver | ↓ Hepatic TAG content ↑ Lipolysis ↑ Mitochondrial proliferation ↑ Mitochondrial oxidative capacity ↑ Expression of pro-inflammatory genes under an HFD ↑ Expression of ER stress-related genes ↑ Lipid peroxidation | [23] |
| Plin5−/− mice | Liver | Under HFD vs. CtrD: ↓ Fat scoring ↓ Ballooning of hepatocytes ↓ Levels of liver damage enzymes ALP, ALT, AST ↓ Bilirubin levels ↓ Cholesterol levels ↑ Levels of mitochondrial structure and trafficking markers ↑ Lipogenesis ↓ Inflammatory markers ↓ Levels of arachidonic acid | [27] |
| MKO mice | Skeletal muscle | ↑ Fat mass ↓ Respiratory exchange ratio ↑ FA oxidation under HFD ↑ Oxidative stress ↑ TAG content ↓ Pro-inflammatory markers | [25] |
| Heart | ↓ TAG content No ER stress, inflammation and oxidative stress | ||
| Hepatocyte-specific Plin5−/− mice | Liver | ↓ FA consumption ↓ FA oxidation ↓ TAG secretion ↓ Lipid peroxidation and oxidative stress Insulin resistance and enhancement under an HFD Glucose intolerance under HFD TAG accumulation under HFD | [21] |
| CM-Plin5 mice | Heart | ↑ Accumulation of TAGs ↑ TAG hydrolytic activities: ↑ ATGL and CGI-58 protein levels Moderately reduced FA oxidizing gene expression levels | [60] |
| MCK-Plin5 mice | Skeletal muscle | ↑ LD formation ↓ Body weight compared to non-transgenic littermates under control and HFD diet ↑ Expression of ER stress markers | [61] |
| Heart | ↑ TAG content | ||
| Diaphragm | ↑ TAG content | ||
| EDL | ↑ TAG content | ||
| Gastrocnemius | ↑ TAG content | ||
| Liver | ↓ Cholesterol levels in an HFD compared to control diet littermates ↓ Lipid uptake ↓ Inflammatory markers |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mass Sanchez, P.B.; Krizanac, M.; Weiskirchen, R.; Asimakopoulos, A. Understanding the Role of Perilipin 5 in Non-Alcoholic Fatty Liver Disease and Its Role in Hepatocellular Carcinoma: A Review of Novel Insights. Int. J. Mol. Sci. 2021, 22, 5284. https://doi.org/10.3390/ijms22105284
Mass Sanchez PB, Krizanac M, Weiskirchen R, Asimakopoulos A. Understanding the Role of Perilipin 5 in Non-Alcoholic Fatty Liver Disease and Its Role in Hepatocellular Carcinoma: A Review of Novel Insights. International Journal of Molecular Sciences. 2021; 22(10):5284. https://doi.org/10.3390/ijms22105284
Chicago/Turabian StyleMass Sanchez, Paola Berenice, Marinela Krizanac, Ralf Weiskirchen, and Anastasia Asimakopoulos. 2021. "Understanding the Role of Perilipin 5 in Non-Alcoholic Fatty Liver Disease and Its Role in Hepatocellular Carcinoma: A Review of Novel Insights" International Journal of Molecular Sciences 22, no. 10: 5284. https://doi.org/10.3390/ijms22105284
APA StyleMass Sanchez, P. B., Krizanac, M., Weiskirchen, R., & Asimakopoulos, A. (2021). Understanding the Role of Perilipin 5 in Non-Alcoholic Fatty Liver Disease and Its Role in Hepatocellular Carcinoma: A Review of Novel Insights. International Journal of Molecular Sciences, 22(10), 5284. https://doi.org/10.3390/ijms22105284