
The Chemistry of the Ketogenic Diet: Updates and Opportunities in Organic Synthesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. History of the Ketogenic Diet
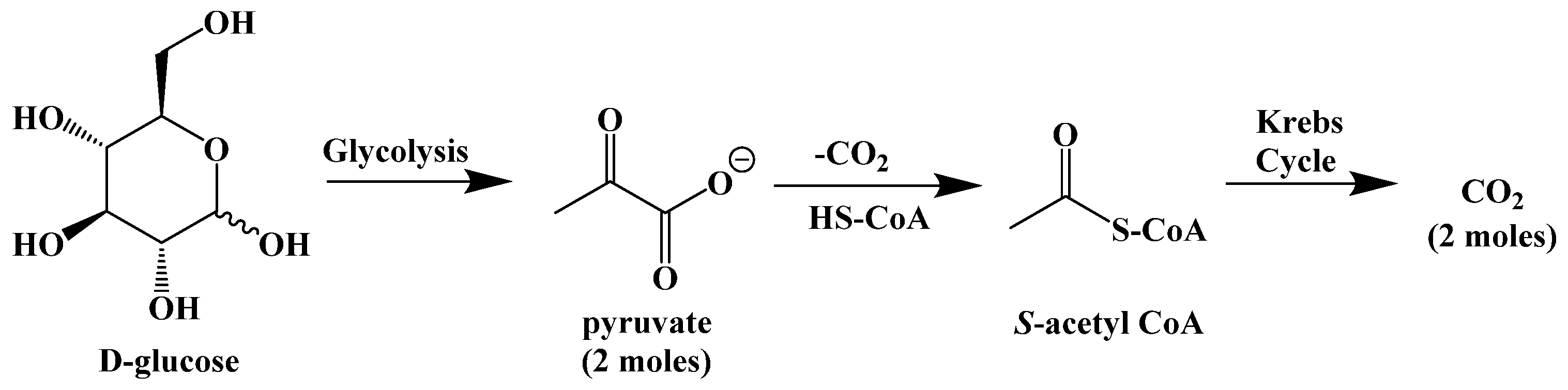
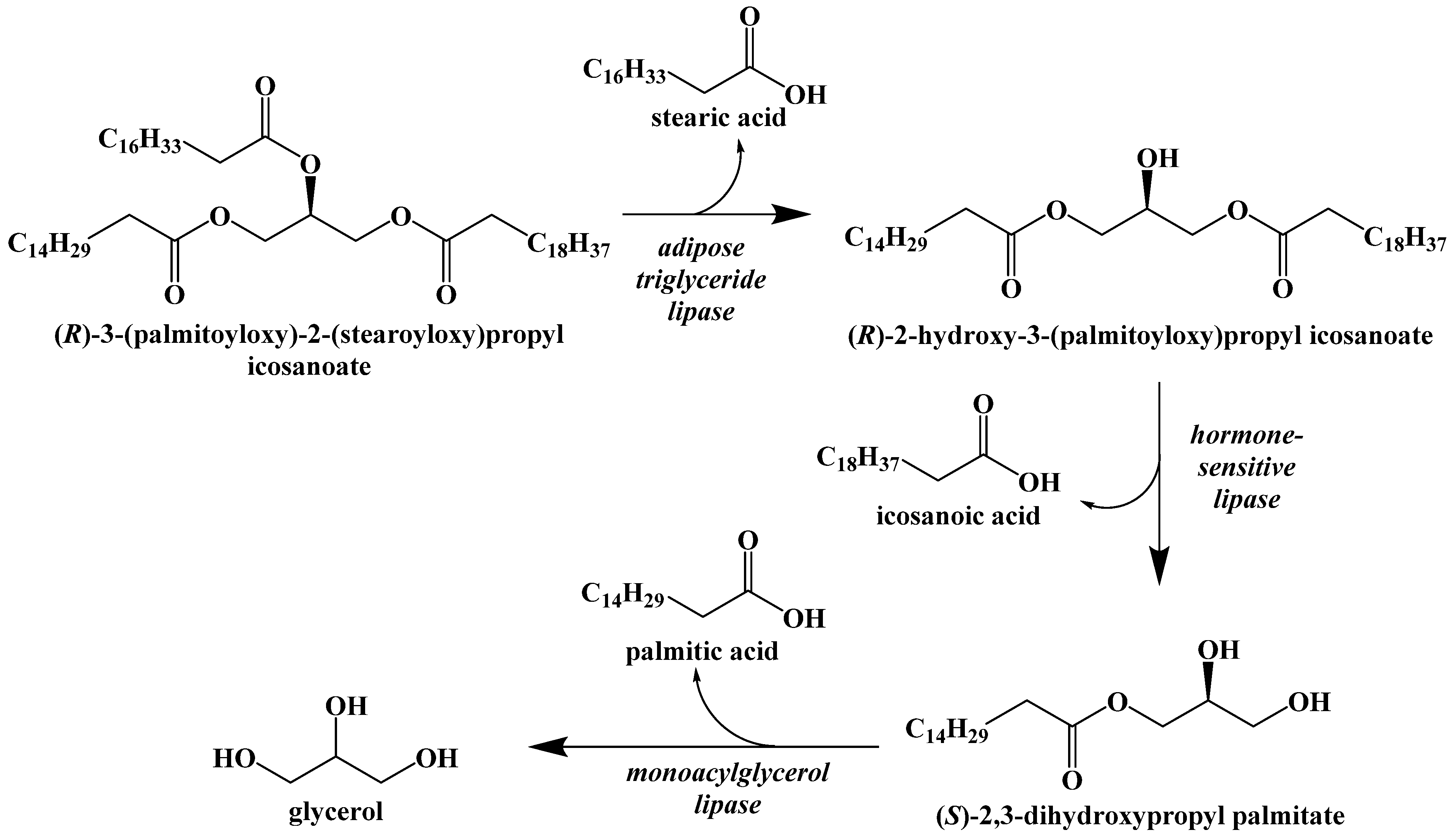
3. Competing Energy Sources in the Body
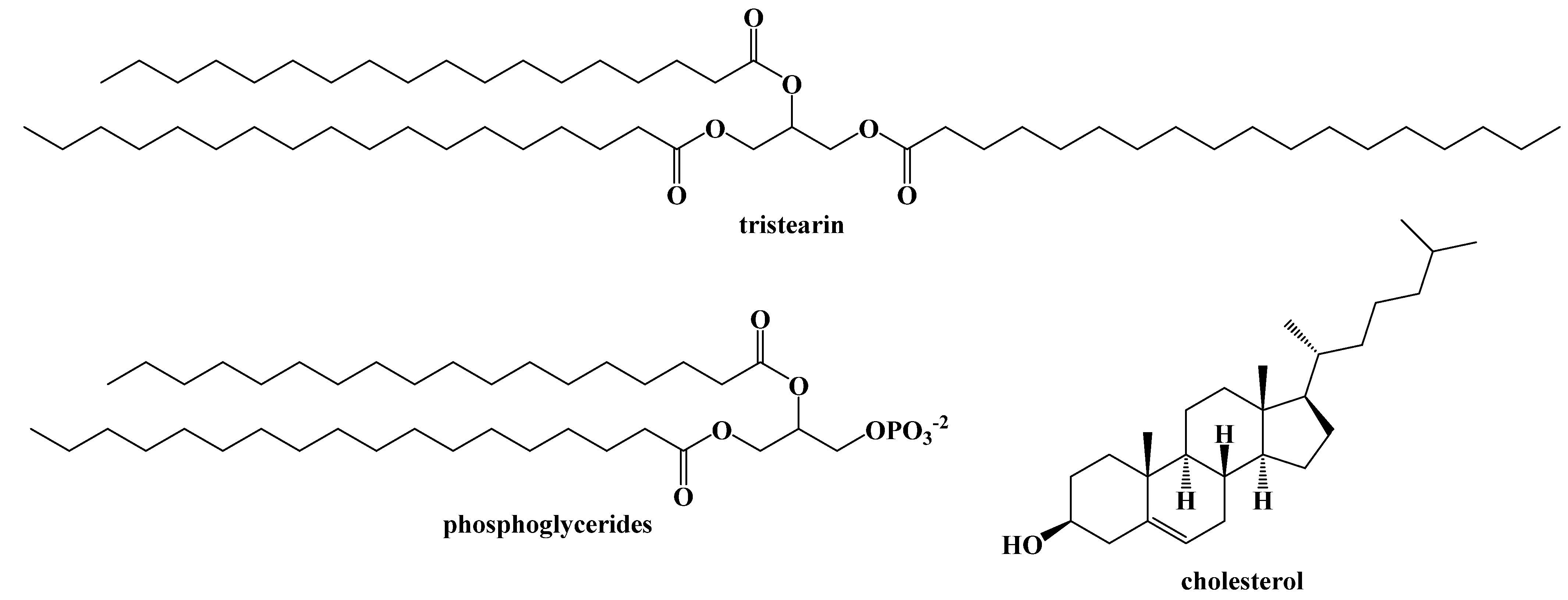
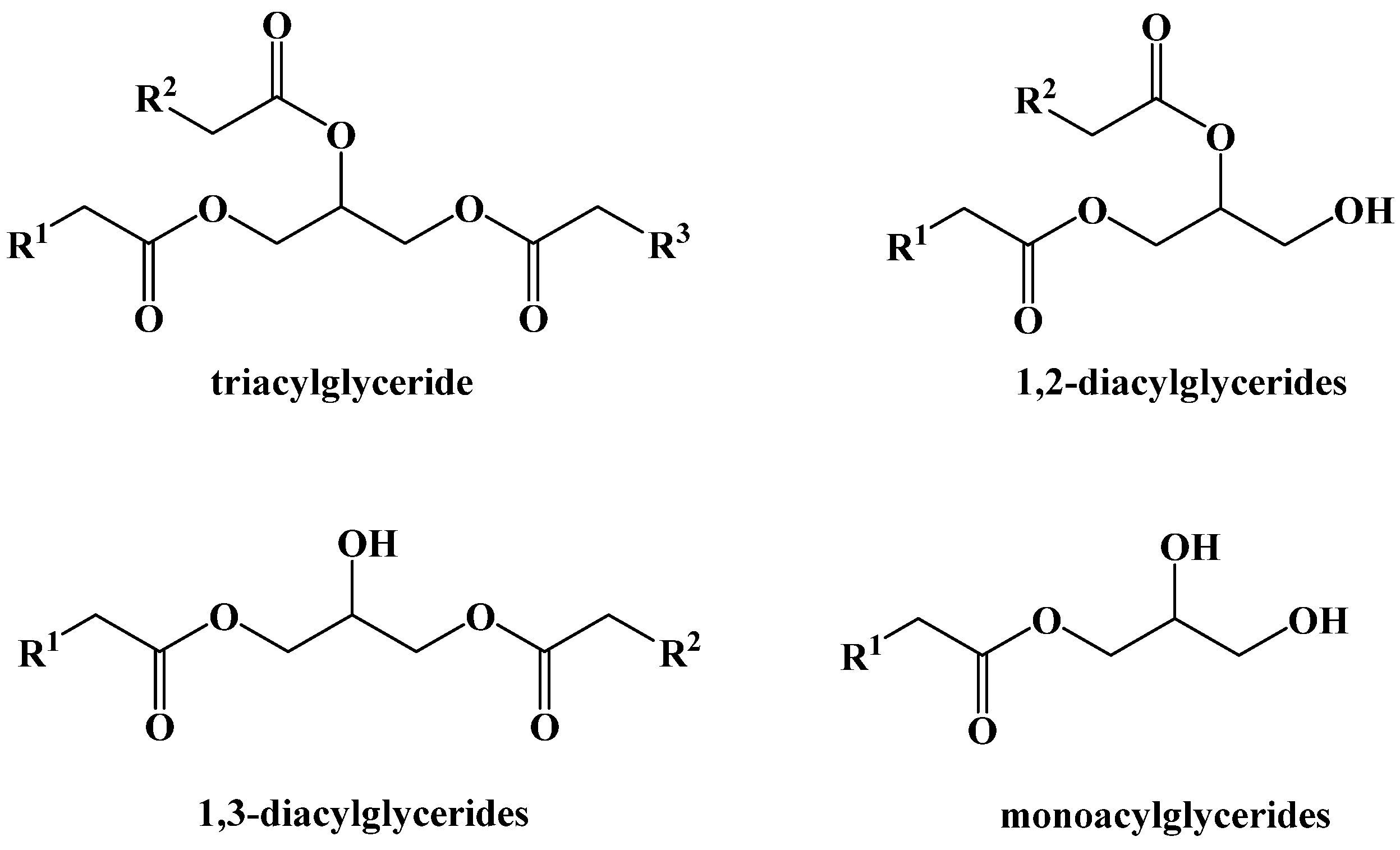
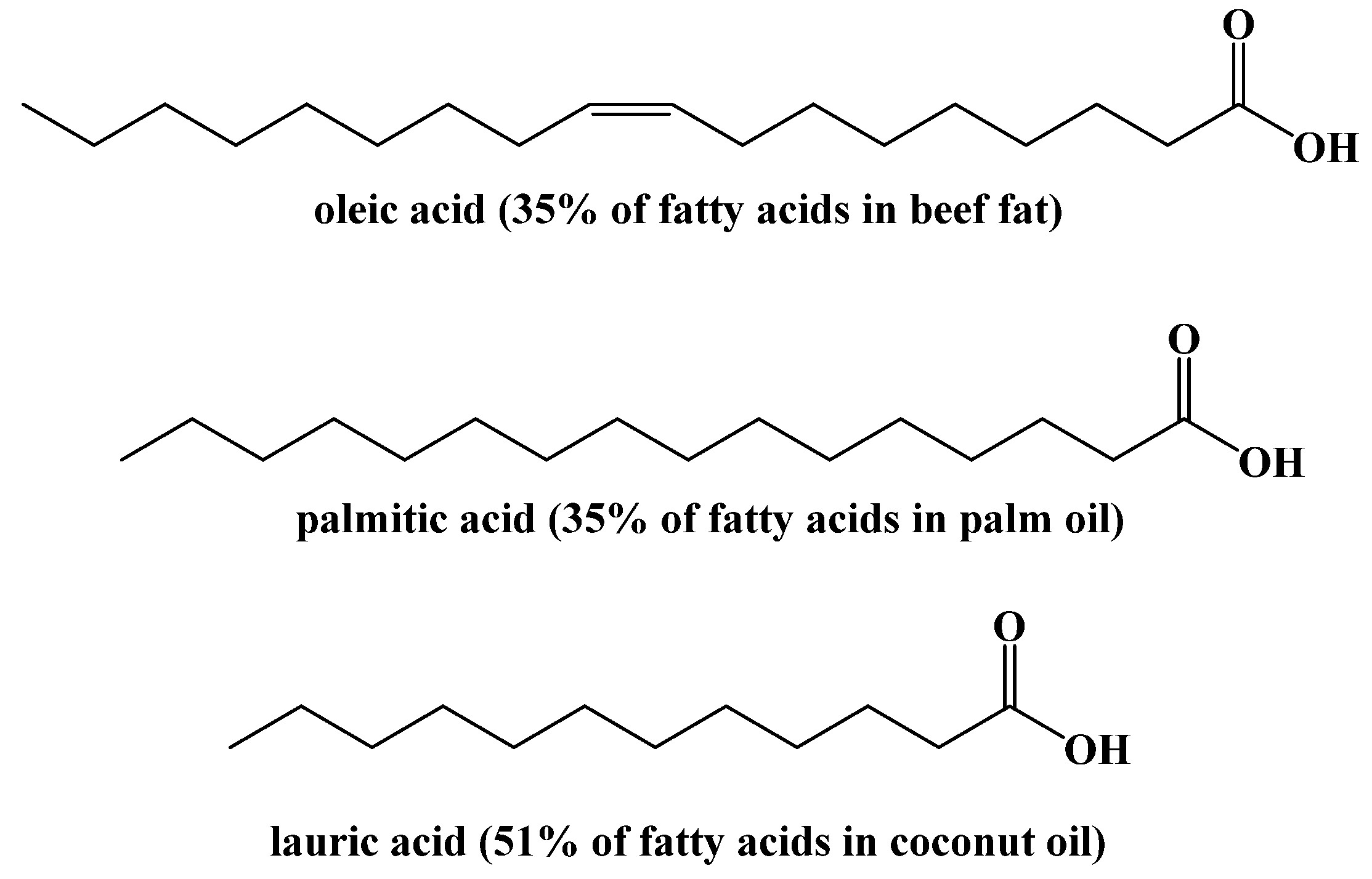
4. Dietary Fats
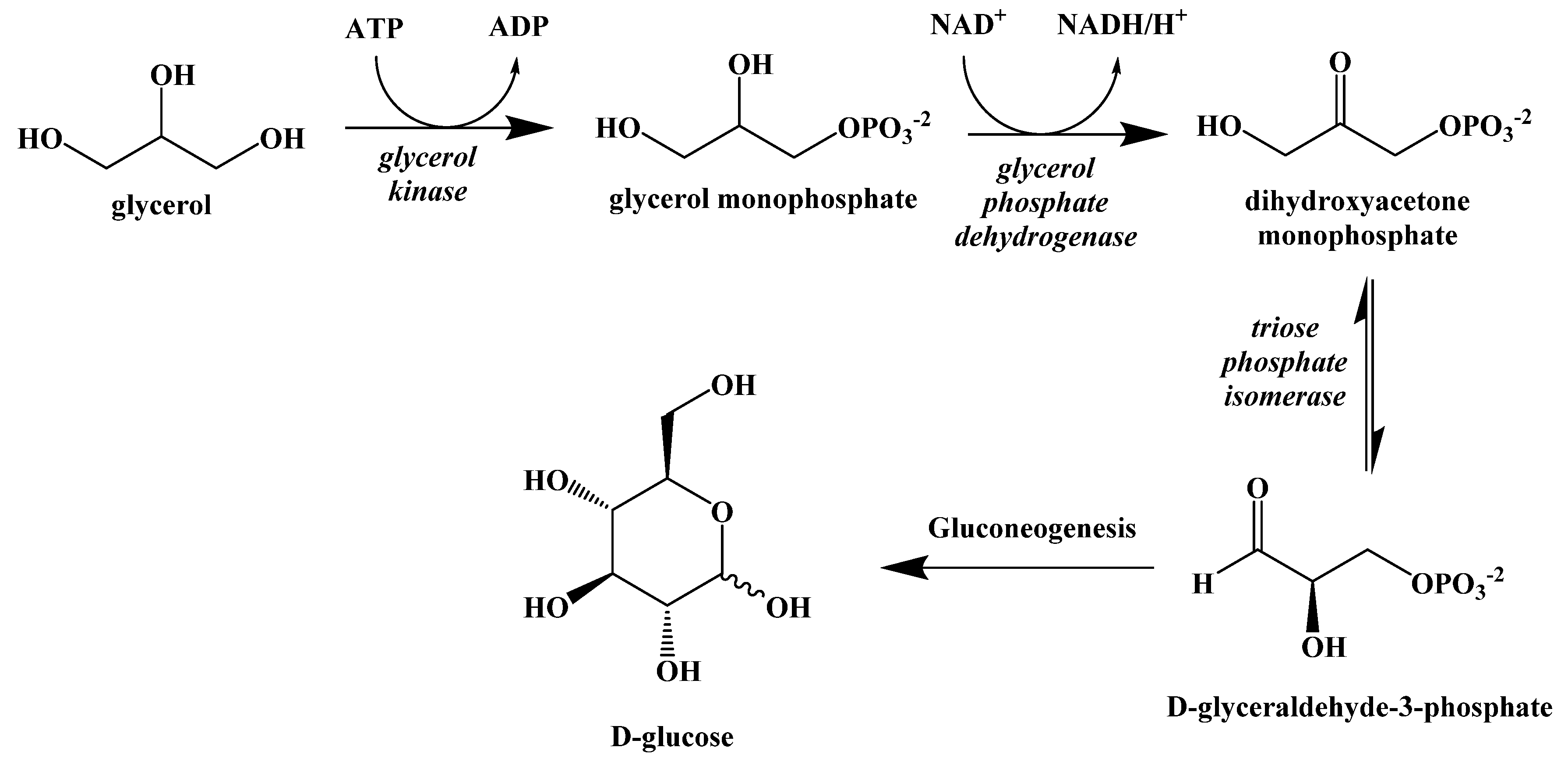
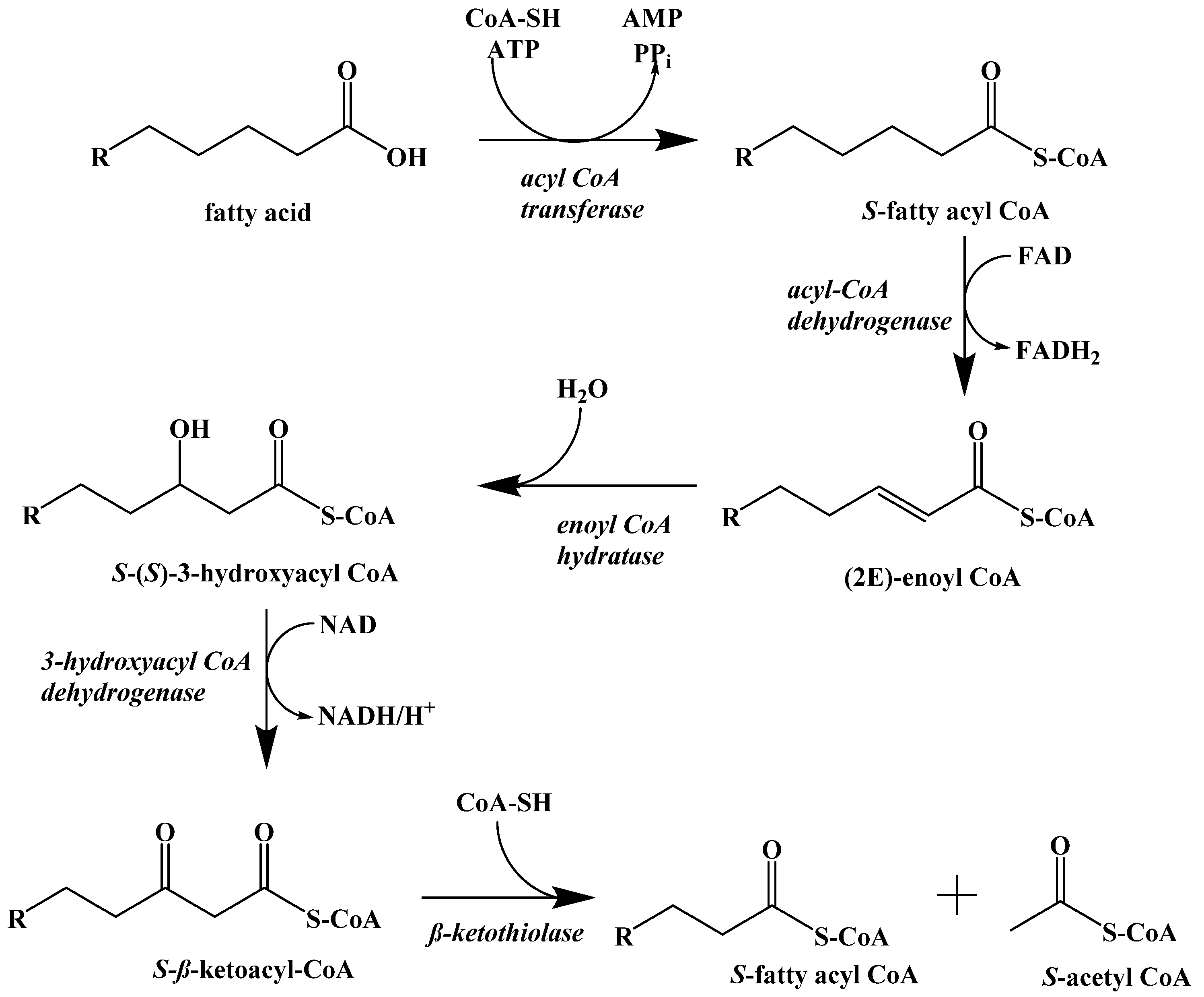
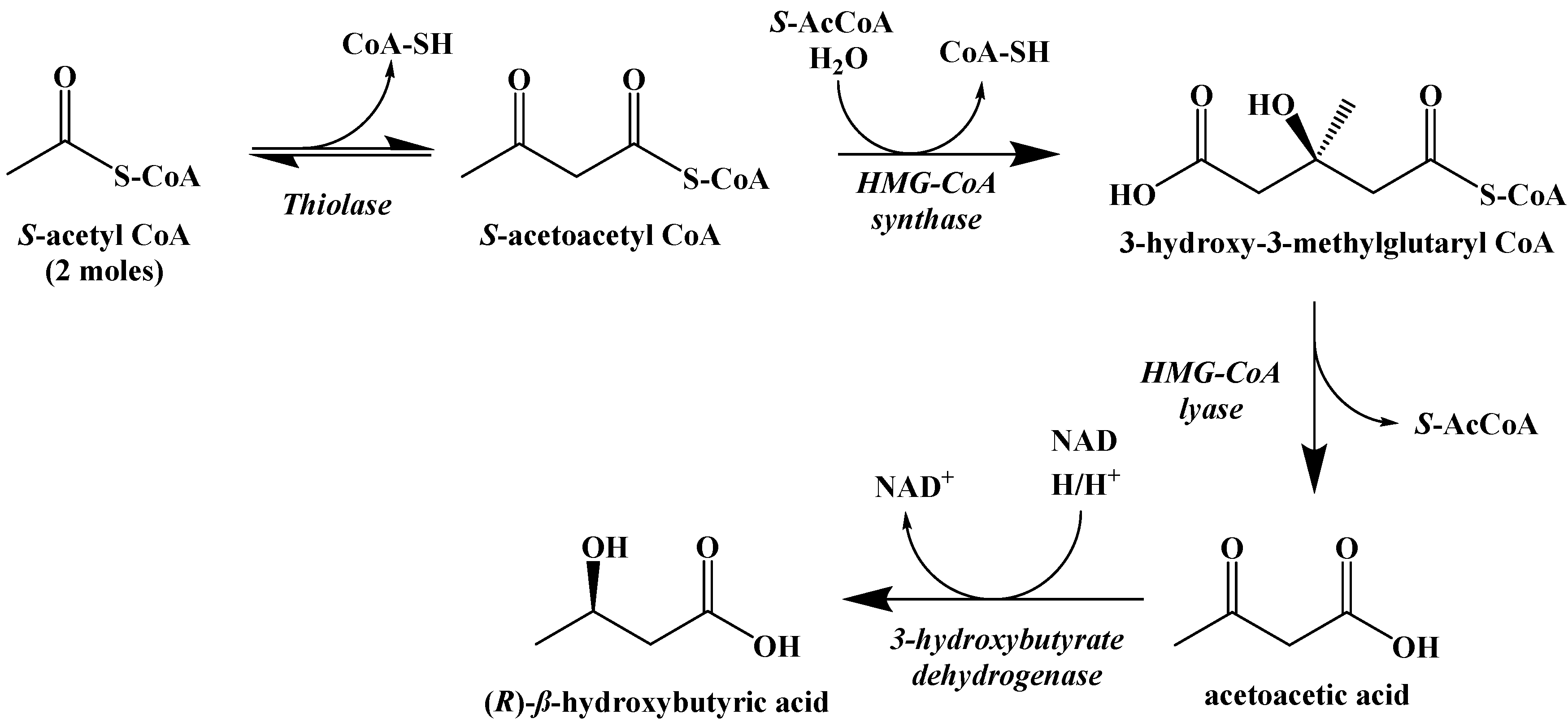
5. Biochemistry of the Ketogenic Diet
6. Metabolic Effect of Ketolysis Versus Glycolysis
7. Benefits of the Ketogenic Diet
8. Limitations of the Ketogenic Diet
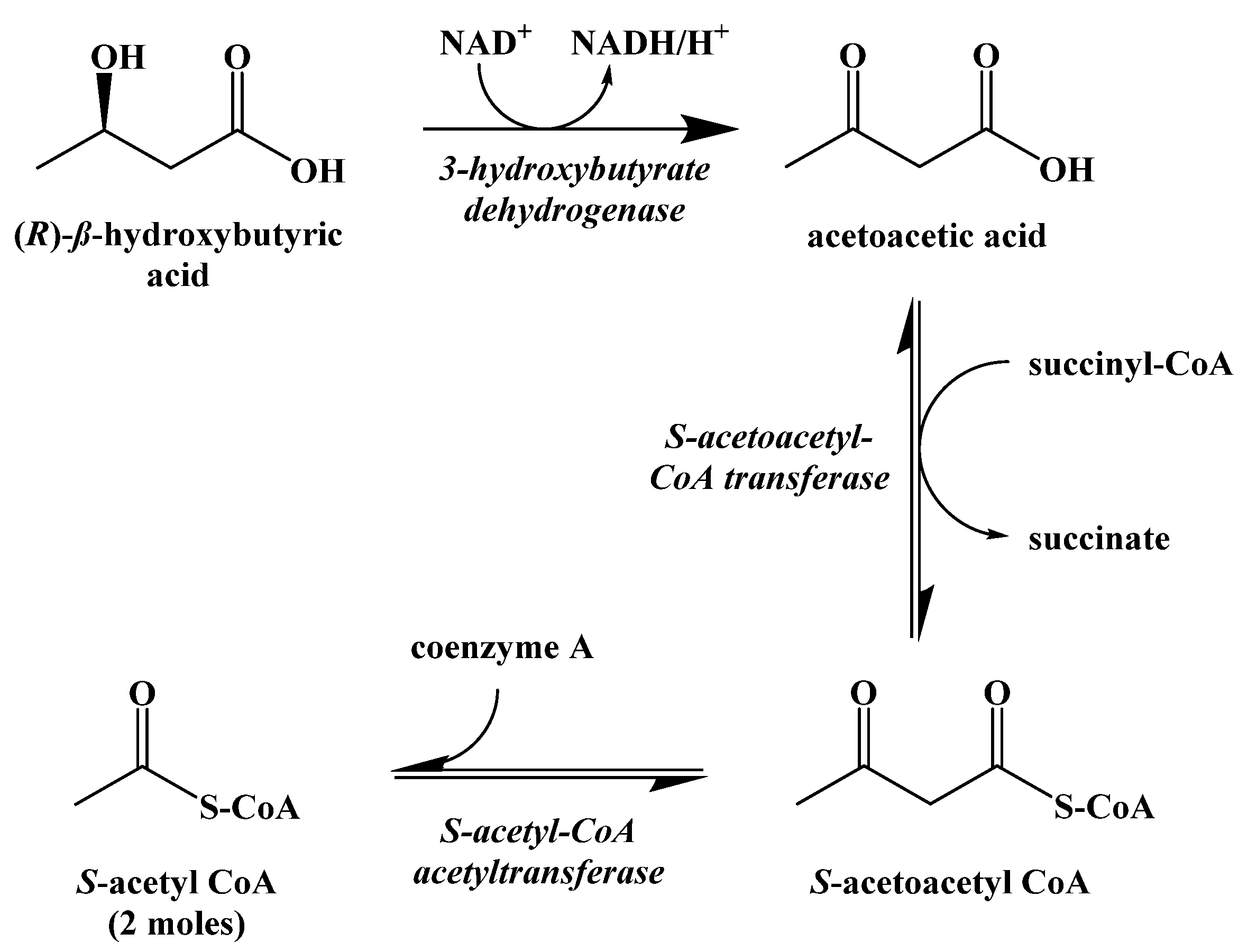
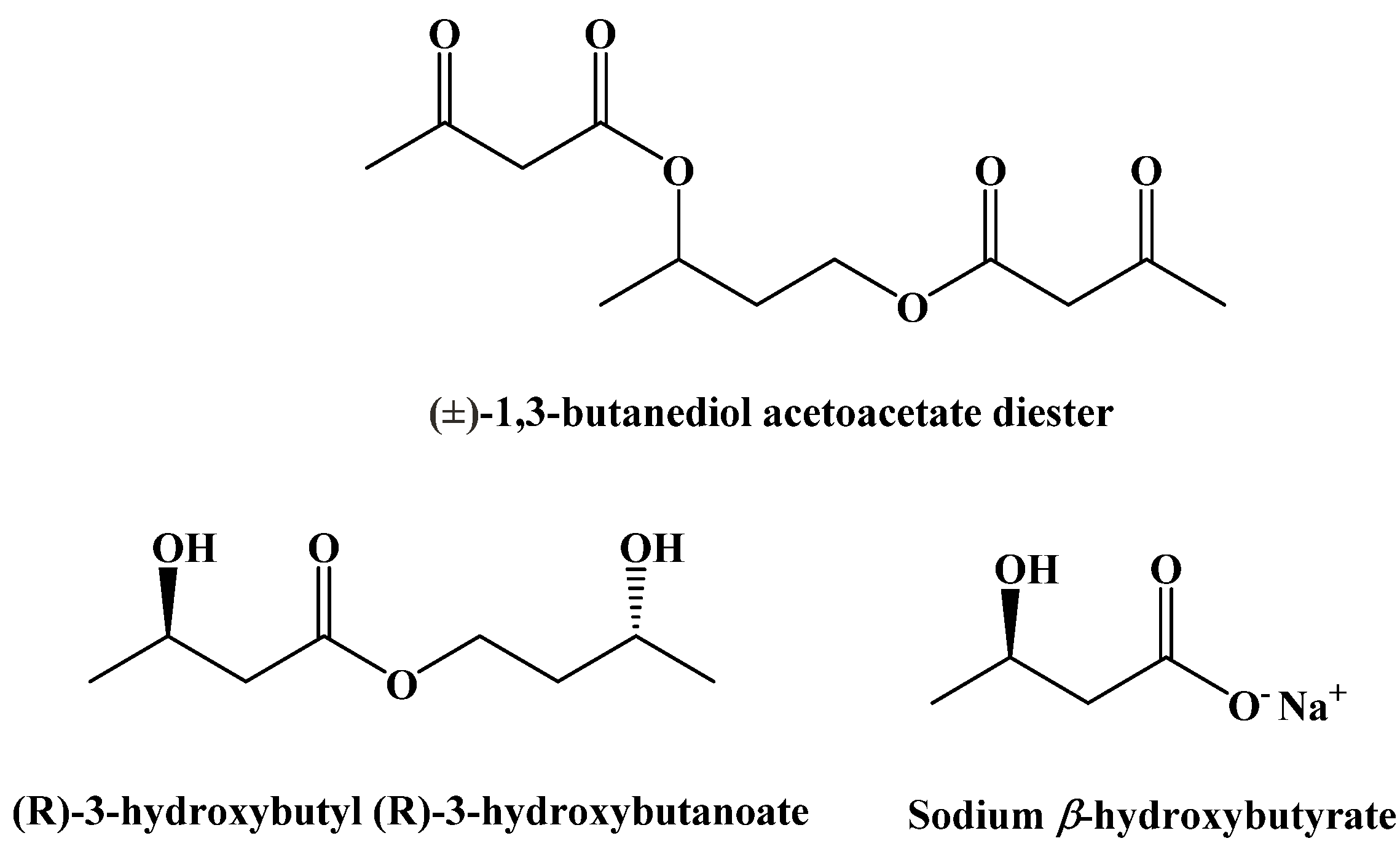
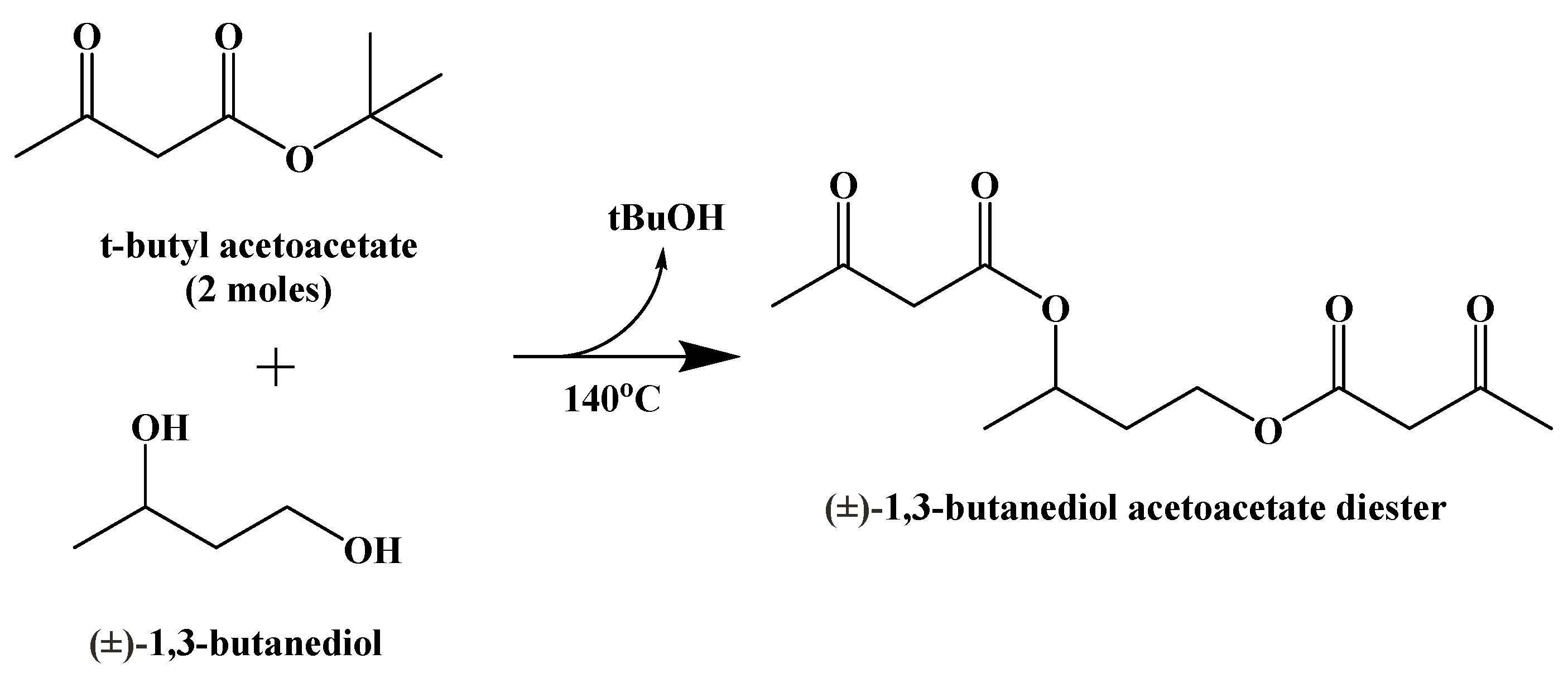
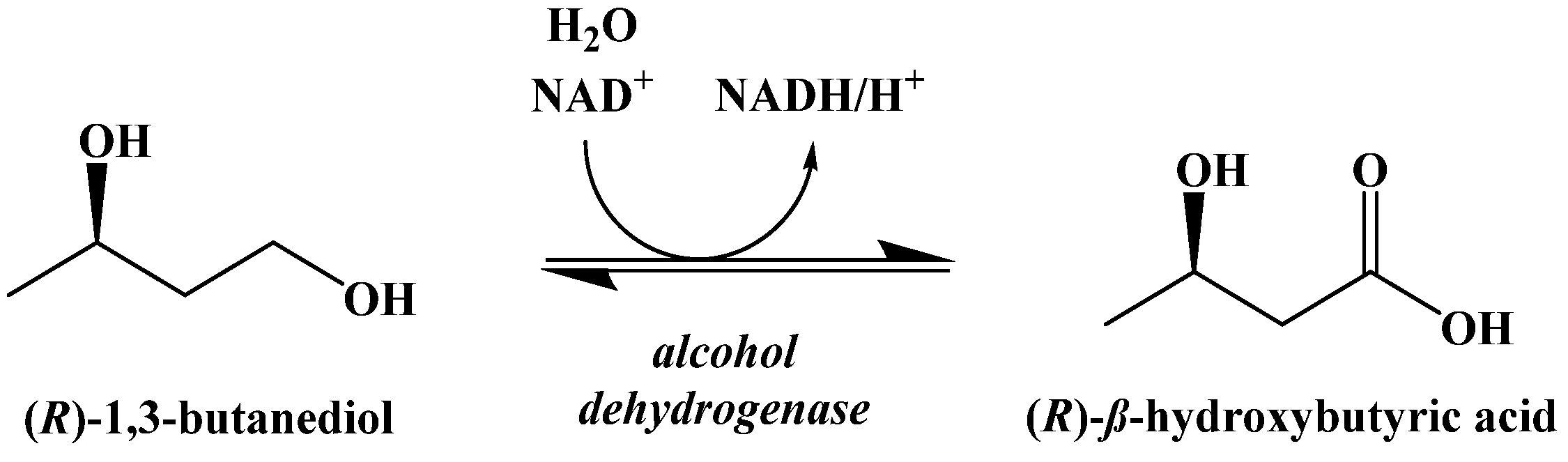
9. Synthetic Ketogenic Compounds as an Alternative Path to Ketosis
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Peterman, M.G. The ketogenic diet in the treatment of epilepsy: A preliminary report. Am. J. Dis. Child. 1924, 28, 28–33. [Google Scholar] [CrossRef]
- Kim, D.Y.; Rho, J.M. The ketogenic diet and epilepsy. Curr. Opin. Clin. Nutr. Metab. Care 2008, 11, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Hartman, A.L.; Gasior, M.; Vining, E.P.G.; Rogawski, M.A. The neuropharmacology of the ketogenic diet. Pediatr. Neurol. 2007, 36, 281–292. [Google Scholar] [CrossRef]
- Evangeliou, A.; Vlachonikolis, I.; Mihailidou, H. Application of a ketogenic diet in children with autistic behavior: Pilot study. J. Child Neurol. 2003, 18, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Poff, A.M.; Ari, C.; Seyfried, T.N.; D’Agostino, D.P. The ketogenic diet and hyperbaric oxygen therapy prolong survival in mice with systematic metastatic cancer. PLoS ONE 2013, 8, e65522. [Google Scholar] [CrossRef]
- Kesl, S.L.; Poff, A.M.; Ward, N.P.; Fiorelli, T.N.; Ari, C.; Van Putten, A.J.; Sherwood, J.W.; Arnold, P.; D’Agostino, D.P. Effects of exogenous ketone supplementation on blood ketone, glucose, triglyceride, and lipoprotein levels in Sprague-Dawley rats. Nutr. Metab. 2016, 13, 9. [Google Scholar] [CrossRef] [PubMed]
- Guelpa, G.; Marie, A. La lutte contre l’epilepsie par la desintoxication et par la reeducation alimentaire. Revue de Therapie Medico-Chirurgicale 1911, 78, 8–13. [Google Scholar]
- Geyelin, H.R. Fasting as method for treating epilepsy. Med. Rec. 1921, 99, 1037–1039. [Google Scholar]
- Newburgh, L.H.; Marsh, P.L. The use of a high fat diet in the treatment of diabetes mellitus first paper. Arch. Intern. Med. 1920, 26, 647–662. [Google Scholar] [CrossRef][Green Version]
- Newburgh, L.H.; Marsh, P.L. The use of a high fat diet in the treatment of diabetes mellitus second paper: Blood sugar. Arch. Intern. Med. 1921, 27, 699–705. [Google Scholar] [CrossRef][Green Version]
- Newburgh, L.H.; Marsh, P.L. Further observations on the use of a high fat diet in the treatment of diabetes mellitus. Arch. Intern. Med. 1923, 31, 455–490. [Google Scholar] [CrossRef]
- Woodyatt, R.T. Objects and methods of diet adjustment in diabetes. Arch. Intern. Med. 1921, 28, 125–141. [Google Scholar] [CrossRef]
- Shaffer, P.A. Antiketogenesis. J. Biol. Chem. 1921, 47, 433–448. [Google Scholar] [CrossRef]
- Gerhardt, C.J.A.C. Ueber diabetes mellitus und aceton. Wiener Medizinische Presse 1865, 28, 675. [Google Scholar]
- Geelmuyden, H.C. Uber die acetonuria bei phlorizinvergiftung. Zeitschr. F. Phys. Chem. 1897, 23, 431. [Google Scholar]
- Banting, F.G.; Best, C.H.; Collip, J.B.; Campbell, W.R.; Fletcher, A.A. Pancreatic extracts in the treatment of diabetes mellitus. Can. Med. Assoc. J. 1922, 12, 141–146. [Google Scholar]
- Wilder, R.M. The effect of ketonemia on the course of epilepsy. Mayo Clin. Bull. 1921, 2, 307. [Google Scholar]
- Wilder, R.M. High fat diets in epilepsy. Mayo Clin. Bull. 1921, 2, 308. [Google Scholar]
- Hohn, S.; Dozieres-Puyravel, B.; Auvin, S. History of dietary treatment from Wilder’s hypothesis to the first open studies in the 1920s. Epilepsy Behav. 2019, 101, 106588. [Google Scholar] [CrossRef]
- Peterman, M.G. The ketogenic diet in epilepsy. JAMA 1925, 84, 1979–1983. [Google Scholar] [CrossRef]
- Talbot, F.B. The treatment of epilepsy of childhood by the ketogenic diet. R. I. Med. J. 1927, 10, 159–162. [Google Scholar]
- Helmholz, H.F. The treatment of epilepsy in childhood—five years’ experience with the ketogenic diet. JAMA 1927, 88, 2028–2032. [Google Scholar] [CrossRef]
- Kwan, P.; Brodie, M.J. Phenobarbital for the treatment of epilepsy in the 21st century: A critical review. Epilepsia 2004, 45, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Millares-Sipin, C.A.; Alafris, A.; Cohen, H. Chapter 15: Phenytoin and fosphenytoin. In Casebook in Clinical Pharmacokinetics and Drug Dosing; McGraw-Hill: New York City, USA, 2015. [Google Scholar]
- Albani, F.; Riva, R.; Baruzzi, A. Carbamazepine clinical pharmacology: A review. Pharmacopsychiatry 1995, 28, 235–244. [Google Scholar] [CrossRef]
- Jeavons, P.M.; Clark, J.E. Sodium valproate in treatment of epilepsy. Br. Med. J. 1974, 2, 584. [Google Scholar] [CrossRef] [PubMed]
- Swink, T.D.; Vining, E.P.; Freeman, J.M. The ketogenic diet: 1997. Adv. Pediatr. 1997, 44, 297–329. [Google Scholar]
- Freeman, J.M.; Vining, E.P.; Pillas, D.J.; Pyzik, P.L.; Casey, J.C.; Kelly, L.M. The efficacy of the ketogenic diet: 1998—a prospective evaluation of intervention in 150 children. Pediatrics 1998, 102, 1358–1363. [Google Scholar] [CrossRef]
- Vining, E.P.; Freeman, J.M.; Ballahn-Gil, K.; Camfield, C.S.; Camfield, P.R.; Holmes, G.L.; Shinnar, S.; Shuman, R.; Trevathan, E.; Wheless, J.W. The ketogenic diet multi-center study group. A multicenter study of the efficacy of the ketogenic diet. Arch. Neurol. 1998, 55, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Phillips, S. reporter. Medical Secrets [Television Broadcast]. Dateline. 26 October 1994; National Broadcasting Company (NBC). [Google Scholar]
- Lekka, V. Towards the twenty-first century. In The Neurological Emergence of Epilepsy; Springer: Cham, Switzerland, 2015. [Google Scholar]
- Paoli, A.; Rubini, A.; Volek, J.S.; Grimaldi, K.A. Beyond weight loss: A review of the therapeutic uses of very-low-carbohydrate (ketogenic) diets. Eur. J. Clin. Nutr. 2013, 67, 789–796. [Google Scholar] [CrossRef]
- Kallio, H.; Yli-Jokipii, K.; Jurvinen, J.P.; Sjovall, O.; Tahvonen, R. Regioisomerism of triacylglycerols in lard, tallow, yolk, chicken skin, palm oil, palm olein, palm stearin, and a transesterified blend of palm stearin and coconut oil analyzed by tandem mass spectrometry. J Agric. Food Chem. 2001, 49, 3363–3369. [Google Scholar] [CrossRef]
- Chowdhury, K.; Banu, L.A.; Khan, S.; Latif, A. Studies on the fatty acid composition of edible oil. Bangladesh J. Sci. Ind. Res. 2007, 42, 311–316. [Google Scholar] [CrossRef]
- Dabbou, S.; Chaieb, I.; Rjiba, I.; Issaoui, M.; Echbili, A.; Nakbi, A.; Gazzah, N.; Hammami, M. Multivariate data analysis of fatty acid content in the classification of olive oils developed through controlled crossbreeding. J. Am. Oil Chem. Soc. 2012, 89, 667–674. [Google Scholar] [CrossRef]
- Hers, H.G.; Hue, L. Gluconeogenesis and related aspects of glycolysis. Ann. Rev. Biochem. 1983, 52, 617–653. [Google Scholar] [CrossRef] [PubMed]
- Roach, P.J. Glycogen and its metabolism. Curr. Mol. Med. 2002, 2, 101–120. [Google Scholar] [CrossRef] [PubMed]
- Duncan, R.E.; Ahmadian, M.; Jaworski, K.; Sarkadi-Nagy, E.; Sul, H.S. Regulation of lipolysis in adipocytes. Annu. Rev. Nutr. 2007, 27, 79–101. [Google Scholar] [CrossRef] [PubMed]
- Eichmann, T.O.; Kumari, M.; Haas, J.T.; Farese, R.V., Jr.; Zimmermann, R.; Lass, A.; Zechner, R. Studies on the substrate and stereo/regioselectivity of adipose triglyceride lipase, hormone-sensitive lipase, and diacylglycerol-O-acyltransferases. J. Biol. Chem. 2012, 287, 41446–41457. [Google Scholar] [CrossRef] [PubMed]
- Owen, O.E.; Morgan, A.P.; Kemp, H.G.; Sullivan, J.M.; Herrera, M.G.; Cahill, G.F., Jr. Brain Metabolism during fasting. J. Clin. Investig. 1967, 46, 1589–1595. [Google Scholar] [CrossRef] [PubMed]
- Fukao, T.; Lopaschuk, G.D.; Mitchell, G.A. Pathways and control of ketone body metabolism: On the fringe of lipid biochemistry. Prostaglandins Leukot. Essent. Fatty Acids 2004, 70, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Hegardt, F.G. Mitochondrial 3-hydroxy-3-methylgutaryl-CoA synthase: A control enzyme in ketogenesis. Biochem. J. 1999, 338, 569–582. [Google Scholar] [CrossRef]
- Miller, V.J.; Villamena, F.A.; Volek, J.S. Nutritional ketosis and mitohormesis: Potential implications for mitochondrial function and human health. J. Nutr. Metab. 2018, 2018, 5157645. [Google Scholar] [CrossRef] [PubMed]
- Mullins, G.; Hallam, C.L.; Broom, I. Ketosis, ketoacidosis and very-low-calorie diets: Putting the record straight. Nutr. Bull. 2011, 36, 397–402. [Google Scholar] [CrossRef]
- Bueno, N.B.; de Melo, I.S.V.; de Oliveira, S.L.; da Rocha Ataide, T. Very-low-carbohydrate ketogenic diet v. low-fat diet for long-term weight loss: A meta-analysis of randomized controlled trials. Br. J. Nutr. 2013, 110, 1178–1187. [Google Scholar] [CrossRef]
- Brehm, B.J.; Seeley, R.J.; Daniels, S.R.; D’Alessio, D.A. A randomized trial comparing a very low carbohydrate diet and a calorie-restricted low fat diet on body weight and cardiovascular risk factors in healthy women. J. Clin. Endocrinol. Metab. 2003, 88, 1617–1623. [Google Scholar] [CrossRef]
- Shai, I.; Schwarzfuchs, D.; Henklin, Y.; Shahar, D.R.; Witkow, S.; Greenberg, I. Weight loss with a low-carbohydrate, Mediterranean, or low-fat diet. N. Engl. J. Med. 2008, 359, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Bistrian, B.R.; Blackburn, G.L.; Flatt, J.P.; Sizer, J.; Scrimshaw, N.S.; Sherman, M. Nitrogen metabolism and insulin requirements in obese diabetic adults on a protein-sparing modified fast. Diabetes 1976, 25, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Gumbiner, B.; Wendel, J.A.; McDermott, M.P. Effects of diet composition and ketosis on glycemia during very-low-energy-diet therapy in obese patients with non-insulin-dependent diabetes mellitus. Am. J. Clin. Nutr. 1996, 63, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Boden, G.; Sargrad, K.; Homko, C.; Mozzoli, M.; Stein, T.P. Effect of a low-carbohydrate diet on appetite, blood glucose levels, and insulin resistance in obese patients with type 2 diabetes. Ann. Intern. Med. 2005, 142, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Dashit, H.M.; Al-Zaid, N.S.; Mathew, T.C.; Al-Mousawi, M.; Talib, H.; Asfar, S.K. Long term effects of ketogenic diet in obese subjects with high cholesterol level. Mol. Cell. Biochem. 2006, 286, 1–9. [Google Scholar] [CrossRef]
- Paoli, A.; Cenci, L.; Fancelli, M.; Parmagnani, A.; Fratter, A.; Cucchi, A. Ketogenic diet and phytoextracts comparison of the efficacy of mediterranean, zone and tisanoreica diet on some health risk factors. Agro Food Ind. Hi-Tech 2010, 21, 24. [Google Scholar]
- Sharman, M.J.; Kraemer, W.J.; Love, D.M.; Avery, N.G.; Gomez, A.L.; Scheett, T.P. A ketogenic diet favorably affects serum biomarkers for cardiovascular disease in normal-weight men. J. Nutr. 2002, 132, 1879–1885. [Google Scholar] [CrossRef] [PubMed]
- Volek, J.S.; Sharman, M.J.; Forsythe, C.E. Modification of lipoproteins by very low-carbohydrate diets. J. Nutr. 2005, 135, 1339–1342. [Google Scholar] [CrossRef] [PubMed]
- Baranano, K.W.; Hartman, A.L. The ketogenic diet: Uses in epilepsy and other neurologic illnesses. Curr. Treat. Options Neurol. 2008, 10, 410–419. [Google Scholar] [CrossRef]
- Stafstrom, C.E.; Rho, J.M. The ketogenic diet as a treatment paradigm for diverse neurological disorders. Front. Pharmacol. 2012, 3, 59. [Google Scholar] [CrossRef] [PubMed]
- Jensen, N.J.; Wodschow, H.Z.; Nilsson, M. Effects of ketone bodies on brain metabolism and function in neurodegenerative diseases. Int. J. Mol. Sci. 2020, 21, 1–17. [Google Scholar] [CrossRef]
- Vanitallie, T.B.; Nonas, C.; Di Rocc, A.; Boyar, K.; Hyams, K.; Heymsfield, S.B. Treatment of Parkinson disease with diet-induced hyperketonemia: A feasibility study. Neurology 2005, 64, 728–730. [Google Scholar] [CrossRef] [PubMed]
- Tieu, K.; Perier, C.; Caspersen, C.; Teismann, P.; Wu, D.; Yan, S.; Naini, A.; Vila, M.; Jackson-Lewis, V.; Ramasamy, R.; et al. D-beta-Hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J. Clin. Investig. 2003, 112, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Kashiwaya, Y.; Takeshima, T.; Mori, N.; Nakashima, K.; Clarke, K.; Veech, R.L. D-beta-Hydroxybutyrate protects neurons in models of Alzheimer’s and Parkinson’s disease. Proc. Natl Acad. Sci. USA 2000, 97, 5440–5444. [Google Scholar] [CrossRef]
- Van der Auwera, I.; Wera, S.; Van Leuven, F.; Henderson, S.T. A ketogenic diet reduces amyloid beta 40 and 42 in a mouse model of Alzheimer’s disease. Nutr. Metab. 2005, 2, 28. [Google Scholar] [CrossRef]
- Zhao, Z.; Lange, D.J.; Voustianiouk, A.; MacGrogan, D.; Ho, L.; Suh, J.; Humala, N.; Thiyagarajan, M.; Wang, J.; Pasinetti, G.M. A ketogenic diet as a potential novel therapeutic intervention in amyotrophic lateral sclerosis. BMC Neurosci. 2006, 7, 29. [Google Scholar]
- Warburg, O. Uber den stoffwechsel der carzinomzelle. Klinische Wochenschrift 1925, 4, 534–536. [Google Scholar] [CrossRef]
- Klement, R.J.; Kammerer, U. Is there a role for carbohydrate restriction in the treatment and prevention of cancer? Nutr. Metab. 2011, 8, 75. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Demetrakopoulos, G.E.; Linn, B.; Amos, H. Rapid loss of ATP by tumor cells deprived of glucose: Contrast to normal cells. Biochem. Biophys. Res. Commun. 1978, 82, 727–794. [Google Scholar] [CrossRef]
- Priebe, A.; Tan, L.; Wahl, H.; Kueck, A.; He, G.; Kowk, R.; Opipari, A.; Liu, J.R. Glucose deprivation activates AMPK and induces cell death through modulation of Akt in ovarian cancer cells. Gynecol. Oncol. 2011, 122, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Shim, H.; Chun, Y.S.; Lewis, B.C.; Dang, C.V. A unique glucose-dependent apoptotic pathway induced by c-Myc. Proc. Natl Acad. Sci. USA 1998, 95, 1511–1516. [Google Scholar] [CrossRef] [PubMed]
- Tisdale, M.J.; Brenna, R.A. Loss of acetoacetate coenzyme A transferase activity in tumours of peripheral tissues. Br. J. Cancer 1983, 47, 293–297. [Google Scholar] [CrossRef]
- Fredericks, M.; Ramsey, R.B. 3-Oxo acid coenzyme A transferase activity in brain and tumors of the nervous system. J. Neurochem. 1978, 31, 1529–1531. [Google Scholar] [CrossRef]
- Otto, C.; Kaemmerer, U.; Illert, B.; Muehling, B.; Pfetzer, N.; Wittig, R.; Voelker, H.U.; Thiede, A.; Coy, J.F. Growth of human gastric cancer cells in nude mice is delayed by a ketogenic diet supplemented with omege-3 fatty acids and medium-chain triglycerides. BMC Cancer 2008, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Stafford, P.; Abdelwahab, M.G.; Kim, D.Y.; Preul, M.C.; Rho, J.M.; Scheck, A.C. The ketogenic diet reverses gene expression patterns and reduces reactive oxygen species levels when used as an adjuvant therapy for glioma. Nutr. Metab. 2010, 7, 74. [Google Scholar] [CrossRef]
- Abdelwahab, M.G.; Fenton, K.E.; Preul, M.C.; Rho, J.M.; Lynch, A.; Stafford, P.; Scheck, A.C. The ketogenic diet is an effective adjuvant to radiation therapy for the treatment of malignant glioma. PLoS ONE 2012, 7, e36197. [Google Scholar] [CrossRef]
- Nebeling, L.C.; Miraldi, F.; Shurin, S.B.; Lerner, E. Effects of a ketogenic diet on tumor metabolism and nutritional status in pediatric oncology patients: Two case reports. J. Am. Coll. Nutr. 1995, 14, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Zuccoli, G.; Marcello, N.; Pisanello, A.; Servadei, F.; Vaccaro, S.; Mukherjee, P.; Seyfried, T.N. Metabolic management of glioblastoma multiforme using standard therapy together with a restricted ketogenic diet: Case report. Nutr. Metab. 2010, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Dekaban, A.S. Plasma lipids in epileptic children treated with the high fat diet. Arch. Neurol. 1966, 15, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Fenton, C.; Chee, C.M.; Bergqvist, A.G.C. Manipulation of types of fats and cholesterol intake can successfully improve the lipid profile while maintaining the efficacy of the ketogenic diet. Infant Child Adolesc. Nutr. 2009, 1, 338–341. [Google Scholar] [CrossRef][Green Version]
- Kielb, S.; Koo, H.P.; Bloom, D.A.; Faerber, G.J. Nephrolithiasis associated with the ketogenic diet. J. Urol. 2000, 164, 464–466. [Google Scholar] [CrossRef]
- Sampath, A.; Kossoff, E.H.; Furth, S.L.; Pyzik, P.L.; Vining, E.P. Kidney stones and the ketogenic diet: Risk factors and prevention. J. Child Neurol. 2007, 22, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Couch, S.C.; Schwarzman, F.; Carroll, J.; Koenigsberger, D.; Nordli, D.R.; Deckelbaum, R.J.; DeFelice, A.R. Growth and nutritional outcomes of children treated with the ketogenic diet. J. Am. Diet. Assoc. 1999, 99, 1573–1575. [Google Scholar] [CrossRef]
- Peterson, S.J.; Tangney, C.C.; Pimentel-Zablah, E.M.; Hjelmgren, B.; Booth, G.; Berry-Kravis, E. Changes in growth and seizure reduction in children on the ketogenic diet as a treatment for intractable epilepsy. J. Am. Diet. Assoc. 2005, 105, 718–725. [Google Scholar] [CrossRef]
- Bergqvist, A.G.; Schall, J.I.; Stallings, V.A.; Zemel, B.S. Progressive bone mineral content loss in children with intractable epilepsy treated with the ketogenic diet. Am. J. Clin. Nutr. 2008, 88, 1678–1684. [Google Scholar] [CrossRef]
- Sheth, R.D.; Binkley, N.; Hermann, B.P. Progressive bone deficit in epilepsy. Neurology 2008, 70, 170–176. [Google Scholar] [CrossRef]
- Halevy, A.; Peleg-Weiss, L.; Cohen, R.; Shuper, A. An update on the ketogenic diet, 2012. Rambam Maimonides Med. J. 2012, 3, e0005. [Google Scholar] [CrossRef] [PubMed]
- Amari, A.; Grace, N.; Fisher, W. Achieving and maintaining compliance with the ketogenic diet. J. Appl. Behav. Anal. 1995, 28, 341–342. [Google Scholar] [CrossRef] [PubMed]
- Bergen, S.S., Jr.; Hashim, S.A.; Van Itallie, T.B. Hyperketonemia induced in man by medium-chain triglyceride. Diabetes 1966, 15, 723–725. [Google Scholar] [CrossRef] [PubMed]
- Huttenlocher, P.R.; Wilbourn, A.J.; Signore, J.M. Medium-chain triglycerides as a therapy for intractable childhood epilepsy. Neurology 1971, 21, 1097–1103. [Google Scholar] [CrossRef]
- Veech, R.L.; Chance, B.; Kashiwaya, Y.; Lardy, H.A.; Cahill, G.F., Jr. Ketone bodies, potential therapeutic uses. IUBMB Life 2001, 51, 241–247. [Google Scholar]
- Veech, R.L. The therapeutic implications of ketone bodies: The effects of ketone bodies in pathological conditions: Ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leukot. Essent. Fatty Acids 2004, 70, 309–319. [Google Scholar] [CrossRef]
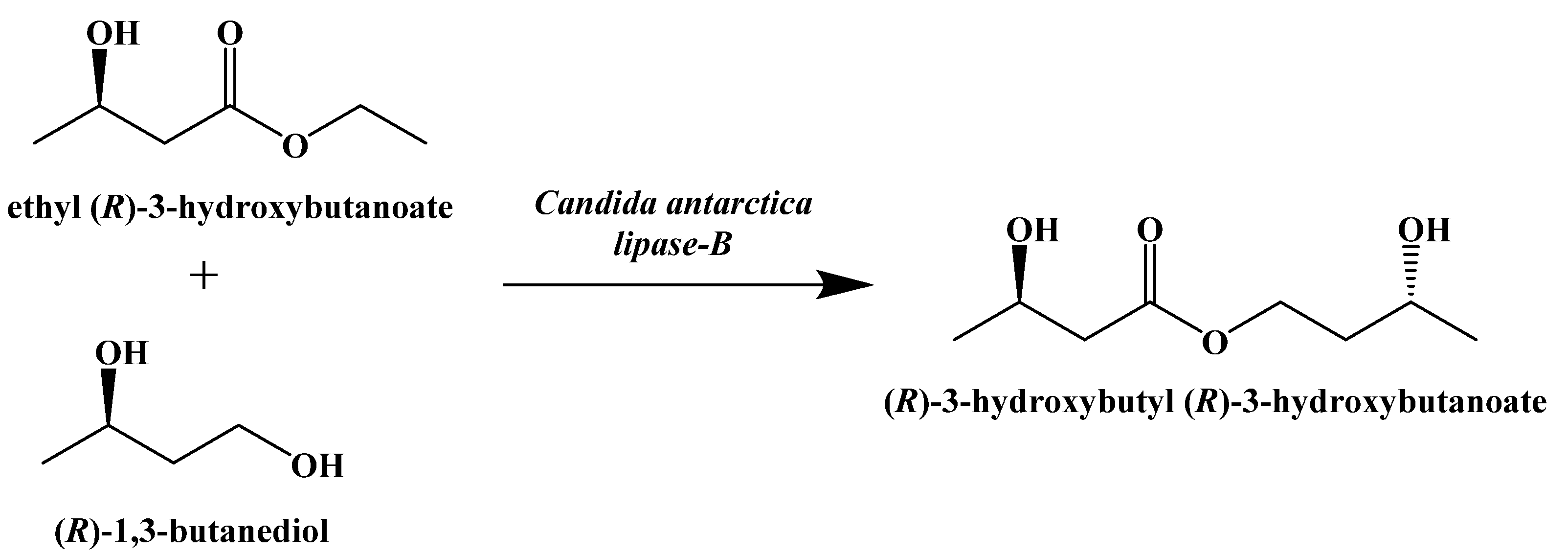
- Desrochers, S.; Quinze, K.; Dugas, H.; Dubreuil, P.; Bomont, C.; David, F.; Agarwal, K.C.; Kumar, A.; Soloviev, M.V.; Powers, L.; et al. R,S-1,3-Butanediol acetoacetate esters, potential alternates to lipid emulsions for total parenteral nutrition. J. Nutr. Biochem. 1995, 6, 111–118. [Google Scholar] [CrossRef]
- Desrochers, S.; Dubreuil, P.; Brunet, J.; Jette, M.; David, F.; Landau, B.R.; Brunengraber, H. Metabolism of (R,S)-1,3-butanediol acetoacetate esters, potential parenteral and enteral nutrients in conscious pigs. Am. J. Physiol. 1995, 268, E660–E667. [Google Scholar] [CrossRef]
- Tate, R.L.; Mehlman, M.A.; Tobin, R.B. Metabolic fate of 1,3-butanediol in the rat: Conversion of β-hydroxybutyrate. J. Nutr. 1971, 101, 1719–1726. [Google Scholar] [CrossRef] [PubMed]
- Lincoln, B.C.; Des Rosiers, C.; Brunengraber, H. Metabolism of S-3-hydroxybutyrate in the perfused rat liver. Arch. Biochem. Biophys. 1987, 259, 149–156. [Google Scholar] [CrossRef]
- Ari, C.; Murdun, C.; Koutnik, A.P.; Goldhagen, C.R.; Rogers, C.; Park, C.; Bharwani, S.; Diamond, D.M.; Kindy, M.S.; D’Agostino, D.P.; et al. Exogenous ketones lower blood glucose level in rested and exercised rodent models. Nutrients 2019, 11, 2330. [Google Scholar] [CrossRef] [PubMed]
- Clarke, K.; Veech, R.L. Preparation of (3R)-hydroxybutyl (3R)-hydroxybutyrate as a drug and food additive. PCT Int. Appl. 2010, 2010021766, 25. [Google Scholar]
- Tobin, R.B.; Garthoff, L.H.; Mehlman, M.A.; Veech, R.L. Metabolite levels, redox states, and gluconeogenic enzyme activities in livers of rats fed diets containing 1,3-butanediol. J. Environ. Pathol. Toxicol. 1978, 2, 389–398. [Google Scholar] [PubMed]
- Clarke, K.; Tchabanenko, K.; Pawlosky, R.; Carter, E.; King, M.T.; Musa-Veloso, K.; Ho, M.; Roberts, A.; Robertson, J.; VanItallie, T.B.; et al. Kinetics, safety and tolerability of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate in healthy adult subjects. Regul. Toxicol. Pharmacol. 2012, 63, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Soto-Mata, A.; Vansant, H.; Evans, R.D.; Clarke, K. Safety and tolerability of sustained exogenous ketosis using ketone monoester drinks for 28 days in healthy adults. Regul. Toxicol. Pharmacol. 2019, 109, 104506. [Google Scholar] [CrossRef]
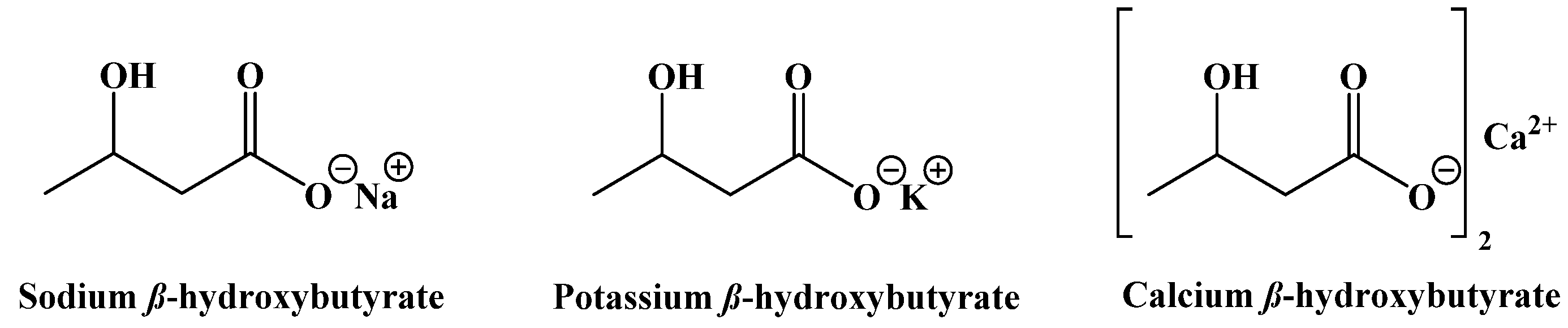
- Fischer, T.; Och, U.; Klawon, I.; Och, T.; Gruneberg, M.; Fobker, M.; Bordewick-Dell, U.; Marquardt, T. Effect of a sodium and calcium DL-β-hydroxybutyrate salt in healthy adults. Nutr. Metab. 2018, 2018, 1–8. [Google Scholar] [CrossRef]
- Kovacs, Z.; D’Agostino, D.P.; Dobolyi, A.; Ari, C. Adenosine A1 receptor antagonism abolished the anti-seizure effects of exogenous ketone supplementation in wistar albino Glaxo Rijswijk rats. Front. Mol. Neurosci. 2017, 10, 235. [Google Scholar] [CrossRef] [PubMed]


















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Williams, M.S.; Turos, E. The Chemistry of the Ketogenic Diet: Updates and Opportunities in Organic Synthesis. Int. J. Mol. Sci. 2021, 22, 5230. https://doi.org/10.3390/ijms22105230
Williams MS, Turos E. The Chemistry of the Ketogenic Diet: Updates and Opportunities in Organic Synthesis. International Journal of Molecular Sciences. 2021; 22(10):5230. https://doi.org/10.3390/ijms22105230
Chicago/Turabian StyleWilliams, Michael Scott, and Edward Turos. 2021. "The Chemistry of the Ketogenic Diet: Updates and Opportunities in Organic Synthesis" International Journal of Molecular Sciences 22, no. 10: 5230. https://doi.org/10.3390/ijms22105230
APA StyleWilliams, M. S., & Turos, E. (2021). The Chemistry of the Ketogenic Diet: Updates and Opportunities in Organic Synthesis. International Journal of Molecular Sciences, 22(10), 5230. https://doi.org/10.3390/ijms22105230