
Artificial Sweeteners Negatively Regulate Pathogenic Characteristics of Two Model Gut Bacteria, E. coli and E. faecalis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
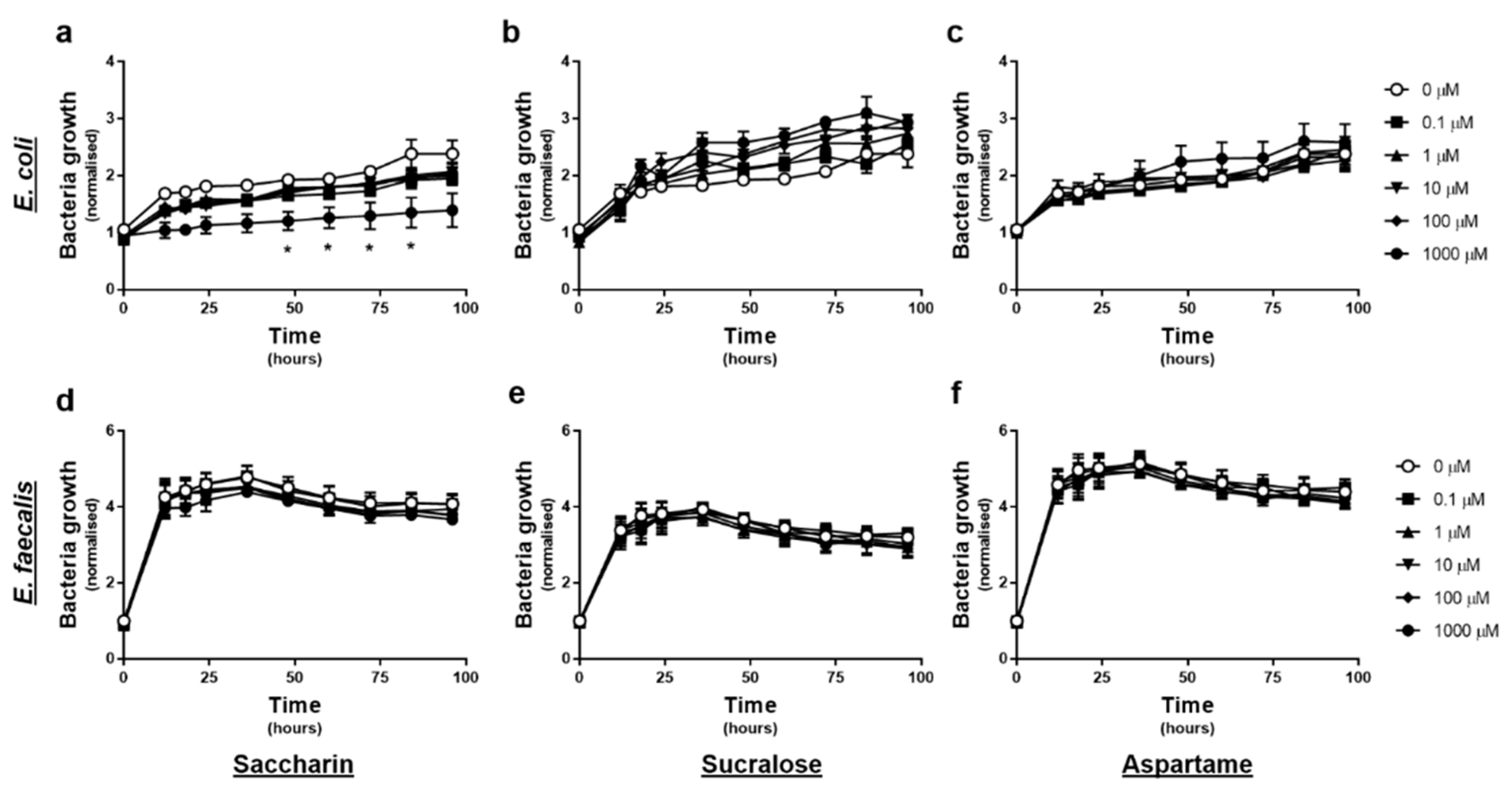
2.1. Only the Artificial Sweetener Saccharin Affects E. coli Model Gut Bacteria Growth at High Concentrations
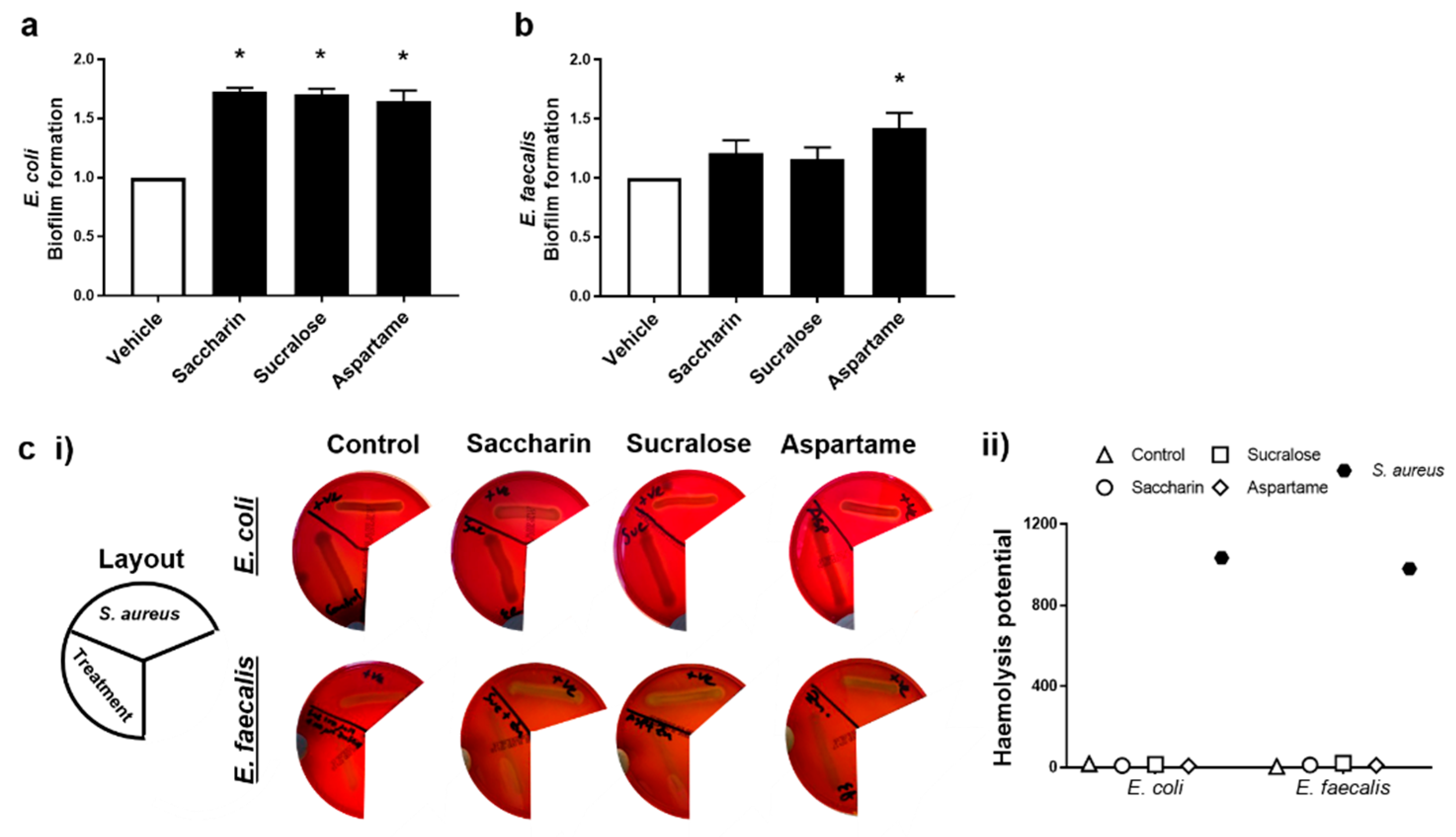
2.2. Artificial Sweeteners Differentially Increase Biofilm Formation, but Not Haemolytic Activity, in the Two Model Gut Bacteria
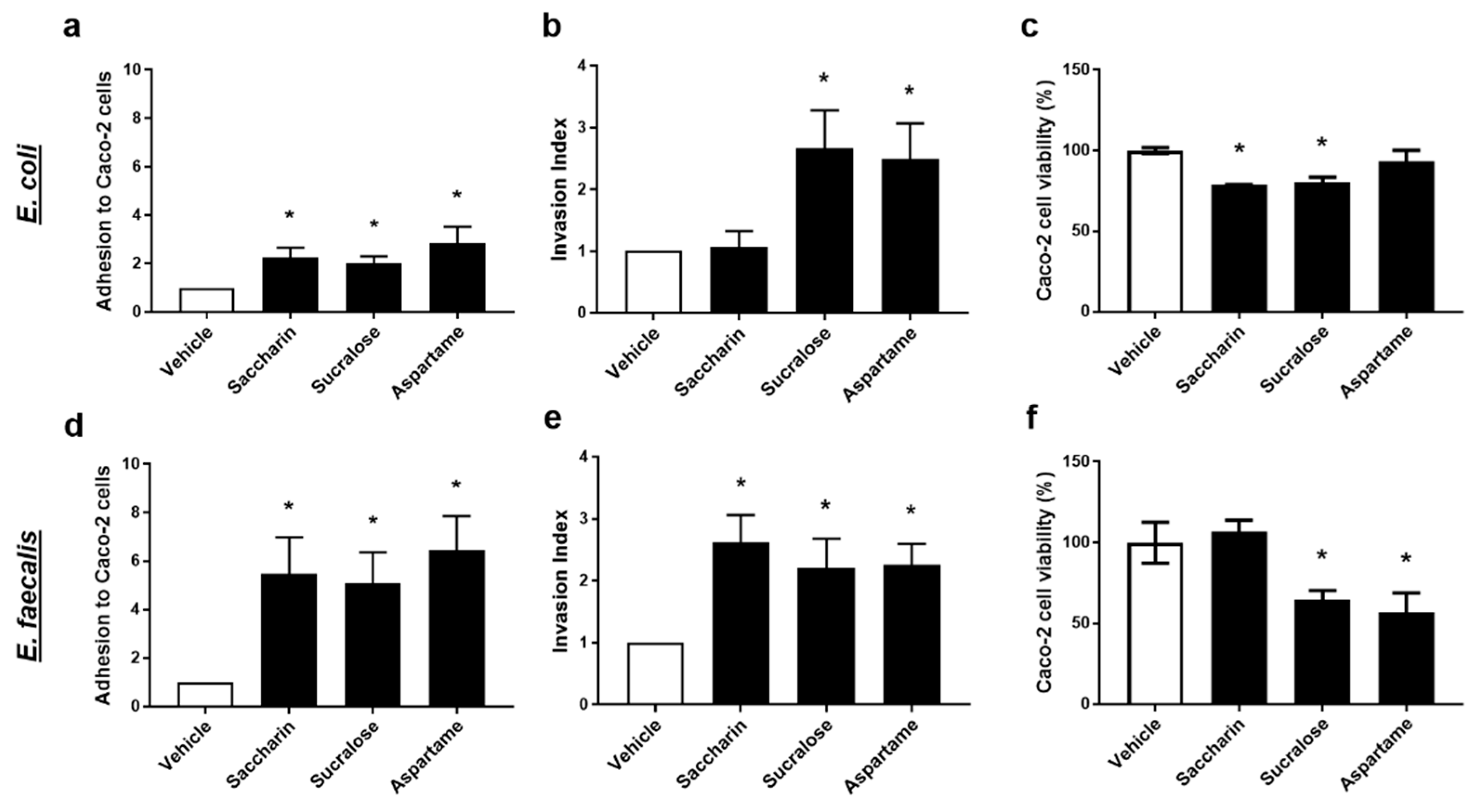
2.3. Artificial Sweeteners Significantly Disrupt the Interaction between Model Gut Bacterial and Intestinal Epithelial Cells
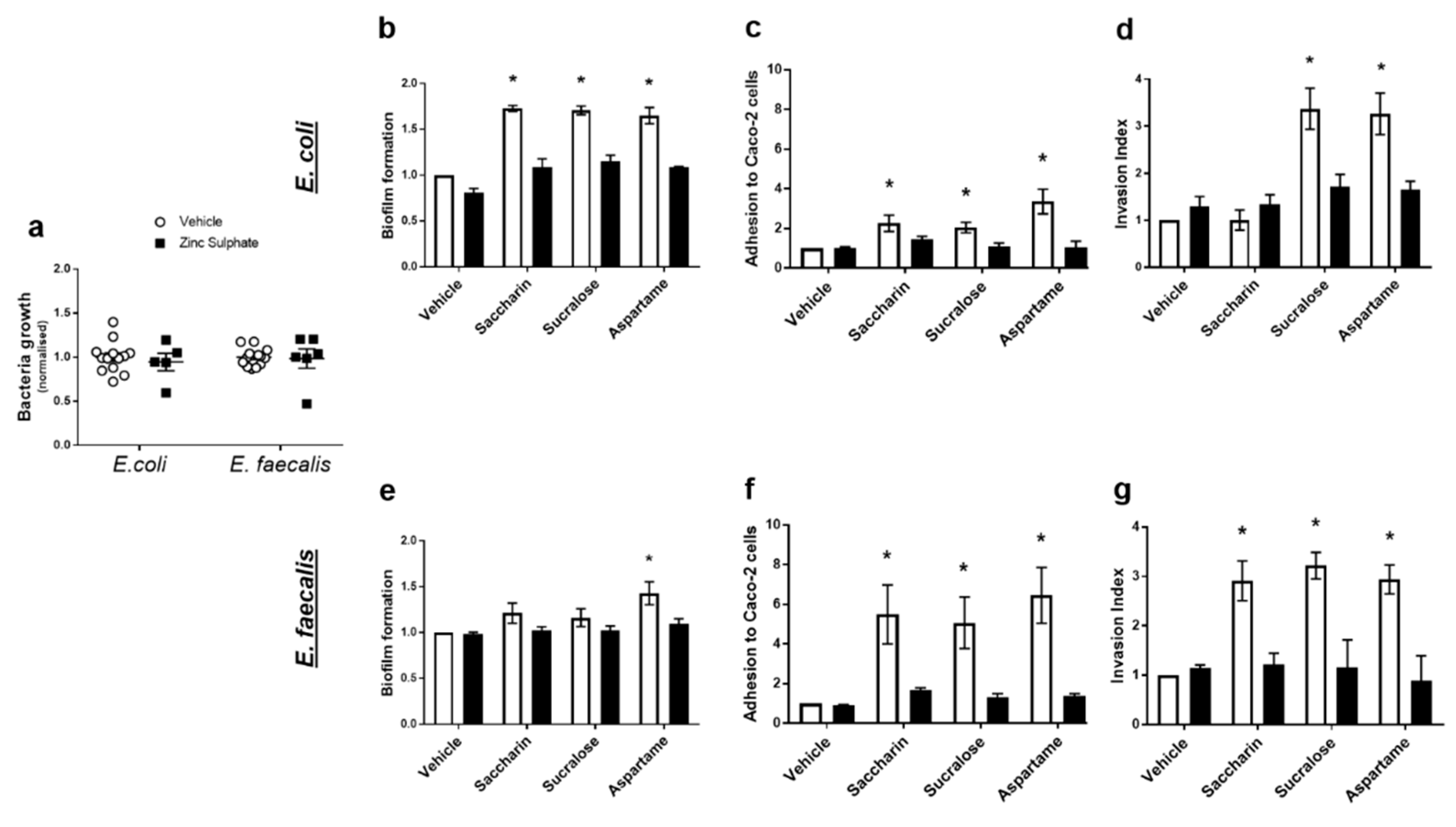
2.4. Artificial Sweeteners Impact Model Gut Bacteria through a Taste Sensing Mechanism
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Bacterial and Mammalian Cell Culture
4.3. Growth Curve Determination
4.4. Biofilm Formation Assay
4.5. Haemolysis Assay Using Blood Agar Plates
4.6. Adhesion Assay
4.7. Invasion Assay
4.8. Cytotoxicity Assay
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ley, R.E.; Peterson, D.A.; Gordon, J.I. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 2006, 124, 837–848. [Google Scholar] [CrossRef]
- Wekerle, H. Brain autoimmunity and intestinal microbiota: 100 trillion game changers. Trends Immunol. 2017, 38, 483–497. [Google Scholar] [CrossRef]
- Li, J.; Jia, H.; Cai, X.; Zhong, H.; Feng, Q.; Sunagawa, S.; Arumugam, M.; Kultima, J.R.; Prifti, E.; Nielsen, T.; et al. An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 2014, 32, 834–841. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Chassaing, B.; Koren, O.; Goodrich, J.K.; Poole, S.; Srinivasan, S.; Ley, R.E.; Gewirtz, A.T. Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature 2015, 519, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Petersson, J.; Schreiber, O.; Hansson, G.C.; Gendler, S.J.; Velcich, A.; Lundberg, J.O.; Roos, S.; Holm, L.; Philipson, M. Importance and regulation of the colonic mucus barrier in a mouse model of colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, 2. [Google Scholar] [CrossRef] [PubMed]
- Vaishnava, S.; Behrendt, C.L.; Ismail, A.S.; Eckmann, L.; Hooper, L.V. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc. Natl. Acad. Sci. USA 2008, 10552, 20858–20863. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.J.; Zheng, L.; Campbell, E.L.; Saeedi, B.; Scholz, C.C.; Bayless, A.J.; Wilson, K.E.; Glover, L.E.; Kominsky, D.J.; Magnuson, A.; et al. Crosstalk between microbiota-derived short-chain fatty acids and intestinal epithelial HIF augments tissue barrier function. Cell Host Microbe 2015, 175, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Iraporda, C.; Errea, A.; Romanin, D.E.; Cayet, D.; Pereyra, E.; Pignataro, O.; Sirard, J.C.; Garrote, G.L.; Abraham, A.G.; Rumbo, M. Lactate and short chain fatty acids produced by microbial fermentation downregulate proinflammatory responses in intestinal epithelial cells and myeloid cells. Immunobiology 2015, 22010, 1161–1169. [Google Scholar] [CrossRef]
- de la Cuesta-Zuluaga, J.; Mueller, N.T.; Alvarez-Quintero, R.; Velasquez-Meija, E.P.; Sierra, J.A.; Corrales-Agudelo, V.; Carmona, J.A.; Abad, J.M.; Escobar, J.S. Higher fecal short-chain fatty acid levels are associated with gut microbiome dysbiosis, obesity, hypertension and cardiometabolic disease risk factors. Nutrients 2018, 11, 51. [Google Scholar] [CrossRef]
- LeBlanc, J.G.; Milani, C.; de Giori, G.S.; Sesma, F.; van Sinderen, D.; Ventura, M. Bacteria as vitamin suppliers to their host: A gut microbiota perspective. Curr. Opin. Biotechnol. 2013, 24, 160–168. [Google Scholar] [CrossRef]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [PubMed]
- Suez, J.; Korem, T.; Zeevi, D.; Zilberman-Schapira, G.; Thaiss, C.A.; Maza, O.; Israeli, D.; Zmora, N.; Gilad, S.; Weinberger, A.; et al. Artificial sweeteners induce glucose intolerance by altering the gut microbiota. Nature 2014, 514, 181–186. [Google Scholar] [CrossRef]
- Gardner, C.; Wylie-Rosett, J.; Gidding, S.S.; Sen, L.M.; Johnson, R.K.; Reader, D.; Lichtenstein, A.H.; Physical Activity and Metabolism American Heart Association Nutrition Committee of the Council on Nutrition and American Diabetes Association. Nonnutritive sweeteners: Current use and health perspectives: A scientific statement from the american heart association and the american diabetes association. Diabetes Care 2012, 35, 1798–1808. [Google Scholar] [CrossRef]
- Blackburn, G.L.; Kanders, B.S.; Lavin, P.T.; Keller, S.D.; Whatley, J. The effect of aspartame as part of a multidisciplinary weight-control program on short- and long-term control of body weight. Am. J. Clin. Nutr. 1997, 65, 409–418. [Google Scholar] [CrossRef]
- Suez, J.; Korem, T.; Zilberman-Schapira, G.; Segal, E.; Elinav, E. Non-caloric artificial sweeteners and the microbiome: Findings and challenges. Gut Microbes 2015, 6, 149–155. [Google Scholar] [CrossRef]
- Frankenfeld, C.L.; Sikaroodi, M.; Lamb, E.; Shoemaker, S.; Gillevet, P.M. High-intensity sweetener consumption and gut microbiome content and predicted gene function in a cross-sectional study of adults in the united states. Ann. Epidemiol. 2015, 25, 736–742. [Google Scholar] [CrossRef]
- Bian, X.; Chi, L.; Gao, B.; Tu, P.; Ru, H.; Lu, K. Gut microbiome response to sucralose and its potential role in inducing liver inflammation in mice. Front. Physiol. 2017, 8, 487. [Google Scholar] [CrossRef]
- Bian, X.; Tu, P.; Chi, L.; Gao, B.; Ru, H.; Lu, K. Saccharin induced liver inflammation in mice by altering the gut microbiota and its metabolic functions. Food Chem. Toxicol. 2017, 107, 530–539. [Google Scholar] [CrossRef] [PubMed]
- Shil, A.; Olusanya, O.; Ghufoor, Z.; Forson, B.; Marks, J.; Chichger, H. Artificial sweeteners disrupt tight junctions and barrier function in the intestinal epithelium through activation of the sweet taste receptor, T1R3. Nutrients 2020, 12, 1862. [Google Scholar] [CrossRef]
- Mohamed, J.A.; Huang, D.B. Biofilm formation by enterococci. J. Med. Microbiol. 2007, 56, 1581–1588. [Google Scholar] [CrossRef] [PubMed]
- Tenaillon, O.; Skurnik, D.; Picard, B.; Denamur, E. The population genetics of commensal Escherichia coli. Nat. Rev. Microbiol. 2010, 8, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, E.; Marin, M.L.; Martin, R.; Odriozola, J.M.; Olivares, M.; Xaus, J.; Fernandez, L.; Rodriguez, J.M. Is meconium from healthy newborns actually sterile? Res. Microbiol. 2008, 159, 187–193. [Google Scholar] [CrossRef]
- Mace, O.J.; Affleck, J.; Patel, N.; Kellett, G.L. Sweet taste receptors in rat small intestine stimulate glucose absorption through apical GLUT2. J. Physiol. 2007, 582, 379–392. [Google Scholar] [CrossRef]
- Keast, R.S.; Canty, T.M.; Breslin, P.A. Oral zinc sulfate solutions inhibit sweet taste perception. Chem. Senses 2004, 29, 513–521. [Google Scholar] [CrossRef]
- Palmnas, M.S.; Cowan, T.E.; Bomhof, M.R.; Su, J.; Reimer, R.A.; Vogel, H.J.; Hittel, D.S.; Shearer, J. Low-dose aspartame consumption differentially affects gut microbiota-host metabolic interactions in the diet-induced obese rat. PLoS ONE 2014, 9, e109841. [Google Scholar] [CrossRef]
- Wang, Q.P.; Browman, D.; Herzog, H.; Neely, G.G. Non-nutritive sweeteners possess a bacteriostatic effect and alter gut microbiota in mice. PLoS ONE 2018, 13, e0199080. [Google Scholar] [CrossRef]
- Ceri, H.; Olson, M.; Morck, D.; Storey, D.; Read, R.; Olson, B. The MBEC assay system: Multiple equivalent biofilms for antibiotic and biocide susceptibility testing. Methods Enzymol. 2001, 337, 377–385. [Google Scholar] [CrossRef]
- Kong, K.F.; Vuong, C.; Otto, M. Staphylococcus quorum sensing in biofilm formation and infection. Int. J. Med. Microbiol. 2006, 296, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Wagner, V.E.; Li, L.L.; Isabella, V.M.; Iglewski, B.H. Analysis of the hierarchy of quorum-sensing regulation in Pseudomonas aeruginosa. Anal. Bioanal. Chem. 2007, 387, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Hyland, R.M.; Sun, J.; Griener, T.P.; Mulvey, G.L.; Klassen, J.S.; Donnerberg, M.S.; Armstrong, G.D. The bundlin pilin protein of enteropathogenic Escherichia coli is an N-acetyllactosamine-specific lectin. Cell Microbiol. 2008, 10, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Oscarsson, J.; Mizunoe, Y.; Li, L.; Lai, X.H.; Wieslander, A.; Uhlin, B.E. Molecular analysis of the cytolytic protein ClyA (SheA) from Escherichia coli. Mol. Microbiol. 1999, 32, 1226–1238. [Google Scholar] [CrossRef] [PubMed]
- Saldana, Z.; Erdem, A.L.; Schuller, S.; Okeke, I.N.; Lucas, M.; Sivananthan, A.; Philips, A.D.; Kaper, J.B.; Puente, J.L.; Giron, J.A. The Escherichia coli common pilus and the bundle-forming pilus act in concert during the formation of localized adherence by enteropathogenic E. coli. J. Bacteriol. 2009, 191, 3451–3461. [Google Scholar] [CrossRef]
- Huycke, M.M.; Joyce, W.A.; Gilmore, M.S. Enterococcus faecalis cytolysin without effect on the intestinal growth of susceptible enterococci in mice. J. Infect. Dis. 1995, 172, 273–276. [Google Scholar] [CrossRef]
- Theilacker, C.; Sanchez-Carballo, P.; Toma, I.; Fabretti, F.; Sava, I.; Kropec, A.; Holst, O.; Huebner, J. Glycolipids are involved in biofilm accumulation and prolonged bacteraemia in Enterococcus faecalis. Mol. Microbiol. 2009, 71, 1055–1069. [Google Scholar] [CrossRef]
- Wells, C.L.; Moore, E.A.; Hoag, J.A.; Hirt, H.; Dunny, G.M.; Erlandsen, S.L. Inducible expression of Enterococcus faecalis aggregation substance surface protein facilitates bacterial internalization by cultured enterocytes. Infect. Immun. 2000, 68, 7190–7194. [Google Scholar] [CrossRef]
- Sartingen, S.; Rozdzinski, E.; Muscholl-Silberhorn, A.; Marre, R. Aggregation substance increases adherence and internalization, but not translocation, of Enterococcus faecalis through different intestinal epithelial cells in vitro. Infect. Immun. 2000, 68, 6044–6047. [Google Scholar] [CrossRef]
- Ludwig, A.; Bauer, S.; Benz, R.; Bergmann, B.; Goebel, W. Analysis of the SlyA-controlled expression, subcellular localization and pore-forming activity of a 34 kDa haemolysin (ClyA) from Escherichia coli K-12. Mol. Microbiol. 1999, 31, 557–567. [Google Scholar] [CrossRef]
- Sheikh, J.; Hicks, S.; Dall’Agnol, M.; Phillips, A.D.; Nataro, J.P. Roles for fis and YafK in biofilm formation by enteroaggregative Escherichia coli. Mol. Microbiol. 2001, 41, 983–997. [Google Scholar] [CrossRef]
- Heikens, E.; Bonten, M.J.; Willems, R.J. Enterococcal surface protein esp is important for biofilm formation of Enterococcus faecium E1162. J. Bacteriol. 2007, 189, 8233–8240. [Google Scholar] [CrossRef]
- Srivastava, M.; Mallard, C.; Barke, T.; Hancock, L.E.; Self, W.T. A selenium-dependent xanthine dehydrogenase triggers biofilm proliferation in Enterococcus faecalis through oxidant production. J. Bacteriol. 2011, 193, 1643–1652. [Google Scholar] [CrossRef] [PubMed]
- Shankar, N.; Baghdayan, A.S.; Gilmore, M.S. Modulation of virulence within a pathogenicity island in vancomycin-resistant Enterococcus faecalis. Nature 2002, 417, 746–750. [Google Scholar] [CrossRef]
- Giaffer, M.H.; Holdsworth, C.D.; Duerden, B.I. Virulence properties of Escherichia coli strains isolated from patients with inflammatory bowel disease. Gut 1992, 33, 646–650. [Google Scholar] [CrossRef]
- Ike, Y.; Hashimoto, H.; Clewell, D.B. High incidence of hemolysin production by Enterococcus (streptococcus) faecalis strains associated with human parenteral infections. J. Clin. Microbiol. 1987, 25, 1524–1528. [Google Scholar] [CrossRef] [PubMed]
- Mirsepasi-Lauridsen, H.C.; Du, Z.; Struve, C.; Charbon, G.; Karczewski, J.; Krogfelt, K.A.; Petersen, A.M.; Wells, J.M. Secretion of alpha-hemolysin by Escherichia coli disrupts tight junctions in ulcerative colitis patients. Clin. Transl. Gastroenterol. 2016, 7, e149. [Google Scholar] [CrossRef] [PubMed]
- Nelson, G.; Hoon, M.A.; Chandrashekar, J.; Zhang, Y.; Ryba, N.J.; Zuker, C.S. Mammalian sweet taste receptors. Cell 2001, 106, 381–390. [Google Scholar] [CrossRef]
- Wee, M.; Tan, V.; Forde, C. A comparison of psychophysical dose-response behaviour across 16 sweeteners. Nutrients 2018, 10, 1632. [Google Scholar] [CrossRef] [PubMed]
- Nijland, R.; Burgess, J.G. Bacterial olfaction. Biotechnol. J. 2010, 5, 974–977. [Google Scholar] [CrossRef]
- Bandyopadhyay, A.; Ghoshal, S.; Mukherjee, A. Genotoxicity testing of low-calorie sweeteners: Aspartame, acesulfame-K., and saccharin. Drug Chem. Toxicol. 2008, 31, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Li, R.; Jiang, M.; Wang, X. Sucralose increases antimicrobial resistance and stimulates recovery of Escherichia coli mutants. Curr. Microbiol. 2017, 74, 885–888. [Google Scholar] [CrossRef] [PubMed]
- Rencuzogullari, E.; Tuylu, B.A.; Topaktas, M.; Ila, H.B.; Kayraldiz, A.; Arslan, M.; Diler, S.B. Genotoxicity of aspartame. Drug Chem. Toxicol. 2004, 27, 257–268. [Google Scholar] [CrossRef]
- Yu, Z.; Wang, Y.; Lu, J.; Bond, P.L.; Guo, J. Nonnutritive sweeteners can promote the dissemination of antibiotic resistance through conjugative gene transfer. ISME J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Frey, R.L.; He, L.; Cui, Y.; Decho, A.W.; Kawaguchi, T.; Fergusonm, P.L.; Ferry, J.L. Reaction of N-acylhomoserine lactones with hydroxyl radicals: Rates, products, and effects on signaling activity. Environ. Sci. Technol. 2010, 44, 7465–7469. [Google Scholar] [CrossRef]
- Rothfork, J.M.; Timmins, G.S.; Harris, M.N.; Chen, X.; Lusis, A.J.; Otto, M.; Cheung, A.L.; Gresham, D. Inactivation of a bacterial virulence pheromone by phagocyte-derived oxidants: New role for the NADPH oxidase in host defense. Proc. Natl. Acad. Sci. USA 2004, 101, 13867–13872. [Google Scholar] [CrossRef]
- Ghoshal, U.C.; Ghoshal, U.; Jain, M.; Kumar, A.; Aggarwal, R.; Misra, A.; Ayyagari, A.; Naik, S.R. Strongyloides stercoralis infestation associated with septicemia due to intestinal transmural migration of bacteria. J. Gastroenterol. Hepatol. 2002, 17, 1331–1333. [Google Scholar] [CrossRef]
- Wells, C.L.; Jechorek, R.P.; Erlandsen, S.L. Evidence for the translocation of Enterococcus faecalis across the mouse intestinal tract. J. Infect. Dis. 1990, 162, 82–90. [Google Scholar] [CrossRef]
- Wells, C.L.; Erlandsen, S.L. Localization of translocating Escherichia coli, proteus mirabilis, and Enterococcus faecalis within cecal and colonic tissues of monoassociated mice. Infect. Immun. 1991, 59, 4693–4697. [Google Scholar] [CrossRef]
- Santos, P.S.; Caria, C.R.P.; Gotardo, E.M.F.; Ribeiro, M.L.; Pedrazzoli, J.; Gambero, A. Artificial sweetener saccharin disrupts intestinal epithelial cells’ barrier function in vitro. Food Funct. 2018, 9, 3815–3822. [Google Scholar] [CrossRef]
- Yang, Y.Y.; Liu, W.R.; Liu, Y.S.; Zhao, J.L.; Zhang, Q.Q.; Zhang, M.; Zhang, J.N.; Jiang, Y.X.; Zhang, L.J.; Ying, G.G. Suitability of pharmaceuticals and personal care products (PPCPs) and artificial sweeteners (ASs) as wastewater indicators in the pearl river delta, south china. Sci. Total Environ. 2017, 590–591, 611–619. [Google Scholar] [CrossRef]
- Tran, N.H.; Hu, J.; Li, J.; Ong, S.L. Suitability of artificial sweeteners as indicators of raw wastewater contamination in surface water and groundwater. Water Res. 2014, 48, 443–456. [Google Scholar] [CrossRef]
- Tran, N.H.; Gan, J.; Nguyen, V.T.; Chen, H.; You, L.; Duarah, A.; Zhang, L.; Gin, K.Y. Sorption and biodegradation of artificial sweeteners in activated sludge processes. Bioresour. Technol. 2015, 197, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Hall-Stoodley, L.; Costerton, J.W.; Stoodley, P. Bacterial biofilms: From the natural environment to infectious diseases. Nat. Rev. Microbiol. 2004, 2, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Lleo, M.; Bonato, B.; Tafi, M.C.; Caburlotto, G.; Benedetti, D.; Canepari, P. Adhesion to medical device materials and biofilm formation capability of some species of enterococci in different physiological states. FEMS Microbiol. Lett. 2007, 274, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Mack, D.; Rohde, H.; Harris, L.G.; Davies, A.P.; Horstkotte, M.A.; Knobloch, J.K. Biofilm formation in medical device-related infection. Int. J. Artif. Organs. 2006, 29, 343–359. [Google Scholar] [CrossRef] [PubMed]
- Braxton, E.E.; Ehrlich, G.D.; Hall-Stoodley, L.; Stoodley, P.; Veeh, R.; Fux, C.; Hu, F.Z.; Quigley, M.; Post, J.C. Role of biofilms in neurosurgical device-related infections. Neurosurg. Rev. 2005, 28, 249–255. [Google Scholar] [CrossRef]
- Burton, E.; Yakandawala, N.; LoVetri, K.; Madhyastha, M.S. A microplate spectrofluorometric assay for bacterial biofilms. J. Ind. Microbiol. Biotechnol. 2007, 34, 1–4. [Google Scholar] [CrossRef]
- Wiseman, G.M. The hemolysins of Staphylococcus aureus. Bacteriol. Rev. 1975, 39, 317–344. [Google Scholar] [CrossRef] [PubMed]
- Darfeuille-Michaud, A.; Aubel, D.; Chauviere, G.; Rich, C.; Bourges, M.; Servin, A.; Joly, B. Adhesion of enterotoxigenic Escherichia coli to the human colon carcinoma cell line caco-2 in culture. Infect. Immun. 1990, 58, 893–902. [Google Scholar] [CrossRef]
- Inaba, H.; Nomura, R.; Kato, Y.; Takeuchi, H.; Amano, A.; Asai, F.; Nakano, K.; Lamont, R.J.; Matsumoto-Nakano, M. Adhesion and invasion of gingival epithelial cells by Porphyromonas gulae. PLoS ONE 2019, 14, e0213309. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, P.M.; Furumura, M.T.; Santos, A.M.; Sousa, A.C.; Kota, D.J.; Levy, C.E.; Yano, T. Cytotoxic activity of clinical Stenotrophomonas maltophilia. Lett. Appl. Microbiol. 2006, 43, 443–449. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shil, A.; Chichger, H. Artificial Sweeteners Negatively Regulate Pathogenic Characteristics of Two Model Gut Bacteria, E. coli and E. faecalis. Int. J. Mol. Sci. 2021, 22, 5228. https://doi.org/10.3390/ijms22105228
Shil A, Chichger H. Artificial Sweeteners Negatively Regulate Pathogenic Characteristics of Two Model Gut Bacteria, E. coli and E. faecalis. International Journal of Molecular Sciences. 2021; 22(10):5228. https://doi.org/10.3390/ijms22105228
Chicago/Turabian StyleShil, Aparna, and Havovi Chichger. 2021. "Artificial Sweeteners Negatively Regulate Pathogenic Characteristics of Two Model Gut Bacteria, E. coli and E. faecalis" International Journal of Molecular Sciences 22, no. 10: 5228. https://doi.org/10.3390/ijms22105228
APA StyleShil, A., & Chichger, H. (2021). Artificial Sweeteners Negatively Regulate Pathogenic Characteristics of Two Model Gut Bacteria, E. coli and E. faecalis. International Journal of Molecular Sciences, 22(10), 5228. https://doi.org/10.3390/ijms22105228