
A Novel Relative High-Density Lipoprotein Index to Predict the Structural Changes in High-Density Lipoprotein and Its Ability to Inhibit Endothelial–Mesenchymal Transition

 ,
, 
 ,
,
Abstract
1. Introduction
2. Results
2.1. TGF-β1 Induces Morphological Changes and EndoMT in EPCs That May Be Inhibited by HDL from Patients with rHDL Index Value Lower Than 2
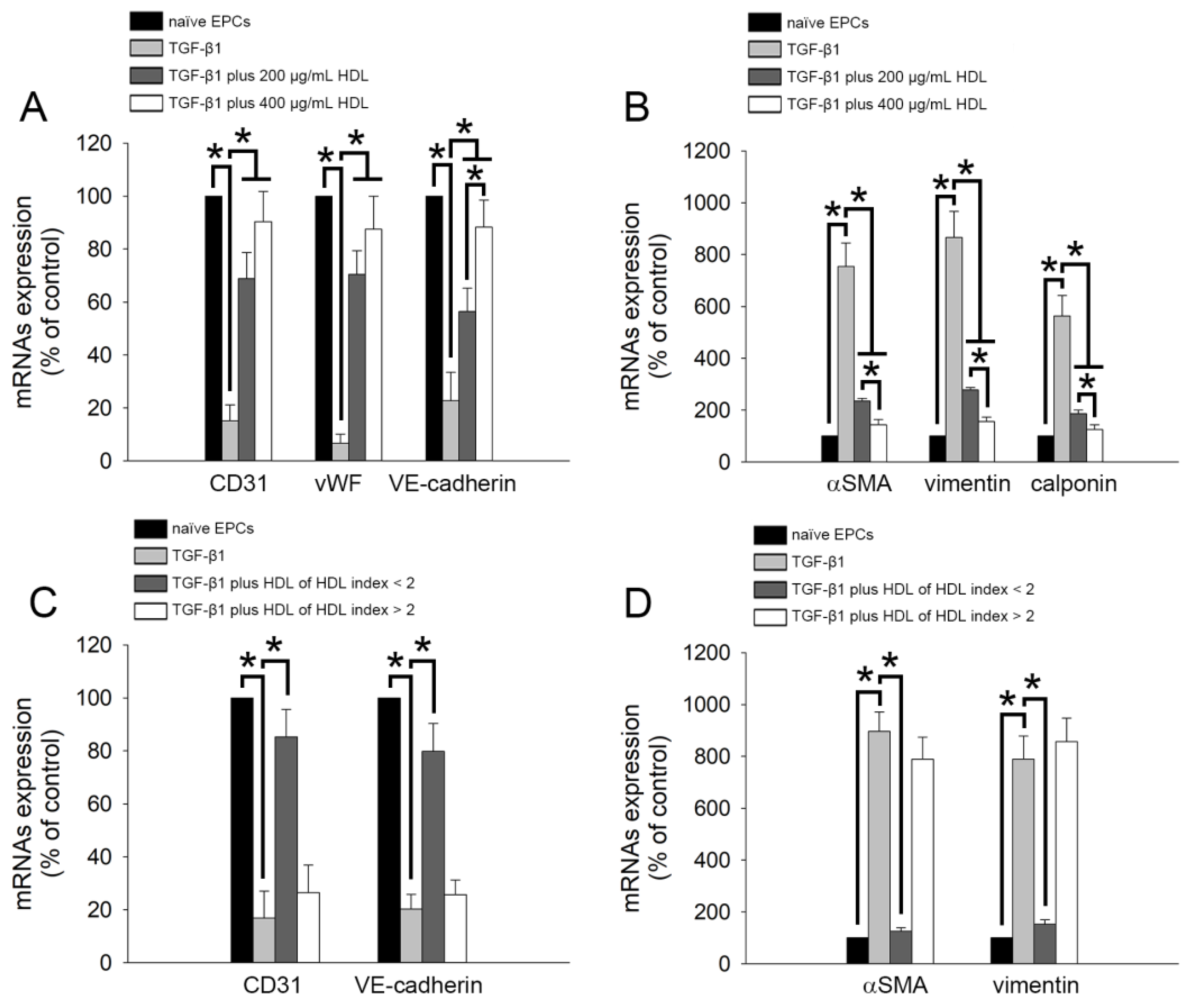
2.2. TGF-β1-Induced Expression of EndoMT-Related Genes Was Regulated by HDL from Healthy Subjects and Patients with Lower rHDL Index
2.3. TGF-β1-Induced EndoMT Was Regulated by HDL from Healthy Subjects and Patients with Lower rHDL Index
2.4. HDL Regulates TGF-β1-Induced EndoMT via Smad- and Snail-Dependent Pathways
2.5. The Clinical Characteristics Did Not Differ between Patients with rHDL Index> and <2.
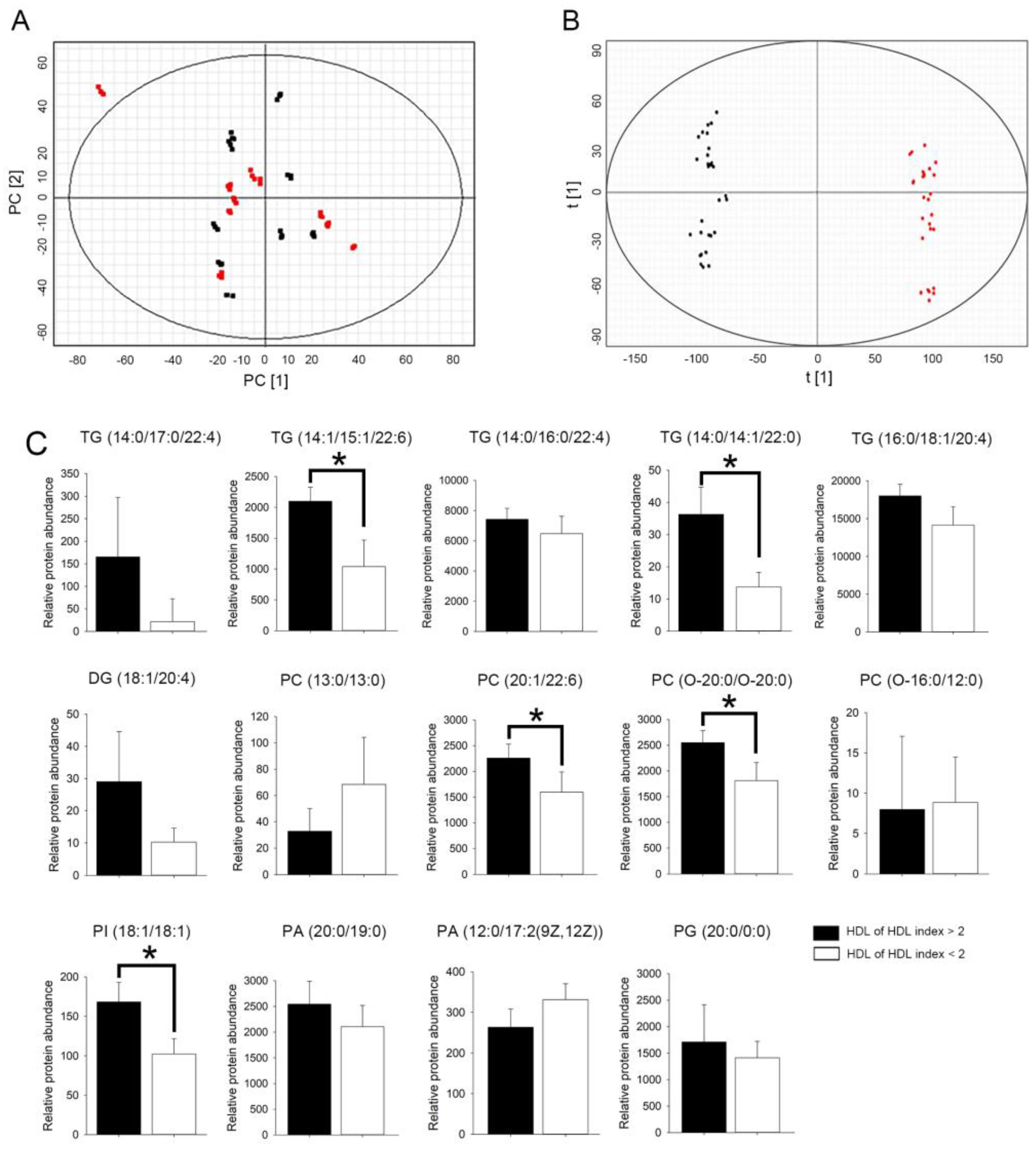
2.6. HDL from Patients with rHDL Index Value > 2 Had Higher Relative Abundance of Glycerolipid in Select Triglyceride (TG), Phosphatidylcholine (PC), and Phosphatidylinositol (PI) Species
3. Discussion
4. Materials and Methods
4.1. Ethics and Patient/Volunteer Demographics
4.2. Preparation of HDL
4.3. EPC Isolation and Cultivation
4.4. Immunofluorescence Staining
4.5. Real-Time Quantitative Polymerase Chain Reactions
4.6. Western Blot Analysis
4.7. DCF Assay for Identification of rHDL Index Value
4.8. Lipidomic Analysis
4.9. Lipid Identification
4.10. Statistics Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Expert Panel on Detection E. Treatment of high blood cholesterol in a. executive summary of the third report of the national cholesterol education program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III). JAMA 2001, 285, 2486–2497. [Google Scholar]
- Zou, T.B.; Zhu, S.S.; Luo, F.; Li, W.Q.; Sun, X.R.; Wu, H.F. Effects of Astaxanthin on Reverse Cholesterol Transport and Atherosclerosis in Mice. Biomed. Res. Int. 2017, 2017, 4625932. [Google Scholar] [CrossRef]
- Vickers, K.C.; Landstreet, S.R.; Levin, M.G.; Shoucri, B.M.; Toth, C.L.; Taylor, R.C.; Palmisano, B.T.; Tabet, F.; Cui, H.L.; Rye, K.A.; et al. MicroRNA-223 coordinates cholesterol homeostasis. Proc. Natl. Acad. Sci. USA 2014, 111, 14518–14523. [Google Scholar] [CrossRef] [PubMed]
- Besler, C.; Luscher, T.F.; Landmesser, U. Molecular mechanisms of vascular effects of High-density lipoprotein: Alterations in cardiovascular disease. EMBO Mol. Med. 2012, 4, 251–268. [Google Scholar] [CrossRef]
- Fogelman, A.M. Further evidence that high-density lipoprotein is a chameleon-like lipoprotein. Eur. Heart J. 2015, 36, 3017–3019. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Barrett, T.J.; Distel, E.; Murphy, A.J.; Hu, J.; Garshick, M.S.; Ogando, Y.; Liu, J.; Vaisar, T.; Heinecke, J.W.; Berger, J.S.; et al. Apolipoprotein AI) Promotes Atherosclerosis Regression in Diabetic Mice by Suppressing Myelopoiesis and Plaque Inflammation. Circulation 2019, 140, 1170–1184. [Google Scholar] [CrossRef]
- van der Steeg, W.A.; Holme, I.; Boekholdt, S.M.; Larsen, M.L.; Lindahl, C.; Stroes, E.S.; Tikkanen, M.J.; Wareham, N.J.; Faergeman, O.; Olsson, A.G.; et al. High-density lipoprotein cholesterol, high-density lipoprotein particle size, and apolipoprotein A-I: Significance for cardiovascular risk: Yhe IDEAL and EPIC-Norfolk studies. J. Am. Coll. Cardiol. 2008, 51, 634–642. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Lundman, P.; Harmer, J.A.; Cutri, B.; Griffiths, K.A.; Rye, K.A.; Barter, P.J.; Celermajer, D.S. Consumption of saturated fat impairs the anti-inflammatory properties of high-density lipoproteins and endothelial function. J. Am. Coll. Cardiol. 2006, 48, 715–720. [Google Scholar] [CrossRef]
- Kosmas, C.E.; Martinez, I.; Sourlas, A.; Bouza, K.V.; Campos, F.N.; Torres, V.; Montan, P.D.; Guzman, E. High-density lipoprotein (HDL) functionality and its relevance to atherosclerotic cardiovascular disease. Drugs Context 2018, 7, 212525. [Google Scholar] [CrossRef]
- Qi, Y.; Fan, J.; Liu, J.; Wang, W.; Wang, M.; Sun, J.; Liu, J.; Xie, W.; Zhao, F.; Li, Y.; et al. Cholesterol-overloaded HDL particles are independently associated with progression of carotid atherosclerosis in a cardiovascular disease-free population: A community-based cohort study. J. Am. Coll. Cardiol. 2015, 65, 355–363. [Google Scholar] [CrossRef]
- Madsen, C.M.; Varbo, A.; Nordestgaard, B.G. Extreme high high-density lipoprotein cholesterol is paradoxically associated with high mortality in men and women: Two prospective cohort studies. Eur. Heart J. 2017, 38, 2478–2486. [Google Scholar] [CrossRef] [PubMed]
- Hirata, A.; Sugiyama, D.; Watanabe, M.; Tamakoshi, A.; Iso, H.; Kotani, K.; Kiyama, M.; Yamada, M.; Ishikawa, S.; Murakami, Y.; et al. Association of extremely high levels of high-density lipoprotein cholesterol with cardiovascular mortality in a pooled analysis of 9 cohort studies including 43,407 individuals: The EPOCH-JAPAN study. J. Clin. Lipidol. 2018, 12, 674–684.e5. [Google Scholar] [CrossRef]
- de Carvalho, J.F.; Bonfa, E.; Borba, E.F. Systemic lupus erythematosus and “lupus dyslipoproteinemia”. Autoimmun. Rev. 2008, 7, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.K.; Vivekanandan-Giri, A.; Tang, C.; Knight, J.S.; Mathew, A.; Padilla, R.L.; Gillespie, B.W.; Carmona-Rivera, C.; Liu, X.; Subramanian, V.; et al. Neutrophil extracellular trap-derived enzymes oxidize high-density lipoprotein: An additional proatherogenic mechanism in systemic lupus erythematosus. Arthritis Rheumatol. 2014, 66, 2532–2544. [Google Scholar] [CrossRef] [PubMed]
- Mu, C.Q.; Wang, S.Q.; Liu, Y.; Li, C.L.; Hu, X.S.; Hua, H. Development of a facescan 3D facial reconstruction technology method for quantitative evaluation of cheilitisgranulomatosa. Sci. Rep. 2017, 7, 1295. [Google Scholar] [CrossRef] [PubMed]
- Besler, C.; Heinrich, K.; Rohrer, L.; Doerries, C.; Riwanto, M.; Shih, D.M.; Chroni, A.; Yonekawa, K.; Stein, S.; Schaefer, N.; et al. Mechanisms underlying adverse effects of HDL on eNOS-activating pathways in patients with coronary artery disease. J. Clin. Investig. 2011, 121, 2693–2708. [Google Scholar] [CrossRef] [PubMed]
- Kratzer, A.; Giral, H.; Landmesser, U. High-density lipoproteins as modulators of endothelial cell functions: Alterations in patients with coronary artery disease. Cardiovasc. Res. 2014, 103, 350–361. [Google Scholar] [CrossRef]
- Shih, C.M.; Lin, F.Y.; Yeh, J.S.; Lin, Y.W.; Loh, S.H.; Tsao, N.W.; Nakagami, H.; Morishita, R.; Sawamura, T.; Li, C.Y.; et al. Dysfunctional high density lipoprotein failed to rescue the function of oxidized low density lipoprotein-treated endothelial progenitor cells: A novel index for the prediction of HDL functionality. Transl. Res. 2019, 205, 17–32. [Google Scholar] [CrossRef]
- Ribatti, D. The discovery of endothelial progenitor cells.An historical review. Leuk. Res. 2007, 31, 439–444. [Google Scholar] [CrossRef]
- Thum, T.; Fraccarollo, D.; Thum, S.; Schultheiss, M.; Daiber, A.; Wenzel, P.; Munzel, T.; Ertl, G.; Bauersachs, J. Differential effects of organic nitrates on endothelial progenitor cells are determined by oxidative stress. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 748–754. [Google Scholar] [CrossRef]
- Ingram, D.A.; Krier, T.R.; Mead, L.E.; McGuire, C.; Prater, D.N.; Bhavsar, J.; Saadatzadeh, M.R.; Bijangi-Vishehsaraei, K.; Li, F.; Yoder, M.C.; et al. Clonogenic endothelial progenitor cells are sensitive to oxidative stress. Stem Cells 2007, 25, 297–304. [Google Scholar] [CrossRef]
- Chen, Y.H.; Lin, S.J.; Lin, F.Y.; Wu, T.C.; Tsao, C.R.; Huang, P.H.; Liu, P.L.; Chen, Y.L.; Chen, J.W. High glucose impairs early and late endothelial progenitor cells by modifying nitric oxide-related but not oxidative stress-mediated mechanisms. Diabetes 2007, 56, 1559–1568. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniam, V.; Mervis, C.F.; Maxey, A.M.; Markham, N.E.; Abman, S.H. Hyperoxia reduces bone marrow, circulating, and lung endothelial progenitor cells in the developing lung: Implications for the pathogenesis of bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, L1073–L1084. [Google Scholar] [CrossRef] [PubMed]
- Gulati, R.; Jevremovic, D.; Peterson, T.E.; Chatterjee, S.; Shah, V.; Vile, R.G.; Simari, R.D. Diverse origin and function of cells with endothelial phenotype obtained from adult human blood. Circ. Res. 2003, 93, 1023–1025. [Google Scholar] [CrossRef]
- Rao, R.M.; Yang, L.; Garcia-Cardena, G.; Luscinskas, F.W. Endothelial-dependent mechanisms of leukocyte recruitment to the vascular wall. Circ. Res. 2007, 101, 234–247. [Google Scholar] [CrossRef] [PubMed]
- Frid, M.G.; Kale, V.A.; Stenmark, K.R. Mature vascular endothelium can give rise to smooth muscle cells via endothelial-mesenchymal transdifferentiation: In vitro analysis. Circ. Res. 2002, 90, 1189–1196. [Google Scholar] [CrossRef]
- Saiura, A.; Sata, M.; Hirata, Y.; Nagai, R.; Makuuchi, M. Circulating smooth muscle progenitor cells contribute to atherosclerosis. Nat. Med. 2001, 7, 382–383. [Google Scholar] [CrossRef] [PubMed]
- Sata, M. Circulating vascular progenitor cells contribute to vascular repair, remodeling, and lesion formation. Trends Cardiovasc. Med. 2003, 13, 249–253. [Google Scholar] [CrossRef]
- Diez, M.; Barbera, J.A.; Ferrer, E.; Fernandez-Lloris, R.; Pizarro, S.; Roca, J.; Peinado, V.I. Plasticity of CD133+ cells: Role in pulmonary vascular remodeling. Cardiovasc. Res. 2007, 76, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Moonen, J.R.; Krenning, G.; Brinker, M.G.; Koerts, J.A.; van Luyn, M.J.; Harmsen, M.C. Endothelial progenitor cells give rise to pro-angiogenic smooth muscle-like progeny. Cardiovasc. Res. 2010, 86, 506–515. [Google Scholar] [CrossRef]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Kitao, A.; Sato, Y.; Sawada-Kitamura, S.; Harada, K.; Sasaki, M.; Morikawa, H.; Shiomi, S.; Honda, M.; Matsui, O.; Nakanuma, Y. Endothelial to mesenchymal transition via transforming growth factor-beta1/Smad activation is associated with portal venous stenosis in idiopathic portal hypertension. Am. J. Pathol. 2009, 175, 616–626. [Google Scholar] [CrossRef]
- Martinez-Estrada, O.M.; Lettice, L.A.; Essafi, A.; Guadix, J.A.; Slight, J.; Velecela, V.; Hall, E.; Reichmann, J.; Devenney, P.S.; Hohenstein, P.; et al. Wt1 is required for cardiovascular progenitor cell formation through transcriptional control of Snail and E-cadherin. Nat. Genet. 2010, 42, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Maleszewska, M.; Moonen, J.R.; Huijkman, N.; van de Sluis, B.; Krenning, G.; Harmsen, M.C. IL-1beta and TGFbeta2 synergistically induce endothelial to mesenchymal transition in an NFkappaB-dependent manner. Immunobiology 2013, 218, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Choi, S.H.; Jin, Y.B.; Lee, H.J.; Ji, Y.H.; Kim, J.; Lee, Y.S.; Lee, Y.J. The effect of oxidized low-density lipoprotein (ox-LDL) on radiation-induced endothelial-to-mesenchymal transition. Int. J. Radiat. Biol. 2013, 89, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Kovacic, J.C.; Mercader, N.; Torres, M.; Boehm, M.; Fuster, V. Epithelial-to-mesenchymal and endothelial-to-mesenchymal transition: From cardiovascular development to disease. Circulation 2012, 125, 1795–1808. [Google Scholar] [CrossRef]
- Piera-Velazquez, S.; Li, Z.; Jimenez, S.A. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am. J. Pathol. 2011, 179, 1074–1080. [Google Scholar] [CrossRef]
- Ma, J.; Liu, T.; Dong, X. Advanced glycation end products of bovine serum albumin-induced endothelial-to-mesenchymal transition in cultured human and monkey endothelial cells via protein kinase B signaling cascades. Mol. Vis. 2010, 16, 2669–2679. [Google Scholar] [PubMed]
- Spillmann, F.; Miteva, K.; Pieske, B.; Tschope, C.; van Linthout, S. High-density lipoproteins reduce endothelial-to-mesenchymal transition. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1774–1777. [Google Scholar] [CrossRef]
- Mahmoud, M.M.; Serbanovic-Canic, J.; Feng, S.; Souilhol, C.; Xing, R.; Hsiao, S.; Mammoto, A.; Chen, J.; Ariaans, M.; Francis, S.E.; et al. Shear stress induces endothelial-to-mesenchymal transition via the transcription factor Snail. Sci. Rep. 2017, 7, 3375. [Google Scholar] [CrossRef]
- Platel, V.; Faure, S.; Corre, I.; Clere, N. Endothelial-to-Mesenchymal Transition (EndoMT): Roles in Tumorigenesis, Metastatic Extravasation and Therapy Resistance. J. Oncol. 2019, 2019, 8361945. [Google Scholar] [CrossRef]
- Rye, K.A.; Barter, P.J. Cardioprotective functions of HDLs. J. Lipid Res. 2014, 55, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W.; Innerarity, T.L.; Rall, S.C., Jr.; Weisgraber, K.H. Plasma lipoproteins: Apolipoprotein structure and function. J. Lipid Res. 1984, 25, 1277–1294. [Google Scholar] [CrossRef]
- Vaughan, A.M.; Oram, J.F. ABCG1 redistributes cell cholesterol to domains removable by high density lipoprotein but not by lipid-depleted apolipoproteins. J. Biol. Chem. 2005, 280, 30150–30157. [Google Scholar] [CrossRef]
- Zhang, Y.; Gordon, S.M.; Xi, H.; Choi, S.; Paz, M.A.; Sun, R.; Yang, W.; Saredy, J.; Khan, M.; Remaley, A.T.; et al. HDL subclass proteomic analysis and functional implication of protein dynamic change during HDL maturation. Redox Biol. 2019, 24, 101222. [Google Scholar] [CrossRef] [PubMed]
- Vickers, K.C.; Palmisano, B.T.; Shoucri, B.M.; Shamburek, R.D.; Remaley, A.T. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat. Cell. Biol. 2011, 13, 423–433. [Google Scholar] [CrossRef]
- Rohatgi, A.; Khera, A.; Berry, J.D.; Givens, E.G.; Ayers, C.R.; Wedin, K.E.; Neeland, I.J.; Yuhanna, I.S.; Rader, D.R.; de Lemos, J.A.; et al. HDL cholesterol efflux capacity and incident cardiovascular events. N. Engl. J. Med. 2014, 371, 2383–2393. [Google Scholar] [CrossRef]
- Khera, A.V.; Cuchel, M.; de la Llera-Moya, M.; Rodrigues, A.; Burke, M.F.; Jafri, K.; French, B.C.; Phillips, J.A.; Mucksavage, M.L.; Wilensky, R.L.; et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N. Engl. J. Med. 2011, 364, 127–135. [Google Scholar] [CrossRef]
- Ogura, M.; Hori, M.; Harada-Shiba, M. Association Between Cholesterol Efflux Capacity and Atherosclerotic Cardiovascular Disease in Patients with Familial Hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 181–188. [Google Scholar] [CrossRef]
- Gille, A.; Easton, R.; D’Andrea, D.; Wright, S.D.; Shear, C.L. CSL112 enhances biomarkers of reverse cholesterol transport after single and multiple infusions in healthy subjects. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2106–2114. [Google Scholar] [CrossRef] [PubMed]
- Lee-Rueckert, M.; Escola-Gil, J.C.; Kovanen, P.T. HDL functionality in reverse cholesterol transport—Challenges in translating data emerging from mouse models to human disease. Biochim. Biophys. Acta 2016, 1861, 566–583. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; DiDonato, J.A.; Levison, B.S.; Schmitt, D.; Li, L.; Wu, Y.; Buffa, J.; Kim, T.; Gerstenecker, G.S.; Gu, X.; et al. An abundant dysfunctional apolipoprotein A1 in human atheroma. Nat. Med. 2014, 20, 193–203. [Google Scholar] [CrossRef]
- Sawada, N.; Obama, T.; Koba, S.; Takaki, T.; Iwamoto, S.; Aiuchi, T.; Kato, R.; Kikuchi, M.; Hamazaki, Y.; Itabe, H. Circulating oxidized LDL, increased in patients with acute myocardial infarction, is accompanied by heavily modified HDL. J. Lipid Res. 2020, 61, 816–829. [Google Scholar] [CrossRef] [PubMed]
- Chiesa, S.T.; Charakida, M. High-Density Lipoprotein Function and Dysfunction in Health and Disease. Cardiovasc. Drugs Ther. 2019, 33, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Rosenson, R.S.; Brewer, H.B., Jr.; Ansell, B.J.; Barter, P.; Chapman, M.J.; Heinecke, J.W.; Kontush, A.; Tall, A.R.; Webb, N.R. Dysfunctional HDL and atherosclerotic cardiovascular disease. Nat. Rev. Cardiol. 2016, 13, 48–60. [Google Scholar] [CrossRef] [PubMed]
- DiDonato, J.A.; Huang, Y.; Aulak, K.S.; Even-Or, O.; Gerstenecker, G.; Gogonea, V.; Wu, Y.; Fox, P.L.; Tang, W.H.; Plow, E.F.; et al. Function and distribution of apolipoprotein A1 in the artery wall are markedly distinct from those in plasma. Circulation 2013, 128, 1644–1655. [Google Scholar] [CrossRef]
- Itabe, H. Oxidative Modification of LDL: Its Pathological Role in Atherosclerosis. Clin. Rev. Allergy Immunol. 2009, 37, 4–11. [Google Scholar] [CrossRef]
- Levitan, I.; Volkov, S.; Subbaiah, P.V. Oxidized LDL: Diversity, patterns of recognition, and pathophysiology. Antioxid. Redox Signal 2010, 13, 39–75. [Google Scholar] [CrossRef]
- Bochkov, V.N.; Oskolkova, O.V.; Birukov, K.G.; Levonen, A.L.; Binder, C.J.; Stockl, J. Generation and biological activities of oxidized phospholipids. Antioxid. Redox Signal 2010, 12, 1009–1059. [Google Scholar] [CrossRef]
- Hazen, S.L. Oxidized phospholipids as endogenous pattern recognition ligands in innate immunity. J. Biol. Chem. 2008, 283, 15527–15531. [Google Scholar] [CrossRef]
- Zhuge, Q.; Zhang, Y.; Liu, B.; Wu, M. Blueberry polyphenols play a preventive effect on alcoholic fatty liver disease C57BL/6 J mice by promoting autophagy to accelerate lipolysis to eliminate excessive TG accumulation in hepatocytes. Ann. Palliat. Med. 2020, 9, 1045–1054. [Google Scholar] [CrossRef]
- Acton, S.; Rigotti, A.; Landschulz, K.T.; Xu, S.; Hobbs, H.H.; Krieger, M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 1996, 271, 518–520. [Google Scholar] [CrossRef]
- Lin, S.J.; Yen, H.T.; Chen, Y.H.; Ku, H.H.; Lin, F.Y.; Chen, Y.L. Expression of interleukin-1 beta and interleukin-1 receptor antagonist in oxLDL-treated human aortic smooth muscle cells and in the neointima of cholesterol-fed endothelia-denuded rabbits. J. Cell Biochem. 2003, 88, 836–847. [Google Scholar] [CrossRef]
- Huang, C.Y.; Lin, F.Y.; Shih, C.M.; Au, H.K.; Chang, Y.J.; Nakagami, H.; Morishita, R.; Chang, N.C.; Shyu, K.G.; Chen, J.W. Moderate to high concentrations of high-density lipoprotein from healthy subjects paradoxically impair human endothelial progenitor cells and related angiogenesis by activating Rho-associated kinase pathways. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2405–2417. [Google Scholar] [CrossRef]
- Hsu, Y.T.; Wolter, K.G.; Youle, R.J. Cytosol-to-membrane redistribution of Bax and Bcl-X(L) during apoptosis. Proc. Natl. Acad. Sci. USA 1997, 94, 3668–3672. [Google Scholar] [CrossRef]
- Navab, M.; Hama, S.Y.; Hough, G.P.; Subbanagounder, G.; Reddy, S.T.; Fogelman, A.M. A cell-free assay for detecting HDL that is dysfunctional in preventing the formation of or inactivating oxidized phospholipids. J. Lipid Res. 2001, 42, 1308–1317. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients with rHDL Index > 2 (n = 10) | Patients with rHDL Index < 2 (n = 10) | |
|---|---|---|
| Gender (Male/Female) | 5/5 | 5/5 |
| Age (years) | 61.5 ± 5.7 | 65.9 ± 4.7 |
| Body weight (kg) | 63.5 ± 15.7 | 65.7 ± 19.4 |
| Body height (cm) | 167.2 ± 13.7 | 164.1 ± 19.2 |
| Smoke (n, %) | 0, 0% | 0, 0% |
| Alcohol drinking (n, %) | 0, 0% | 0, 0% |
| Hypertension (n, %) | 8, 80% | 9, 90% |
| Hypercholesterolemia (n, %) | 10,100% | 10,100% |
| Diabetes mellitus (n, %) | 2, 20% | 3, 30% |
| Peripheral vascular disease (n, %) | 0, 0% | 0, 0% |
| Chronic obstructive pulmonary disease (n, %) | 0, 0% | 0, 0% |
| Old stroke (n, %) | 0, 0% | 0, 0% |
| Prior myocardial infarction (n, %) | 0, 0% | 0, 0% |
| Autoimmune disorders | 0, 0% | 0, 0% |
| Rheumatoid arthritis | 0, 0% | 0, 0% |
| Asthma | 0, 0% | 0, 0% |
| Chronic kidney deficiency | 0, 0% | 0, 0% |
| Cancer | 0, 0% | 0, 0% |
| NO | Formula | Description (Lipid) | Neutral Mass | m/z | Retention Time (min) | p Value | Fold Change | VIP Value |
|---|---|---|---|---|---|---|---|---|
| 1 | C56H100O6 | TG(14:0/17:0/22:4)[iso6] | 840.721 | 1779.520 | 16.068 | 0.02 | 4.50 | 2.12 |
| 2 | C54H88O6 | TG(14:1/15:1/22:6)[iso6] | 832.658 | 853.642 | 11.413 | <0.01 | 2.19 | 1.70 |
| 3 | C55H98O6 | TG(14:0/16:0/22:4)[iso6] | 854.736 | 1710.471 | 16.414 | <0.01 | 2.67 | 2.08 |
| 4 | C53H100O6 | TG(14:0/14:1/22:0)[iso6] | 832.752 | 1683.528 | 16.697 | <0.01 | 2.18 | 1.98 |
| 5 | C53H98O6 | TG(14:0/14:1/22:1)[iso6] | 830.734 | 1679.501 | 16.414 | 0.01 | 2.77 | 1.82 |
| 6 | C50H83N3O15P2 | DG(18:1/20:4) | 1027.530 | 1060.566 | 4.086 | <0.01 | 2.37 | 1.75 |
| 7 | C34H68NO8P | PC(13:0/13:0) | 649.468 | 970.594 | 4.107 | 0.02 | 2.33 | 1.73 |
| 8 | C50H86NO8P | PC(20:1/22:6) | 859.609 | 896.564 | 10.776 | <0.01 | 4.08 | 1.7 |
| 9 | C48H100NO6P | PC(O-20:0/O-20:0) | 831.708 | 1653.485 | 16.393 | 0.05 | 3.15 | 1.82 |
| 10 | C36H74NO7P | PC(O-16:0/12:0) | 633.502 | 708.489 | 2.723 | 0.02 | 2.11 | 1.61 |
| 11 | C45H83O13P | PI(18:1/18:1) | 862.557 | 1743.145 | 9.821 | <0.01 | 2.70 | 2.08 |
| 12 | C42H83O8P | PA(20:0/19:0) | 746.583 | 1535.212 | 16.047 | 0.01 | 2.13 | 1.82 |
| 13 | C32H59O8P | PA(12:0/17:2(9Z,12Z)) | 602.395 | 616.413 | 4.598 | 0.02 | 2.14 | 1.51 |
| 14 | C26H53O9P | PG(20:0/0:0) | 540.343 | 1098.708 | 4.148 | 0.05 | 2.26 | 1.8 |
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| CD31 | 5′-GTG AAG TCC GGA AAG CTG TCC-3′ | 5′-GGG CAG GTT CAT AAA TAA GTG CAC-3′ |
| vWF | 5′-GGC TGC AGT ATG TCA AGG TGG-3′ | 5′-AGA GCC ATT GGT GCA GTG CAG-3′ |
| VE-cadherin | 5′-AGA CAA TGG GAT GCC AAG TCB-3′ | 5′-AAG ATG AGC AGG GTG ATC ACT G-3′ |
| calponin | 5′-ACC TCT ACG ACC CCA AGC TG-3′ | 5′-GAC ATT GAG CGT GTC GCA GTG-3′ |
| αSMA | 5′-CTA TCA GGG GGC ACC ACT ATG-3′ | 5′-CCG ATC CAG ACA GAG TAT TTG CG-3′ |
| vimentin | 5′-AGG CAA AGC AGG AGT CCA CTG A-3′ | 5′-ATC TGG CGT TCC AGG GAC TCAT -3′ |
| GAPDH | 5′-TGC CCC CTC TGC TGA TGC C-3′ | 5′-CCT CCG ACG CCT GCT TCA CCA C-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, F.-Y.; Lin, Y.-W.; Shih, C.-M.; Lin, S.-J.; Tung, Y.-T.; Li, C.-Y.; Chen, Y.-H.; Lin, C.-Y.; Tsai, Y.-T.; Huang, C.-Y. A Novel Relative High-Density Lipoprotein Index to Predict the Structural Changes in High-Density Lipoprotein and Its Ability to Inhibit Endothelial–Mesenchymal Transition. Int. J. Mol. Sci. 2021, 22, 5210. https://doi.org/10.3390/ijms22105210
Lin F-Y, Lin Y-W, Shih C-M, Lin S-J, Tung Y-T, Li C-Y, Chen Y-H, Lin C-Y, Tsai Y-T, Huang C-Y. A Novel Relative High-Density Lipoprotein Index to Predict the Structural Changes in High-Density Lipoprotein and Its Ability to Inhibit Endothelial–Mesenchymal Transition. International Journal of Molecular Sciences. 2021; 22(10):5210. https://doi.org/10.3390/ijms22105210
Chicago/Turabian StyleLin, Feng-Yen, Yi-Wen Lin, Chun-Ming Shih, Shing-Jong Lin, Yu-Tang Tung, Chi-Yuan Li, Yung-Hsiang Chen, Cheng-Yen Lin, Yi-Ting Tsai, and Chun-Yao Huang. 2021. "A Novel Relative High-Density Lipoprotein Index to Predict the Structural Changes in High-Density Lipoprotein and Its Ability to Inhibit Endothelial–Mesenchymal Transition" International Journal of Molecular Sciences 22, no. 10: 5210. https://doi.org/10.3390/ijms22105210
APA StyleLin, F.-Y., Lin, Y.-W., Shih, C.-M., Lin, S.-J., Tung, Y.-T., Li, C.-Y., Chen, Y.-H., Lin, C.-Y., Tsai, Y.-T., & Huang, C.-Y. (2021). A Novel Relative High-Density Lipoprotein Index to Predict the Structural Changes in High-Density Lipoprotein and Its Ability to Inhibit Endothelial–Mesenchymal Transition. International Journal of Molecular Sciences, 22(10), 5210. https://doi.org/10.3390/ijms22105210