
Human Platelet Lysate for Good Manufacturing Practice-Compliant Cell Production

 , and
, and
Abstract
1. Introduction
2. Safety of Blood Products
2.1. Donor Eligibility
2.2. Donor Blood Group Testing and Antibody Screening
2.3. Donor Screening for Potential Transfusion-Transmitted Diseases
3. Whole Blood Processing and Production of Platelet Concentrates
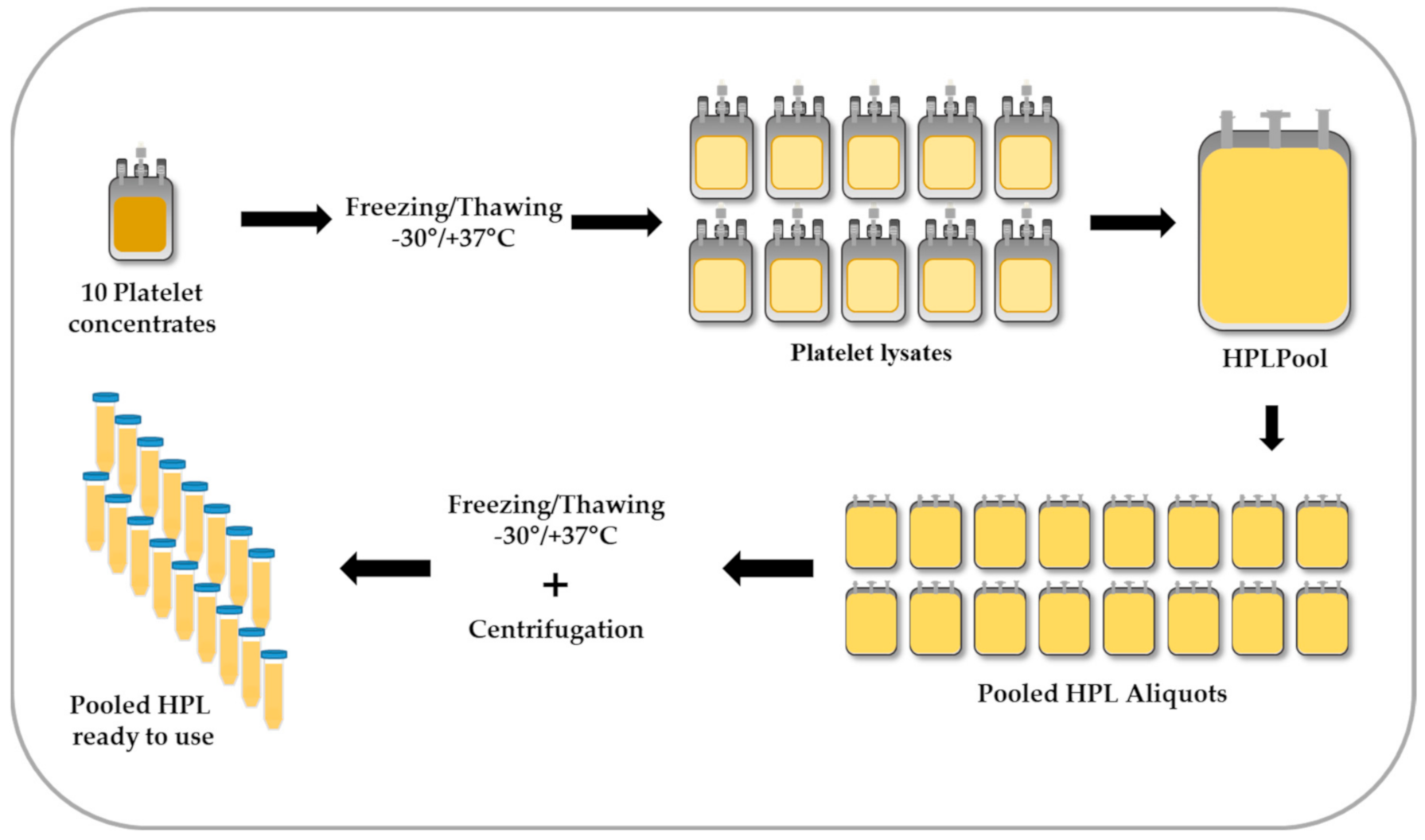
4. Preparation of HPL
5. Quality Control and Release Criteria of HPL
5.1. Screening for Bacterial and Fungal Contamination
5.2. Testing for Mycoplasma
5.3. Endotoxin Test
5.4. Biochemical Analysis
5.5. Testing of Isoagglutinin Titer
5.6. Performance Testing
5.7. Platelet-Derived Growth Factors, Cytokines and Chemokines
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ATP | adenosine triphosphate |
| BDNF | brain-derived neurotrophic factor |
| bFGF | basic fibroblast growth factor |
| CCL5/RANTES | CC-chemokine ligand 5 |
| CD40L | CD40 ligand |
| CFU | colony-forming units |
| cMYC | myc proto-oncogene protein |
| DENV | dengue virus |
| DNA | deoxyribonucleic acid |
| ECFCs | endothelial colony-forming progenitor cells |
| EGF | epidermal growth factor |
| EIA/ECLIA | enzyme- or electro-chemiluminiscence immunoassays |
| ELISA | enzyme-linked immunosorbent assay |
| EMA | European Medicine Agency |
| EU | endotoxin unit |
| FBS | fetal bovine serum |
| FDA | U.S. Food and Drug Administration |
| GF | growth factor |
| GMP | good manufacturing practice |
| HAV | hepatitis A virus |
| HBV | hepatitis B virus |
| HCV | hepatitis C virus |
| HEV | hepatitis E virus |
| HGF | hepatocyte growth factor |
| HIV-1/2 | human immunodeficiency virus type 1 or type 2 |
| HPL | human platelet lysate |
| IAT | indirect antiglobulin test |
| ICAM-1 | intercellular adhesion molecule 1 |
| IGF-1 | insulin-like growth factor 1 |
| IU | international units |
| KLF4 | Kruppel-like factor 4 |
| LAL | limulus amebocyte lysate |
| MPCs | myoprogenitor cells |
| MSCs | mesenchymal stromal cells |
| NAT | nucleic acid amplification technology |
| PB19 | parvovirus B19 |
| PCR | polymerase chain reaction |
| PCR-SSP | polymerase chain reaction-single specific primer |
| PDGF | platelet-derived growth factor |
| PR | pathogen reduction |
| PRP | platelet rich plasma |
| rh | recombinant human |
| RNA | ribonucleic acid |
| S/D | solvent/detergent |
| SOX2 | sex-determining region Y (SRY)- box 2 |
| SPC | statistical process control |
| TGF-β | transforming growth factor β |
| U.S. | United States |
| USP | U.S. Pharmacopeia |
| UV | ultraviolet |
| VCAM-1 | vascular cell adhesion molecule-1 |
| VEGF | vascular endothelial growth factor |
| WNV | West Nile virus |
| ZIKV | Zika virus |
References
- Squillaro, T.; Peluso, G.; Galderisi, U. Clinical Trials With Mesenchymal Stem Cells: An Update. Cell Transplant. 2016, 25, 829–848. [Google Scholar] [CrossRef] [PubMed]
- Pamies, D.; Bal-Price, A.; Simeonov, A.; Tagle, D.; Allen, D.; Gerhold, D.; Yin, D.; Pistollato, F.; Inutsuka, T.; Sullivan, K.; et al. Good Cell Culture Practice for stem cells and stem-cell-derived models. ALTEX 2017, 34, 95–132. [Google Scholar] [CrossRef] [PubMed]
- Henschler, R.; Gabriel, C.; Schallmoser, K.; Burnouf, T.; Koh, M.B.C. Human platelet lysate current standards and future developments. Transfusion 2019, 59, 1407–1413. [Google Scholar] [CrossRef] [PubMed]
- Bieback, K.; Fernandez-Munoz, B.; Pati, S.; Schafer, R. Gaps in the knowledge of human platelet lysate as a cell culture supplement for cell therapy: A joint publication from the AABB and the International Society for Cell & Gene Therapy. Transfusion 2019, 59, 3448–3460. [Google Scholar] [CrossRef]
- Mendicino, M.; Bailey, A.M.; Wonnacott, K.; Puri, R.K.; Bauer, S.R. MSC-based product characterization for clinical trials: An FDA perspective. Cell Stem Cell 2014, 14, 141–145. [Google Scholar] [CrossRef]
- van der Valk, J.; Bieback, K.; Buta, C.; Cochrane, B.; Dirks, W.G.; Fu, J.; Hickman, J.J.; Hohensee, C.; Kolar, R.; Liebsch, M.; et al. Fetal Bovine Serum (FBS): Past-Present-Future. ALTEX 2018, 35, 99–118. [Google Scholar] [CrossRef]
- Trento, C.; Bernardo, M.E.; Nagler, A.; Kuci, S.; Bornhauser, M.; Kohl, U.; Strunk, D.; Galleu, A.; Sanchez-Guijo, F.; Gaipa, G.; et al. Manufacturing Mesenchymal Stromal Cells for the Treatment of Graft-versus-Host Disease: A Survey among Centers Affiliated with the European Society for Blood and Marrow Transplantation. Biol. Blood Marrow Transpl. 2018, 24, 2365–2370. [Google Scholar] [CrossRef]
- Macy, E.; Bulpitt, K.; Champlin, R.E.; Saxon, A. Anaphylaxis to infusion of autologous bone marrow: An apparent reaction to self, mediated by IgE antibody to bovine serum albumin. J. Allergy Clin. Immunol. 1989, 83, 871–875. [Google Scholar] [CrossRef]
- Selvaggi, T.A.; Walker, R.E.; Fleisher, T.A. Development of antibodies to fetal calf serum with arthus-like reactions in human immunodeficiency virus-infected patients given syngeneic lymphocyte infusions. Blood 1997, 89, 776–779. [Google Scholar] [CrossRef]
- Mackensen, A.; Drager, R.; Schlesier, M.; Mertelsmann, R.; Lindemann, A. Presence of IgE antibodies to bovine serum albumin in a patient developing anaphylaxis after vaccination with human peptide-pulsed dendritic cells. Cancer Immunol. Immunother. 2000, 49, 152–156. [Google Scholar]
- Tuschong, L.; Soenen, S.L.; Blaese, R.M.; Candotti, F.; Muul, L.M. Immune response to fetal calf serum by two adenosine deaminase-deficient patients after T cell gene therapy. Hum. Gene Ther. 2002, 13, 1605–1610. [Google Scholar] [CrossRef]
- Hemeda, H.; Giebel, B.; Wagner, W. Evaluation of human platelet lysate versus fetal bovine serum for culture of mesenchymal stromal cells. Cytotherapy 2014, 16, 170–180. [Google Scholar] [CrossRef]
- Schallmoser, K.; Strunk, D. Generation of a pool of human platelet lysate and efficient use in cell culture. Methods Mol. Biol. 2013, 946, 349–362. [Google Scholar] [CrossRef]
- Doucet, C.; Ernou, I.; Zhang, Y.; Llense, J.R.; Begot, L.; Holy, X.; Lataillade, J.J. Platelet lysates promote mesenchymal stem cell expansion: A safety substitute for animal serum in cell-based therapy applications. J. Cell. Physiol. 2005, 205, 228–236. [Google Scholar] [CrossRef]
- Schallmoser, K.; Bartmann, C.; Rohde, E.; Reinisch, A.; Kashofer, K.; Stadelmeyer, E.; Drexler, C.; Lanzer, G.; Linkesch, W.; Strunk, D. Human platelet lysate can replace fetal bovine serum for clinical-scale expansion of functional mesenchymal stromal cells. Transfusion 2007, 47, 1436–1446. [Google Scholar] [CrossRef]
- Bieback, K.; Hecker, A.; Kocaomer, A.; Lannert, H.; Schallmoser, K.; Strunk, D.; Kluter, H. Human alternatives to fetal bovine serum for the expansion of mesenchymal stromal cells from bone marrow. Stem Cells 2009, 27, 2331–2341. [Google Scholar] [CrossRef]
- Reinisch, A.; Hofmann, N.A.; Obenauf, A.C.; Kashofer, K.; Rohde, E.; Schallmoser, K.; Flicker, K.; Lanzer, G.; Linkesch, W.; Speicher, M.R.; et al. Humanized large-scale expanded endothelial colony-forming cells function in vitro and in vivo. Blood 2009, 113, 6716–6725. [Google Scholar] [CrossRef]
- Burnouf, T.; Strunk, D.; Koh, M.B.; Schallmoser, K. Human platelet lysate: Replacing fetal bovine serum as a gold standard for human cell propagation? Biomaterials 2016, 76, 371–387. [Google Scholar] [CrossRef]
- Strunk, D.; Lozano, M.; Marks, D.C.; Loh, Y.S.; Gstraunthaler, G.; Schennach, H.; Rohde, E.; Laner-Plamberger, S.; Oller, M.; Nystedt, J.; et al. International Forum on GMP-grade human platelet lysate for cell propagation: Summary. Vox Sang. 2018, 113, 80–87. [Google Scholar] [CrossRef]
- Laner-Plamberger, S.; Oeller, M.; Mrazek, C.; Hartl, A.; Sonderegger, A.; Rohde, E.; Strunk, D.; Schallmoser, K. Upregulation of mitotic bookmarking factors during enhanced proliferation of human stromal cells in human platelet lysate. J. Transl. Med. 2019, 17, 432. [Google Scholar] [CrossRef]
- Schallmoser, K.; Henschler, R.; Gabriel, C.; Koh, M.B.C.; Burnouf, T. Production and Quality Requirements of Human Platelet Lysate: A Position Statement from the Working Party on Cellular Therapies of the International Society of Blood Transfusion. Trends Biotechnol. 2020, 38, 13–23. [Google Scholar] [CrossRef] [PubMed]
- European Directorate for the Quality of Medicines & HealthCare: Guide to the Preparation, Use and Quality Assurance of Blood Components, 20th ed.; Council of Europe: Strasbourg, France, 2020; Chapter 5; pp. 223–245.
- Schallmoser, K.; Rohde, E.; Reinisch, A.; Bartmann, C.; Thaler, D.; Drexler, C.; Obenauf, A.C.; Lanzer, G.; Linkesch, W.; Strunk, D. Rapid large-scale expansion of functional mesenchymal stem cells from unmanipulated bone marrow without animal serum. Tissue Eng. Part C Methods 2008, 14, 185–196. [Google Scholar] [CrossRef]
- Oeller, M.; Laner-Plamberger, S.; Hochmann, S.; Ketterl, N.; Feichtner, M.; Brachtl, G.; Hochreiter, A.; Scharler, C.; Bieler, L.; Romanelli, P.; et al. Selection of Tissue Factor-Deficient Cell Transplants as a Novel Strategy for Improving Hemocompatibility of Human Bone Marrow Stromal Cells. Theranostics 2018, 8, 1421–1434. [Google Scholar] [CrossRef] [PubMed]
- Uccelli, A.; Laroni, A.; Brundin, L.; Clanet, M.; Fernandez, O.; Nabavi, S.M.; Muraro, P.A.; Oliveri, R.S.; Radue, E.W.; Sellner, J.; et al. MEsenchymal StEm cells for Multiple Sclerosis (MESEMS): A randomized, double blind, cross-over phase I/II clinical trial with autologous mesenchymal stem cells for the therapy of multiple sclerosis. Trials 2019, 20, 263. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, N.A.; Reinisch, A.; Strunk, D. Isolation and large scale expansion of adult human endothelial colony forming progenitor cells. J. Vis. Exp. 2009. [Google Scholar] [CrossRef] [PubMed]
- American Association of Blood Banks. Standards for Blood Banks and Transfusion Services, 32nd ed.; American Association of Blood Banks: Bethesda, MD, USA, 2020. [Google Scholar]
- Williams, L.A.; Snyder, E.L. Transfusion-Related Adverse Events, Reference Module in Biomedical Sciences; Elsevier: Amsterdam, The Netherlands, 2014; ISBN 9780128012383. Available online: https://www.sciencedirect.com/science/article/pii/B9780128012383000751 (accessed on 10 April 2021). [CrossRef]
- Castillo, B.; Dasgupta, A.; Klein, K.; Tint, H.; Wahed, A. (Eds.) Chapter 3—Transfusion reactions. In Transfusion Medicine for Pathologists; Elsevier: Amsterdam, The Netherlands, 2018; pp. 37–49. ISBN 9780128143131. [Google Scholar]
- Strobel, E. Hemolytic Transfusion Reactions. Transfus. Med. Hemother. 2008, 35, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Harewood, J.; Ramsey, A.; Master, S.R. Hemolytic Transfusion Reaction; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Dean, L. Blood Groups and Red Cell Antigens; The ABO Blood Group-Blood Groups and Red Cell Antigens-NCBI Bookshelf (nih.gov); National Center for Biotechnology Information: Bethesda, MD, USA, 2005.
- Busch, M.P.; Bloch, E.M.; Kleinman, S. Prevention of transfusion-transmitted infections. Blood 2019, 133, 1854–1864. [Google Scholar] [CrossRef]
- Burnouf, T.; Barro, L.; Nebie, O.; Wu, Y.W.; Goubran, H.; Knutson, F.; Seghatchian, J. Viral safety of human platelet lysate for cell therapy and regenerative medicine: Moving forward, yes, but without forgetting the past. Transfus. Apher. Sci. 2019, 58, 102674. [Google Scholar] [CrossRef]
- Roth, W.K. History and Future of Nucleic Acid Amplification Technology Blood Donor Testing. Transfus. Med. Hemother. 2019, 46, 67–75. [Google Scholar] [CrossRef]
- Gallian, P.; Barlet, V.; Mouna, L.; Gross, S.; Lecam, S.; Ricard, C.; Wind, F.; Pouchol, E.; Fabra, C.; Flan, B.; et al. Hepatitis A: An epidemiological survey in blood donors, France 2015 to 2017. Eurosurveillance 2018, 23. [Google Scholar] [CrossRef]
- Roth, W.K.; Busch, M.P.; Schuller, A.; Ismay, S.; Cheng, A.; Seed, C.R.; Jungbauer, C.; Minsk, P.M.; Sondag-Thull, D.; Wendel, S.; et al. International survey on NAT testing of blood donations: Expanding implementation and yield from 1999 to 2009. Vox Sang. 2012, 102, 82–90. [Google Scholar] [CrossRef]
- Marano, G.; Vaglio, S.; Pupella, S.; Facco, G.; Calizzani, G.; Candura, F.; Liumbruno, G.M.; Grazzini, G. Human Parvovirus B19 and blood product safety: A tale of twenty years of improvements. Blood Transfus. 2015, 13, 184–196. [Google Scholar] [CrossRef]
- Domanovic, D.; Gossner, C.M.; Lieshout-Krikke, R.; Mayr, W.; Baroti-Toth, K.; Dobrota, A.M.; Escoval, M.A.; Henseler, O.; Jungbauer, C.; Liumbruno, G.; et al. West Nile and Usutu Virus Infections and Challenges to Blood Safety in the European Union. Emerg. Infect. Dis. 2019, 25, 1050–1057. [Google Scholar] [CrossRef]
- Groves, J.A.; Foster, G.A.; Dodd, R.Y.; Stramer, S.L. West Nile virus activity in United States blood donors and optimizing detection strategies: 2014–2018. Transfusion 2020, 60, 94–105. [Google Scholar] [CrossRef]
- Stanley, J.; Chongkolwatana, V.; Duong, P.T.; Kitpoka, P.; Stramer, S.L.; Dung, N.T.T.; Grimm, K.E.; Pojanasingchod, A.; Suksomboonvong, P.; Galel, S.A. Detection of dengue, chikungunya, and Zika RNA in blood donors from Southeast Asia. Transfusion 2021, 61, 134–143. [Google Scholar] [CrossRef]
- Liu, R.; Wang, X.; Ma, Y.; Wu, J.; Mao, C.; Yuan, L.; Lu, J. Prevalence of Zika virus in blood donations: A systematic review and meta-analysis. BMC Infect. Dis. 2019, 19, 590. [Google Scholar] [CrossRef]
- Williamson, P.C.; Biggerstaff, B.J.; Simmons, G.; Stone, M.; Winkelman, V.; Latoni, G.; Alsina, J.; Bakkour, S.; Newman, C.; Pate, L.L.; et al. Evolving viral and serological stages of Zika virus RNA-positive blood donors and estimation of incidence of infection during the 2016 Puerto Rican Zika epidemic: An observational cohort study. Lancet Infect. Dis. 2020, 20, 1437–1445. [Google Scholar] [CrossRef]
- Vollmer, T.; Diekmann, J.; Knabbe, C.; Dreier, J. Hepatitis E virus blood donor NAT screening: As much as possible or as much as needed? Transfusion 2019, 59, 612–622. [Google Scholar] [CrossRef]
- Bi, H.; Yang, R.; Wu, C.; Xia, J. Hepatitis E virus and blood transfusion safety. Epidemiol. Infect. 2020, 148, e158. [Google Scholar] [CrossRef]
- Blood Safety and Availability. Available online: https://www.who.int/news-room/fact-sheets/detail/blood-safety-and-availability (accessed on 10 April 2021).
- Schrezenmeier, H.; Seifried, E. Buffy-coat-derived pooled platelet concentrates and apheresis platelet concentrates: Which product type should be preferred? Vox Sang. 2010, 99, 1–15. [Google Scholar] [CrossRef]
- Greening, D.W.; Simpson, R.J.; Sparrow, R.L. Preparation of Platelet Concentrates for Research and Transfusion Purposes. Methods Mol. Biol. 2017, 1619, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Geilenkeuser, W.J.; Sireis, W.; Seifried, E.; Hourfar, K. Emerging Pathogens—How Safe is Blood? Transfus. Med. Hemother. 2014, 41, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Stramer, S.L.; Hollinger, F.B.; Katz, L.M.; Kleinman, S.; Metzel, P.S.; Gregory, K.R.; Dodd, R.Y. Emerging infectious disease agents and their potential threat to transfusion safety. Transfusion 2009, 49 (Suppl. 2), 1S–29S. [Google Scholar] [CrossRef] [PubMed]
- Schlenke, P. Pathogen inactivation technologies for cellular blood components: An update. Transfus. Med. Hemother. 2014, 41, 309–325. [Google Scholar] [CrossRef]
- Kaiser-Guignard, J.; Canellini, G.; Lion, N.; Abonnenc, M.; Osselaer, J.C.; Tissot, J.D. The clinical and biological impact of new pathogen inactivation technologies on platelet concentrates. Blood Rev. 2014, 28, 235–241. [Google Scholar] [CrossRef]
- Fazzina, R.; Iudicone, P.; Mariotti, A.; Fioravanti, D.; Procoli, A.; Cicchetti, E.; Scambia, G.; Bonanno, G.; Pierelli, L. Culture of human cell lines by a pathogen-inactivated human platelet lysate. Cytotechnology 2016, 68, 1185–1195. [Google Scholar] [CrossRef][Green Version]
- Viau, S.; Chabrand, L.; Eap, S.; Lorant, J.; Rouger, K.; Goudaliez, F.; Sumian, C.; Delorme, B. Pathogen reduction through additive-free short-wave UV light irradiation retains the optimal efficacy of human platelet lysate for the expansion of human bone marrow mesenchymal stem cells. PLoS ONE 2017, 12, e0181406. [Google Scholar] [CrossRef]
- Viau, S.; Eap, S.; Chabrand, L.; Lagrange, A.; Delorme, B. Viral inactivation of human platelet lysate by gamma irradiation preserves its optimal efficiency in the expansion of human bone marrow mesenchymal stromal cells. Transfusion 2019, 59, 1069–1079. [Google Scholar] [CrossRef]
- Christensen, C.; Jonsdottir-Buch, S.M.; Sigurjonsson, O.E. Effects of amotosalen treatment on human platelet lysate bioactivity: A proof-of-concept study. PLoS ONE 2020, 15, e0220163. [Google Scholar] [CrossRef]
- Jonsdottir-Buch, S.M.; Sigurgrimsdottir, H.; Lieder, R.; Sigurjonsson, O.E. Expired and Pathogen-Inactivated Platelet Concentrates Support Differentiation and Immunomodulation of Mesenchymal Stromal Cells in Culture. Cell Transplant. 2015, 24, 1545–1554. [Google Scholar] [CrossRef]
- Schubert, P.; Johnson, L.; Marks, D.C.; Devine, D.V. Ultraviolet-Based Pathogen Inactivation Systems: Untangling the Molecular Targets Activated in Platelets. Front. Med. 2018, 5, 129. [Google Scholar] [CrossRef]
- Osman, A.; Hitzler, W.E.; Provost, P. Peculiarities of studying the effects of pathogen reduction technologies on platelets. Proteom. Clin. Appl. 2016, 10, 805–815. [Google Scholar] [CrossRef]
- Stroncek, D.F.; Rebulla, P. Platelet transfusions. Lancet 2007, 370, 427–438. [Google Scholar] [CrossRef]
- Jonsdottir-Buch, S.M.; Lieder, R.; Sigurjonsson, O.E. Platelet lysates produced from expired platelet concentrates support growth and osteogenic differentiation of mesenchymal stem cells. PLoS ONE 2013, 8, e68984. [Google Scholar] [CrossRef]
- Bieback, K. Platelet lysate as replacement for fetal bovine serum in mesenchymal stromal cell cultures. Transfus. Med. Hemother. 2013, 40, 326–335. [Google Scholar] [CrossRef]
- Dessels, C.; Durandt, C.; Pepper, M.S. Comparison of human platelet lysate alternatives using expired and freshly isolated platelet concentrates for adipose-derived stromal cell expansion. Platelets 2019, 30, 356–367. [Google Scholar] [CrossRef]
- Bernardi, M.; Albiero, E.; Alghisi, A.; Chieregato, K.; Lievore, C.; Madeo, D.; Rodeghiero, F.; Astori, G. Production of human platelet lysate by use of ultrasound for ex vivo expansion of human bone marrow-derived mesenchymal stromal cells. Cytotherapy 2013, 15, 920–929. [Google Scholar] [CrossRef]
- Shih, D.T.; Burnouf, T. Preparation, quality criteria, and properties of human blood platelet lysate supplements for ex vivo stem cell expansion. New Biotechnol. 2015, 32, 199–211. [Google Scholar] [CrossRef]
- Copland, I.B.; Garcia, M.A.; Waller, E.K.; Roback, J.D.; Galipeau, J. The effect of platelet lysate fibrinogen on the functionality of MSCs in immunotherapy. Biomaterials 2013, 34, 7840–7850. [Google Scholar] [CrossRef]
- Kocaoemer, A.; Kern, S.; Kluter, H.; Bieback, K. Human AB serum and thrombin-activated platelet-rich plasma are suitable alternatives to fetal calf serum for the expansion of mesenchymal stem cells from adipose tissue. Stem Cells 2007, 25, 1270–1278. [Google Scholar] [CrossRef]
- Agostini, F.; Polesel, J.; Battiston, M.; Lombardi, E.; Zanolin, S.; Da Ponte, A.; Astori, G.; Durante, C.; Mazzucato, M. Standardization of platelet releasate products for clinical applications in cell therapy: A mathematical approach. J. Transl. Med. 2017, 15, 107. [Google Scholar] [CrossRef] [PubMed]
- Horn, P.; Bokermann, G.; Cholewa, D.; Bork, S.; Walenda, T.; Koch, C.; Drescher, W.; Hutschenreuther, G.; Zenke, M.; Ho, A.D.; et al. Impact of individual platelet lysates on isolation and growth of human mesenchymal stromal cells. Cytotherapy 2010, 12, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Pierce, J.; Benedetti, E.; Preslar, A.; Jacobson, P.; Jin, P.; Stroncek, D.F.; Reems, J.A. Comparative analyses of industrial-scale human platelet lysate preparations. Transfusion 2017, 57, 2858–2869. [Google Scholar] [CrossRef] [PubMed]
- Viau, S.; Lagrange, A.; Chabrand, L.; Lorant, J.; Charrier, M.; Rouger, K.; Alvarez, I.; Eap, S.; Delorme, B. A highly standardized and characterized human platelet lysate for efficient and reproducible expansion of human bone marrow mesenchymal stromal cells. Cytotherapy 2019, 21, 738–754. [Google Scholar] [CrossRef]
- Raw materials of biological origin for the production of cell-based and gene therapy medicinal products. In European Pharmacopoeia, 10th ed.; Council of Europe: Strasbourg, France, 2019; Chapter 5.2.12.
- Schafer, R.; Schnaidt, M.; Klaffschenkel, R.A.; Siegel, G.; Schule, M.; Radlein, M.A.; Hermanutz-Klein, U.; Ayturan, M.; Buadze, M.; Gassner, C.; et al. Expression of blood group genes by mesenchymal stem cells. Br. J. Haematol. 2011, 153, 520–528. [Google Scholar] [CrossRef][Green Version]
- Jaffe, E.A.; Nachman, R.L.; Becker, C.G.; Minick, C.R. Culture of human endothelial cells derived from umbilical veins. Identification by morphologic and immunologic criteria. J. Clin. Investig. 1973, 52, 2745–2756. [Google Scholar] [CrossRef]
- Solomon, J.; Csontos, L.; Clarke, D.; Bonyhadi, M.; Zylberberg, C.; McNiece, I.; Kurtzberg, J.; Bell, R.; Deans, R. Current perspectives on the use of ancillary materials for the manufacture of cellular therapies. Cytotherapy 2016, 18, 1–12. [Google Scholar] [CrossRef]
- General chapter <1046> Cellular and Tissue-based Products. USP 34-NF 29. In U.S. Pharmacopeia; United States Pharmacopoeia Commission: North Bethesda, ML, USA, 2011.
- General chapter <1043> Ancillary Materials. USP 29-NF 24. In U.S. Pharmacopeia; United States Pharmacopoeia Commission: North Bethesda, ML, USA, 2006.
- Sterility. In European Pharmacopoeia, 5th ed.; Council of Europe: Strasbourg, France, 2005; Chapter 2.6.1.
- Gunther, S.K.; Geiss, C.; Kaiser, S.J.; Mutters, N.T.; Gunther, F. Microbiological Control of Cellular Products: The Relevance of the Cellular Matrix, Incubation Temperature, and Atmosphere for the Detection Performance of Automated Culture Systems. Transfus. Med. Hemother. 2020, 47, 254–263. [Google Scholar] [CrossRef]
- Alternative methods for control of microbiological quality. In European Pharmacopoeia, 10th ed.; Council of Europe: Strasbourg, France, 2019; Chapter 5.1.6.
- Bacterial endotoxins. In European Pharmacopoeia, 5th ed.; Council of Europe: Strasbourg, France, 2005; Chapter 2.6.14.
- Mycoplasmas. In European Pharmacopoeia, 5th ed.; Council of Europe: Strasbourg, France, 2005; Chapter 2.6.7.
- Volokhov, D.V.; Graham, L.J.; Brorson, K.A.; Chizhikov, V.E. Mycoplasma testing of cell substrates and biologics: Review of alternative non-microbiological techniques. Mol. Cell. Probes 2011, 25, 69–77. [Google Scholar] [CrossRef]
- Drexler, H.G.; Uphoff, C.C. Mycoplasma contamination of cell cultures: Incidence, sources, effects, detection, elimination, prevention. Cytotechnology 2002, 39, 75–90. [Google Scholar] [CrossRef]
- Schneier, M.; Razdan, S.; Miller, A.M.; Briceno, M.E.; Barua, S. Current technologies to endotoxin detection and removal for biopharmaceutical purification. Biotechnol. Bioeng. 2020, 117, 2588–2609. [Google Scholar] [CrossRef]
- Test for bacterial endotoxins with recombinant factor C (rFC). In European Pharmacopoeia, 10th ed.; Council of Europe: Strasbourg, France, 2019; Chapter 2.6.32.
- Burnouf, T.; Lee, C.Y.; Luo, C.W.; Kuo, Y.P.; Chou, M.L.; Wu, Y.W.; Tseng, Y.H.; Su, C.Y. Human blood-derived fibrin releasates: Composition and use for the culture of cell lines and human primary cells. Biologicals 2012, 40, 21–30. [Google Scholar] [CrossRef]
- Laner-Plamberger, S.; Lener, T.; Schmid, D.; Streif, D.A.; Salzer, T.; Oller, M.; Hauser-Kronberger, C.; Fischer, T.; Jacobs, V.R.; Schallmoser, K.; et al. Mechanical fibrinogen-depletion supports heparin-free mesenchymal stem cell propagation in human platelet lysate. J. Transl. Med. 2015, 13, 354. [Google Scholar] [CrossRef]
- Mojica-Henshaw, M.P.; Jacobson, P.; Morris, J.; Kelley, L.; Pierce, J.; Boyer, M.; Reems, J.A. Serum-converted platelet lysate can substitute for fetal bovine serum in human mesenchymal stromal cell cultures. Cytotherapy 2013, 15, 1458–1468. [Google Scholar] [CrossRef]
- Fekete, N.; Gadelorge, M.; Furst, D.; Maurer, C.; Dausend, J.; Fleury-Cappellesso, S.; Mailander, V.; Lotfi, R.; Ignatius, A.; Sensebe, L.; et al. Platelet lysate from whole blood-derived pooled platelet concentrates and apheresis-derived platelet concentrates for the isolation and expansion of human bone marrow mesenchymal stromal cells: Production process, content and identification of active components. Cytotherapy 2012, 14, 540–554. [Google Scholar] [CrossRef]


{kind=link}
| Parameters | Range of Specification | Frequency of Testing | |
|---|---|---|---|
| Markers for transfusion-transmissible infections | Anti-HIV-1/2, anti-HCV, HBV surface antigen; NAT for HIV-1/2, HBV and HCV, syphilis and others as required | Negative by approved test system | All blood donations |
| Sterility | Bacteria and fungi | Negative by approved test system | All units |
| Final volume | >40 mL per 0.6 × 1011 platelets | According to SPC | |
| Biochemical analysis | pH | >6.4 at the end of storage | According to SPC |
| Cell amount | Platelets * | ≥2 × 1011/unit | According to SPC |
| Residual leukocytes * | <1 × 106/unit | According to SPC |
| Parameter | Range of Specification | Test Method | Frequency of Testing | |
|---|---|---|---|---|
| Pool size | Number of blood donations | 10–16 * | - | - |
| Sterility | Endotoxin | <0.5 EU/mL | LAL endotoxin test [81] | Each batch |
| Bacteria and fungi | Negative | Automated microbial detection system, PCR or ELISA | ||
| Mycoplasma | Negative | Culture [82] or PCR | ||
| Biochemical analysis | pH Osmolality Total protein | According to the range of standard blood values | pH-meter Osmometer Biuret protein assay | Each batch (for 100% plasma HPL) |
| Immunology | Isoagglutinin titer | Depending on the cell type in culture | IAT | Randomly |
| Stability | Shelf life | To be validated | Performance testing GF analysis | Randomly |
| Potency | Cell proliferation | To be validated | Performance testing | Randomly |
| Platelet-derived growth factors | To be validated | Multiplex assays, ELISA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oeller, M.; Laner-Plamberger, S.; Krisch, L.; Rohde, E.; Strunk, D.; Schallmoser, K. Human Platelet Lysate for Good Manufacturing Practice-Compliant Cell Production. Int. J. Mol. Sci. 2021, 22, 5178. https://doi.org/10.3390/ijms22105178
Oeller M, Laner-Plamberger S, Krisch L, Rohde E, Strunk D, Schallmoser K. Human Platelet Lysate for Good Manufacturing Practice-Compliant Cell Production. International Journal of Molecular Sciences. 2021; 22(10):5178. https://doi.org/10.3390/ijms22105178
Chicago/Turabian StyleOeller, Michaela, Sandra Laner-Plamberger, Linda Krisch, Eva Rohde, Dirk Strunk, and Katharina Schallmoser. 2021. "Human Platelet Lysate for Good Manufacturing Practice-Compliant Cell Production" International Journal of Molecular Sciences 22, no. 10: 5178. https://doi.org/10.3390/ijms22105178
APA StyleOeller, M., Laner-Plamberger, S., Krisch, L., Rohde, E., Strunk, D., & Schallmoser, K. (2021). Human Platelet Lysate for Good Manufacturing Practice-Compliant Cell Production. International Journal of Molecular Sciences, 22(10), 5178. https://doi.org/10.3390/ijms22105178