
PD-1/PD-L1 in Cancer: Pathophysiological, Diagnostic and Therapeutic Aspects


 , ,
, ,  ,
, 
 and
and 
Abstract
1. Introduction
2. PD-1 Expression in Antitumor Effector T and NK Cells
3. Mechanisms of PD-1 Expression
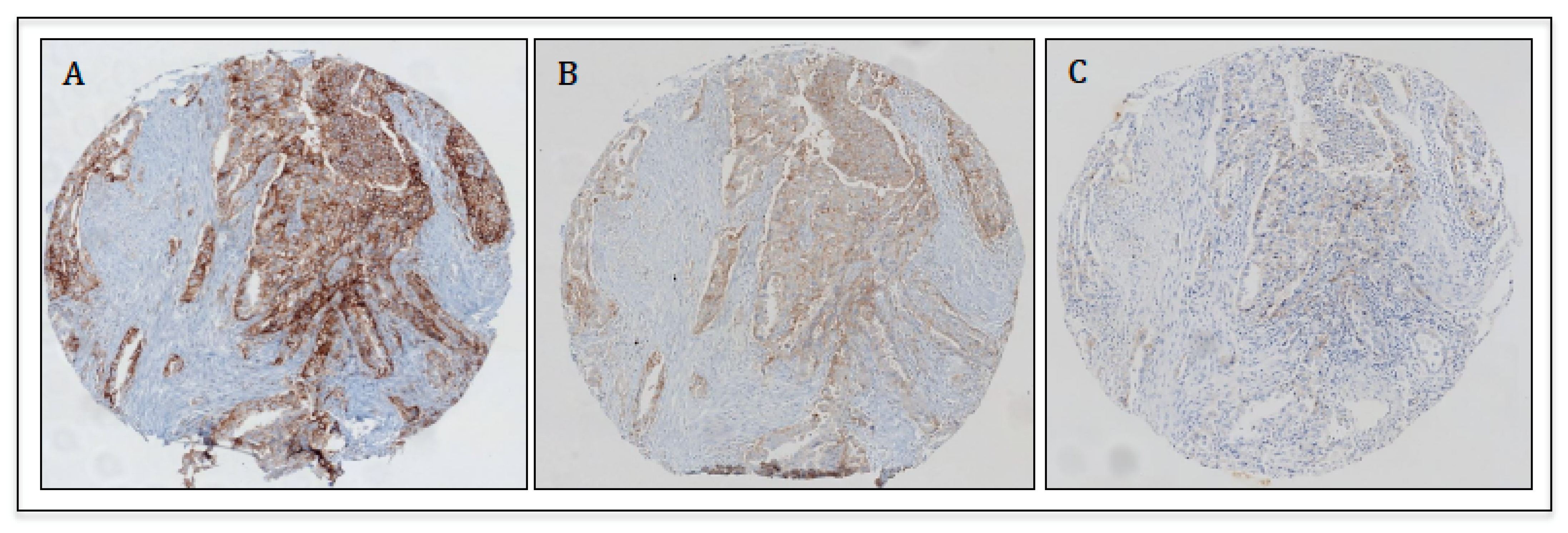
4. Applications of PD-L1 Immunohistochemistry
5. Additional and Novel Strategies to Improve Patient Selection: Beyond PD-L1
5.1. Tumor Mutational Burden
5.2. Mismatch Repair Deficiency (dMMR)/Microsatellite Instability (MSI)
5.3. Tumor-Infiltrating Lymphocytes (TILs)
6. Disruption of PD1/PD-L1 Axis in Cancer Therapy
Neuroblastoma and Other Pediatric Tumors
7. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Minato, N.; Nakano, T.; Honjo, T. Immunological studies on PD-1-deficient mice: Implication of PD-1 as a negative regulator for B cell responses. Int. Immunol. 1998, 10, 1563–1572. [Google Scholar] [CrossRef]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 1999, 11, 141–151. [Google Scholar] [CrossRef]
- Boussiotis, V.A.; Chatterjee, P.; Li, L. Biochemical signaling of PD-1 on T cells and its functional implications. Cancer J. USA 2014, 20, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Chamoto, K.; Al-Habsi, M.; Honjo, T. Role of PD-1 in immunity and diseases. In Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2017; Volume 410, pp. 75–97. [Google Scholar]
- Huang, A.C.; Postow, M.A.; Orlowski, R.J.; Mick, R.; Bengsch, B.; Manne, S.; Xu, W.; Harmon, S.; Giles, J.R.; Wenz, B.; et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 2017, 545, 60–65. [Google Scholar] [CrossRef]
- Pesce, S.; Greppi, M.; Tabellini, G.; Rampinelli, F.; Parolini, S.; Olive, D.; Moretta, L.; Moretta, A.; Marcenaro, E. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: A phenotypic and functional characterization. J. Allergy Clin. Immunol. 2017, 139, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Liang, S.C.; Guleria, I.; Latchman, Y.E.; Qipo, A.; Albacker, L.A.; Koulmanda, M.; Freeman, G.J.; Sayegh, M.H.; Sharpe, A.H. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J. Exp. Med. 2006, 203, 883–895. [Google Scholar] [CrossRef]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef]
- Simon, S.; Labarriere, N. PD-1 expression on tumor-specific T cells: Friend or foe for immunotherapy? Oncoimmunology 2018, 7, e1364828. [Google Scholar] [CrossRef]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef]
- Dondero, A.; Pastorino, F.; Della Chiesa, M.; Corrias, M.V.; Morandi, F.; Pistoia, V.; Olive, D.; Bellora, F.; Locatelli, F.; Castellano, A.; et al. PD-L1 expression in metastatic neuroblastoma as an additional mechanism for limiting immune surveillance. Oncoimmunology 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Brody, R.; Zhang, Y.; Ballas, M.; Siddiqui, M.K.; Gupta, P.; Barker, C.; Midha, A.; Walker, J. PD-L1 expression in advanced NSCLC: Insights into risk stratification and treatment selection from a systematic literature review. Lung Cancer 2017, 112, 200–215. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.H.; Chan, L.C.; Li, C.W.; Hsu, J.L.; Hung, M.C. Mechanisms controlling PD-L1 expression in cancer. Mol. Cell 2019, 76, 359–370. [Google Scholar] [CrossRef]
- Chiossone, L.; Vivier, E. Immune checkpoints on innate lymphoid cells. J. Exp. Med. 2017, 214, 1561–1563. [Google Scholar] [CrossRef]
- Davis, A.A.; Patel, V.G. The role of PD-L1 expression as a predictive biomarker: An analysis of all US food and drug administration (FDA) approvals of immune checkpoint inhibitors. J. Immunother. Cancer 2019, 7. [Google Scholar] [CrossRef]
- Mahoney, K.M.; Freeman, G.J.; McDermott, D.F. The next immune-checkpoint inhibitors: Pd-1/pd-l1 blockade in melanoma. Clin. Ther. 2015, 37, 764–782. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Thommen, D.S.; Schreiner, J.; Müller, P.; Herzig, P.; Roller, A.; Belousov, A.; Umana, P.; Pisa, P.; Klein, C.; Bacac, M.; et al. Progression of lung cancer is associated with increased dysfunction of T cells defined by coexpression of multiple inhibitory receptors. Cancer Immunol. Res. 2015, 3, 1344–1354. [Google Scholar] [CrossRef]
- Chapon, M.; Randriamampita, C.; Maubec, E.; Badoual, C.; Fouquet, S.; Wang, S.-F.; Marinho, E.; Farhi, D.; Garcette, M.; Jacobelli, S.; et al. Progressive upregulation of PD-1 in primary and metastatic melanomas associated with blunted TCR signaling in infiltrating T lymphocytes. J. Investig. Derm. 2011, 131, 1300–1307. [Google Scholar] [CrossRef]
- Gros, A.; Robbins, P.F.; Yao, X.; Li, Y.F.; Turcotte, S.; Tran, E.; Wunderlich, J.R.; Mixon, A.; Farid, S.; Dudley, M.E.; et al. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J. Clin. Investig. 2014, 124, 2246–2259. [Google Scholar] [CrossRef]
- Montler, R.; Bell, R.B.; Thalhofer, C.; Leidner, R.; Feng, Z.; Fox, B.A.; Cheng, A.C.; Bui, T.G.; Tucker, C.; Hoen, H.; et al. OX40, PD-1 and CTLA-4 are selectively expressed on tumor-infiltrating T cells in head and neck cancer. Clin. Transl. Immunol. 2016, 5, e70. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.D.; Song, G.W.; Park, S.; Jung, M.K.; Kim, M.H.; Kang, H.J.; Yoo, C.; Yi, K.; Kim, K.H.; Eo, S.; et al. Association between expression level of PD1 by tumor-infiltrating CD8+ T cells and features of hepatocellular carcinoma. Gastroenterology 2018, 155, 1936–1950. [Google Scholar] [CrossRef]
- Zhao, Y.J.; Zhang, J.; Shi, F.; Hu, Z.P.; Wu, J.P.; Wu, G.J.; Wang, R.B.; Zhou, Q.; Chang, H.; Li, Y.N.; et al. Expression of PD-1 on CD4+ tumor-infiltrating lymphocytes in tumor microenvironment associated with pathological characteristics of breast cancer. J. Immunol. Res. 2018, 2018, 5690258. [Google Scholar] [CrossRef]
- Kumagai, S.; Togashi, Y.; Kamada, T.; Sugiyama, E.; Nishinakamura, H.; Takeuchi, Y.; Vitaly, K.; Itahashi, K.; Maeda, Y.; Matsui, S.; et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat. Immunol. 2020, 21, 1346–1358. [Google Scholar] [CrossRef]
- Mazzaschi, G.; Madeddu, D.; Falco, A.; Bocchialini, G.; Goldoni, M.; Sogni, F.; Armani, G.; Lagrasta, C.A.; Lorusso, B.; Mangiaracina, C.; et al. Low PD-1 expression in cytotoxic CD8 + tumor-infiltrating lymphocytes confers an immune-privileged tissue microenvironment in NSCLC with a prognostic and predictive value. Clin. Cancer Res. 2018, 24, 407–419. [Google Scholar] [CrossRef]
- Cader, F.Z.; Schackmann, R.C.J.; Hu, X.; Wienand, K.; Redd, R.; Chapuy, B.; Ouyang, J.; Paul, N.; Gjini, E.; Lipschitz, M.; et al. Mass cytometry of Hodgkin lymphoma reveals a CD41 regulatory T-cell–rich and exhausted T-effector microenvironment. Blood 2018, 132, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Myklebust, J.H.; Irish, J.M.; Brody, J.; Czerwinski, D.K.; Houot, R.; Kohrt, H.E.; Timmerman, J.; Said, J.; Green, M.R.; Delabie, J.; et al. High PD-1 expression and suppressed cytokine signaling distinguish T cells infiltrating follicular lymphoma tumors from peripheral T cells. Blood 2013, 121, 1367–1376. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, R.; Fan, P.; Yao, X.; Qin, L.; Peng, Y.; Ma, M.; Asley, N.; Chang, X.; Feng, Y.; et al. A comprehensive analysis of key immune checkpoint receptors on tumor-infiltrating t cells from multiple types of cancer. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef]
- Tassi, E.; Grazia, G.; Vegetti, C.; Bersani, I.; Bertolini, G.; Molla, A.; Baldassari, P.; Andriani, F.; Roz, L.; Sozzi, G.; et al. Early effector T lymphocytes coexpress multiple inhibitory receptors in primary non-small cell lung cancer. Cancer Res. 2017, 77, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Sprengers, D.; Boor, P.P.C.; Doukas, M.; Schutz, H.; Mancham, S.; Pedroza-Gonzalez, A.; Polak, W.G.; de Jonge, J.; Gaspersz, M.; et al. Antibodies against immune checkpoint molecules restore functions of tumor-infiltrating t cells in hepatocellular carcinomas. Gastroenterology 2017, 153, 1107–1119. [Google Scholar] [CrossRef] [PubMed]
- Jinhua, X.; Ji, W.; Shouliang, C.; Liangfeng, Z.; Feiyue, J.; Lin, Y.; Yan, Z.; Haoming, J. Expression of immune checkpoints in T cells of esophageal cancer patients. Oncotarget 2016, 7, 63669–63678. [Google Scholar] [CrossRef]
- Egelston, C.A.; Avalos, C.; Tu, T.Y.; Simons, D.L.; Jimenez, G.; Jung, J.Y.; Melstrom, L.; Margolin, K.; Yim, J.H.; Kruper, L.; et al. Human breast tumor-infiltrating CD8+ T cells retain polyfunctionality despite PD-1 expression. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Benson, D.M.; Bakan, C.E.; Mishra, A.; Hofmeister, C.C.; Efebera, Y.; Becknell, B.; Baiocchi, R.A.; Zhang, J.; Yu, J.; Smith, M.K.; et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: A therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood 2010, 116, 2286–2294. [Google Scholar] [CrossRef]
- Liu, Y.; Cheng, Y.; Xu, Y.; Wang, Z.; Du, X.; Li, C.; Peng, J.; Gao, L.; Liang, X.; Ma, C. Increased expression of programmed cell death protein 1 on NK cells inhibits NK-cell-mediated anti-tumor function and indicates poor prognosis in digestive cancers. Oncogene 2017, 36, 6143–6153. [Google Scholar] [CrossRef]
- Beldi-Ferchiou, A.; Lambert, M.; Dogniaux, S.; Vély, F.; Vivier, E.; Olive, D.; Dupuy, S.; Levasseur, F.; Zucman, D.; Lebbé, C.; et al. PD-1 mediates functional exhaustion of activated NK cells in patients with Kaposi sarcoma. Oncotarget 2016, 7, 72961–72977. [Google Scholar] [CrossRef]
- Vari, F.; Arpon, D.; Keane, C.; Hertzberg, M.S.; Talaulikar, D.; Jain, S.; Cui, Q.; Han, E.; Tobin, J.; Bird, R.; et al. Immune evasion via PD-1/PD-L1 on NK cells and monocyte/macrophages is more prominent in Hodgkin lymphoma than DLBCL. Blood 2018, 131, 1809–1819. [Google Scholar] [CrossRef]
- MacFarlane, A.W.; Jillab, M.; Plimack, E.R.; Hudes, G.R.; Uzzo, R.G.; Litwin, S.; Dulaimi, E.; Al-Saleem, T.; Campbell, K.S. PD-1 expression on peripheral blood cells increases with stage in renal cell carcinoma patients and is rapidly reduced after surgical tumor resection. Cancer Immunol. Res. 2014, 2, 320–331. [Google Scholar] [CrossRef]
- Hsu, J.; Hodgins, J.J.; Marathe, M.; Nicolai, C.J.; Bourgeois-Daigneault, M.C.; Trevino, T.N.; Azimi, C.S.; Scheer, A.K.; Randolph, H.E.; Thompson, T.W.; et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J. Clin. Investig. 2018, 128, 4654–4668. [Google Scholar] [CrossRef]
- Niu, C.; Li, M.; Zhu, S.; Chen, Y.; Zhou, L.; Xu, D.; Xu, J.; Li, Z.; Li, W.; Cui, J. Pd-1-positive natural killer cells have a weaker antitumor function than that of pd-1-negative natural killer cells in lung cancer. Int. J. Med. Sci. 2020, 17, 1964–1973. [Google Scholar] [CrossRef] [PubMed]
- Trefny, M.P.; Kaiser, M.; Stanczak, M.A.; Herzig, P.; Savic, S.; Wiese, M.; Lardinois, D.; Läubli, H.; Uhlenbrock, F.; Zippelius, A. PD-1+ natural killer cells in human non-small cell lung cancer can be activated by PD-1/PD-L1 blockade. Cancer Immunol. Immunother. 2020, 69, 1505–1517. [Google Scholar] [CrossRef]
- Tumino, N.; Martini, S.; Munari, E.; Scordamaglia, F.; Besi, F.; Mariotti, F.R.; Bogina, G.; Mingari, M.C.; Vacca, P.; Moretta, L. Presence of innate lymphoid cells in pleural effusions of primary and metastatic tumors: Functional analysis and expression of PD-1 receptor. Int. J. Cancer 2019, 145, 1660–1668. [Google Scholar] [CrossRef] [PubMed]
- Salimi, M.; Wang, R.; Yao, X.; Li, X.; Wang, X.; Hu, Y.; Chang, X.; Fan, P.; Dong, T.; Ogg, G. Activated innate lymphoid cell populations accumulate in human tumour tissues. BMC Cancer 2018, 18. [Google Scholar] [CrossRef]
- Mallett, G.; Laurence, A.; Amarnath, S. Programmed cell death-1 receptor (Pd-1)-mediated regulation of innate lymphoid cells. Int. J. Mol. Sci. 2019, 20, 2836. [Google Scholar] [CrossRef]
- Quatrini, L.; Vacca, P.; Tumino, N.; Besi, F.; Di Pace, A.L.; Scordamaglia, F.; Martini, S.; Munari, E.; Mingari, M.C.; Ugolini, S.; et al. Glucocorticoids and the cytokines IL-12, IL-15, and IL-18 present in the tumor microenvironment induce PD-1 expression on human natural killer cells. J. Allergy Clin. Immunol. 2021, 147, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Quatrini, L.; Wieduwild, E.; Escaliere, B.; Filtjens, J.; Chasson, L.; Laprie, C.; Vivier, E.; Ugolini, S. Endogenous glucocorticoids control host resistance to viral infection through the tissue-specific regulation of PD-1 expression on NK cells. Nat. Immunol. 2018, 19, 954–962. [Google Scholar] [CrossRef]
- Xing, K.; Gu, B.; Zhang, P.; Wu, X. Dexamethasone enhances programmed cell death 1 (PD-1) expression during T cell activation: An insight into the optimum application of glucocorticoids in anti-cancer therapy. BMC Immunol. 2015, 16. [Google Scholar] [CrossRef]
- Maeda, N.; Maruhashi, T.; Sugiura, D.; Shimizu, K.; Okazaki, I.M.; Okazaki, T. Glucocorticoids potentiate the inhibitory capacity of programmed cell death 1 by up-regulating its expression on T cells. J. Biol. Chem. 2019, 294, 19896–19906. [Google Scholar] [CrossRef]
- Acharya, N.; Madi, A.; Zhang, H.; Klapholz, M.; Escobar, G.; Dulberg, S.; Christian, E.; Ferreira, M.; Dixon, K.O.; Fell, G.; et al. Endogenous glucocorticoid signaling regulates CD8+ T cell differentiation and development of dysfunction in the tumor microenvironment. Immunity 2020, 53, 658–671. [Google Scholar] [CrossRef]
- Gaucher, L.; Adda, L.; Séjourné, A.; Joachim, C.; Chaby, G.; Poulet, C.; Liabeuf, S.; Gras-Champel, V.; Masmoudi, K.; Moreira, A.; et al. Impact of the corticosteroid indication and administration route on overall survival and the tumor response after immune checkpoint inhibitor initiation. Ther. Adv. Med. Oncol. 2021, 13. [Google Scholar] [CrossRef]
- Garon, E.B.; Hellmann, M.D.; Rizvi, N.A.; Carcereny, E.; Leighl, N.B.; Ahn, M.-J.; Eder, J.P.; Balmanoukian, A.S.; Aggarwal, C.; Horn, L.; et al. Five-year overall survival for patients with advanced non‒small-cell lung cancer treated with pembrolizumab: Results from the phase I KEYNOTE-001 study. J. Clin. Oncol. 2019, 37, 2518–2527. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Govindan, R.; Anders, R.A.; Antonia, S.J.; Sagorsky, S.; Davies, M.J.; Dubinett, S.M.; Ferris, A.; Gandhi, L.; Garon, E.B.; et al. The society for immunotherapy of cancer consensus statement on immunotherapy for the treatment of non-small cell lung cancer (NSCLC). J. Immunother. Cancer 2018, 6, 75. [Google Scholar] [CrossRef] [PubMed]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.; Weber, J.S.; et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann. Oncol. 2019, 30, 582–588. [Google Scholar] [CrossRef]
- US Food and Drug Administration. List of Cleared or Approved Companion Diagnostic Devices (In Vitro and Imaging Tools); US Food and Drug Administration: Silver Spring, MD, USA, 2021. [Google Scholar]
- US Food and Drug Administration. Durvalumab (Imfinzi); US Food and Drug Administration: Silver Spring, MD, USA, 2017. [Google Scholar]
- Doroshow, D.B.; Bhalla, S.; Beasley, M.B.; Sholl, L.M.; Kerr, K.M.; Gnjatic, S.; Wistuba, I.I.; Rimm, D.L.; Tsao, M.S.; Hirsch, F.R. PD-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2021. [Google Scholar] [CrossRef]
- Hirsch, F.R.; McElhinny, A.; Stanforth, D.; Ranger-Moore, J.; Jansson, M.; Kulangara, K.; Richardson, W.; Towne, P.; Hanks, D.; Vennapusa, B.; et al. PD-L1 Immunohistochemistry assays for lung cancer: Results from Phase 1 of the blueprint PD-L1 IHC assay comparison project. J. Thorac. Oncol. 2017, 12, 208–222. [Google Scholar] [CrossRef]
- Rimm, D.L.; Han, G.; Taube, J.M.; Yi, E.S.; Bridge, J.A.; Flieder, D.B.; Homer, R.; West, W.W.; Wu, H.; Roden, A.C.; et al. A prospective, multi-institutional, pathologist-based assessment of 4 immunohistochemistry assays for PD-L1 expression in non–small cell lung cancer. JAMA Oncol. 2017, 3, 1051–1058. [Google Scholar] [CrossRef]
- Grote, H.J.; Feng, Z.; Schlichting, M.; Helwig, C.; Ruisi, M.; Jin, H.; Scheuenpflug, J.; Gann, C.-N.; Su, Z.; Reck, M.; et al. Programmed death-ligand 1 immunohistochemistry assay comparison studies in NSCLC: Characterization of the 73-10 assay. J. Thorac. Oncol. 2020, 15, 1306–1316. [Google Scholar] [CrossRef]
- Munari, E.; Rossi, G.; Zamboni, G.; Lunardi, G.; Marconi, M.; Sommaggio, M.; Netto, G.J.; Hoque, M.O.; Brunelli, M.; Martignoni, G.; et al. PD-L1 assays 22C3 and SP263 are not interchangeable in non–small cell lung cancer when considering clinically relevant cutoffs. Am. J. Surg. Pathol. 2018, 42, 1384–1389. [Google Scholar] [CrossRef]
- Torlakovic, E.; Lim, H.J.; Adam, J.; Barnes, P.; Bigras, G.; Chan, A.W.H.; Cheung, C.C.; Chung, J.-H.; Couture, C.; Fiset, P.O.; et al. “Interchangeability” of PD-L1 immunohistochemistry assays: A meta-analysis of diagnostic accuracy. Mod. Pathol. 2020, 33, 4–17. [Google Scholar] [CrossRef]
- Munari, E.; Zamboni, G.; Lunardi, G.; Marconi, M.; Brunelli, M.; Martignoni, G.; Netto, G.J.; Quatrini, L.; Vacca, P.; Moretta, L.; et al. PD-L1 expression in non–small cell lung cancer: Evaluation of the diagnostic accuracy of a laboratory-developed test using clone E1L3N in comparison with 22C3 and SP263 assays. Hum. Pathol. 2019, 90, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Kerr, K.M.; Tsao, M.-S.; Nicholson, A.G.; Yatabe, Y.; Wistuba, I.I.; Hirsch, F.R. Programmed death-ligand 1 immunohistochemistry in lung cancer: In what state is this art? J. Thorac. Oncol. 2015, 10, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Kerr, K.M.; Hirsch, F.R. Programmed death ligand-1 immunohistochemistry: Friend or foe? Arch. Pathol. Lab. Med. 2016, 140, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Fehrenbacher, L.; Spira, A.; Ballinger, M.; Kowanetz, M.; Vansteenkiste, J.; Mazieres, J.; Park, K.; Smith, D.; Artal-Cortes, A.; Lewanski, C.; et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): A multicentre, open-label, phase 2 randomised controlled trial. Lancet 2016, 387, 1837–1846. [Google Scholar] [CrossRef]
- Vennapusa, B.; Baker, B.; Kowanetz, M.; Boone, J.; Menzl, I.; Bruey, J.-M.; Fine, G.; Mariathasan, S.; McCaffery, I.; Mocci, S.; et al. Development of a PD-L1 Complementary Diagnostic Immunohistochemistry Assay (SP142) for Atezolizumab. Appl. Immunohistochem. Mol. Morphol. 2019, 27, 92–100. [Google Scholar] [CrossRef]
- Tsao, M.S.; Kerr, K.M.; Kockx, M.; Beasley, M.-B.; Borczuk, A.C.; Botling, J.; Bubendorf, L.; Chirieac, L.; Chen, G.; Chou, T.-Y.; et al. PD-L1 immunohistochemistry comparability study in real-life clinical samples: Results of blueprint phase 2 project. J. Thorac. Oncol. 2018, 13, 1302–1311. [Google Scholar] [CrossRef] [PubMed]
- Cooper, W.A.; Russell, P.A.; Cherian, M.; Duhig, E.E.; Godbolt, D.; Jessup, P.J.; Khoo, C.; Leslie, C.; Mahar, A.; Moffat, D.F.; et al. Intra- and interobserver reproducibility assessment of PD-L1 biomarker in non–small cell lung cancer. Clin. Cancer Res. 2017, 23, 4569–4577. [Google Scholar] [CrossRef]
- Kulangara, K.; Zhang, N.; Corigliano, E.; Guerrero, L.; Waldroup, S.; Jaiswal, D.; MS, M.J.; Shah, S.; Hanks, D.; Wang, J.; et al. Clinical utility of the combined positive score for programmed death ligand-1 expression and the approval of pembrolizumab for treatment of gastric cancer. Arch. Pathol. Lab. Med. 2019, 143, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Koh, J.; Na, H.Y.; Kwak, Y.; Lee, K.-W.; Ahn, S.-H.; Park, D.J.; Kim, H.-H.; Lee, H.S. PD-L1 Testing in gastric cancer by the combined positive score of the 22C3 PharmDx and SP263 assay with clinically relevant cut-offs. Cancer Res. Treat. 2020, 52, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Munari, E.; Zamboni, G.; Marconi, M.; Sommaggio, M.; Brunelli, M.; Martignoni, G.; Netto, G.J.; Moretta, F.; Mingari, M.C.; Salgarello, M.; et al. PD-L1 expression heterogeneity in non-small cell lung cancer: Evaluation of small biopsies reliability. Oncotarget 2017, 8, 90123–90131. [Google Scholar] [CrossRef]
- Noll, B.; Wang, W.-L.; Gong, Y.; Zhao, J.; Kalhor, N.; Prieto, V.; Staerkel, G.; Roy-Chowdhuri, S. Programmed death ligand 1 testing in non-small cell lung carcinoma cytology cell block and aspirate smear preparations. Cancer Cytopathol. 2018, 126, 342–352. [Google Scholar] [CrossRef]
- Munari, E.; Zamboni, G.; Lunardi, G.; Marchionni, L.; Marconi, M.; Sommaggio, M.; Brunelli, M.; Martignoni, G.; Netto, G.J.; Hoque, M.O.; et al. PD-L1 expression heterogeneity in non–small cell lung cancer: Defining criteria for harmonization between biopsy specimens and whole sections. J. Thorac. Oncol. 2018, 13, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Munari, E.; Zamboni, G.; Sighele, G.; Marconi, M.; Sommaggio, M.; Lunardi, G.; Rossi, G.; Cavazza, A.; Moretta, F.; Gilioli, E.; et al. Expression of programmed cell death ligand 1 in non–small cell lung cancer: Comparison between cytologic smears, core biopsies, and whole sections using the SP263 assay. Cancer Cytopathol. 2019, 127, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Ratcliffe, M.J.; Sharpe, A.; Midha, A.; Barker, C.; Scott, M.; Scorer, P.; Al-Masri, H.; Rebelatto, M.C.; Walker, J. Agreement between programmed cell death ligand-1 diagnostic assays across multiple protein expression cutoffs in non–small cell lung cancer. Clin. Cancer Res. 2017, 23, 3585–3591. [Google Scholar] [CrossRef] [PubMed]
- Aeffner, F.; Adissu, H.A.; Boyle, M.C.; Cardiff, R.D.; Hagendorn, E.; Hoenerhoff, M.J.; Klopfleisch, R.; Newbigging, S.; Schaudien, D.; Turner, O.; et al. Digital microscopy, image analysis, and virtual slide repository. ILAR J. 2018, 59, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Samstein, R.M.; Lee, C.-H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Roszik, J.; Haydu, L.E.; Hess, K.R.; Oba, J.; Joon, A.Y.; Siroy, A.E.; Karpinets, T.V.; Stingo, F.C.; Baladandayuthapani, V.; Tetzlaff, M.T.; et al. Novel algorithmic approach predicts tumor mutation load and correlates with immunotherapy clinical outcomes using a defined gene mutation set. BMC Med. 2016, 14, 168. [Google Scholar] [CrossRef]
- US Food and Drug Administration. FDA Approves Pembrolizumab for Adults and Children With TMB-H Solid Tumors; US Food and Drug Administration: Silver Spring, MD, USA, 2020. [Google Scholar]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor mutational burden and response rate to PD-1 inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Lee, V.; Murphy, A.; Le, D.T.; Diaz, L.A. Mismatch repair deficiency and response to immune checkpoint blockade. Oncologist 2016, 21, 1200–1211. [Google Scholar] [CrossRef]
- Cortes-Ciriano, I.; Lee, S.; Park, W.-Y.; Kim, T.-M.; Park, P.J. A molecular portrait of microsatellite instability across multiple cancers. Nat. Commun. 2017, 8, 15180. [Google Scholar] [CrossRef]
- US Food and Drug Administration. FDA Grants Accelerated Approval to Pembrolizumab for First Tissue/Site Agnostic Indication; US Food and Drug Administration: Silver Spring, MD, USA, 2017. [Google Scholar]
- Thomas, N.E.; Busam, K.J.; From, L.; Kricker, A.; Armstrong, B.K.; Anton-Culver, H.; Gruber, S.B.; Gallagher, R.P.; Zanetti, R.; Rosso, S.; et al. Tumor-infiltrating lymphocyte grade in primary melanomas is independently associated with melanoma-specific survival in the population-based genes, environment and melanoma study. J. Clin. Oncol. 2013, 31, 4252–4259. [Google Scholar] [CrossRef]
- Brambilla, E.; Le Teuff, G.; Marguet, S.; Lantuejoul, S.; Dunant, A.; Graziano, S.; Pirker, R.; Douillard, J.-Y.; Le Chevalier, T.; Filipits, M.; et al. Prognostic effect of tumor lymphocytic infiltration in resectable non–small-cell lung cancer. J. Clin. Oncol. 2016, 34, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Wang, Z.; Qu, X.; Zhang, Z. Prognostic value of tumor-infiltrating lymphocytes in patients with triple-negative breast cancer: A systematic review and meta-analysis. BMC Cancer 2020, 20, 179. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Soria, J.-C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef]
- Galon, J.; Mlecnik, B.; Bindea, G.; Angell, H.K.; Berger, A.; Lagorce, C.; Lugli, A.; Zlobec, I.; Hartmann, A.; Bifulco, C.; et al. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J. Pathol. 2014, 232, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Lanzi, A. Immunoscore and its introduction in clinical practice. Q. J. Nucl. Med. Mol. Imaging 2020, 64, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.S.; D’Angelo, S.P.; Minor, D.; Hodi, F.S.; Gutzmer, R.; Neyns, B.; Hoeller, C.; Khushalani, N.I.; Miller, W.H.; Lao, C.D.; et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): A randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015, 16, 375–384. [Google Scholar] [CrossRef]
- Robert, C.; Ribas, A.; Wolchok, J.D.; Hodi, F.S.; Hamid, O.; Kefford, R.; Weber, J.S.; Joshua, A.M.; Hwu, W.-J.; Gangadhar, T.C.; et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: A randomised dose-comparison cohort of a phase 1 trial. Lancet 2014, 384, 1109–1117. [Google Scholar] [CrossRef]
- Mok, T.S.K.; Wu, Y.-L.; Kudaba, I.; Kowalski, D.M.; Cho, B.C.; Turna, H.Z.; Castro, G.; Srimuninnimit, V.; Laktionov, K.K.; Bondarenko, I.; et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): A randomised, open-label, controlled, phase 3 trial. Lancet 2019, 393, 1819–1830. [Google Scholar] [CrossRef]
- Herbst, R.S.; Baas, P.; Kim, D.-W.; Felip, E.; Pérez-Gracia, J.L.; Han, J.-Y.; Molina, J.; Kim, J.-H.; Arvis, C.D.; Ahn, M.-J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet 2016, 387, 1540–1550. [Google Scholar] [CrossRef]
- Langer, C.J.; Gadgeel, S.M.; Borghaei, H.; Papadimitrakopoulou, V.A.; Patnaik, A.; Powell, S.F.; Gentzler, R.D.; Martins, R.G.; Stevenson, J.P.; Jalal, S.I.; et al. Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: A randomised, phase 2 cohort of the open-label KEYNOTE-021 study. Lancet Oncol. 2016, 17, 1497–1508. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Luft, A.; Vicente, D.; Tafreshi, A.; Gümüş, M.; Mazières, J.; Hermes, B.; Çay Şenler, F.; Csőszi, T.; Fülöp, A.; et al. Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. N. Engl. J. Med. 2018, 379, 2040–2051. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez–Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Updated analysis of KEYNOTE-024: Pembrolizumab versus platinum-based chemotherapy for advanced non–small-cell lung cancer with PD-l1 tumor proportion score of 50% or greater. J. Clin. Oncol. 2019, 37, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Von Pawel, J.; Bordoni, R.; Satouchi, M.; Fehrenbacher, L.; Cobo, M.; Han, J.Y.; Hida, T.; Moro-Sibilot, D.; Conkling, P.; Gandara, D.R.; et al. Long-term survival in patients with advanced non–small-cell lung cancer treated with atezolizumab versus docetaxel: Results from the randomised phase III OAK study. Eur. J. Cancer 2019, 107, 124–132. [Google Scholar] [CrossRef]
- Reck, M.; Mok, T.S.K.; Nishio, M.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; et al. Atezolizumab plus bevacizumab and chemotherapy in non-small-cell lung cancer (IMpower150): Key subgroup analyses of patients with EGFR mutations or baseline liver metastases in a randomised, open-label phase 3 trial. Lancet Respir. Med. 2019, 7, 387–401. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus docetaxel in advanced nonsquamous non–small-cell lung cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Kurata, T.; Chiappori, A.; Lee, K.H.; De Wit, M.; et al. Overall survival with durvalumab after chemoradiotherapy in stage III NSCLC. N. Engl. J. Med. 2018, 379, 2342–2350. [Google Scholar] [CrossRef]
- Horn, L.; Mansfield, A.S.; Szczęsna, A.; Havel, L.; Krzakowski, M.; Hochmair, M.J.; Huemer, F.; Losonczy, G.; Johnson, M.L.; Nishio, M.; et al. First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N. Engl. J. Med. 2018, 379, 2220–2229. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Dvorkin, M.; Chen, Y.; Reinmuth, N.; Hotta, K.; Trukhin, D.; Statsenko, G.; Hochmair, M.J.; Özgüroğlu, M.; Ji, J.H.; et al. Durvalumab plus platinum–etoposide versus platinum–etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): A randomised, controlled, open-label, phase 3 trial. Lancet 2019, 394, 1929–1939. [Google Scholar] [CrossRef]
- Antonia, S.J.; López-Martin, J.A.; Bendell, J.; Ott, P.A.; Taylor, M.; Eder, J.P.; Jäger, D.; Pietanza, M.C.; Le, D.T.; de Braud, F.; et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): A multicentre, open-label, phase 1/2 trial. Lancet Oncol. 2016, 17, 883–895. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.-A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in advanced triple-negative breast cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Weber, J.; Mandala, M.; Del Vecchio, M.; Gogas, H.J.; Arance, A.M.; Cowey, C.L.; Dalle, S.; Schenker, M.; Chiarion-Sileni, V.; Marquez-Rodas, I.; et al. Adjuvant nivolumab versus ipilimumab in resected stage III or IV melanoma. N. Engl. J. Med. 2017, 377, 1824–1835. [Google Scholar] [CrossRef]
- Eggermont, A.M.M.; Chiarion-sileni, V.; Grob, J.-J.; Dummer, R.; Wolchok, J.D.; Schmidt, H.; Hamid, O.; Robert, C.; Ascierto, P.A.; Richards, J.M.; et al. Prolonged survival in stage III melanoma with ipilimumab adjuvant therapy. N. Engl. J. Med. 2016, 375, 1845–1855. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Puzanov, I.; Dummer, R.; Schadendorf, D.; Hamid, O.; Robert, C.; Hodi, F.S.; Schachter, J.; Pavlick, A.C.; Lewis, K.D.; et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): A randomised, controlled, phase 2 trial. Lancet Oncol. 2015, 16, 908–918. [Google Scholar] [CrossRef]
- Nghiem, P.; Bhatia, S.; Lipson, E.J.; Sharfman, W.H.; Kudchadkar, R.R.; Brohl, A.S.; Friedlander, P.A.; Daud, A.; Kluger, H.M.; Reddy, S.A.; et al. Durable tumor regression and overall survival in patients with advanced merkel cell carcinoma receiving pembrolizumab as first-line therapy. J. Clin. Oncol. 2019, 37, 693–702. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Russell, J.; Hamid, O.; Bhatia, S.; Terheyden, P.; D’Angelo, S.P.; Shih, K.C.; Lebbé, C.; Linette, G.P.; Milella, M.; et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: A multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 1374–1385. [Google Scholar] [CrossRef]
- Migden, M.R.; Rischin, D.; Schmults, C.D.; Guminski, A.; Hauschild, A.; Lewis, K.D.; Chung, C.H.; Hernandez-Aya, L.; Lim, A.M.; Chang, A.L.S.; et al. PD-1 blockade with cemiplimab in advanced cutaneous squamous-cell carcinoma. N. Engl. J. Med. 2018, 379, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Arén Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B.; et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Necchi, A.; Joseph, R.W.; Loriot, Y.; Hoffman-Censits, J.; Perez-Gracia, J.L.; Petrylak, D.P.; Derleth, C.L.; Tayama, D.; Zhu, Q.; Ding, B.; et al. Atezolizumab in platinum-treated locally advanced or metastatic urothelial carcinoma: Post-progression outcomes from the phase II IMvigor210 study. Ann. Oncol. 2017, 28, 3044–3050. [Google Scholar] [CrossRef] [PubMed]
- Balar, A.V.; Castellano, D.; O’Donnell, P.H.; Grivas, P.; Vuky, J.; Powles, T.; Plimack, E.R.; Hahn, N.M.; de Wit, R.; Pang, L.; et al. First-line pembrolizumab in cisplatin-ineligible patients with locally advanced and unresectable or metastatic urothelial cancer (KEYNOTE-052): A multicentre, single-arm, phase 2 study. Lancet Oncol. 2017, 18, 1483–1492. [Google Scholar] [CrossRef]
- Sharma, P.; Retz, M.; Siefker-Radtke, A.; Baron, A.; Necchi, A.; Bedke, J.; Plimack, E.R.; Vaena, D.; Grimm, M.-O.; Bracarda, S.; et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): A multicentre, single-arm, phase 2 trial. Lancet Oncol. 2017, 18, 312–322. [Google Scholar] [CrossRef]
- Massard, C.; Gordon, M.S.; Sharma, S.; Rafii, S.; Wainberg, Z.A.; Luke, J.; Curiel, T.J.; Colon-Otero, G.; Hamid, O.; Sanborn, R.E.; et al. Safety and efficacy of durvalumab (MEDI4736), an anti-programmed cell death ligand-1 immune checkpoint inhibitor, in patients with advanced urothelial bladder cancer. J. Clin. Oncol. 2016, 34, 3119–3125. [Google Scholar] [CrossRef]
- Keilholz, U.; Mehnert, J.M.; Bauer, S.; Bourgeois, H.; Patel, M.R.; Gravenor, D.; Nemunaitis, J.J.; Taylor, M.H.; Wyrwicz, L.; Lee, K.-W.; et al. Avelumab in patients with previously treated metastatic melanoma: Phase 1b results from the JAVELIN solid tumor trial. J. Immunother. Cancer 2019, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Bellmunt, J.; De Wit, R.; Vaughn, D.J.; Fradet, Y.; Lee, J.-L.; Fong, L.; Vogelzang, N.J.; Climent, M.A.; Petrylak, D.P.; Choueiri, T.K.; et al. Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N. Engl. J. Med. 2017, 376, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.C.; Ros, W.; Delord, J.P.; Perets, R.; Italiano, A.; Shapira-Frommer, R.; Manzuk, L.; Piha-Paul, S.A.; Xu, L.; Zeigenfuss, S.; et al. Efficacy and safety of pembrolizumab in previously treated advanced cervical cancer: Results from the phase II KEYNOTE-158 study. J. Clin. Oncol. 2019, 37, 1470–1478. [Google Scholar] [CrossRef] [PubMed]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Kojima, T.; Muro, K.; Francois, E.; Hsu, C.-H.; Moriwaki, T.; Kim, S.-B.; Lee, S.-H.; Bennouna, J.; Kato, K.; Lin, S.; et al. Pembrolizumab versus chemotherapy as second-line therapy for advanced esophageal cancer: Phase III KEYNOTE-181 study. J. Clin. Oncol. 2019, 37, 2. [Google Scholar] [CrossRef]
- Fuchs, C.S.; Doi, T.; Jang, R.W.; Muro, K.; Satoh, T.; Machado, M.; Sun, W.; Jalal, S.I.; Shah, M.A.; Metges, J.-P.; et al. Safety and efficacy of pembrolizumab monotherapy in patients with previously treated advanced gastric and gastroesophageal junction cancer. JAMA Oncol. 2018, 4, e180013. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; de Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/ mismatch repair–deficient cancer: Results from the phase II KEYNOTE-158 study. J. Clin. Oncol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Rischin, D.; Harrington, K.J.; Greil, R.; Soulieres, D.; Tahara, M.; de Castro, G.; Psyrri, A.; Baste, N.; Neupane, P.C.; Bratland, A.; et al. Protocol-specified final analysis of the phase 3 KEYNOTE-048 trial of pembrolizumab (pembro) as first-line therapy for recurrent/metastatic head and neck squamous cell carcinoma (R/M HNSCC). J. Clin. Oncol. 2019, 37, 6000. [Google Scholar] [CrossRef]
- Seiwert, T.Y.; Burtness, B.; Mehra, R.; Weiss, J.; Berger, R.; Eder, J.P.; Heath, K.; McClanahan, T.; Lunceford, J.; Gause, C.; et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): An open-label, multicentre, phase 1b trial. Lancet Oncol. 2016, 17, 956–965. [Google Scholar] [CrossRef]
- Ferris, R.L.; Blumenschein, G.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef]
- Chen, R.; Zinzani, P.L.; Fanale, M.A.; Armand, P.; Johnson, N.A.; Brice, P.; Radford, J.; Ribrag, V.; Molin, D.; Vassilakopoulos, T.P.; et al. Phase II study of the efficacy and safety of pembrolizumab for relapsed/refractory classic Hodgkin Lymphoma. J. Clin. Oncol. 2017, 35, 2125–2132. [Google Scholar] [CrossRef]
- Zinzani, P.; Thieblemont, C.; Melnichenko, V.; Osmanov, D.; Bouabdallah, K.; Walewski, J.; Majlis, A.; Fogliatto, L.; Caballero Barrigón, M.D.; Christian, B.; et al. Efficacy and safety of pembrolizumab in relapsed/refractory primary mediastinal large B-cell lymphoma (rrPMBCL): Interim analysis of the KEYNOTE-170 phase 2 trial. Hematol. Oncol. 2017, 35, 62–63. [Google Scholar] [CrossRef]
- Andorsky, D.J.; Yamada, R.E.; Said, J.; Pinkus, G.S.; Betting, D.J.; Timmerman, J.M. Programmed death ligand 1 is expressed by non-Hodgkin lymphomas and inhibits the activity of tumor-associated T cells. Clin. Cancer Res. 2011, 17, 4232–4244. [Google Scholar] [CrossRef]
- Chen, X.; Liu, S.; Wang, L.; Zhang, W.; Ji, Y.; Ma, X. Clinical significance of B7-H1 (PD-L1) expression in human acute leukemia. Cancer Biol. Ther. 2008, 7, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Tao, R.; Wang, X.; Wang, Y.; Mao, Y.; Zhou, L.F. B7-H1 is correlated with malignancy-grade gliomas but is not expressed exclusively on tumor stem-like cells. Neuro. Oncol. 2009, 11, 757–766. [Google Scholar] [CrossRef]
- Kabir, T.F.; Chauhan, A.; Anthony, L.; Hildebrandt, G.C. Immune checkpoint inhibitors in pediatric solid tumors: Status in 2018. Ochsner J. 2018, 18, 370–376. [Google Scholar] [CrossRef]
- Chowdhury, F.; Dunn, S.; Mitchell, S.; Mellows, T.; Ashton-Key, M.; Gray, J.C. PD-L1 and CD8 + PD1 + lymphocytes exist as targets in the pediatric tumor microenvironment for immunomodulatory therapy. Oncoimmunology 2015, 4, e1029701. [Google Scholar] [CrossRef]
- Wagner, L.M.; Adams, V.R. Targeting the PD-1 pathway in pediatric solid tumors and brain tumors. Onco. Targets. Ther. 2017, 10, 2097–2106. [Google Scholar] [CrossRef] [PubMed]
- Park, J.A.; Cheung, N.K.V. Targets and antibody formats for immunotherapy of neuroblastoma. J. Clin. Oncol. 2020, 38, 1836–1848. [Google Scholar] [CrossRef]
- Davis, K.L.; Fox, E.; Merchant, M.S.; Reid, J.M.; Kudgus, R.A.; Liu, X.; Minard, C.G.; Voss, S.; Berg, S.L.; Weigel, B.J.; et al. Nivolumab in children and young adults with relapsed or refractory solid tumours or lymphoma (ADVL1412): A multicentre, open-label, single-arm, phase 1–2 trial. Lancet Oncol. 2020, 21, 541–550. [Google Scholar] [CrossRef]
- Hutzen, B.; Paudel, S.N.; Naeimi Kararoudi, M.; Cassady, K.A.; Lee, D.A.; Cripe, T.P. Immunotherapies for pediatric cancer: Current landscape and future perspectives. Cancer Metastasis Rev. 2019, 38, 573–594. [Google Scholar] [CrossRef]
- Azzaoui, I.; Uhel, F.; Rossille, D.; Pangault, C.; Dulong, J.; Le Priol, J.; Lamy, T.; Houot, R.; Le Gouill, S.; Cartron, G.; et al. T-cell defect in diffuse large B-cell lymphomas involves expansion of myeloid-derived suppressor cells. Blood 2016, 128, 1081–1092. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Redd, P.S.; Lee, J.R.; Savage, N.; Liu, K. The expression profiles and regulation of PD-L1 in tumor-induced myeloid-derived suppressor cells. Oncoimmunology 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Iwata, T.; Kondo, Y.; Kimura, O.; Morosawa, T.; Fujisaka, Y.; Umetsu, T.; Kogure, T.; Inoue, J.; Nakagome, Y.; Shimosegawa, T. PD-L1+ MDSCs are increased in HCC patients and induced by soluble factor in the tumor microenvironment. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Stiff, A.; Trikha, P.; Mundy-Bosse, B.; McMichael, E.; Mace, T.A.; Benner, B.; Kendra, K.; Campbell, A.; Gautam, S.; Abood, D.; et al. Nitric oxide production by myeloid-derived suppressor cells plays a role in impairing Fc receptor–mediated natural killer cell function. Clin. Cancer Res. 2018, 24, 1891–1904. [Google Scholar] [CrossRef]
- Tumino, N.; Di Pace, A.L.; Besi, F.; Quatrini, L.; Vacca, P.; Moretta, L. Interaction between MDSC and NK cells in solid and hematological malignancies: Impact on HSCT. Front. Immunol. 2021, 12, 638841. [Google Scholar] [CrossRef]
- Locati, M.; Curtale, G.; Mantovani, A. Diversity, mechanisms, and significance of macrophage plasticity. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 123–147. [Google Scholar] [CrossRef]


{kind=link}
| Type of Tumors | Cell Subsets | References |
|---|---|---|
| Breast Cancer (BC) | ||
| Invasive ductal BC | CD4+ TILs | [26] |
| Primary BC | CD8+ TILs | [35] |
| Melanoma | ||
| Metastatic melanoma lesions | CD4+, CD8+ TILs | [12] |
| Metastatic melanoma | CD8+ TILs | [23] |
| Malignant melanoma (MM) | CD8+ TILs | [27] |
| Follicular lymphoma (FL) | CD4+ TILs | [30] |
| Hodgkin Lymphoma (HL) | ||
| Primary classical HL | CD4+ TILs | [29] |
| HL and DLBCL | NK PB and Intratumoural | [39] |
| Ovarian carcinoma | NK peritoneal fluid/ascites | [7] |
| Karposi sarcoma | NK PB | [38] |
| Renal cell carcinoma (RCC) | NK PB | [40] |
| Lung cancers | ||
| Primary and metastatic | ILC3 PE | [44] |
| Lung cancer and ADC | NK PE | [47] |
| NK PB | [42] | |
| NSCLC (ADC and SCC) | CD8+ TILs | [28] |
| Advanced and primary NSCLC | CD8+, CD4+ TILs | [27,32] |
| NSCLC | NK Intratumoural | [43] |
| Digestive Cancers | ||
| Gastric cancer | CD8+ TILs | [27] |
| Gastrointestinal (oesophageal, gastric, colon, rectal tumors) | ILC2, ILC3, NK intratumoral | [45] |
| Hepatocellular carcinoma (HCC) | CD8+ TILs | [25] |
| CD4+, CD8+ TILs | [33] | |
| ESCC, HCC, colorectal cancer and biliary cancer | NK Intratumoural and PB | [37] |
| Oesophageal cancer | CD4+ CD8+ PB and TILs | [34] |
| Multiple Myeloma | NK PB | [36] |
| Type | Treatment | Indications (Ref) |
|---|---|---|
| NSCLC | Pembrolizumab | I line (ALK and EGFR wt, TPS ≥ 1%) [95] II line (TPS ≥ 1%) [96] |
| Pembrolizumab + chemotherapy | I line [97,98] | |
| Atezolizumab | I line (ALK and EGFR wt, TPS ≥ 50% and/or IC ≥ 10%) [99] II line [100] | |
| Atezolizumab + chemotherapy | I line in non-squamous histology, ALK and EGFR wt [101] | |
| Nivolumab | II line [102] | |
| Durvalumab | After chemoradiation for unresectable stage III NSCLC [103] | |
| SCLC | Atezolizumab + chemotherapy | I line [104] |
| Durvalumab + chemotherapy | I line [105] | |
| Nivolumab | II line [106] | |
| TNBC | Atezolizumab + chemotherapy | I line (IC ≥ 1%) [107] |
| Melanoma | Pembrolizumab, Nivolumab | Adjuvant treatment after radical surgery [108,109]; I line [110,111]; II line [93,112] |
| Merkel cell carcinoma | Pembrolizumab | I line [113] |
| Avelumab | II line [114] | |
| CSCC | Cemiplimab | I line [115] |
| RCC | Nivolumab + ipilimumab | I-II line [116,117] |
| Pembrolizumab/avelumab + axitinib | I line [118,119] | |
| UC | Pembrolizumab | I line in patients ineligible for cisplatin-containing therapy (CPS ≥ 10%), or patients unfit for platinum-containing chemotherapy [120] |
| Atezolizumab | I line in patients ineligible for cisplatin-containing therapy (IC ≥ 5%), or patients unfit for platinum-containing chemotherapy [121] | |
| Atezolizumab, Nivolumab, Durvalumab, Avelumab, Pembrolizumab | Disease progression during or after platinum-based chemotherapy or within one year after adjuvant or neoadjuvant chemotherapy [120,122,123,124,125] | |
| Cervical cancer | Pembrolizumab | II line (CPS ≥ 1%) [126] |
| HCC | Nivolumab/Pembrolizumab | II line [127,128] |
| Esophageal cancer | Pembrolizumab | II line (CPS ≥ 10%) [129] |
| Gastric/GEJ adenocarcinoma | Pembrolizumab | II line (CPS ≥ 1%) [130] |
| MSI-H dMMR cancers | Pembrolizumab | II line irrespective of primary location [131] |
| HNSCC | Pembrolizumab | I line (CPS ≥ 1%) [132] |
| Pembrolizumab/Nivolumab | II line [133,134] | |
| HL | Pembrolizumab | II line [135] |
| PMLBCL | Pembrolizumab | II line [136] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Munari, E.; Mariotti, F.R.; Quatrini, L.; Bertoglio, P.; Tumino, N.; Vacca, P.; Eccher, A.; Ciompi, F.; Brunelli, M.; Martignoni, G.; et al. PD-1/PD-L1 in Cancer: Pathophysiological, Diagnostic and Therapeutic Aspects. Int. J. Mol. Sci. 2021, 22, 5123. https://doi.org/10.3390/ijms22105123
Munari E, Mariotti FR, Quatrini L, Bertoglio P, Tumino N, Vacca P, Eccher A, Ciompi F, Brunelli M, Martignoni G, et al. PD-1/PD-L1 in Cancer: Pathophysiological, Diagnostic and Therapeutic Aspects. International Journal of Molecular Sciences. 2021; 22(10):5123. https://doi.org/10.3390/ijms22105123
Chicago/Turabian StyleMunari, Enrico, Francesca R. Mariotti, Linda Quatrini, Pietro Bertoglio, Nicola Tumino, Paola Vacca, Albino Eccher, Francesco Ciompi, Matteo Brunelli, Guido Martignoni, and et al. 2021. "PD-1/PD-L1 in Cancer: Pathophysiological, Diagnostic and Therapeutic Aspects" International Journal of Molecular Sciences 22, no. 10: 5123. https://doi.org/10.3390/ijms22105123
APA StyleMunari, E., Mariotti, F. R., Quatrini, L., Bertoglio, P., Tumino, N., Vacca, P., Eccher, A., Ciompi, F., Brunelli, M., Martignoni, G., Bogina, G., & Moretta, L. (2021). PD-1/PD-L1 in Cancer: Pathophysiological, Diagnostic and Therapeutic Aspects. International Journal of Molecular Sciences, 22(10), 5123. https://doi.org/10.3390/ijms22105123