
Biology of the mRNA Splicing Machinery and Its Dysregulation in Cancer Providing Therapeutic Opportunities

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
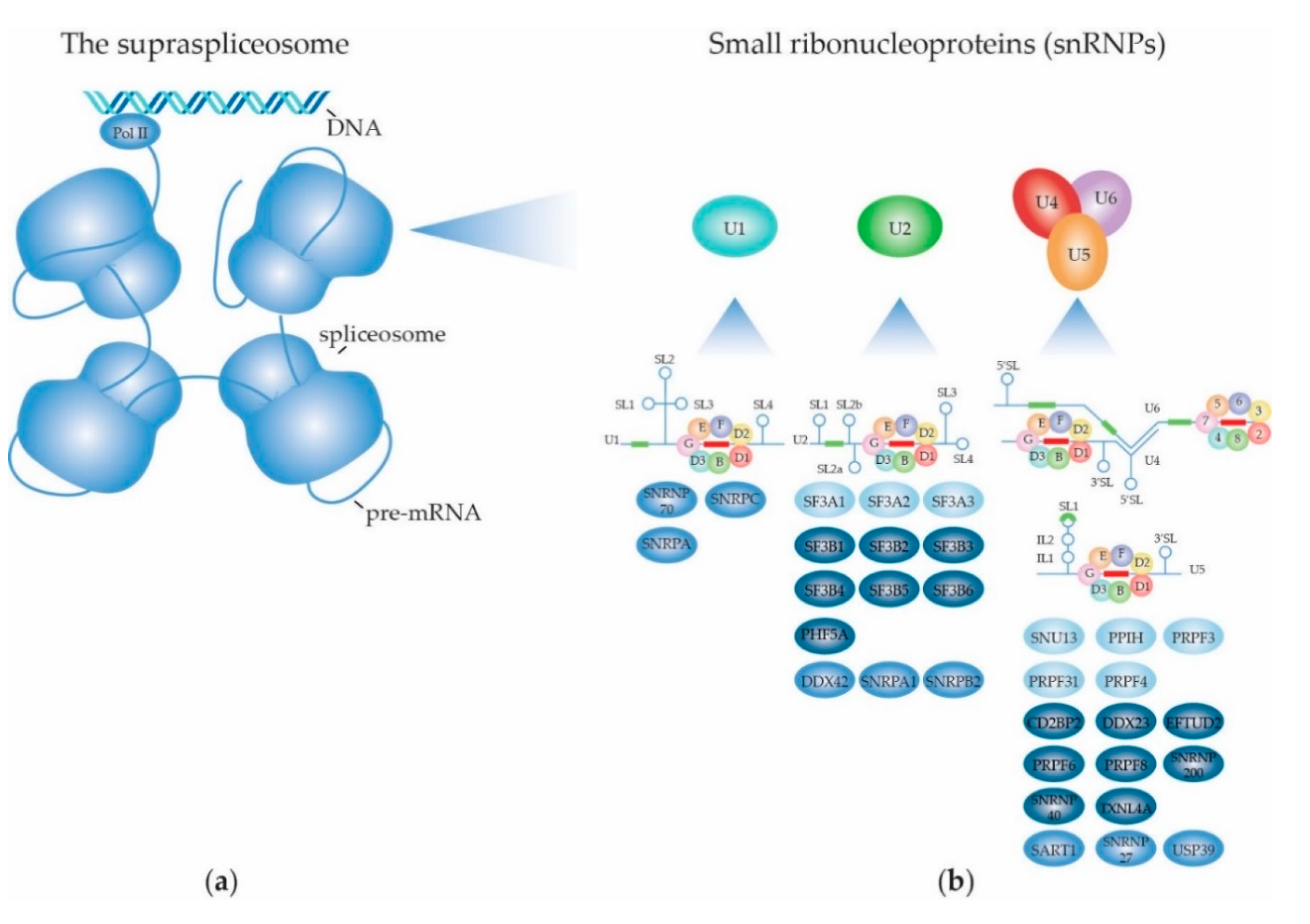
2. Structure and Function of the (Supra) Spliceosome
2.1. The RNA Splicing Machinery and the Splicing Reaction
2.1.1. Biogenesis of the Spliceosome: Assembly and Transport of Sm-snRNA Complexes
2.1.2. Biogenesis of the Spliceosome: Structure and Assembly of the U1 snRNP
2.1.3. Biogenesis of the Spliceosome: Structure and Assembly of the Other snRNPs
2.1.4. Dynamic Composition of the Spliceosome: Assembly on the pre-mRNA Substrate
2.1.5. Dynamic Composition of the Spliceosome: Activation and Catalytic Steps
2.2. Regulation of (Alternative) mRNA Splicing
2.2.1. Trans-Acting mRNA Splicing Factors
2.2.2. Effect of Secondary mRNA Structure
2.2.3. Effect of mRNA Elongation Rate
2.2.4. Effect of Chromatin Structure
2.3. Other RNA Processing Activities in the Supraspliceosome
2.3.1. mRNA 5′ Capping
2.3.2. mRNA 3′ End Cleavage and Polyadenylation
2.3.3. mRNA Internal Adenosine Methylation
2.3.4. mRNA Base Editing
2.3.5. Intronic Pri-miRNAs
2.3.6. mRNA Transport
3. RNA Splicing Dysregulation in Lung Cancer
3.1. Alterations in Cis-Acting Elements on Target Pre-mRNAs
3.1.1. ARID1A
3.1.2. TP53
3.1.3. METΔ14
3.1.4. BIM
3.1.5. AIMP2
3.2. Alterations in Trans-Acting mRNA Splicing Factors
3.2.1. U2AF35
3.2.2. RBM Proteins
3.2.3. QKI-5
3.2.4. SR and SR-like Proteins
3.2.5. hnRNPs
3.2.6. Dysregulated Phosphorylation of Splicing Factors
4. Therapeutic Targeting of Dysregulated mRNA Splicing in Cancer
4.1. Target Discovery
4.1.1. Splicing Factor Regulatory Kinases
4.1.2. Splicing Factors
4.1.3. RNA Helicases
4.1.4. U2 snRNP Components
4.1.5. Other Spliceosome Structural Components
4.1.6. Sm Proteins
4.2. Drug Development
4.2.1. Phenotypic Screening to Discover Spliceosome Targeting Drugs
4.2.2. Screens for Inhibition of pre-mRNA Splicing
4.2.3. Screens for Stalling of Spliceosome Assembly
4.2.4. Hypothesis-Driven Identification of Spliceosome Inhibitors
4.2.5. Discovery of Inhibitors of the U2 SF3b Complex
4.2.6. Discovery of Inhibitors of Later Spliceosome Assembly and Activation Steps
4.2.7. Discovery of Inhibitors of Spliceosome Catalytic Activity
4.2.8. Discovery of Inhibitors of mRNA Splicing Factors and Regulatory Kinases
4.2.9. Discovery of Inhibitors of Sm-snRNP Core Complex Assembly
5. Concluding Remarks and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Berget, S.M.; Moore, C.; Sharp, P.A. Spliced segments at the 5′ terminus of adenovirus 2 late mRNA. Proc. Natl. Acad. Sci. USA 1977, 74, 3171–3175. [Google Scholar] [CrossRef]
- Chow, L.T.; Gelinas, R.E.; Broker, T.R.; Roberts, R.J. An amazing sequence arrangement at the 5′ ends of adenovirus 2 messenger RNA. Cell 1977, 12, 1–8. [Google Scholar] [CrossRef]
- Bai, R.; Yan, C.; Wan, R.; Lei, J.; Shi, Y. Structure of the Post-catalytic Spliceosome from Saccharomyces cerevisiae. Cell 2017, 171, 1589–1598.e8. [Google Scholar] [CrossRef]
- Wan, R.; Yan, C.; Bai, R.; Huang, G.; Shi, Y. Structure of a yeast catalytic step I spliceosome at 3.4 A resolution. Science 2016, 353, 895–904. [Google Scholar] [CrossRef]
- Yan, C.; Hang, J.; Wan, R.; Huang, M.; Wong, C.C.; Shi, Y. Structure of a yeast spliceosome at 3.6-angstrom resolution. Science 2015, 349, 1182–1191. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Yan, C.; Zhang, X.; Lei, J.; Shi, Y. Structure of a human catalytic step I spliceosome. Science 2018, 359, 537–545. [Google Scholar] [CrossRef]
- Bentley, D.L. Rules of engagement: Co-transcriptional recruitment of pre-mRNA processing factors. Curr. Opin. Cell Biol. 2005, 17, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Azubel, M.; Habib, N.; Sperling, R.; Sperling, J. Native spliceosomes assemble with pre-mRNA to form supraspliceosomes. J. Mol. Biol. 2006, 356, 955–966. [Google Scholar] [CrossRef]
- Muller, S.; Wolpensinger, B.; Angenitzki, M.; Engel, A.; Sperling, J.; Sperling, R. A supraspliceosome model for large nuclear ribonucleoprotein particles based on mass determinations by scanning transmission electron microscopy. J. Mol. Biol. 1998, 283, 383–394. [Google Scholar] [CrossRef]
- Raitskin, O.; Angenitzki, M.; Sperling, J.; Sperling, R. Large nuclear RNP particles—The nuclear pre-mRNA processing machine. J. Struct. Biol. 2002, 140, 123–130. [Google Scholar] [CrossRef]
- Raitskin, O.; Cho, D.S.; Sperling, J.; Nishikura, K.; Sperling, R. RNA editing activity is associated with splicing factors in lnRNP particles: The nuclear pre-mRNA processing machinery. Proc. Natl. Acad. Sci. USA 2001, 98, 6571–6576. [Google Scholar] [CrossRef] [PubMed]
- Girard, C.; Will, C.L.; Peng, J.; Makarov, E.M.; Kastner, B.; Lemm, I.; Urlaub, H.; Hartmuth, K.; Luhrmann, R. Post-transcriptional spliceosomes are retained in nuclear speckles until splicing completion. Nat. Commun. 2012, 3, 994. [Google Scholar] [CrossRef]
- Shefer, K.; Sperling, J.; Sperling, R. The Supraspliceosome—A Multi-Task Machine for Regulated Pre-mRNA Processing in the Cell Nucleus. Comput. Struct. Biotechnol. J. 2014, 11, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Roca, X.; Krainer, A.R.; Eperon, I.C. Pick one, but be quick: 5′ splice sites and the problems of too many choices. Genes Dev. 2013, 27, 129–144. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.S.; Kinney, J.B.; Krainer, A.R. Quantitative Activity Profile and Context Dependence of All Human 5′ Splice Sites. Mol. Cell 2018, 71, 1012–1026.e3. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef]
- Tung, K.F.; Pan, C.Y.; Chen, C.H.; Lin, W.C. Top-ranked expressed gene transcripts of human protein-coding genes investigated with GTEx dataset. Sci. Rep. 2020, 10, 16245. [Google Scholar] [CrossRef]
- Fiszbein, A.; Kornblihtt, A.R. Alternative splicing switches: Important players in cell differentiation. Bioessays 2017, 39. [Google Scholar] [CrossRef]
- Maniatis, T.; Tasic, B. Alternative pre-mRNA splicing and proteome expansion in metazoans. Nature 2002, 418, 236–243. [Google Scholar] [CrossRef]
- Noh, S.J.; Lee, K.; Paik, H.; Hur, C.G. TISA: Tissue-specific alternative splicing in human and mouse genes. DNA Res. 2006, 13, 229–243. [Google Scholar] [CrossRef][Green Version]
- Singh, R.K.; Cooper, T.A. Pre-mRNA splicing in disease and therapeutics. Trends Mol. Med. 2012, 18, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Kahles, A.; Lehmann, K.V.; Toussaint, N.C.; Huser, M.; Stark, S.G.; Sachsenberg, T.; Stegle, O.; Kohlbacher, O.; Sander, C.; The Cancer Genome Atlas Research Network; et al. Comprehensive Analysis of Alternative Splicing Across Tumors from 8705 Patients. Cancer Cell 2018, 34, 211–224.e6. [Google Scholar] [CrossRef] [PubMed]
- Sperling, R.; Koster, A.J.; Melamed-Bessudo, C.; Rubinstein, A.; Angenitzki, M.; Berkovitch-Yellin, Z.; Sperling, J. Three-dimensional image reconstruction of large nuclear RNP (lnRNP) particles by automated electron tomography. J. Mol. Biol. 1997, 267, 570–583. [Google Scholar] [CrossRef]
- Kotzer-Nevo, H.; de Lima Alves, F.; Rappsilber, J.; Sperling, J.; Sperling, R. Supraspliceosomes at defined functional states portray the pre-assembled nature of the pre-mRNA processing machine in the cell nucleus. Int. J. Mol. Sci. 2014, 15, 11637–11664. [Google Scholar] [CrossRef]
- Cohen-Krausz, S.; Sperling, R.; Sperling, J. Exploring the architecture of the intact supraspliceosome using electron microscopy. J. Mol. Biol. 2007, 368, 319–327. [Google Scholar] [CrossRef]
- Frankenstein, Z.; Sperling, J.; Sperling, R.; Eisenstein, M. A unique spatial arrangement of the snRNPs within the native spliceosome emerges from in silico studies. Structure 2012, 20, 1097–1106. [Google Scholar] [CrossRef][Green Version]
- Sharp, P.A.; Burge, C.B. Classification of introns: U2-type or U12-type. Cell 1997, 91, 875–879. [Google Scholar] [CrossRef]
- Fischer, U.; Englbrecht, C.; Chari, A. Biogenesis of spliceosomal small nuclear ribonucleoproteins. Wiley Interdiscip. Rev. RNA 2011, 2, 718–731. [Google Scholar] [CrossRef]
- Kiss, T. Biogenesis of small nuclear RNPs. J. Cell Sci. 2004, 117, 5949–5951. [Google Scholar] [CrossRef]
- Matera, A.G.; Wang, Z. A day in the life of the spliceosome. Nat. Rev. Mol. Cell Biol. 2014, 15, 108–121. [Google Scholar] [CrossRef]
- Coady, T.H.; Lorson, C.L. SMN in spinal muscular atrophy and snRNP biogenesis. Wiley Interdiscip. Rev. RNA 2011, 2, 546–564. [Google Scholar] [CrossRef] [PubMed]
- Galganski, L.; Urbanek, M.O.; Krzyzosiak, W.J. Nuclear speckles: Molecular organization, biological function and role in disease. Nucleic Acids Res. 2017, 45, 10350–10368. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.P.; Hall, L.L.; Lawrence, J.B. Nuclear hubs built on RNAs and clustered organization of the genome. Curr. Opin. Cell Biol. 2020, 64, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Ghaemi, Z.; Peterson, J.R.; Gruebele, M.; Luthey-Schulten, Z. An in-silico human cell model reveals the influence of spatial organization on RNA splicing. PLoS Comput. Biol. 2020, 16, e1007717. [Google Scholar] [CrossRef]
- Veretnik, S.; Wills, C.; Youkharibache, P.; Valas, R.E.; Bourne, P.E. Sm/Lsm genes provide a glimpse into the early evolution of the spliceosome. PLoS Comput. Biol. 2009, 5, e1000315. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Lin, Y.S.; Deng, Y.F.; Hsu, W.T.; Shen, C.C.; Cheng, Y.H.; Huang, Y.T.; Li, C. Modulation of alternative splicing by expression of small nuclear ribonucleoprotein polypeptide N. FEBS J. 2014, 281, 5194–5207. [Google Scholar] [CrossRef]
- Hermann, H.; Fabrizio, P.; Raker, V.A.; Foulaki, K.; Hornig, H.; Brahms, H.; Luhrmann, R. snRNP Sm proteins share two evolutionarily conserved sequence motifs which are involved in Sm protein-protein interactions. EMBO J. 1995, 14, 2076–2088. [Google Scholar] [CrossRef]
- Li, J.; Leung, A.K.; Kondo, Y.; Oubridge, C.; Nagai, K. Re-refinement of the spliceosomal U4 snRNP core-domain structure. Acta Crystallogr. D Struct. Biol. 2016, 72, 131–146. [Google Scholar] [CrossRef]
- Raker, V.A.; Hartmuth, K.; Kastner, B.; Luhrmann, R. Spliceosomal U snRNP core assembly: Sm proteins assemble onto an Sm site RNA nonanucleotide in a specific and thermodynamically stable manner. Mol. Cell. Biol. 1999, 19, 6554–6565. [Google Scholar] [CrossRef]
- Neuenkirchen, N.; Englbrecht, C.; Ohmer, J.; Ziegenhals, T.; Chari, A.; Fischer, U. Reconstitution of the human U snRNP assembly machinery reveals stepwise Sm protein organization. EMBO J. 2015, 34, 1925–1941. [Google Scholar] [CrossRef]
- Prusty, A.B.; Meduri, R.; Prusty, B.K.; Vanselow, J.; Schlosser, A.; Fischer, U. Impaired spliceosomal UsnRNP assembly leads to Sm mRNA down-regulation and Sm protein degradation. J. Cell Biol. 2017, 216, 2391–2407. [Google Scholar] [CrossRef]
- Deng, X.; Lu, T.; Wang, L.; Gu, L.; Sun, J.; Kong, X.; Liu, C.; Cao, X. Recruitment of the NineTeen Complex to the activated spliceosome requires AtPRMT5. Proc. Natl. Acad. Sci. USA 2016, 113, 5447–5452. [Google Scholar] [CrossRef]
- Gonsalvez, G.B.; Tian, L.; Ospina, J.K.; Boisvert, F.M.; Lamond, A.I.; Matera, A.G. Two distinct arginine methyltransferases are required for biogenesis of Sm-class ribonucleoproteins. J. Cell Biol. 2007, 178, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Grimm, C.; Chari, A.; Pelz, J.P.; Kuper, J.; Kisker, C.; Diederichs, K.; Stark, H.; Schindelin, H.; Fischer, U. Structural basis of assembly chaperone- mediated snRNP formation. Mol. Cell 2013, 49, 692–703. [Google Scholar] [CrossRef]
- Yi, H.; Mu, L.; Shen, C.; Kong, X.; Wang, Y.; Hou, Y.; Zhang, R. Negative cooperativity between Gemin2 and RNA provides insights into RNA selection and the SMN complex’s release in snRNP assembly. Nucleic Acids Res. 2020, 48, 895–911. [Google Scholar] [CrossRef]
- Ma, Y.; Dostie, J.; Dreyfuss, G.; Van Duyne, G.D. The Gemin6-Gemin7 heterodimer from the survival of motor neurons complex has an Sm protein-like structure. Structure 2005, 13, 883–892. [Google Scholar] [CrossRef]
- Burghes, A.H.; Beattie, C.E. Spinal muscular atrophy: Why do low levels of survival motor neuron protein make motor neurons sick? Nat. Rev. Neurosci. 2009, 10, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Zhang, B.; Ziegenhals, T.; Prusty, A.B.; Frohler, S.; Grimm, C.; Hu, Y.; Schaefke, B.; Fang, L.; Zhang, M.; et al. A missense mutation in SNRPE linked to non-syndromal microcephaly interferes with U snRNP assembly and pre-mRNA splicing. PLoS Genet. 2019, 15, e1008460. [Google Scholar] [CrossRef]
- Jin, W.; Wang, Y.; Liu, C.P.; Yang, N.; Jin, M.; Cong, Y.; Wang, M.; Xu, R.M. Structural basis for snRNA recognition by the double-WD40 repeat domain of Gemin5. Genes Dev. 2016, 30, 2391–2403. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Bharath, S.R.; Piao, S.; Tan, V.Q.; Bowler, M.W.; Song, H. Structural basis for specific recognition of pre-snRNA by Gemin5. Cell Res. 2016, 26, 1353–1356. [Google Scholar] [CrossRef]
- Xu, C.; Ishikawa, H.; Izumikawa, K.; Li, L.; He, H.; Nobe, Y.; Yamauchi, Y.; Shahjee, H.M.; Wu, X.H.; Yu, Y.T.; et al. Structural insights into Gemin5-guided selection of pre-snRNAs for snRNP assembly. Genes Dev. 2016, 30, 2376–2390. [Google Scholar] [CrossRef]
- Yong, J.; Kasim, M.; Bachorik, J.L.; Wan, L.; Dreyfuss, G. Gemin5 delivers snRNA precursors to the SMN complex for snRNP biogenesis. Mol. Cell 2010, 38, 551–562. [Google Scholar] [CrossRef] [PubMed]
- So, B.R.; Wan, L.; Zhang, Z.; Li, P.; Babiash, E.; Duan, J.; Younis, I.; Dreyfuss, G. A U1 snRNP-specific assembly pathway reveals the SMN complex as a versatile hub for RNP exchange. Nat. Struct. Mol. Biol. 2016, 23, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Weber, G.; Trowitzsch, S.; Kastner, B.; Luhrmann, R.; Wahl, M.C. Functional organization of the Sm core in the crystal structure of human U1 snRNP. EMBO J. 2010, 29, 4172–4184. [Google Scholar] [CrossRef]
- Lund, E.; Dahlberg, J.E. Cyclic 2′,3′-phosphates and nontemplated nucleotides at the 3′ end of spliceosomal U6 small nuclear RNA’s. Science 1992, 255, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Sontheimer, E.J.; Steitz, J.A. Three novel functional variants of human U5 small nuclear RNA. Mol. Cell Biol. 1992, 12, 734–746. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Urlaub, H.; Raker, V.A.; Kostka, S.; Luhrmann, R. Sm protein-Sm site RNA interactions within the inner ring of the spliceosomal snRNP core structure. EMBO J. 2001, 20, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Schwer, B.; Kruchten, J.; Shuman, S. Structure-function analysis and genetic interactions of the SmG, SmE, and SmF subunits of the yeast Sm protein ring. RNA 2016, 22, 1320–1328. [Google Scholar] [CrossRef] [PubMed]
- Mouaikel, J.; Narayanan, U.; Verheggen, C.; Matera, A.G.; Bertrand, E.; Tazi, J.; Bordonne, R. Interaction between the small-nuclear-RNA cap hypermethylase and the spinal muscular atrophy protein, survival of motor neuron. EMBO Rep. 2003, 4, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Huber, J.; Dickmanns, A.; Luhrmann, R. The importin-beta binding domain of snurportin1 is responsible for the Ran- and energy-independent nuclear import of spliceosomal U snRNPs in vitro. J. Cell Biol. 2002, 156, 467–479. [Google Scholar] [CrossRef]
- Roithova, A.; Klimesova, K.; Panek, J.; Will, C.L.; Luhrmann, R.; Stanek, D.; Girard, C. The Sm-core mediates the retention of partially-assembled spliceosomal snRNPs in Cajal bodies until their full maturation. Nucleic Acids Res. 2018, 46, 3774–3790. [Google Scholar] [CrossRef]
- Aoyama, T.; Yamashita, S.; Tomita, K. Mechanistic insights into m6A modification of U6 snRNA by human METTL16. Nucleic Acids Res. 2020, 48, 5157–5168. [Google Scholar] [CrossRef] [PubMed]
- Goh, Y.T.; Koh, C.W.Q.; Sim, D.Y.; Roca, X.; Goh, W.S.S. METTL4 catalyzes m6Am methylation in U2 snRNA to regulate pre-mRNA splicing. Nucleic Acids Res. 2020, 48, 9250–9261. [Google Scholar] [CrossRef]
- Mauer, J.; Sindelar, M.; Despic, V.; Guez, T.; Hawley, B.R.; Vasseur, J.J.; Rentmeister, A.; Gross, S.S.; Pellizzoni, L.; Debart, F.; et al. FTO controls reversible m(6)Am RNA methylation during snRNA biogenesis. Nat. Chem. Biol. 2019, 15, 340–347. [Google Scholar] [CrossRef]
- Pomeranz Krummel, D.A.; Oubridge, C.; Leung, A.K.; Li, J.; Nagai, K. Crystal structure of human spliceosomal U1 snRNP at 5.5 A resolution. Nature 2009, 458, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Ling, S.C.; Qiu, J.; Albuquerque, C.P.; Zhou, Y.; Tokunaga, S.; Li, H.; Qiu, H.; Bui, A.; Yeo, G.W.; et al. ALS-causative mutations in FUS/TLS confer gain and loss of function by altered association with SMN and U1-snRNP. Nat. Commun. 2015, 6, 6171. [Google Scholar] [CrossRef]
- Li, X.; Liu, S.; Jiang, J.; Zhang, L.; Espinosa, S.; Hill, R.C.; Hansen, K.C.; Zhou, Z.H.; Zhao, R. CryoEM structure of Saccharomyces cerevisiae U1 snRNP offers insight into alternative splicing. Nat. Commun. 2017, 8, 1035. [Google Scholar] [CrossRef]
- Nelissen, R.L.; Will, C.L.; van Venrooij, W.J.; Luhrmann, R. The association of the U1-specific 70K and C proteins with U1 snRNPs is mediated in part by common U snRNP proteins. EMBO J. 1994, 13, 4113–4125. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Oubridge, C.; van Roon, A.M.; Nagai, K. Crystal structure of human U1 snRNP, a small nuclear ribonucleoprotein particle, reveals the mechanism of 5′ splice site recognition. Elife 2015, 4. [Google Scholar] [CrossRef]
- Rosel-Hillgartner, T.D.; Hung, L.H.; Khrameeva, E.; Le Querrec, P.; Gelfand, M.S.; Bindereif, A. A novel intra-U1 snRNP cross-regulation mechanism: Alternative splicing switch links U1C and U1-70K expression. PLoS Genet. 2013, 9, e1003856. [Google Scholar] [CrossRef]
- Zhang, Z.; Will, C.L.; Bertram, K.; Dybkov, O.; Hartmuth, K.; Agafonov, D.E.; Hofele, R.; Urlaub, H.; Kastner, B.; Luhrmann, R.; et al. Molecular architecture of the human 17S U2 snRNP. Nature 2020, 583, 310–313. [Google Scholar] [CrossRef] [PubMed]
- Martelly, W.; Fellows, B.; Senior, K.; Marlowe, T.; Sharma, S. Identification of a noncanonical RNA binding domain in the U2 snRNP protein SF3A1. RNA 2019, 25, 1509–1521. [Google Scholar] [CrossRef] [PubMed]
- Crisci, A.; Raleff, F.; Bagdiul, I.; Raabe, M.; Urlaub, H.; Rain, J.C.; Kramer, A. Mammalian splicing factor SF1 interacts with SURP domains of U2 snRNP-associated proteins. Nucleic Acids Res. 2015, 43, 10456–10473. [Google Scholar] [CrossRef] [PubMed]
- Cretu, C.; Schmitzova, J.; Ponce-Salvatierra, A.; Dybkov, O.; De Laurentiis, E.I.; Sharma, K.; Will, C.L.; Urlaub, H.; Luhrmann, R.; Pena, V. Molecular Architecture of SF3b and Structural Consequences of Its Cancer-Related Mutations. Mol. Cell 2016, 64, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Kfir, N.; Lev-Maor, G.; Glaich, O.; Alajem, A.; Datta, A.; Sze, S.K.; Meshorer, E.; Ast, G. SF3B1 association with chromatin determines splicing outcomes. Cell Rep. 2015, 11, 618–629. [Google Scholar] [CrossRef] [PubMed]
- Sander, B.; Golas, M.M.; Makarov, E.M.; Brahms, H.; Kastner, B.; Luhrmann, R.; Stark, H. Organization of core spliceosomal components U5 snRNA loop I and U4/U6 Di-snRNP within U4/U6.U5 Tri-snRNP as revealed by electron cryomicroscopy. Mol. Cell 2006, 24, 267–278. [Google Scholar] [CrossRef]
- Haselbach, D.; Komarov, I.; Agafonov, D.E.; Hartmuth, K.; Graf, B.; Dybkov, O.; Urlaub, H.; Kastner, B.; Luhrmann, R.; Stark, H. Structure and Conformational Dynamics of the Human Spliceosomal B(act) Complex. Cell 2018, 172, 454–464.e11. [Google Scholar] [CrossRef]
- Agafonov, D.E.; Kastner, B.; Dybkov, O.; Hofele, R.V.; Liu, W.T.; Urlaub, H.; Luhrmann, R.; Stark, H. Molecular architecture of the human U4/U6.U5 tri-snRNP. Science 2016, 351, 1416–1420. [Google Scholar] [CrossRef]
- Hardin, J.W.; Warnasooriya, C.; Kondo, Y.; Nagai, K.; Rueda, D. Assembly and dynamics of the U4/U6 di-snRNP by single-molecule FRET. Nucleic Acids Res. 2015, 43, 10963–10974. [Google Scholar] [CrossRef]
- Cloutier, P.; Poitras, C.; Durand, M.; Hekmat, O.; Fiola-Masson, E.; Bouchard, A.; Faubert, D.; Chabot, B.; Coulombe, B. R2TP/Prefoldin-like component RUVBL1/RUVBL2 directly interacts with ZNHIT2 to regulate assembly of U5 small nuclear ribonucleoprotein. Nat. Commun. 2017, 8, 15615. [Google Scholar] [CrossRef]
- Malinova, A.; Cvackova, Z.; Mateju, D.; Horejsi, Z.; Abeza, C.; Vandermoere, F.; Bertrand, E.; Stanek, D.; Verheggen, C. Assembly of the U5 snRNP component PRPF8 is controlled by the HSP90/R2TP chaperones. J. Cell Biol. 2017, 216, 1579–1596. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, S.; Takagi, Y.; Nagaike, T.; Tomita, K. Crystal structures of U6 snRNA-specific terminal uridylyltransferase. Nat. Commun. 2017, 8, 15788. [Google Scholar] [CrossRef]
- Achsel, T.; Brahms, H.; Kastner, B.; Bachi, A.; Wilm, M.; Luhrmann, R. A doughnut-shaped heteromer of human Sm-like proteins binds to the 3′-end of U6 snRNA, thereby facilitating U4/U6 duplex formation in vitro. EMBO J. 1999, 18, 5789–5802. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Hang, J.; Zhou, Y.; Wan, R.; Lu, G.; Yin, P.; Yan, C.; Shi, Y. Crystal structures of the Lsm complex bound to the 3′ end sequence of U6 small nuclear RNA. Nature 2014, 506, 116–120. [Google Scholar] [CrossRef]
- Montemayor, E.J.; Didychuk, A.L.; Yake, A.D.; Sidhu, G.K.; Brow, D.A.; Butcher, S.E. Architecture of the U6 snRNP reveals specific recognition of 3′-end processed U6 snRNA. Nat. Commun. 2018, 9, 1749. [Google Scholar] [CrossRef] [PubMed]
- Mund, M.; Neu, A.; Ullmann, J.; Neu, U.; Sprangers, R. Structure of the LSm657 complex: An assembly intermediate of the LSm1-7 and LSm2-8 rings. J. Mol. Biol. 2011, 414, 165–176. [Google Scholar] [CrossRef]
- Brahms, H.; Meheus, L.; de Brabandere, V.; Fischer, U.; Luhrmann, R. Symmetrical dimethylation of arginine residues in spliceosomal Sm protein B/B’ and the Sm-like protein LSm4, and their interaction with the SMN protein. RNA 2001, 7, 1531–1542. [Google Scholar] [CrossRef]
- Shi, Y. Mechanistic insights into precursor messenger RNA splicing by the spliceosome. Nat. Rev. Mol. Cell Biol. 2017, 18, 655–670. [Google Scholar] [CrossRef]
- Wan, R.; Bai, R.; Shi, Y. Molecular choreography of pre-mRNA splicing by the spliceosome. Curr. Opin. Struct. Biol. 2019, 59, 124–133. [Google Scholar] [CrossRef]
- Will, C.L.; Luhrmann, R. Spliceosome structure and function. Cold Spring Harb. Perspect Biol. 2011, 3. [Google Scholar] [CrossRef]
- Robberson, B.L.; Cote, G.J.; Berget, S.M. Exon definition may facilitate splice site selection in RNAs with multiple exons. Mol. Cell Biol. 1990, 10, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Schwer, B.; Shuman, S. Structure-function analysis of the Yhc1 subunit of yeast U1 snRNP and genetic interactions of Yhc1 with Mud2, Nam8, Mud1, Tgs1, U1 snRNA, SmD3 and Prp28. Nucleic Acids Res. 2014, 42, 4697–4711. [Google Scholar] [CrossRef]
- Das, R.; Yu, J.; Zhang, Z.; Gygi, M.P.; Krainer, A.R.; Gygi, S.P.; Reed, R. SR proteins function in coupling RNAP II transcription to pre-mRNA splicing. Mol. Cell 2007, 26, 867–881. [Google Scholar] [CrossRef]
- Li, X.; Liu, S.; Zhang, L.; Issaian, A.; Hill, R.C.; Espinosa, S.; Shi, S.; Cui, Y.; Kappel, K.; Das, R.; et al. A unified mechanism for intron and exon definition and back-splicing. Nature 2019, 573, 375–380. [Google Scholar] [CrossRef]
- Lockhart, S.R.; Rymond, B.C. Commitment of yeast pre-mRNA to the splicing pathway requires a novel U1 small nuclear ribonucleoprotein polypeptide, Prp39p. Mol. Cell. Biol. 1994, 14, 3623–3633. [Google Scholar] [CrossRef] [PubMed]
- Briese, M.; Haberman, N.; Sibley, C.R.; Faraway, R.; Elser, A.S.; Chakrabarti, A.M.; Wang, Z.; Konig, J.; Perera, D.; Wickramasinghe, V.O.; et al. A systems view of spliceosomal assembly and branchpoints with iCLIP. Nat. Struct. Mol. Biol. 2019, 26, 930–940. [Google Scholar] [CrossRef] [PubMed]
- Zarnack, K.; Konig, J.; Tajnik, M.; Martincorena, I.; Eustermann, S.; Stevant, I.; Reyes, A.; Anders, S.; Luscombe, N.M.; Ule, J. Direct competition between hnRNP C and U2AF65 protects the transcriptome from the exonization of Alu elements. Cell 2013, 152, 453–466. [Google Scholar] [CrossRef]
- Plaschka, C.; Lin, P.C.; Charenton, C.; Nagai, K. Prespliceosome structure provides insights into spliceosome assembly and regulation. Nature 2018, 559, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kohlstaedt, L.A.; Damianov, A.; Rio, D.C.; Black, D.L. Polypyrimidine tract binding protein controls the transition from exon definition to an intron defined spliceosome. Nat. Struct. Mol. Biol. 2008, 15, 183–191. [Google Scholar] [CrossRef]
- Charenton, C.; Wilkinson, M.E.; Nagai, K. Mechanism of 5′ splice site transfer for human spliceosome activation. Science 2019, 364, 362–367. [Google Scholar] [CrossRef]
- Huang, Y.H.; Chung, C.S.; Kao, D.I.; Kao, T.C.; Cheng, S.C. Sad1 counteracts Brr2-mediated dissociation of U4/U6.U5 in tri-snRNP homeostasis. Mol. Cell. Biol. 2014, 34, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Boesler, C.; Rigo, N.; Anokhina, M.M.; Tauchert, M.J.; Agafonov, D.E.; Kastner, B.; Urlaub, H.; Ficner, R.; Will, C.L.; Luhrmann, R. A spliceosome intermediate with loosely associated tri-snRNP accumulates in the absence of Prp28 ATPase activity. Nat. Commun. 2016, 7, 11997. [Google Scholar] [CrossRef]
- de Almeida, R.A.; O’Keefe, R.T. The NineTeen Complex (NTC) and NTC-associated proteins as targets for spliceosomal ATPase action during pre-mRNA splicing. RNA Biol. 2015, 12, 109–114. [Google Scholar] [CrossRef]
- Hogg, R.; McGrail, J.C.; O’Keefe, R.T. The function of the NineTeen Complex (NTC) in regulating spliceosome conformations and fidelity during pre-mRNA splicing. Biochem. Soc. Trans. 2010, 38, 1110–1115. [Google Scholar] [CrossRef]
- Nguyen, T.H.; Galej, W.P.; Bai, X.C.; Savva, C.G.; Newman, A.J.; Scheres, S.H.; Nagai, K. The architecture of the spliceosomal U4/U6.U5 tri-snRNP. Nature 2015, 523, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Zahler, A.M.; Rogel, L.E.; Glover, M.L.; Yitiz, S.; Ragle, J.M.; Katzman, S. SNRP-27, the C. elegans homolog of the tri-snRNP 27K protein, has a role in 5′ splice site positioning in the spliceosome. RNA 2018, 24, 1314–1325. [Google Scholar] [CrossRef]
- Townsend, C.; Leelaram, M.N.; Agafonov, D.E.; Dybkov, O.; Will, C.L.; Bertram, K.; Urlaub, H.; Kastner, B.; Stark, H.; Luhrmann, R. Mechanism of protein-guided folding of the active site U2/U6 RNA during spliceosome activation. Science 2020, 370. [Google Scholar] [CrossRef]
- Hang, J.; Wan, R.; Yan, C.; Shi, Y. Structural basis of pre-mRNA splicing. Science 2015, 349, 1191–1198. [Google Scholar] [CrossRef] [PubMed]
- Rauhut, R.; Fabrizio, P.; Dybkov, O.; Hartmuth, K.; Pena, V.; Chari, A.; Kumar, V.; Lee, C.T.; Urlaub, H.; Kastner, B.; et al. Molecular architecture of the Saccharomyces cerevisiae activated spliceosome. Science 2016, 353, 1399–1405. [Google Scholar] [CrossRef]
- Sun, C.; Rigo, N.; Fabrizio, P.; Kastner, B.; Luhrmann, R. A protein map of the yeast activated spliceosome as obtained by electron microscopy. RNA 2016, 22, 1427–1440. [Google Scholar] [CrossRef]
- Bao, P.; Boon, K.L.; Will, C.L.; Hartmuth, K.; Luhrmann, R. Multiple RNA-RNA tertiary interactions are dispensable for formation of a functional U2/U6 RNA catalytic core in the spliceosome. Nucleic Acids Res. 2018, 46, 12126–12138. [Google Scholar] [CrossRef] [PubMed]
- Galej, W.P.; Wilkinson, M.E.; Fica, S.M.; Oubridge, C.; Newman, A.J.; Nagai, K. Cryo-EM structure of the spliceosome immediately after branching. Nature 2016, 537, 197–201. [Google Scholar] [CrossRef]
- Zhang, X.; Yan, C.; Zhan, X.; Li, L.; Lei, J.; Shi, Y. Structure of the human activated spliceosome in three conformational states. Cell Res. 2018, 28, 307–322. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Wan, R.; Bai, R.; Huang, G.; Shi, Y. Structure of a yeast step II catalytically activated spliceosome. Science 2017, 355, 149–155. [Google Scholar] [CrossRef]
- Wan, R.; Bai, R.; Yan, C.; Lei, J.; Shi, Y. Structures of the Catalytically Activated Yeast Spliceosome Reveal the Mechanism of Branching. Cell 2019, 177, 339–351.e13. [Google Scholar] [CrossRef]
- Chung, C.S.; Tseng, C.K.; Lai, Y.H.; Wang, H.F.; Newman, A.J.; Cheng, S.C. Dynamic protein-RNA interactions in mediating splicing catalysis. Nucleic Acids Res. 2019, 47, 899–910. [Google Scholar] [CrossRef]
- Gehring, N.H.; Lamprinaki, S.; Hentze, M.W.; Kulozik, A.E. The hierarchy of exon-junction complex assembly by the spliceosome explains key features of mammalian nonsense-mediated mRNA decay. PLoS Biol. 2009, 7, e1000120. [Google Scholar] [CrossRef] [PubMed]
- Schlautmann, L.P.; Gehring, N.H. A Day in the Life of the Exon Junction Complex. Biomolecules 2020, 10, 866. [Google Scholar] [CrossRef]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef]
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef]
- Kornblihtt, A.R.; Schor, I.E.; Allo, M.; Dujardin, G.; Petrillo, E.; Munoz, M.J. Alternative splicing: A pivotal step between eukaryotic transcription and translation. Nat. Rev. Mol. Cell Biol. 2013, 14, 153–165. [Google Scholar] [CrossRef]
- Ule, J.; Blencowe, B.J. Alternative Splicing Regulatory Networks: Functions, Mechanisms, and Evolution. Mol. Cell 2019, 76, 329–345. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Mattox, W. Activation and repression functions of an SR splicing regulator depend on exonic versus intronic-binding position. Nucleic Acids Res. 2012, 40, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Bradley, T.; Cook, M.E.; Blanchette, M. SR proteins control a complex network of RNA-processing events. RNA 2015, 21, 75–92. [Google Scholar] [CrossRef]
- Pandit, S.; Zhou, Y.; Shiue, L.; Coutinho-Mansfield, G.; Li, H.; Qiu, J.; Huang, J.; Yeo, G.W.; Ares, M., Jr.; Fu, X.D. Genome-wide analysis reveals SR protein cooperation and competition in regulated splicing. Mol. Cell 2013, 50, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Deininger, P. Alu elements: Know the SINEs. Genome Biol. 2011, 12, 236. [Google Scholar] [CrossRef]
- Payer, L.M.; Steranka, J.P.; Ardeljan, D.; Walker, J.; Fitzgerald, K.C.; Calabresi, P.A.; Cooper, T.A.; Burns, K.H. Alu insertion variants alter mRNA splicing. Nucleic Acids Res. 2019, 47, 421–431. [Google Scholar] [CrossRef]
- Loh, T.J.; Cho, S.; Moon, H.; Jang, H.N.; Williams, D.R.; Jung, D.W.; Kim, I.C.; Ghigna, C.; Biamonti, G.; Zheng, X.; et al. hnRNP L inhibits CD44 V10 exon splicing through interacting with its upstream intron. Biochim. Biophys. Acta 2015, 1849, 743–750. [Google Scholar] [CrossRef]
- Ji, X.; Park, J.W.; Bahrami-Samani, E.; Lin, L.; Duncan-Lewis, C.; Pherribo, G.; Xing, Y.; Liebhaber, S.A. alphaCP binding to a cytosine-rich subset of polypyrimidine tracts drives a novel pathway of cassette exon splicing in the mammalian transcriptome. Nucleic Acids Res. 2016, 44, 2283–2297. [Google Scholar] [CrossRef]
- Xiao, X.; Wang, Z.; Jang, M.; Nutiu, R.; Wang, E.T.; Burge, C.B. Splice site strength-dependent activity and genetic buffering by poly-G runs. Nat. Struct Mol. Biol. 2009, 16, 1094–1100. [Google Scholar] [CrossRef]
- Chen, C.D.; Kobayashi, R.; Helfman, D.M. Binding of hnRNP H to an exonic splicing silencer is involved in the regulation of alternative splicing of the rat beta-tropomyosin gene. Genes Dev. 1999, 13, 593–606. [Google Scholar] [CrossRef]
- Jamison, S.F.; Pasman, Z.; Wang, J.; Will, C.; Luhrmann, R.; Manley, J.L.; Garcia-Blanco, M.A. U1 snRNP-ASF/SF2 interaction and 5′ splice site recognition: Characterization of required elements. Nucleic Acids Res. 1995, 23, 3260–3267. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cho, S.; Hoang, A.; Sinha, R.; Zhong, X.Y.; Fu, X.D.; Krainer, A.R.; Ghosh, G. Interaction between the RNA binding domains of Ser-Arg splicing factor 1 and U1-70K snRNP protein determines early spliceosome assembly. Proc. Natl. Acad. Sci. USA 2011, 108, 8233–8238. [Google Scholar] [CrossRef] [PubMed]
- Jobbins, A.M.; Reichenbach, L.F.; Lucas, C.M.; Hudson, A.J.; Burley, G.A.; Eperon, I.C. The mechanisms of a mammalian splicing enhancer. Nucleic Acids Res. 2018, 46, 2145–2158. [Google Scholar] [CrossRef] [PubMed]
- Aubol, B.E.; Wu, G.; Keshwani, M.M.; Movassat, M.; Fattet, L.; Hertel, K.J.; Fu, X.D.; Adams, J.A. Release of SR Proteins from CLK1 by SRPK1: A Symbiotic Kinase System for Phosphorylation Control of Pre-mRNA Splicing. Mol. Cell 2016, 63, 218–228. [Google Scholar] [CrossRef]
- Keiper, S.; Papasaikas, P.; Will, C.L.; Valcarcel, J.; Girard, C.; Luhrmann, R. Smu1 and RED are required for activation of spliceosomal B complexes assembled on short introns. Nat. Commun. 2019, 10, 3639. [Google Scholar] [CrossRef]
- Papasaikas, P.; Tejedor, J.R.; Vigevani, L.; Valcarcel, J. Functional splicing network reveals extensive regulatory potential of the core spliceosomal machinery. Mol. Cell 2015, 57, 7–22. [Google Scholar] [CrossRef]
- Subramania, S.; Gagne, L.M.; Campagne, S.; Fort, V.; O’Sullivan, J.; Mocaer, K.; Feldmuller, M.; Masson, J.Y.; Allain, F.H.T.; Hussein, S.M.; et al. SAM68 interaction with U1A modulates U1 snRNP recruitment and regulates mTor pre-mRNA splicing. Nucleic Acids Res. 2019, 47, 4181–4197. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.M.; Coates, M.N.; Jackson, S.R.; Jurica, M.S.; Davis, T.L. Nuclear cyclophilins affect spliceosome assembly and function in vitro. Biochem. J. 2015, 469, 223–233. [Google Scholar] [CrossRef]
- Bartys, N.; Kierzek, R.; Lisowiec-Wachnicka, J. The regulation properties of RNA secondary structure in alternative splicing. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 194401. [Google Scholar] [CrossRef] [PubMed]
- Warf, M.B.; Diegel, J.V.; von Hippel, P.H.; Berglund, J.A. The protein factors MBNL1 and U2AF65 bind alternative RNA structures to regulate splicing. Proc. Natl. Acad. Sci. USA 2009, 106, 9203–9208. [Google Scholar] [CrossRef]
- von Hacht, A.; Seifert, O.; Menger, M.; Schutze, T.; Arora, A.; Konthur, Z.; Neubauer, P.; Wagner, A.; Weise, C.; Kurreck, J. Identification and characterization of RNA guanine-quadruplex binding proteins. Nucleic Acids Res. 2014, 42, 6630–6644. [Google Scholar] [CrossRef] [PubMed]
- Conlon, E.G.; Lu, L.; Sharma, A.; Yamazaki, T.; Tang, T.; Shneider, N.A.; Manley, J.L. The C9ORF72 GGGGCC expansion forms RNA G-quadruplex inclusions and sequesters hnRNP H to disrupt splicing in ALS brains. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhang, J.; Harvey, S.E.; Hu, X.; Cheng, C. RNA G-quadruplex secondary structure promotes alternative splicing via the RNA-binding protein hnRNPF. Genes Dev. 2017, 31, 2296–2309. [Google Scholar] [CrossRef]
- Baraniak, A.P.; Lasda, E.L.; Wagner, E.J.; Garcia-Blanco, M.A. A stem structure in fibroblast growth factor receptor 2 transcripts mediates cell-type-specific splicing by approximating intronic control elements. Mol. Cell. Biol. 2003, 23, 9327–9337. [Google Scholar] [CrossRef]
- Munding, E.M.; Shiue, L.; Katzman, S.; Donohue, J.P.; Ares, M., Jr. Competition between pre-mRNAs for the splicing machinery drives global regulation of splicing. Mol. Cell 2013, 51, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Elowitz, M.B. Constitutive splicing and economies of scale in gene expression. Nat. Struct. Mol. Biol. 2019, 26, 424–432. [Google Scholar] [CrossRef]
- Alexander, R.D.; Innocente, S.A.; Barrass, J.D.; Beggs, J.D. Splicing-dependent RNA polymerase pausing in yeast. Mol. Cell 2010, 40, 582–593. [Google Scholar] [CrossRef]
- Chen, F.X.; Smith, E.R.; Shilatifard, A. Born to run: Control of transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2018, 19, 464–478. [Google Scholar] [CrossRef]
- Naftelberg, S.; Schor, I.E.; Ast, G.; Kornblihtt, A.R. Regulation of alternative splicing through coupling with transcription and chromatin structure. Annu. Rev. Biochem 2015, 84, 165–198. [Google Scholar] [CrossRef] [PubMed]
- Fong, N.; Kim, H.; Zhou, Y.; Ji, X.; Qiu, J.; Saldi, T.; Diener, K.; Jones, K.; Fu, X.D.; Bentley, D.L. Pre-mRNA splicing is facilitated by an optimal RNA polymerase II elongation rate. Genes Dev. 2014, 28, 2663–2676. [Google Scholar] [CrossRef]
- Baluapuri, A.; Hofstetter, J.; Dudvarski Stankovic, N.; Endres, T.; Bhandare, P.; Vos, S.M.; Adhikari, B.; Schwarz, J.D.; Narain, A.; Vogt, M.; et al. MYC Recruits SPT5 to RNA Polymerase II to Promote Processive Transcription Elongation. Mol. Cell 2019, 74, 674–687.e11. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Loven, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012, 151, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Luco, R.F.; Allo, M.; Schor, I.E.; Kornblihtt, A.R.; Misteli, T. Epigenetics in alternative pre-mRNA splicing. Cell 2011, 144, 16–26. [Google Scholar] [CrossRef]
- Narayanan, S.P.; Singh, S.; Shukla, S. A saga of cancer epigenetics: Linking epigenetics to alternative splicing. Biochem. J. 2017, 474, 885–896. [Google Scholar] [CrossRef]
- Schwartz, S.; Meshorer, E.; Ast, G. Chromatin organization marks exon-intron structure. Nat. Struct. Mol. Biol. 2009, 16, 990–995. [Google Scholar] [CrossRef]
- Spies, N.; Nielsen, C.B.; Padgett, R.A.; Burge, C.B. Biased chromatin signatures around polyadenylation sites and exons. Mol. Cell 2009, 36, 245–254. [Google Scholar] [CrossRef]
- Sims, R.J., 3rd; Millhouse, S.; Chen, C.F.; Lewis, B.A.; Erdjument-Bromage, H.; Tempst, P.; Manley, J.L.; Reinberg, D. Recognition of trimethylated histone H3 lysine 4 facilitates the recruitment of transcription postinitiation factors and pre-mRNA splicing. Mol. Cell 2007, 28, 665–676. [Google Scholar] [CrossRef]
- Luco, R.F.; Pan, Q.; Tominaga, K.; Blencowe, B.J.; Pereira-Smith, O.M.; Misteli, T. Regulation of alternative splicing by histone modifications. Science 2010, 327, 996–1000. [Google Scholar] [CrossRef]
- Ramanathan, A.; Robb, G.B.; Chan, S.H. mRNA capping: Biological functions and applications. Nucleic Acids Res. 2016, 44, 7511–7526. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Clerici, M.; Muckenfuss, L.M.; Passmore, L.A.; Jinek, M. Mechanistic insights into mRNA 3′-end processing. Curr. Opin. Struct. Biol. 2019, 59, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.I.; Moore, R.E.; Ge, H.Y.; Young, M.K.; Lee, T.D.; Stevens, S.W. Proteomic analysis of in vivo-assembled pre-mRNA splicing complexes expands the catalog of participating factors. Nucleic Acids Res. 2007, 35, 3928–3944. [Google Scholar] [CrossRef] [PubMed]
- Berg, M.G.; Singh, L.N.; Younis, I.; Liu, Q.; Pinto, A.M.; Kaida, D.; Zhang, Z.; Cho, S.; Sherrill-Mix, S.; Wan, L.; et al. U1 snRNP determines mRNA length and regulates isoform expression. Cell 2012, 150, 53–64. [Google Scholar] [CrossRef]
- Kaida, D.; Berg, M.G.; Younis, I.; Kasim, M.; Singh, L.N.; Wan, L.; Dreyfuss, G. U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature 2010, 468, 664–668. [Google Scholar] [CrossRef]
- He, P.C.; He, C. m(6) A RNA methylation: From mechanisms to therapeutic potential. EMBO J. 2021, 40, e105977. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Kong, S.; Tao, M.; Ju, S. The potential role of RNA N6-methyladenosine in Cancer progression. Mol. Cancer 2020, 19, 88. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Dai, Q.; Zheng, G.; He, C.; Parisien, M.; Pan, T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 2015, 518, 560–564. [Google Scholar] [CrossRef]
- Liu, N.; Zhou, K.I.; Parisien, M.; Dai, Q.; Diatchenko, L.; Pan, T. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. 2017, 45, 6051–6063. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, Y.; Sun, B.F.; Shi, Y.; Yang, X.; Xiao, W.; Hao, Y.J.; Ping, X.L.; Chen, Y.S.; Wang, W.J.; et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 2014, 24, 1403–1419. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Dahl, J.A.; Niu, Y.; Fedorcsak, P.; Huang, C.M.; Li, C.J.; Vagbo, C.B.; Shi, Y.; Wang, W.L.; Song, S.H.; et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 2013, 49, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Adhikari, S.; Dahal, U.; Chen, Y.S.; Hao, Y.J.; Sun, B.F.; Sun, H.Y.; Li, A.; Ping, X.L.; Lai, W.Y.; et al. Nuclear m(6)A Reader YTHDC1 Regulates mRNA Splicing. Mol. Cell 2016, 61, 507–519. [Google Scholar] [CrossRef]
- Samuel, C.E. RNA Editing. In Reference Module in Biomedical Sciences; Caplan, M., Ed.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 1–6. [Google Scholar] [CrossRef]
- Phelps, K.J.; Tran, K.; Eifler, T.; Erickson, A.I.; Fisher, A.J.; Beal, P.A. Recognition of duplex RNA by the deaminase domain of the RNA editing enzyme ADAR2. Nucleic Acids Res. 2015, 43, 1123–1132. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Severi, F.; Conticello, S.G. Flow-cytometric visualization of C>U mRNA editing reveals the dynamics of the process in live cells. RNA Biol. 2015, 12, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Levanon, E.Y.; Eisenberg, E.; Yelin, R.; Nemzer, S.; Hallegger, M.; Shemesh, R.; Fligelman, Z.Y.; Shoshan, A.; Pollock, S.R.; Sztybel, D.; et al. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat. Biotechnol. 2004, 22, 1001–1005. [Google Scholar] [CrossRef] [PubMed]
- Rueter, S.M.; Dawson, T.R.; Emeson, R.B. Regulation of alternative splicing by RNA editing. Nature 1999, 399, 75–80. [Google Scholar] [CrossRef]
- Hsiao, Y.E.; Bahn, J.H.; Yang, Y.; Lin, X.; Tran, S.; Yang, E.W.; Quinones-Valdez, G.; Xiao, X. RNA editing in nascent RNA affects pre-mRNA splicing. Genome Res. 2018, 28, 812–823. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Kapeli, K.; Jin, W.; Wong, Y.P.; Arumugam, T.V.; Koh, J.H.; Srimasorn, S.; Mallilankaraman, K.; Chua, J.J.E.; Yeo, G.W.; et al. Tissue-selective restriction of RNA editing of CaV1.3 by splicing factor SRSF9. Nucleic Acids Res. 2018, 46, 7323–7338. [Google Scholar] [CrossRef]
- Tang, S.J.; Shen, H.; An, O.; Hong, H.; Li, J.; Song, Y.; Han, J.; Tay, D.J.T.; Ng, V.H.E.; Bellido Molias, F.; et al. Cis- and trans-regulations of pre-mRNA splicing by RNA editing enzymes influence cancer development. Nat. Commun. 2020, 11, 799. [Google Scholar] [CrossRef]
- Agranat, L.; Sperling, J.; Sperling, R. A novel tissue-specific alternatively spliced form of the A-to-I RNA editing enzyme ADAR2. RNA Biol. 2010, 7, 253–262. [Google Scholar] [CrossRef]
- Agranat-Tamir, L.; Shomron, N.; Sperling, J.; Sperling, R. Interplay between pre-mRNA splicing and microRNA biogenesis within the supraspliceosome. Nucleic Acids Res. 2014, 42, 4640–4651. [Google Scholar] [CrossRef]
- Mahlab-Aviv, S.; Boulos, A.; Peretz, A.R.; Eliyahu, T.; Carmel, L.; Sperling, R.; Linial, M. Small RNA sequences derived from pre-microRNAs in the supraspliceosome. Nucleic Acids Res. 2018, 46, 11014–11029. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Lu, J.Y.; Zhang, X.; Shao, W.; Xu, Y.; Li, P.; Hong, Y.; Cui, L.; Shan, G.; Tian, B.; et al. U1 snRNP regulates chromatin retention of noncoding RNAs. Nature 2020, 580, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Azam, S.; Hou, S.; Zhu, B.; Wang, W.; Hao, T.; Bu, X.; Khan, M.; Lei, H. Nuclear retention element recruits U1 snRNP components to restrain spliced lncRNAs in the nucleus. RNA Biol. 2019, 16, 1001–1009. [Google Scholar] [CrossRef]
- Wang, K.; Yin, C.; Du, X.; Chen, S.; Wang, J.; Zhang, L.; Wang, L.; Yu, Y.; Chi, B.; Shi, M.; et al. A U2-snRNP-independent role of SF3b in promoting mRNA export. Proc. Natl. Acad. Sci. USA 2019, 116, 7837–7846. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; He, X.; Liu, J.; Duan, Z.; Kim, T.; Gerard, J.; Kim, B.; Pillai, M.M.; Lane, W.S.; Noble, W.S.; et al. Systematic proteomics of endogenous human cohesin reveals an interaction with diverse splicing factors and RNA-binding proteins required for mitotic progression. J. Biol. Chem. 2019, 294, 8760–8772. [Google Scholar] [CrossRef] [PubMed]
- Sveen, A.; Kilpinen, S.; Ruusulehto, A.; Lothe, R.A.; Skotheim, R.I. Aberrant RNA splicing in cancer; expression changes and driver mutations of splicing factor genes. Oncogene 2016, 35, 2413–2427. [Google Scholar] [CrossRef]
- Liu, J.; Lee, W.; Jiang, Z.; Chen, Z.; Jhunjhunwala, S.; Haverty, P.M.; Gnad, F.; Guan, Y.; Gilbert, H.N.; Stinson, J.; et al. Genome and transcriptome sequencing of lung cancers reveal diverse mutational and splicing events. Genome Res. 2012, 22, 2315–2327. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.N.; Roberts, C.W. ARID1A mutations in cancer: Another epigenetic tumor suppressor? Cancer Discov. 2013, 3, 35–43. [Google Scholar] [CrossRef]
- Hung, Y.P.; Redig, A.; Hornick, J.L.; Sholl, L.M. ARID1A mutations and expression loss in non-small cell lung carcinomas: Clinicopathologic and molecular analysis. Mod. Pathol. 2020, 33, 2256–2268. [Google Scholar] [CrossRef]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.S.; Ju, Y.S.; Lee, W.C.; Shin, J.Y.; Lee, J.K.; Bleazard, T.; Lee, J.; Jung, Y.J.; Kim, J.O.; Shin, J.Y.; et al. The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome Res. 2012, 22, 2109–2119. [Google Scholar] [CrossRef]
- Jang, S.H.; Lee, J.H.; Lee, H.J.; Cho, H.; Ahn, H.; Song, I.H.; Oh, M.H. Loss of ARID1A expression is associated with poor prognosis in non-small cell lung cancer. Pathol. Res. Pract. 2020, 216, 153156. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Lee, D.; Lee, J.; Park, D.; Kim, Y.J.; Park, W.Y.; Hong, D.; Park, P.J.; Lee, E. Intron retention is a widespread mechanism of tumor-suppressor inactivation. Nat. Genet. 2015, 47, 1242–1248. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, Y.; Kataoka, K.; Chiba, K.; Okada, A.; Kogure, Y.; Tanaka, H.; Ogawa, S.; Miyano, S. A comprehensive characterization of cis-acting splicing-associated variants in human cancer. Genome Res. 2018, 28, 1111–1125. [Google Scholar] [CrossRef]
- Kim, E.K.; Kim, K.A.; Lee, C.Y.; Kim, S.; Chang, S.; Cho, B.C.; Shim, H.S. Molecular Diagnostic Assays and Clinicopathologic Implications of MET Exon 14 Skipping Mutation in Non-small-cell Lung Cancer. Clin. Lung Cancer 2019, 20, e123–e132. [Google Scholar] [CrossRef]
- Tong, J.H.; Yeung, S.F.; Chan, A.W.; Chung, L.Y.; Chau, S.L.; Lung, R.W.; Tong, C.Y.; Chow, C.; Tin, E.K.; Yu, Y.H.; et al. MET Amplification and Exon 14 Splice Site Mutation Define Unique Molecular Subgroups of Non-Small Cell Lung Carcinoma with Poor Prognosis. Clin. Cancer Res. 2016, 22, 3048–3056. [Google Scholar] [CrossRef] [PubMed]
- Kong-Beltran, M.; Seshagiri, S.; Zha, J.; Zhu, W.; Bhawe, K.; Mendoza, N.; Holcomb, T.; Pujara, K.; Stinson, J.; Fu, L.; et al. Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res. 2006, 66, 283–289. [Google Scholar] [CrossRef]
- Lee, G.D.; Lee, S.E.; Oh, D.Y.; Yu, D.B.; Jeong, H.M.; Kim, J.; Hong, S.; Jung, H.S.; Oh, E.; Song, J.Y.; et al. MET Exon 14 Skipping Mutations in Lung Adenocarcinoma: Clinicopathologic Implications and Prognostic Values. J. Thorac. Oncol. 2017, 12, 1233–1246. [Google Scholar] [CrossRef] [PubMed]
- Frampton, G.M.; Ali, S.M.; Rosenzweig, M.; Chmielecki, J.; Lu, X.; Bauer, T.M.; Akimov, M.; Bufill, J.A.; Lee, C.; Jentz, D.; et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 2015, 5, 850–859. [Google Scholar] [CrossRef] [PubMed]
- Paik, P.K.; Felip, E.; Veillon, R.; Sakai, H.; Cortot, A.B.; Garassino, M.C.; Mazieres, J.; Viteri, S.; Senellart, H.; Van Meerbeeck, J.; et al. Tepotinib in Non-Small-Cell Lung Cancer with MET Exon 14 Skipping Mutations. N. Engl. J. Med. 2020, 383, 931–943. [Google Scholar] [CrossRef]
- Wolf, J.; Seto, T.; Han, J.Y.; Reguart, N.; Garon, E.B.; Groen, H.J.M.; Tan, D.S.W.; Hida, T.; de Jonge, M.; Orlov, S.V.; et al. Capmatinib in MET Exon 14-Mutated or MET-Amplified Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 944–957. [Google Scholar] [CrossRef]
- Drilon, A.; Clark, J.W.; Weiss, J.; Ou, S.I.; Camidge, D.R.; Solomon, B.J.; Otterson, G.A.; Villaruz, L.C.; Riely, G.J.; Heist, R.S.; et al. Antitumor activity of crizotinib in lung cancers harboring a MET exon 14 alteration. Nat. Med. 2020, 26, 47–51. [Google Scholar] [CrossRef]
- Suzawa, K.; Offin, M.; Schoenfeld, A.J.; Plodkowski, A.J.; Odintsov, I.; Lu, D.; Lockwood, W.W.; Arcila, M.E.; Rudin, C.M.; Drilon, A.; et al. Acquired MET Exon 14 Alteration Drives Secondary Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor in EGFR-Mutated Lung Cancer. JCO Precis. Oncol. 2019, 3. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.P.; Hillmer, A.M.; Chuah, C.T.; Juan, W.C.; Ko, T.K.; Teo, A.S.; Ariyaratne, P.N.; Takahashi, N.; Sawada, K.; Fei, Y.; et al. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat. Med. 2012, 18, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Juan, W.C.; Roca, X.; Ong, S.T. Identification of cis-acting elements and splicing factors involved in the regulation of BIM Pre-mRNA splicing. PLoS ONE 2014, 9, e95210. [Google Scholar] [CrossRef]
- Choi, J.W.; Kim, D.G.; Park, M.C.; Um, J.Y.; Han, J.M.; Park, S.G.; Choi, E.C.; Kim, S. AIMP2 promotes TNFalpha-dependent apoptosis via ubiquitin-mediated degradation of TRAF2. J. Cell Sci. 2009, 122, 2710–2715. [Google Scholar] [CrossRef]
- Han, J.M.; Park, B.J.; Park, S.G.; Oh, Y.S.; Choi, S.J.; Lee, S.W.; Hwang, S.K.; Chang, S.H.; Cho, M.H.; Kim, S. AIMP2/p38, the scaffold for the multi-tRNA synthetase complex, responds to genotoxic stresses via p53. Proc. Natl. Acad. Sci. USA 2008, 105, 11206–11211. [Google Scholar] [CrossRef] [PubMed]
- Yum, M.K.; Kang, J.S.; Lee, A.E.; Jo, Y.W.; Seo, J.Y.; Kim, H.A.; Kim, Y.Y.; Seong, J.; Lee, E.B.; Kim, J.H.; et al. AIMP2 Controls Intestinal Stem Cell Compartments and Tumorigenesis by Modulating Wnt/beta-Catenin Signaling. Cancer Res. 2016, 76, 4559–4568. [Google Scholar] [CrossRef]
- Choi, J.W.; Kim, D.G.; Lee, A.E.; Kim, H.R.; Lee, J.Y.; Kwon, N.H.; Shin, Y.K.; Hwang, S.K.; Chang, S.H.; Cho, M.H.; et al. Cancer-associated splicing variant of tumor suppressor AIMP2/p38: Pathological implication in tumorigenesis. PLoS Genet. 2011, 7, e1001351. [Google Scholar] [CrossRef]
- Scott, L.M.; Rebel, V.I. Acquired mutations that affect pre-mRNA splicing in hematologic malignancies and solid tumors. J. Natl. Cancer Inst. 2013, 105, 1540–1549. [Google Scholar] [CrossRef]
- Coomer, A.O.; Black, F.; Greystoke, A.; Munkley, J.; Elliott, D.J. Alternative splicing in lung cancer. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 194388. [Google Scholar] [CrossRef] [PubMed]
- Fei, D.L.; Motowski, H.; Chatrikhi, R.; Prasad, S.; Yu, J.; Gao, S.; Kielkopf, C.L.; Bradley, R.K.; Varmus, H. Wild-Type U2AF1 Antagonizes the Splicing Program Characteristic of U2AF1-Mutant Tumors and Is Required for Cell Survival. PLoS Genet. 2016, 12, e1006384. [Google Scholar] [CrossRef] [PubMed]
- Esfahani, M.S.; Lee, L.J.; Jeon, Y.J.; Flynn, R.A.; Stehr, H.; Hui, A.B.; Ishisoko, N.; Kildebeck, E.; Newman, A.M.; Bratman, S.V.; et al. Functional significance of U2AF1 S34F mutations in lung adenocarcinomas. Nat. Commun. 2019, 10, 5712. [Google Scholar] [CrossRef]
- Graubert, T.A.; Shen, D.; Ding, L.; Okeyo-Owuor, T.; Lunn, C.L.; Shao, J.; Krysiak, K.; Harris, C.C.; Koboldt, D.C.; Larson, D.E.; et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat. Genet. 2011, 44, 53–57. [Google Scholar] [CrossRef]
- Ilagan, J.O.; Ramakrishnan, A.; Hayes, B.; Murphy, M.E.; Zebari, A.S.; Bradley, P.; Bradley, R.K. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res. 2015, 25, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Przychodzen, B.; Jerez, A.; Guinta, K.; Sekeres, M.A.; Padgett, R.; Maciejewski, J.P.; Makishima, H. Patterns of missplicing due to somatic U2AF1 mutations in myeloid neoplasms. Blood 2013, 122, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Sebestyen, E.; Singh, B.; Minana, B.; Pages, A.; Mateo, F.; Pujana, M.A.; Valcarcel, J.; Eyras, E. Large-scale analysis of genome and transcriptome alterations in multiple tumors unveils novel cancer-relevant splicing networks. Genome Res. 2016, 26, 732–744. [Google Scholar] [CrossRef]
- Wang, Y.; Gogol-Doring, A.; Hu, H.; Frohler, S.; Ma, Y.; Jens, M.; Maaskola, J.; Murakawa, Y.; Quedenau, C.; Landthaler, M.; et al. Integrative analysis revealed the molecular mechanism underlying RBM10-mediated splicing regulation. EMBO Mol. Med. 2013, 5, 1431–1442. [Google Scholar] [CrossRef] [PubMed]
- Bechara, E.G.; Sebestyen, E.; Bernardis, I.; Eyras, E.; Valcarcel, J. RBM5, 6, and 10 differentially regulate NUMB alternative splicing to control cancer cell proliferation. Mol. Cell 2013, 52, 720–733. [Google Scholar] [CrossRef]
- Sun, Y.; Bao, Y.; Han, W.; Song, F.; Shen, X.; Zhao, J.; Zuo, J.; Saffen, D.; Chen, W.; Wang, Z.; et al. Autoregulation of RBM10 and cross-regulation of RBM10/RBM5 via alternative splicing-coupled nonsense-mediated decay. Nucleic Acids Res. 2017, 45, 8524–8540. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, D.; Qian, H.; Tsai, Y.S.; Shao, S.; Liu, Q.; Dominguez, D.; Wang, Z. The splicing factor RBM4 controls apoptosis, proliferation, and migration to suppress tumor progression. Cancer Cell 2014, 26, 374–389. [Google Scholar] [CrossRef]
- de Miguel, F.J.; Pajares, M.J.; Martinez-Terroba, E.; Ajona, D.; Morales, X.; Sharma, R.D.; Pardo, F.J.; Rouzaut, A.; Rubio, A.; Montuenga, L.M.; et al. A large-scale analysis of alternative splicing reveals a key role of QKI in lung cancer. Mol. Oncol. 2016, 10, 1437–1449. [Google Scholar] [CrossRef] [PubMed]
- Zong, F.Y.; Fu, X.; Wei, W.J.; Luo, Y.G.; Heiner, M.; Cao, L.J.; Fang, Z.; Fang, R.; Lu, D.; Ji, H.; et al. The RNA-binding protein QKI suppresses cancer-associated aberrant splicing. PLoS Genet. 2014, 10, e1004289. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef]
- Hall, M.P.; Nagel, R.J.; Fagg, W.S.; Shiue, L.; Cline, M.S.; Perriman, R.J.; Donohue, J.P.; Ares, M., Jr. Quaking and PTB control overlapping splicing regulatory networks during muscle cell differentiation. RNA 2013, 19, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Dickson, E.J.; Jensen, J.B.; Vivas, O.; Kruse, M.; Traynor-Kaplan, A.E.; Hille, B. Dynamic formation of ER-PM junctions presents a lipid phosphatase to regulate phosphoinositides. J. Cell Biol 2016, 213, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, Y.; Li, X.; Bennett, V. Adducin: Structure, function and regulation. Cell. Mol. Life Sci. 2000, 57, 884–895. [Google Scholar] [CrossRef] [PubMed]
- Langer, W.; Sohler, F.; Leder, G.; Beckmann, G.; Seidel, H.; Grone, J.; Hummel, M.; Sommer, A. Exon array analysis using re-defined probe sets results in reliable identification of alternatively spliced genes in non-small cell lung cancer. BMC Genom. 2010, 11, 676. [Google Scholar] [CrossRef]
- Wang, J.Z.; Fu, X.; Fang, Z.; Liu, H.; Zong, F.Y.; Zhu, H.; Yu, Y.F.; Zhang, X.Y.; Wang, S.F.; Huang, Y.; et al. QKI-5 regulates the alternative splicing of cytoskeletal gene ADD3 in lung cancer. J. Mol. Cell Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Karni, R.; de Stanchina, E.; Lowe, S.W.; Sinha, R.; Mu, D.; Krainer, A.R. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat. Struct. Mol. Biol. 2007, 14, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Anczukow, O.; Akerman, M.; Krainer, A.R. Oncogenic splicing factor SRSF1 is a critical transcriptional target of MYC. Cell Rep. 2012, 1, 110–117. [Google Scholar] [CrossRef]
- Jia, R.; Li, C.; McCoy, J.P.; Deng, C.X.; Zheng, Z.M. SRp20 is a proto-oncogene critical for cell proliferation and tumor induction and maintenance. Int. J. Biol. Sci. 2010, 6, 806–826. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Horikawa, I.; Ajiro, M.; Robles, A.I.; Fujita, K.; Mondal, A.M.; Stauffer, J.K.; Zheng, Z.M.; Harris, C.C. Downregulation of splicing factor SRSF3 induces p53beta, an alternatively spliced isoform of p53 that promotes cellular senescence. Oncogene 2013, 32, 2792–2798. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Eliav, M.; Golan-Gerstl, R.; Siegfried, Z.; Andersen, C.L.; Thorsen, K.; Orntoft, T.F.; Mu, D.; Karni, R. The splicing factor SRSF6 is amplified and is an oncoprotein in lung and colon cancers. J. Pathol. 2013, 229, 630–639. [Google Scholar] [CrossRef]
- Ji, L.; Ni, T.; Shen, Y.; Xue, Q.; Liu, Y.; Chen, B.; Cui, X.; Lv, L.; Yu, X.; Cui, Y.; et al. Transformer 2beta (Tra2beta/SFRS10) positively regulates the progression of NSCLC via promoting cell proliferation. J. Mol. Histol. 2014, 45, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Tacke, R.; Tohyama, M.; Ogawa, S.; Manley, J.L. Human Tra2 proteins are sequence-specific activators of pre-mRNA splicing. Cell 1998, 93, 139–148. [Google Scholar] [CrossRef]
- Hofmann, Y.; Lorson, C.L.; Stamm, S.; Androphy, E.J.; Wirth, B. Htra2-beta 1 stimulates an exonic splicing enhancer and can restore full-length SMN expression to survival motor neuron 2 (SMN2). Proc. Natl. Acad. Sci. USA 2000, 97, 9618–9623. [Google Scholar] [CrossRef] [PubMed]
- Lareau, L.F.; Inada, M.; Green, R.E.; Wengrod, J.C.; Brenner, S.E. Unproductive splicing of SR genes associated with highly conserved and ultraconserved DNA elements. Nature 2007, 446, 926–929. [Google Scholar] [CrossRef]
- Li, H.; Liu, J.; Shen, S.; Dai, D.; Cheng, S.; Dong, X.; Sun, L.; Guo, X. Pan-cancer analysis of alternative splicing regulator heterogeneous nuclear ribonucleoproteins (hnRNPs) family and their prognostic potential. J. Cell. Mol. Med. 2020, 24, 11111–11119. [Google Scholar] [CrossRef]
- Boukakis, G.; Patrinou-Georgoula, M.; Lekarakou, M.; Valavanis, C.; Guialis, A. Deregulated expression of hnRNP A/B proteins in human non-small cell lung cancer: Parallel assessment of protein and mRNA levels in paired tumour/non-tumour tissues. BMC Cancer 2010, 10, 434. [Google Scholar] [CrossRef]
- Zerbe, L.K.; Pino, I.; Pio, R.; Cosper, P.F.; Dwyer-Nield, L.D.; Meyer, A.M.; Port, J.D.; Montuenga, L.M.; Malkinson, A.M. Relative amounts of antagonistic splicing factors, hnRNP A1 and ASF/SF2, change during neoplastic lung growth: Implications for pre-mRNA processing. Mol. Carcinog. 2004, 41, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Goehe, R.W.; Shultz, J.C.; Murudkar, C.; Usanovic, S.; Lamour, N.F.; Massey, D.H.; Zhang, L.; Camidge, D.R.; Shay, J.W.; Minna, J.D.; et al. hnRNP L regulates the tumorigenic capacity of lung cancer xenografts in mice via caspase-9 pre-mRNA processing. J. Clin. Investig. 2010, 120, 3923–3939. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Li, Y.; Ning, J.; Sun, D.; Lin, L.; Liu, X. HnRNP A1/A2 and SF2/ASF regulate alternative splicing of interferon regulatory factor-3 and affect immunomodulatory functions in human non-small cell lung cancer cells. PLoS ONE 2013, 8, e62729. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Shen, L.; Huang, L.; Lei, S.; Cai, X.; Breitzig, M.; Zhang, B.; Yang, A.; Ji, W.; Huang, M.; et al. PTBP1 enhances exon11a skipping in Mena pre-mRNA to promote migration and invasion in lung carcinoma cells. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 858–869. [Google Scholar] [CrossRef] [PubMed]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef]
- David, C.J.; Chen, M.; Assanah, M.; Canoll, P.; Manley, J.L. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 2010, 463, 364–368. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, S.; Liu, J.; Tian, Y.; Ma, B.; Xu, S.; Fu, Y.; Luo, Y. Secreted Pyruvate Kinase M2 Promotes Lung Cancer Metastasis through Activating the Integrin Beta1/FAK Signaling Pathway. Cell Rep. 2020, 30, 1780–1797.e6. [Google Scholar] [CrossRef] [PubMed]
- Allemand, E.; Guil, S.; Myers, M.; Moscat, J.; Caceres, J.F.; Krainer, A.R. Regulation of heterogenous nuclear ribonucleoprotein A1 transport by phosphorylation in cells stressed by osmotic shock. Proc. Natl. Acad. Sci. USA 2005, 102, 3605–3610. [Google Scholar] [CrossRef]
- Caceres, J.F.; Screaton, G.R.; Krainer, A.R. A specific subset of SR proteins shuttles continuously between the nucleus and the cytoplasm. Genes Dev. 1998, 12, 55–66. [Google Scholar] [CrossRef]
- Li, P.; Zhou, L.; Zhao, T.; Liu, X.; Zhang, P.; Liu, Y.; Zheng, X.; Li, Q. Caspase-9: Structure, mechanisms and clinical application. Oncotarget 2017, 8, 23996–24008. [Google Scholar] [CrossRef]
- Vu, N.T.; Park, M.A.; Shultz, J.C.; Goehe, R.W.; Hoeferlin, L.A.; Shultz, M.D.; Smith, S.A.; Lynch, K.W.; Chalfant, C.E. hnRNP U enhances caspase-9 splicing and is modulated by AKT-dependent phosphorylation of hnRNP L. J. Biol. Chem. 2013, 288, 8575–8584. [Google Scholar] [CrossRef] [PubMed]
- Shultz, J.C.; Goehe, R.W.; Murudkar, C.S.; Wijesinghe, D.S.; Mayton, E.K.; Massiello, A.; Hawkins, A.J.; Mukerjee, P.; Pinkerman, R.L.; Park, M.A.; et al. SRSF1 regulates the alternative splicing of caspase 9 via a novel intronic splicing enhancer affecting the chemotherapeutic sensitivity of non-small cell lung cancer cells. Mol. Cancer Res. 2011, 9, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.D.; Lee, N.H. Aberrant RNA Splicing in Cancer and Drug Resistance. Cancers 2018, 10, 458. [Google Scholar] [CrossRef] [PubMed]
- Giroux, V.; Iovanna, J.; Dagorn, J.C. Probing the human kinome for kinases involved in pancreatic cancer cell survival and gemcitabine resistance. FASEB J. 2006, 20, 1982–1991. [Google Scholar] [CrossRef]
- Ngo, V.N.; Davis, R.E.; Lamy, L.; Yu, X.; Zhao, H.; Lenz, G.; Lam, L.T.; Dave, S.; Yang, L.; Powell, J.; et al. A loss-of-function RNA interference screen for molecular targets in cancer. Nature 2006, 441, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Schlabach, M.R.; Luo, J.; Solimini, N.L.; Hu, G.; Xu, Q.; Li, M.Z.; Zhao, Z.; Smogorzewska, A.; Sowa, M.E.; Ang, X.L.; et al. Cancer proliferation gene discovery through functional genomics. Science 2008, 319, 620–624. [Google Scholar] [CrossRef]
- Becerra, S.; Andres-Leon, E.; Prieto-Sanchez, S.; Hernandez-Munain, C.; Sune, C. Prp40 and early events in splice site definition. Wiley Interdiscip. Rev. RNA 2016, 7, 17–32. [Google Scholar] [CrossRef]
- Duan, Z.; Weinstein, E.J.; Ji, D.; Ames, R.Y.; Choy, E.; Mankin, H.; Hornicek, F.J. Lentiviral short hairpin RNA screen of genes associated with multidrug resistance identifies PRP-4 as a new regulator of chemoresistance in human ovarian cancer. Mol. Cancer Ther. 2008, 7, 2377–2385. [Google Scholar] [CrossRef]
- Sakuma, K.; Sasaki, E.; Kimura, K.; Komori, K.; Shimizu, Y.; Yatabe, Y.; Aoki, M. HNRNPLL, a newly identified colorectal cancer metastasis suppressor, modulates alternative splicing of CD44 during epithelial-mesenchymal transition. Gut 2018, 67, 1103–1111. [Google Scholar] [CrossRef]
- Georgilis, A.; Klotz, S.; Hanley, C.J.; Herranz, N.; Weirich, B.; Morancho, B.; Leote, A.C.; D’Artista, L.; Gallage, S.; Seehawer, M.; et al. PTBP1-Mediated Alternative Splicing Regulates the Inflammatory Secretome and the Pro-tumorigenic Effects of Senescent Cells. Cancer Cell 2018, 34, 85–102.e9. [Google Scholar] [CrossRef] [PubMed]
- Lefave, C.V.; Squatrito, M.; Vorlova, S.; Rocco, G.L.; Brennan, C.W.; Holland, E.C.; Pan, Y.X.; Cartegni, L. Splicing factor hnRNPH drives an oncogenic splicing switch in gliomas. EMBO J. 2011, 30, 4084–4097. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, R.; Li, X.; Yu, L.; Hua, D.; Sun, C.; Shi, C.; Luo, W.; Rao, C.; Jiang, Z.; et al. Splicing factor SRSF1 promotes gliomagenesis via oncogenic splice-switching of MYO1B. J. Clin. Investig. 2019, 129, 676–693. [Google Scholar] [CrossRef] [PubMed]
- Adesso, L.; Calabretta, S.; Barbagallo, F.; Capurso, G.; Pilozzi, E.; Geremia, R.; Delle Fave, G.; Sette, C. Gemcitabine triggers a pro-survival response in pancreatic cancer cells through activation of the MNK2/eIF4E pathway. Oncogene 2013, 32, 2848–2857. [Google Scholar] [CrossRef]
- Nakata, D.; Nakao, S.; Nakayama, K.; Araki, S.; Nakayama, Y.; Aparicio, S.; Hara, T.; Nakanishi, A. The RNA helicase DDX39B and its paralog DDX39A regulate androgen receptor splice variant AR-V7 generation. Biochem. Biophys. Res. Commun. 2017, 483, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Tiedemann, R.E.; Zhu, Y.X.; Schmidt, J.; Shi, C.X.; Sereduk, C.; Yin, H.; Mousses, S.; Stewart, A.K. Identification of molecular vulnerabilities in human multiple myeloma cells by RNA interference lethality screening of the druggable genome. Cancer Res. 2012, 72, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Siebring-van Olst, E.; Blijlevens, M.; de Menezes, R.X.; van der Meulen-Muileman, I.H.; Smit, E.F.; van Beusechem, V.W. A genome-wide siRNA screen for regulators of tumor suppressor p53 activity in human non-small cell lung cancer cells identifies components of the RNA splicing machinery as targets for anticancer treatment. Mol. Oncol. 2017, 11, 534–551. [Google Scholar] [CrossRef] [PubMed]
- Hubert, C.G.; Bradley, R.K.; Ding, Y.; Toledo, C.M.; Herman, J.; Skutt-Kakaria, K.; Girard, E.J.; Davison, J.; Berndt, J.; Corrin, P.; et al. Genome-wide RNAi screens in human brain tumor isolates reveal a novel viability requirement for PHF5A. Genes Dev. 2013, 27, 1032–1045. [Google Scholar] [CrossRef]
- Grohar, P.J.; Kim, S.; Rangel Rivera, G.O.; Sen, N.; Haddock, S.; Harlow, M.L.; Maloney, N.K.; Zhu, J.; O’Neill, M.; Jones, T.L.; et al. Functional Genomic Screening Reveals Splicing of the EWS-FLI1 Fusion Transcript as a Vulnerability in Ewing Sarcoma. Cell Rep. 2016, 14, 598–610. [Google Scholar] [CrossRef]
- Gamberi, G.; Cocchi, S.; Benini, S.; Magagnoli, G.; Morandi, L.; Kreshak, J.; Gambarotti, M.; Picci, P.; Zanella, L.; Alberghini, M. Molecular diagnosis in Ewing family tumors: The Rizzoli experience—222 consecutive cases in four years. J. Mol. Diagn. 2011, 13, 313–324. [Google Scholar] [CrossRef]
- Li, J.; Cheng, D.; Zhu, M.; Yu, H.; Pan, Z.; Liu, L.; Geng, Q.; Pan, H.; Yan, M.; Yao, M. OTUB2 stabilizes U2AF2 to promote the Warburg effect and tumorigenesis via the AKT/mTOR signaling pathway in non-small cell lung cancer. Theranostics 2019, 9, 179–195. [Google Scholar] [CrossRef] [PubMed]
- Blijlevens, M.; Komor, M.A.; Sciarrillo, R.; Smit, E.F.; Fijneman, R.J.A.; van Beusechem, V.W. Silencing Core Spliceosome Sm Gene Expression Induces a Cytotoxic Splicing Switch in the Proteasome Subunit Beta 3 mRNA in Non-Small Cell Lung Cancer Cells. Int. J. Mol. Sci. 2020, 21, 4192. [Google Scholar] [CrossRef] [PubMed]
- Blijlevens, M.; van der Meulen-Muileman, I.H.; de Menezes, R.X.; Smit, E.F.; van Beusechem, V.W. High-throughput RNAi screening reveals cancer-selective lethal targets in the RNA spliceosome. Oncogene 2019, 38, 4142–4153. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Derti, A.; Ruddy, D.; Rakiec, D.; Kao, I.; Lira, M.; Gibaja, V.; Chan, H.; Yang, Y.; Min, J.; et al. A chemical genetics approach for the functional assessment of novel cancer genes. Cancer Res. 2015, 75, 1949–1958. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.; Sridhar, P.; Kirchner, R.; Lock, Y.J.; Herbert, Z.; Buonamici, S.; Smith, P.; Lieberman, J.; Petrocca, F. Basal-A Triple-Negative Breast Cancer Cells Selectively Rely on RNA Splicing for Survival. Mol. Cancer Ther. 2017, 16, 2849–2861. [Google Scholar] [CrossRef]
- Laetsch, T.W.; Liu, X.; Vu, A.; Sliozberg, M.; Vido, M.; Elci, O.U.; Goldsmith, K.C.; Hogarty, M.D. Multiple components of the spliceosome regulate Mcl1 activity in neuroblastoma. Cell Death Dis. 2014, 5, e1072. [Google Scholar] [CrossRef]
- Park, S.; Han, S.H.; Kim, H.G.; Jeong, J.; Choi, M.; Kim, H.Y.; Kim, M.G.; Park, J.K.; Han, J.E.; Cho, G.J.; et al. PRPF4 is a novel therapeutic target for the treatment of breast cancer by influencing growth, migration, invasion, and apoptosis of breast cancer cells via p38 MAPK signaling pathway. Mol. Cell. Probes 2019, 47, 101440. [Google Scholar] [CrossRef]
- Tanaka, I.; Chakraborty, A.; Saulnier, O.; Benoit-Pilven, C.; Vacher, S.; Labiod, D.; Lam, E.W.F.; Bieche, I.; Delattre, O.; Pouzoulet, F.; et al. ZRANB2 and SYF2-mediated splicing programs converging on ECT2 are involved in breast cancer cell resistance to doxorubicin. Nucleic Acids Res. 2020, 48, 2676–2693. [Google Scholar] [CrossRef]
- Quidville, V.; Alsafadi, S.; Goubar, A.; Commo, F.; Scott, V.; Pioche-Durieu, C.; Girault, I.; Baconnais, S.; Le Cam, E.; Lazar, V.; et al. Targeting the deregulated spliceosome core machinery in cancer cells triggers mTOR blockade and autophagy. Cancer Res. 2013, 73, 2247–2258. [Google Scholar] [CrossRef] [PubMed]
- Young, J.H.; Peyton, M.; Seok Kim, H.; McMillan, E.; Minna, J.D.; White, M.A.; Marcotte, E.M. Computational discovery of pathway-level genetic vulnerabilities in non-small-cell lung cancer. Bioinformatics 2016, 32, 1373–1379. [Google Scholar] [CrossRef] [PubMed]
- Braun, C.J.; Stanciu, M.; Boutz, P.L.; Patterson, J.C.; Calligaris, D.; Higuchi, F.; Neupane, R.; Fenoglio, S.; Cahill, D.P.; Wakimoto, H.; et al. Coordinated Splicing of Regulatory Detained Introns within Oncogenic Transcripts Creates an Exploitable Vulnerability in Malignant Glioma. Cancer Cell 2017, 32, 411–426.e11. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, K.; Matlin, A.J.; Lowell, A.M.; Moore, M.J. The biflavonoid isoginkgetin is a general inhibitor of Pre-mRNA splicing. J. Biol. Chem. 2008, 283, 33147–33154. [Google Scholar] [CrossRef]
- Vanzyl, E.J.; Rick, K.R.C.; Blackmore, A.B.; MacFarlane, E.M.; McKay, B.C. Flow cytometric analysis identifies changes in S and M phases as novel cell cycle alterations induced by the splicing inhibitor isoginkgetin. PLoS ONE 2018, 13, e0191178. [Google Scholar] [CrossRef] [PubMed]
- Pawellek, A.; Ryder, U.; Tammsalu, T.; King, L.J.; Kreinin, H.; Ly, T.; Hay, R.T.; Hartley, R.C.; Lamond, A.I. Characterisation of the biflavonoid hinokiflavone as a pre-mRNA splicing modulator that inhibits SENP. Elife 2017, 6. [Google Scholar] [CrossRef]
- Zhou, J.; Zhao, R.; Ye, T.; Yang, S.; Li, Y.; Yang, F.; Wang, G.; Xie, Y.; Li, Q. Antitumor activity in colorectal cancer induced by hinokiflavone. J. Gastroenterol. Hepatol. 2019, 34, 1571–1580. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Liu, C.; Liu, F.; Liu, Z.; Lai, G.; Yi, J. Hinokiflavone induces apoptosis and inhibits migration of breast cancer cells via EMT signalling pathway. Cell Biochem. Funct. 2020, 38, 249–256. [Google Scholar] [CrossRef]
- Makarov, E.M.; Owen, N.; Bottrill, A.; Makarova, O.V. Functional mammalian spliceosomal complex E contains SMN complex proteins in addition to U1 and U2 snRNPs. Nucleic Acids Res. 2012, 40, 2639–2652. [Google Scholar] [CrossRef] [PubMed]
- Stoilov, P.; Lin, C.H.; Damoiseaux, R.; Nikolic, J.; Black, D.L. A high-throughput screening strategy identifies cardiotonic steroids as alternative splicing modulators. Proc. Natl. Acad. Sci. USA 2008, 105, 11218–11223. [Google Scholar] [CrossRef]
- Berg, M.G.; Wan, L.; Younis, I.; Diem, M.D.; Soo, M.; Wang, C.; Dreyfuss, G. A quantitative high-throughput in vitro splicing assay identifies inhibitors of spliceosome catalysis. Mol. Cell. Biol. 2012, 32, 1271–1283. [Google Scholar] [CrossRef]
- Brezak, M.C.; Quaranta, M.; Contour-Galcera, M.O.; Lavergne, O.; Mondesert, O.; Auvray, P.; Kasprzyk, P.G.; Prevost, G.P.; Ducommun, B. Inhibition of human tumor cell growth in vivo by an orally bioavailable inhibitor of CDC25 phosphatases. Mol. Cancer Ther. 2005, 4, 1378–1387. [Google Scholar] [CrossRef] [PubMed]
- Cazales, M.; Boutros, R.; Brezak, M.C.; Chaumeron, S.; Prevost, G.; Ducommun, B. Pharmacologic inhibition of CDC25 phosphatases impairs interphase microtubule dynamics and mitotic spindle assembly. Mol. Cancer Ther. 2007, 6, 318–325. [Google Scholar] [CrossRef]
- Effenberger, K.A.; James, R.C.; Urabe, V.K.; Dickey, B.J.; Linington, R.G.; Jurica, M.S. The Natural Product N-Palmitoyl-l-leucine Selectively Inhibits Late Assembly of Human Spliceosomes. J. Biol. Chem. 2015, 290, 27524–27531. [Google Scholar] [CrossRef] [PubMed]
- Effenberger, K.A.; Perriman, R.J.; Bray, W.M.; Lokey, R.S.; Ares, M., Jr.; Jurica, M.S. A high-throughput splicing assay identifies new classes of inhibitors of human and yeast spliceosomes. J. BioMol. Screen. 2013, 18, 1110–1120. [Google Scholar] [CrossRef]
- Shoemaker, R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Vellanki, S.H.; Cruz, R.G.B.; Richards, C.E.; Smith, Y.E.; Hudson, L.; Jahns, H.; Hopkins, A.M. Antibiotic Tetrocarcin-A Down-regulates JAM-A, IAPs and Induces Apoptosis in Triple-negative Breast Cancer Models. Anticancer. Res. 2019, 39, 1197–1204. [Google Scholar] [CrossRef]
- Younis, I.; Berg, M.; Kaida, D.; Dittmar, K.; Wang, C.; Dreyfuss, G. Rapid-response splicing reporter screens identify differential regulators of constitutive and alternative splicing. Mol. Cell. Biol. 2010, 30, 1718–1728. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Bray, W.; Smith, A.J.; Zhou, W.; Calaoagan, J.; Lagisetti, C.; Sambucetti, L.; Crews, P.; Lokey, R.S.; Webb, T.R. An exon skipping screen identifies antitumor drugs that are potent modulators of pre-mRNA splicing, suggesting new therapeutic applications. PLoS ONE 2020, 15, e0233672. [Google Scholar] [CrossRef]
- Shi, Y.; Park, J.; Lagisetti, C.; Zhou, W.; Sambucetti, L.C.; Webb, T.R. A triple exon-skipping luciferase reporter assay identifies a new CLK inhibitor pharmacophore. Bioorg. Med. Chem. Lett. 2017, 27, 406–412. [Google Scholar] [CrossRef]
- Samatov, T.R.; Wolf, A.; Odenwalder, P.; Bessonov, S.; Deraeve, C.; Bon, R.S.; Waldmann, H.; Luhrmann, R. Psoromic acid derivatives: A new family of small-molecule pre-mRNA splicing inhibitors discovered by a stage-specific high-throughput in vitro splicing assay. Chembiochem 2012, 13, 640–644. [Google Scholar] [CrossRef][Green Version]
- Pawellek, A.; McElroy, S.; Samatov, T.; Mitchell, L.; Woodland, A.; Ryder, U.; Gray, D.; Luhrmann, R.; Lamond, A.I. Identification of small molecule inhibitors of pre-mRNA splicing. J. Biol. Chem. 2014, 289, 34683–34698. [Google Scholar] [CrossRef]
- Sidarovich, A.; Will, C.L.; Anokhina, M.M.; Ceballos, J.; Sievers, S.; Agafonov, D.E.; Samatov, T.; Bao, P.; Kastner, B.; Urlaub, H.; et al. Identification of a small molecule inhibitor that stalls splicing at an early step of spliceosome activation. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Borsi, L.; Balza, E.; Gaggero, B.; Allemanni, G.; Zardi, L. The alternative splicing pattern of the tenascin-C pre-mRNA is controlled by the extracellular pH. J. Biol. Chem. 1995, 270, 6243–6245. [Google Scholar] [CrossRef] [PubMed]
- Yuo, C.Y.; Lin, H.H.; Chang, Y.S.; Yang, W.K.; Chang, J.G. 5-(N-ethyl-N-isopropyl)-amiloride enhances SMN2 exon 7 inclusion and protein expression in spinal muscular atrophy cells. Ann. Neurol. 2008, 63, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.H.; Liu, T.C.; Yang, W.K.; Lee, C.C.; Lin, Y.H.; Chen, T.Y.; Chang, J.G. Amiloride modulates alternative splicing in leukemic cells and resensitizes Bcr-AblT315I mutant cells to imatinib. Cancer Res. 2011, 71, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Chang, W.H.; Chang, Y.S.; Yang, J.M.; Chang, C.S.; Hsu, K.C.; Chen, Y.T.; Liu, T.Y.; Chen, Y.C.; Lin, S.Y.; et al. Alternative splicing in human cancer cells is modulated by the amiloride derivative 3,5-diamino-6-chloro-N-(N-(2,6-dichlorobenzoyl)carbamimidoyl)pyrazine-2-carboxide. Mol. Oncol. 2019, 13, 1744–1762. [Google Scholar] [CrossRef]
- Bonnal, S.; Vigevani, L.; Valcarcel, J. The spliceosome as a target of novel antitumour drugs. Nat. Rev. Drug Discov. 2012, 11, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Webb, T.R.; Joyner, A.S.; Potter, P.M. The development and application of small molecule modulators of SF3b as therapeutic agents for cancer. Drug Discov. Today 2013, 18, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Kakeya, H.; Kaida, D.; Sekiya, H.; Nagai, K.; Yoshida, M.; Osada, H. RQN-18690A (18-deoxyherboxidiene) targets SF3b, a spliceosome component, and inhibits angiogenesis. J. Antibiot. 2016, 69, 121–123. [Google Scholar] [CrossRef] [PubMed]
- Chung, F.F.; Tan, P.F.; Raja, V.J.; Tan, B.S.; Lim, K.H.; Kam, T.S.; Hii, L.W.; Tan, S.H.; See, S.J.; Tan, Y.F.; et al. Jerantinine A induces tumor-specific cell death through modulation of splicing factor 3b subunit 1 (SF3B1). Sci. Rep. 2017, 7, 42504. [Google Scholar] [CrossRef]
- Liu, X.; Biswas, S.; Berg, M.G.; Antapli, C.M.; Xie, F.; Wang, Q.; Tang, M.C.; Tang, G.L.; Zhang, L.; Dreyfuss, G.; et al. Genomics-guided discovery of thailanstatins A, B, and C As pre-mRNA splicing inhibitors and antiproliferative agents from Burkholderia thailandensis MSMB43. J. Nat. Prod. 2013, 76, 685–693. [Google Scholar] [CrossRef]
- Puthenveetil, S.; Loganzo, F.; He, H.; Dirico, K.; Green, M.; Teske, J.; Musto, S.; Clark, T.; Rago, B.; Koehn, F.; et al. Natural Product Splicing Inhibitors: A New Class of Antibody-Drug Conjugate (ADC) Payloads. Bioconjug. Chem. 2016, 27, 1880–1888. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Rhoades, D.; Kumar, S.M. Total Syntheses of Thailanstatins A-C, Spliceostatin D, and Analogues Thereof. Stereodivergent Synthesis of Tetrasubstituted Dihydro- and Tetrahydropyrans and Design, Synthesis, Biological Evaluation, and Discovery of Potent Antitumor Agents. J. Am. Chem. Soc. 2018, 140, 8303–8320. [Google Scholar] [CrossRef]
- Corrionero, A.; Minana, B.; Valcarcel, J. Reduced fidelity of branch point recognition and alternative splicing induced by the anti-tumor drug spliceostatin A. Genes Dev. 2011, 25, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Cretu, C.; Agrawal, A.A.; Cook, A.; Will, C.L.; Fekkes, P.; Smith, P.G.; Luhrmann, R.; Larsen, N.; Buonamici, S.; Pena, V. Structural Basis of Splicing Modulation by Antitumor Macrolide Compounds. Mol. Cell 2018, 70, 265–273.e8. [Google Scholar] [CrossRef]
- Finci, L.I.; Zhang, X.; Huang, X.; Zhou, Q.; Tsai, J.; Teng, T.; Agrawal, A.; Chan, B.; Irwin, S.; Karr, C.; et al. The cryo-EM structure of the SF3b spliceosome complex bound to a splicing modulator reveals a pre-mRNA substrate competitive mechanism of action. Genes Dev. 2018, 32, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Folco, E.G.; Coil, K.E.; Reed, R. The anti-tumor drug E7107 reveals an essential role for SF3b in remodeling U2 snRNP to expose the branch point-binding region. Genes Dev. 2011, 25, 440–444. [Google Scholar] [CrossRef]
- Hasegawa, M.; Miura, T.; Kuzuya, K.; Inoue, A.; Won Ki, S.; Horinouchi, S.; Yoshida, T.; Kunoh, T.; Koseki, K.; Mino, K.; et al. Identification of SAP155 as the target of GEX1A (Herboxidiene), an antitumor natural product. ACS Chem. Biol. 2011, 6, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Kaida, D.; Motoyoshi, H.; Tashiro, E.; Nojima, T.; Hagiwara, M.; Ishigami, K.; Watanabe, H.; Kitahara, T.; Yoshida, T.; Nakajima, H.; et al. Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat. Chem. Biol. 2007, 3, 576–583. [Google Scholar] [CrossRef]
- Roybal, G.A.; Jurica, M.S. Spliceostatin A inhibits spliceosome assembly subsequent to prespliceosome formation. Nucleic Acids Res. 2010, 38, 6664–6672. [Google Scholar] [CrossRef]
- Effenberger, K.A.; Urabe, V.K.; Prichard, B.E.; Ghosh, A.K.; Jurica, M.S. Interchangeable SF3B1 inhibitors interfere with pre-mRNA splicing at multiple stages. RNA 2016, 22, 350–359. [Google Scholar] [CrossRef]
- Teng, T.; Tsai, J.H.; Puyang, X.; Seiler, M.; Peng, S.; Prajapati, S.; Aird, D.; Buonamici, S.; Caleb, B.; Chan, B.; et al. Splicing modulators act at the branch point adenosine binding pocket defined by the PHF5A-SF3b complex. Nat. Commun. 2017, 8, 15522. [Google Scholar] [CrossRef]
- Hansen, S.R.; Nikolai, B.J.; Spreacker, P.J.; Carrocci, T.J.; Hoskins, A.A. Chemical Inhibition of Pre-mRNA Splicing in Living Saccharomyces cerevisiae. Cell Chem. Biol. 2019, 26, 443–448.e3. [Google Scholar] [CrossRef]
- Seiler, M.; Yoshimi, A.; Darman, R.; Chan, B.; Keaney, G.; Thomas, M.; Agrawal, A.A.; Caleb, B.; Csibi, A.; Sean, E.; et al. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat. Med. 2018, 24, 497–504. [Google Scholar] [CrossRef]
- Vigevani, L.; Gohr, A.; Webb, T.; Irimia, M.; Valcarcel, J. Molecular basis of differential 3′ splice site sensitivity to anti-tumor drugs targeting U2 snRNP. Nat. Commun. 2017, 8, 2100. [Google Scholar] [CrossRef]
- Yoshimoto, R.; Kaida, D.; Furuno, M.; Burroughs, A.M.; Noma, S.; Suzuki, H.; Kawamura, Y.; Hayashizaki, Y.; Mayeda, A.; Yoshida, M. Global analysis of pre-mRNA subcellular localization following splicing inhibition by spliceostatin A. RNA 2017, 23, 47–57. [Google Scholar] [CrossRef]
- Carvalho, T.; Martins, S.; Rino, J.; Marinho, S.; Carmo-Fonseca, M. Pharmacological inhibition of the spliceosome subunit SF3b triggers exon junction complex-independent nonsense-mediated decay. J. Cell Sci. 2017, 130, 1519–1531. [Google Scholar] [CrossRef]
- Convertini, P.; Shen, M.; Potter, P.M.; Palacios, G.; Lagisetti, C.; de la Grange, P.; Horbinski, C.; Fondufe-Mittendorf, Y.N.; Webb, T.R.; Stamm, S. Sudemycin E influences alternative splicing and changes chromatin modifications. Nucleic Acids Res. 2014, 42, 4947–4961. [Google Scholar] [CrossRef]
- Gao, Y.; Koide, K. Chemical perturbation of Mcl-1 pre-mRNA splicing to induce apoptosis in cancer cells. ACS Chem. Biol. 2013, 8, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, M.K.; Kumar, D.; Villa, R.; La Clair, J.J.; Benner, C.; Sasik, R.; Jones, H.; Ghia, E.M.; Rassenti, L.Z.; Kipps, T.J.; et al. Targeting the spliceosome in chronic lymphocytic leukemia with the macrolides FD-895 and pladienolide-B. Haematologica 2015, 100, 945–954. [Google Scholar] [CrossRef]
- Larrayoz, M.; Blakemore, S.J.; Dobson, R.C.; Blunt, M.D.; Rose-Zerilli, M.J.; Walewska, R.; Duncombe, A.; Oscier, D.; Koide, K.; Forconi, F.; et al. The SF3B1 inhibitor spliceostatin A (SSA) elicits apoptosis in chronic lymphocytic leukaemia cells through downregulation of Mcl-1. Leukemia 2016, 30, 351–360. [Google Scholar] [CrossRef]
- Makowski, K.; Vigevani, L.; Albericio, F.; Valcarcel, J.; Alvarez, M. Sudemycin K: A Synthetic Antitumor Splicing Inhibitor Variant with Improved Activity and Versatile Chemistry. ACS Chem. Biol. 2017, 12, 163–173. [Google Scholar] [CrossRef]
- Sciarrillo, R.; Wojtuszkiewicz, A.; El Hassouni, B.; Funel, N.; Gandellini, P.; Lagerweij, T.; Buonamici, S.; Blijlevens, M.; van der Laan, E.A.Z.; Zaffaroni, N.; et al. Splicing modulation as novel therapeutic strategy against diffuse malignant peritoneal mesothelioma. EBioMedicine 2019, 39, 215–225. [Google Scholar] [CrossRef]
- Fan, L.; Lagisetti, C.; Edwards, C.C.; Webb, T.R.; Potter, P.M. Sudemycins, novel small molecule analogues of FR901464, induce alternative gene splicing. ACS Chem. Biol. 2011, 6, 582–589. [Google Scholar] [CrossRef]
- Ten Hacken, E.; Valentin, R.; Regis, F.F.D.; Sun, J.; Yin, S.; Werner, L.; Deng, J.; Gruber, M.; Wong, J.; Zheng, M.; et al. Splicing modulation sensitizes chronic lymphocytic leukemia cells to venetoclax by remodeling mitochondrial apoptotic dependencies. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Gao, Y.; Trivedi, S.; Ferris, R.L.; Koide, K. Regulation of HPV16 E6 and MCL1 by SF3B1 inhibitor in head and neck cancer cells. Sci. Rep. 2014, 4, 6098. [Google Scholar] [CrossRef]
- Effenberger, K.A.; Anderson, D.D.; Bray, W.M.; Prichard, B.E.; Ma, N.; Adams, M.S.; Ghosh, A.K.; Jurica, M.S. Coherence between cellular responses and in vitro splicing inhibition for the anti-tumor drug pladienolide B and its analogs. J. Biol. Chem. 2014, 289, 1938–1947. [Google Scholar] [CrossRef]
- Lagisetti, C.; Yermolina, M.V.; Sharma, L.K.; Palacios, G.; Prigaro, B.J.; Webb, T.R. Pre-mRNA splicing-modulatory pharmacophores: The total synthesis of herboxidiene, a pladienolide-herboxidiene hybrid analog and related derivatives. ACS Chem. Biol. 2014, 9, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Han, C.; Wang, E.; Lorch, A.H.; Serafin, V.; Cho, B.K.; Gutierrez Diaz, B.T.; Calvo, J.; Fang, C.; Khodadadi-Jamayran, A.; et al. Posttranslational Regulation of the Exon Skipping Machinery Controls Aberrant Splicing in Leukemia. Cancer Discov. 2020, 10, 1388–1409. [Google Scholar] [CrossRef]
- Eskens, F.A.; Ramos, F.J.; Burger, H.; O’Brien, J.P.; Piera, A.; de Jonge, M.J.; Mizui, Y.; Wiemer, E.A.; Carreras, M.J.; Baselga, J.; et al. Phase I pharmacokinetic and pharmacodynamic study of the first-in-class spliceosome inhibitor E7107 in patients with advanced solid tumors. Clin. Cancer Res. 2013, 19, 6296–6304. [Google Scholar] [CrossRef]
- Hong, D.S.; Kurzrock, R.; Naing, A.; Wheler, J.J.; Falchook, G.S.; Schiffman, J.S.; Faulkner, N.; Pilat, M.J.; O’Brien, J.; LoRusso, P. A phase I, open-label, single-arm, dose-escalation study of E7107, a precursor messenger ribonucleic acid (pre-mRNA) splicesome inhibitor administered intravenously on days 1 and 8 every 21 days to patients with solid tumors. Investig. New Drugs 2014, 32, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Diouf, B.; Lin, W.; Goktug, A.; Grace, C.R.R.; Waddell, M.B.; Bao, J.; Shao, Y.; Heath, R.J.; Zheng, J.J.; Shelat, A.A.; et al. Alteration of RNA Splicing by Small-Molecule Inhibitors of the Interaction between NHP2L1 and U4. SLAS Discov. 2018, 23, 164–173. [Google Scholar] [CrossRef]
- Rossi, F.; Labourier, E.; Forne, T.; Divita, G.; Derancourt, J.; Riou, J.F.; Antoine, E.; Cathala, G.; Brunel, C.; Tazi, J. Specific phosphorylation of SR proteins by mammalian DNA topoisomerase I. Nature 1996, 381, 80–82. [Google Scholar] [CrossRef]
- Pilch, B.; Allemand, E.; Facompre, M.; Bailly, C.; Riou, J.F.; Soret, J.; Tazi, J. Specific inhibition of serine- and arginine-rich splicing factors phosphorylation, spliceosome assembly, and splicing by the antitumor drug NB-506. Cancer Res. 2001, 61, 6876–6884. [Google Scholar] [PubMed]
- Tazi, J.; Bakkour, N.; Soret, J.; Zekri, L.; Hazra, B.; Laine, W.; Baldeyrou, B.; Lansiaux, A.; Bailly, C. Selective inhibition of topoisomerase I and various steps of spliceosome assembly by diospyrin derivatives. Mol. Pharmacol. 2005, 67, 1186–1194. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Krzysko, K.A.; Kowalska-Loth, B.; Skrajna, A.M.; Czubaty, A.; Girstun, A.; Cieplak, M.K.; Lesyng, B.; Staron, K. Activities of topoisomerase I in its complex with SRSF1. Biochemistry 2012, 51, 1803–1816. [Google Scholar] [CrossRef] [PubMed]
- Ajuh, P.; Lamond, A.I. Identification of peptide inhibitors of pre-mRNA splicing derived from the essential interaction domains of CDC5L and PLRG1. Nucleic Acids Res. 2003, 31, 6104–6116. [Google Scholar] [CrossRef]
- Grillari, J.; Loscher, M.; Denegri, M.; Lee, K.; Fortschegger, K.; Eisenhaber, F.; Ajuh, P.; Lamond, A.I.; Katinger, H.; Grillari-Voglauer, R. Blom7alpha is a novel heterogeneous nuclear ribonucleoprotein K homology domain protein involved in pre-mRNA splicing that interacts with SNEVPrp19-Pso4. J. Biol. Chem. 2009, 284, 29193–29204. [Google Scholar] [CrossRef]
- Loscher, M.; Schosserer, M.; Dausse, E.; Lee, K.; Ajuh, P.; Grillari-Voglauer, R.; Lamond, A.I.; Toulme, J.J.; Grillari, J. Inhibition of pre-mRNA splicing by a synthetic Blom7alpha-interacting small RNA. PLoS ONE 2012, 7, e47497. [Google Scholar] [CrossRef]
- Lallena, M.J.; Chalmers, K.J.; Llamazares, S.; Lamond, A.I.; Valcarcel, J. Splicing regulation at the second catalytic step by Sex-lethal involves 3′ splice site recognition by SPF45. Cell 2002, 109, 285–296. [Google Scholar] [CrossRef]
- Corsini, L.; Bonnal, S.; Basquin, J.; Hothorn, M.; Scheffzek, K.; Valcarcel, J.; Sattler, M. U2AF-homology motif interactions are required for alternative splicing regulation by SPF45. Nat. Struct. Mol. Biol. 2007, 14, 620–629. [Google Scholar] [CrossRef]
- Sampath, J.; Long, P.R.; Shepard, R.L.; Xia, X.; Devanarayan, V.; Sandusky, G.E.; Perry, W.L., 3rd; Dantzig, A.H.; Williamson, M.; Rolfe, M.; et al. Human SPF45, a splicing factor, has limited expression in normal tissues, is overexpressed in many tumors, and can confer a multidrug-resistant phenotype to cells. Am. J. Pathol. 2003, 163, 1781–1790. [Google Scholar] [CrossRef]
- Lu, J.; Li, Q.; Cai, L.; Zhu, Z.; Guan, J.; Wang, C.; Xia, J.; Xia, L.; Wen, M.; Zheng, W.; et al. RBM17 controls apoptosis and proliferation to promote Glioma progression. Biochem. Biophys. Res. Commun. 2018, 505, 20–28. [Google Scholar] [CrossRef]
- Jagtap, P.K.; Garg, D.; Kapp, T.G.; Will, C.L.; Demmer, O.; Luhrmann, R.; Kessler, H.; Sattler, M. Rational Design of Cyclic Peptide Inhibitors of U2AF Homology Motif (UHM) Domains To Modulate Pre-mRNA Splicing. J. Med. Chem. 2016, 59, 10190–10197. [Google Scholar] [CrossRef]
- Jagtap, P.K.A.; Kubelka, T.; Soni, K.; Will, C.L.; Garg, D.; Sippel, C.; Kapp, T.G.; Potukuchi, H.K.; Schorpp, K.; Hadian, K.; et al. Identification of phenothiazine derivatives as UHM-binding inhibitors of early spliceosome assembly. Nat. Commun. 2020, 11, 5621. [Google Scholar] [CrossRef]
- Tari, M.; Manceau, V.; de Matha Salone, J.; Kobayashi, A.; Pastre, D.; Maucuer, A. U2AF(65) assemblies drive sequence-specific splice site recognition. EMBO Rep. 2019, 20, e47604. [Google Scholar] [CrossRef]
- Mercier, I.; Casimiro, M.C.; Zhou, J.; Wang, C.; Plymire, C.; Bryant, K.G.; Daumer, K.M.; Sotgia, F.; Bonuccelli, G.; Witkiewicz, A.K.; et al. Genetic ablation of caveolin-1 drives estrogen-hypersensitivity and the development of DCIS-like mammary lesions. Am. J. Pathol. 2009, 174, 1172–1190. [Google Scholar] [CrossRef] [PubMed]
- Sillars-Hardebol, A.H.; Carvalho, B.; Tijssen, M.; Belien, J.A.; de Wit, M.; Delis-van Diemen, P.M.; Ponten, F.; van de Wiel, M.A.; Fijneman, R.J.; Meijer, G.A. TPX2 and AURKA promote 20q amplicon-driven colorectal adenoma to carcinoma progression. Gut 2012, 61, 1568–1575. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Lu, S.X.; Pastore, A.; Chen, X.; Imig, J.; Chun-Wei Lee, S.; Hockemeyer, K.; Ghebrechristos, Y.E.; Yoshimi, A.; Inoue, D.; et al. Targeting an RNA-Binding Protein Network in Acute Myeloid Leukemia. Cancer Cell 2019, 35, 369–384.e7. [Google Scholar] [CrossRef] [PubMed]
- Ellis, J.D.; Lleres, D.; Denegri, M.; Lamond, A.I.; Caceres, J.F. Spatial mapping of splicing factor complexes involved in exon and intron definition. J. Cell Biol. 2008, 181, 921–934. [Google Scholar] [CrossRef] [PubMed]
- Prigge, J.R.; Iverson, S.V.; Siders, A.M.; Schmidt, E.E. Interactome for auxiliary splicing factor U2AF(65) suggests diverse roles. Biochim. Biophys. Acta 2009, 1789, 487–492. [Google Scholar] [CrossRef]
- Stepanyuk, G.A.; Serrano, P.; Peralta, E.; Farr, C.L.; Axelrod, H.L.; Geralt, M.; Das, D.; Chiu, H.J.; Jaroszewski, L.; Deacon, A.M.; et al. UHM-ULM interactions in the RBM39-U2AF65 splicing-factor complex. Acta Crystallogr. D Struct. Biol. 2016, 72, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Loerch, S.; Maucuer, A.; Manceau, V.; Green, M.R.; Kielkopf, C.L. Cancer-relevant splicing factor CAPERalpha engages the essential splicing factor SF3b155 in a specific ternary complex. J. Biol. Chem. 2014, 289, 17325–17337. [Google Scholar] [CrossRef] [PubMed]
- Uehara, T.; Minoshima, Y.; Sagane, K.; Sugi, N.H.; Mitsuhashi, K.O.; Yamamoto, N.; Kamiyama, H.; Takahashi, K.; Kotake, Y.; Uesugi, M.; et al. Selective degradation of splicing factor CAPERalpha by anticancer sulfonamides. Nat. Chem. Biol. 2017, 13, 675–680. [Google Scholar] [CrossRef]
- Carabet, L.A.; Leblanc, E.; Lallous, N.; Morin, H.; Ghaidi, F.; Lee, J.; Rennie, P.S.; Cherkasov, A. Computer-Aided Discovery of Small Molecules Targeting the RNA Splicing Activity of hnRNP A1 in Castration-Resistant Prostate Cancer. Molecules 2019, 24, 763. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Iwatani, M.; Yamamoto, T.; Tanaka, T.; Kawamoto, T.; Morishita, D.; Nakanishi, A.; Maezaki, H. Discovery of spiro[indole-3,2′-pyrrolidin]-2(1H)-one based inhibitors targeting Brr2, a core component of the U5 snRNP. Bioorg Med. Chem. 2017, 25, 4753–4767. [Google Scholar] [CrossRef]
- Iwatani-Yoshihara, M.; Ito, M.; Klein, M.G.; Yamamoto, T.; Yonemori, K.; Tanaka, T.; Miwa, M.; Morishita, D.; Endo, S.; Tjhen, R.; et al. Discovery of Allosteric Inhibitors Targeting the Spliceosomal RNA Helicase Brr2. J. Med. Chem. 2017, 60, 5759–5771. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, T.; Hosoya, T.; Shimizu, S.; Sumi, K.; Oshiro, T.; Yoshinaka, Y.; Suzuki, M.; Yamamoto, N.; Herzenberg, L.A.; Herzenberg, L.A.; et al. Utilization of host SR protein kinases and RNA-splicing machinery during viral replication. Proc. Natl. Acad. Sci. USA 2006, 103, 11329–11333. [Google Scholar] [CrossRef]
- Siqueira, R.P.; Barbosa Ede, A.; Poleto, M.D.; Righetto, G.L.; Seraphim, T.V.; Salgado, R.L.; Ferreira, J.G.; Barros, M.V.; de Oliveira, L.L.; Laranjeira, A.B.; et al. Potential Antileukemia Effect and Structural Analyses of SRPK Inhibition by N-(2-(Piperidin-1-yl)-5-(Trifluoromethyl)Phenyl)Isonicotinamide (SRPIN340). PLoS ONE 2015, 10, e0134882. [Google Scholar] [CrossRef]
- Gammons, M.V.; Lucas, R.; Dean, R.; Coupland, S.E.; Oltean, S.; Bates, D.O. Targeting SRPK1 to control VEGF-mediated tumour angiogenesis in metastatic melanoma. Br. J. Cancer 2014, 111, 477–485. [Google Scholar] [CrossRef]
- Siqueira, R.P.; Barros, M.V.A.; Barbosa, E.A.A.; Onofre, T.S.; Goncalves, V.H.S.; Pereira, H.S.; Silva Junior, A.; de Oliveira, L.L.; Almeida, M.R.; Fietto, J.L.R.; et al. Trifluoromethyl arylamides with antileukemia effect and intracellular inhibitory activity over serine/arginine-rich protein kinases (SRPKs). Eur. J. Med. Chem. 2017, 134, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Gammons, M.V.; Fedorov, O.; Ivison, D.; Du, C.; Clark, T.; Hopkins, C.; Hagiwara, M.; Dick, A.D.; Cox, R.; Harper, S.J.; et al. Topical antiangiogenic SRPK1 inhibitors reduce choroidal neovascularization in rodent models of exudative AMD. Investig. Ophthalmol. Vis. Sci. 2013, 54, 6052–6062. [Google Scholar] [CrossRef] [PubMed]
- Batson, J.; Toop, H.D.; Redondo, C.; Babaei-Jadidi, R.; Chaikuad, A.; Wearmouth, S.F.; Gibbons, B.; Allen, C.; Tallant, C.; Zhang, J.; et al. Development of Potent, Selective SRPK1 Inhibitors as Potential Topical Therapeutics for Neovascular Eye Disease. ACS Chem. Biol. 2017, 12, 825–832. [Google Scholar] [CrossRef]
- Hatcher, J.M.; Wu, G.; Zeng, C.; Zhu, J.; Meng, F.; Patel, S.; Wang, W.; Ficarro, S.B.; Leggett, A.L.; Powell, C.E.; et al. SRPKIN-1: A Covalent SRPK1/2 Inhibitor that Potently Converts VEGF from Pro-angiogenic to Anti-angiogenic Isoform. Cell Chem. Biol. 2018, 25, 460–470.e6. [Google Scholar] [CrossRef]
- Nayler, O.; Stamm, S.; Ullrich, A. Characterization and comparison of four serine- and arginine-rich (SR) protein kinases. Biochem. J. 1997, 326 Pt 3, 693–700. [Google Scholar] [CrossRef]
- Muraki, M.; Ohkawara, B.; Hosoya, T.; Onogi, H.; Koizumi, J.; Koizumi, T.; Sumi, K.; Yomoda, J.; Murray, M.V.; Kimura, H.; et al. Manipulation of alternative splicing by a newly developed inhibitor of Clks. J. Biol. Chem. 2004, 279, 24246–24254. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, O.; Huber, K.; Eisenreich, A.; Filippakopoulos, P.; King, O.; Bullock, A.N.; Szklarczyk, D.; Jensen, L.J.; Fabbro, D.; Trappe, J.; et al. Specific CLK inhibitors from a novel chemotype for regulation of alternative splicing. Chem. Biol. 2011, 18, 67–76. [Google Scholar] [CrossRef]
- Boutz, P.L.; Bhutkar, A.; Sharp, P.A. Detained introns are a novel, widespread class of post-transcriptionally spliced introns. Genes Dev. 2015, 29, 63–80. [Google Scholar] [CrossRef]
- Iwai, K.; Yaguchi, M.; Nishimura, K.; Yamamoto, Y.; Tamura, T.; Nakata, D.; Dairiki, R.; Kawakita, Y.; Mizojiri, R.; Ito, Y.; et al. Anti-tumor efficacy of a novel CLK inhibitor via targeting RNA splicing and MYC-dependent vulnerability. EMBO Mol. Med. 2018, 10. [Google Scholar] [CrossRef]
- Riggs, J.R.; Nagy, M.; Elsner, J.; Erdman, P.; Cashion, D.; Robinson, D.; Harris, R.; Huang, D.; Tehrani, L.; Deyanat-Yazdi, G.; et al. The Discovery of a Dual TTK Protein Kinase/CDC2-Like Kinase (CLK2) Inhibitor for the Treatment of Triple Negative Breast Cancer Initiated from a Phenotypic Screen. J. Med. Chem. 2017, 60, 8989–9002. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Xu, S.; Deyanat-Yazdi, G.; Peng, S.X.; Barnes, L.A.; Narla, R.K.; Tran, T.; Mikolon, D.; Ning, Y.; Shi, T.; et al. Synthetic Lethal Strategy Identifies a Potent and Selective TTK and CLK1/2 Inhibitor for Treatment of Triple-Negative Breast Cancer with a Compromised G1-S Checkpoint. Mol. Cancer Ther. 2018, 17, 1727–1738. [Google Scholar] [CrossRef]
- Araki, S.; Dairiki, R.; Nakayama, Y.; Murai, A.; Miyashita, R.; Iwatani, M.; Nomura, T.; Nakanishi, O. Inhibitors of CLK protein kinases suppress cell growth and induce apoptosis by modulating pre-mRNA splicing. PLoS ONE 2015, 10, e0116929. [Google Scholar] [CrossRef]
- Funnell, T.; Tasaki, S.; Oloumi, A.; Araki, S.; Kong, E.; Yap, D.; Nakayama, Y.; Hughes, C.S.; Cheng, S.G.; Tozaki, H.; et al. CLK-dependent exon recognition and conjoined gene formation revealed with a novel small molecule inhibitor. Nat. Commun. 2017, 8, 7. [Google Scholar] [CrossRef]
- Tam, B.Y.; Chiu, K.; Chung, H.; Bossard, C.; Nguyen, J.D.; Creger, E.; Eastman, B.W.; Mak, C.C.; Ibanez, M.; Ghias, A.; et al. The CLK inhibitor SM08502 induces anti-tumor activity and reduces Wnt pathway gene expression in gastrointestinal cancer models. Cancer Lett. 2020, 473, 186–197. [Google Scholar] [CrossRef]
- Bezzi, M.; Teo, S.X.; Muller, J.; Mok, W.C.; Sahu, S.K.; Vardy, L.A.; Bonday, Z.Q.; Guccione, E. Regulation of constitutive and alternative splicing by PRMT5 reveals a role for Mdm4 pre-mRNA in sensing defects in the spliceosomal machinery. Genes Dev. 2013, 27, 1903–1916. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Gao, S.; Zhang, F.; Wang, Z.; Ma, W.; Davis, R.E.; Wang, Z. Protein arginine methyltransferase 5 is essential for growth of lung cancer cells. Biochem. J. 2012, 446, 235–241. [Google Scholar] [CrossRef]
- Smil, D.; Eram, M.S.; Li, F.; Kennedy, S.; Szewczyk, M.M.; Brown, P.J.; Barsyte-Lovejoy, D.; Arrowsmith, C.H.; Vedadi, M.; Schapira, M. Discovery of a Dual PRMT5-PRMT7 Inhibitor. ACS Med. Chem. Lett. 2015, 6, 408–412. [Google Scholar] [CrossRef] [PubMed]
- Chan-Penebre, E.; Kuplast, K.G.; Majer, C.R.; Boriack-Sjodin, P.A.; Wigle, T.J.; Johnston, L.D.; Rioux, N.; Munchhof, M.J.; Jin, L.; Jacques, S.L.; et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat. Chem. Biol. 2015, 11, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Duncan, K.W.; Rioux, N.; Boriack-Sjodin, P.A.; Munchhof, M.J.; Reiter, L.A.; Majer, C.R.; Jin, L.; Johnston, L.D.; Chan-Penebre, E.; Kuplast, K.G.; et al. Structure and Property Guided Design in the Identification of PRMT5 Tool Compound EPZ015666. ACS Med. Chem. Lett. 2016, 7, 162–166. [Google Scholar] [CrossRef]
- Kong, G.M.; Yu, M.; Gu, Z.; Chen, Z.; Xu, R.M.; O’Bryant, D.; Wang, Z. Selective small-chemical inhibitors of protein arginine methyltransferase 5 with anti-lung cancer activity. PLoS ONE 2017, 12, e0181601. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, L.; Chen, L.; Wei, H.; Demir, O.; Safa, A.; Zeng, L.; Amaro, R.E.; O’Neil, B.H.; Zhang, Z.Y.; Lu, T. Development of an AlphaLISA high throughput technique to screen for small molecule inhibitors targeting protein arginine methyltransferases. Mol. Biosyst. 2017, 13, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Fong, J.Y.; Pignata, L.; Goy, P.A.; Kawabata, K.C.; Lee, S.C.; Koh, C.M.; Musiani, D.; Massignani, E.; Kotini, A.G.; Penson, A.; et al. Therapeutic Targeting of RNA Splicing Catalysis through Inhibition of Protein Arginine Methylation. Cancer Cell 2019, 36, 194–209.e9. [Google Scholar] [CrossRef]
- AbuHammad, S.; Cullinane, C.; Martin, C.; Bacolas, Z.; Ward, T.; Chen, H.; Slater, A.; Ardley, K.; Kirby, L.; Chan, K.T.; et al. Regulation of PRMT5-MDM4 axis is critical in the response to CDK4/6 inhibitors in melanoma. Proc. Natl. Acad. Sci. USA 2019, 116, 17990–18000. [Google Scholar] [CrossRef] [PubMed]
- Bidwell, G.L., 3rd; Whittom, A.A.; Thomas, E.; Lyons, D.; Hebert, M.D.; Raucher, D. A thermally targeted peptide inhibitor of symmetrical dimethylation inhibits cancer-cell proliferation. Peptides 2010, 31, 834–841. [Google Scholar] [CrossRef][Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Godfrey, C.; Desviat, L.R.; Smedsrod, B.; Pietri-Rouxel, F.; Denti, M.A.; Disterer, P.; Lorain, S.; Nogales-Gadea, G.; Sardone, V.; Anwar, R.; et al. Delivery is key: Lessons learnt from developing splice-switching antisense therapies. EMBO Mol. Med. 2017, 9, 545–557. [Google Scholar] [CrossRef]
- Luo, J.; Solimini, N.L.; Elledge, S.J. Principles of cancer therapy: Oncogene and non-oncogene addiction. Cell 2009, 136, 823–837. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blijlevens, M.; Li, J.; van Beusechem, V.W. Biology of the mRNA Splicing Machinery and Its Dysregulation in Cancer Providing Therapeutic Opportunities. Int. J. Mol. Sci. 2021, 22, 5110. https://doi.org/10.3390/ijms22105110
Blijlevens M, Li J, van Beusechem VW. Biology of the mRNA Splicing Machinery and Its Dysregulation in Cancer Providing Therapeutic Opportunities. International Journal of Molecular Sciences. 2021; 22(10):5110. https://doi.org/10.3390/ijms22105110
Chicago/Turabian StyleBlijlevens, Maxime, Jing Li, and Victor W. van Beusechem. 2021. "Biology of the mRNA Splicing Machinery and Its Dysregulation in Cancer Providing Therapeutic Opportunities" International Journal of Molecular Sciences 22, no. 10: 5110. https://doi.org/10.3390/ijms22105110
APA StyleBlijlevens, M., Li, J., & van Beusechem, V. W. (2021). Biology of the mRNA Splicing Machinery and Its Dysregulation in Cancer Providing Therapeutic Opportunities. International Journal of Molecular Sciences, 22(10), 5110. https://doi.org/10.3390/ijms22105110