
Deregulation of Ca2+-Signaling Systems in White Adipocytes, Manifested as the Loss of Rhythmic Activity, Underlies the Development of Multiple Hormonal Resistance at Obesity and Type 2 Diabetes


Abstract
1. Introduction
- -
- short PFL based on CICR encoded by inositol-3-phosphate (IP3)-dependent Ca2+-receptor (IP3R):Ca2+ → IP3R → Ca2+
- -
- long PFL based on feedback activation of phospholipase C (PLC) by Ca2+ and subsequent activation of IP3R by the product of PLC reaction IP3:Ca2+ → PLC → IP3 → IP3R → Ca2+
2. Results
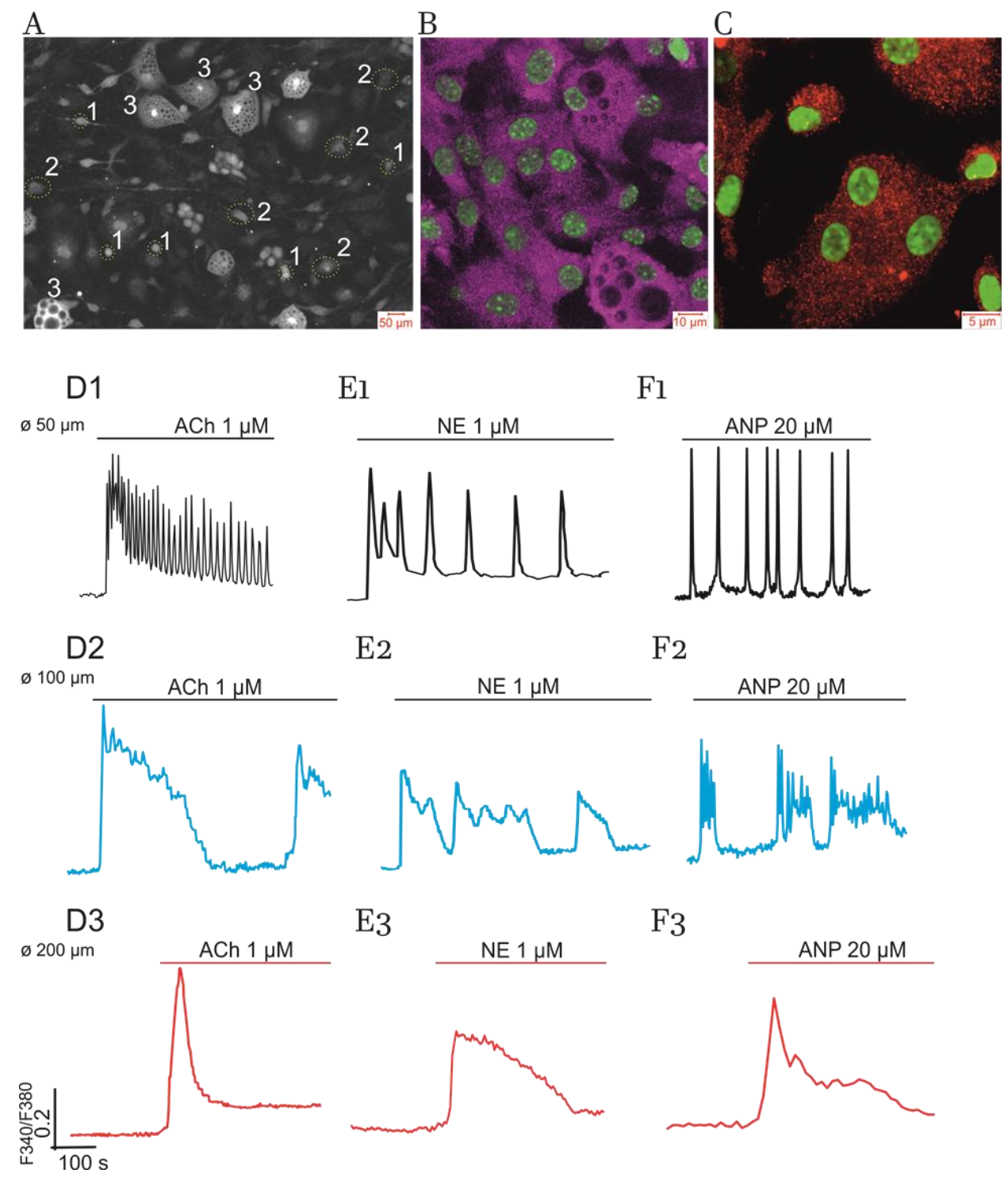
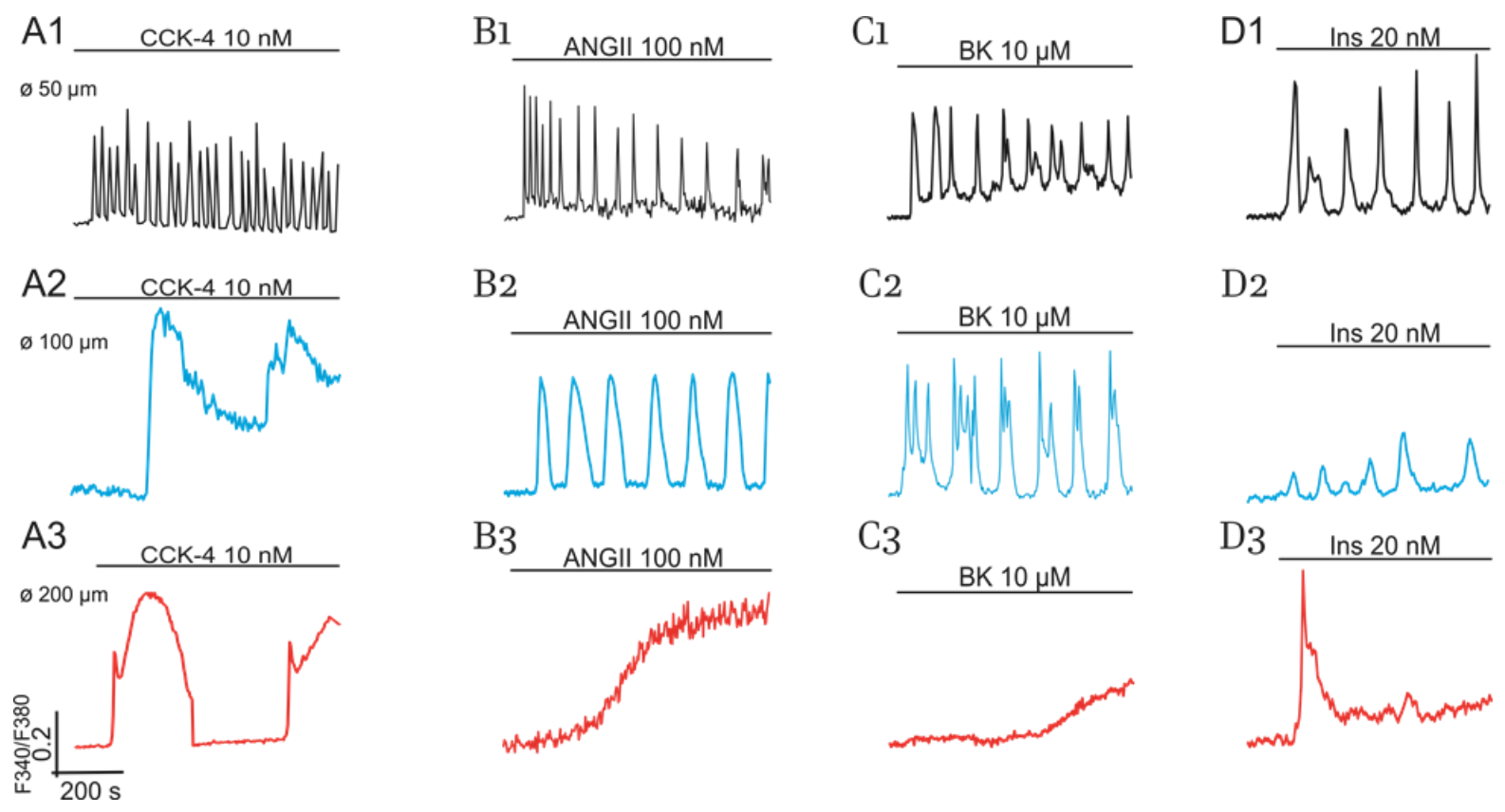
2.1. Diversity of Ca2+-Responses Evoked by Various Hormones in Cultured Adipocytes
2.1.1. Impact of Cell Size
- (1)
- in total, 10% to 15% of all cells in culture having small lipid inclusions (discernible with specific staining by Oil Red) and size (Ø) equal to or higher than 50 µM (Ø ≥ 50 µM);
- (2)
- in total, 50% to 60% of cells having several lipid droplets and Ø ≥ 100 µM;
- (3)
- the rest of the cells (15–20%) representing hypertrophied “obese” adipocytes completely filled with numerous lipid inclusions and having Ø ≥ 200 µM.
2.1.2. Impact of GPCR and RTK and Cultured Cells’ State on Switching on of PLC-G and NOS-G
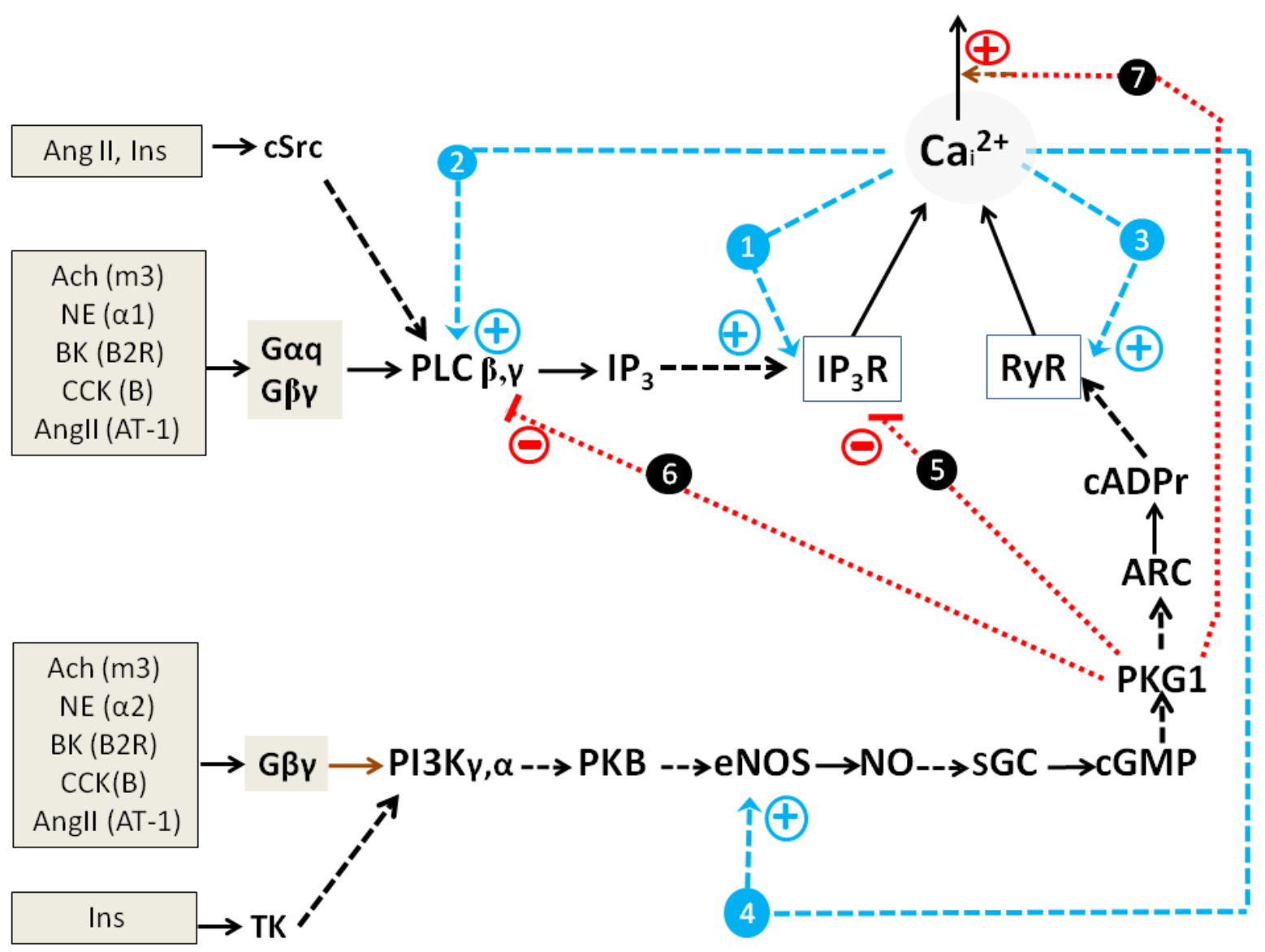
2.1.3. Simplified Kinetic Model of NOS-G and PLC-G with Input Signaling Axes
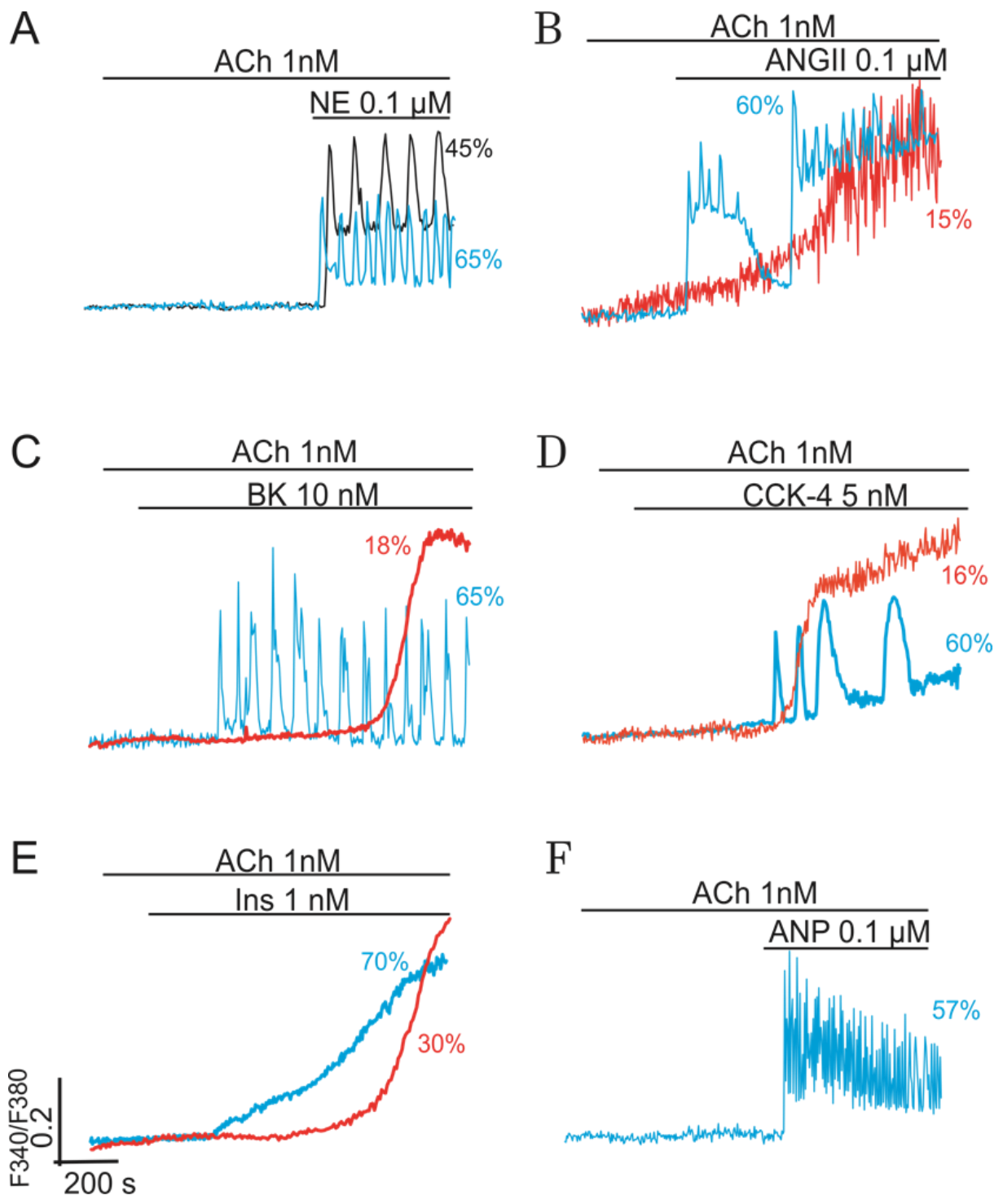
2.2. Combined Synergistic Action of Hormones Implicated in Parametric Control of NOS-RG
2.2.1. Gβγ-Subunits Interplay
2.2.2. Gβγ/TK Interplay
2.2.3. Gβγ/Gα Proteins Interplay
2.2.4. Gβγ/Gs Proteins Interplay
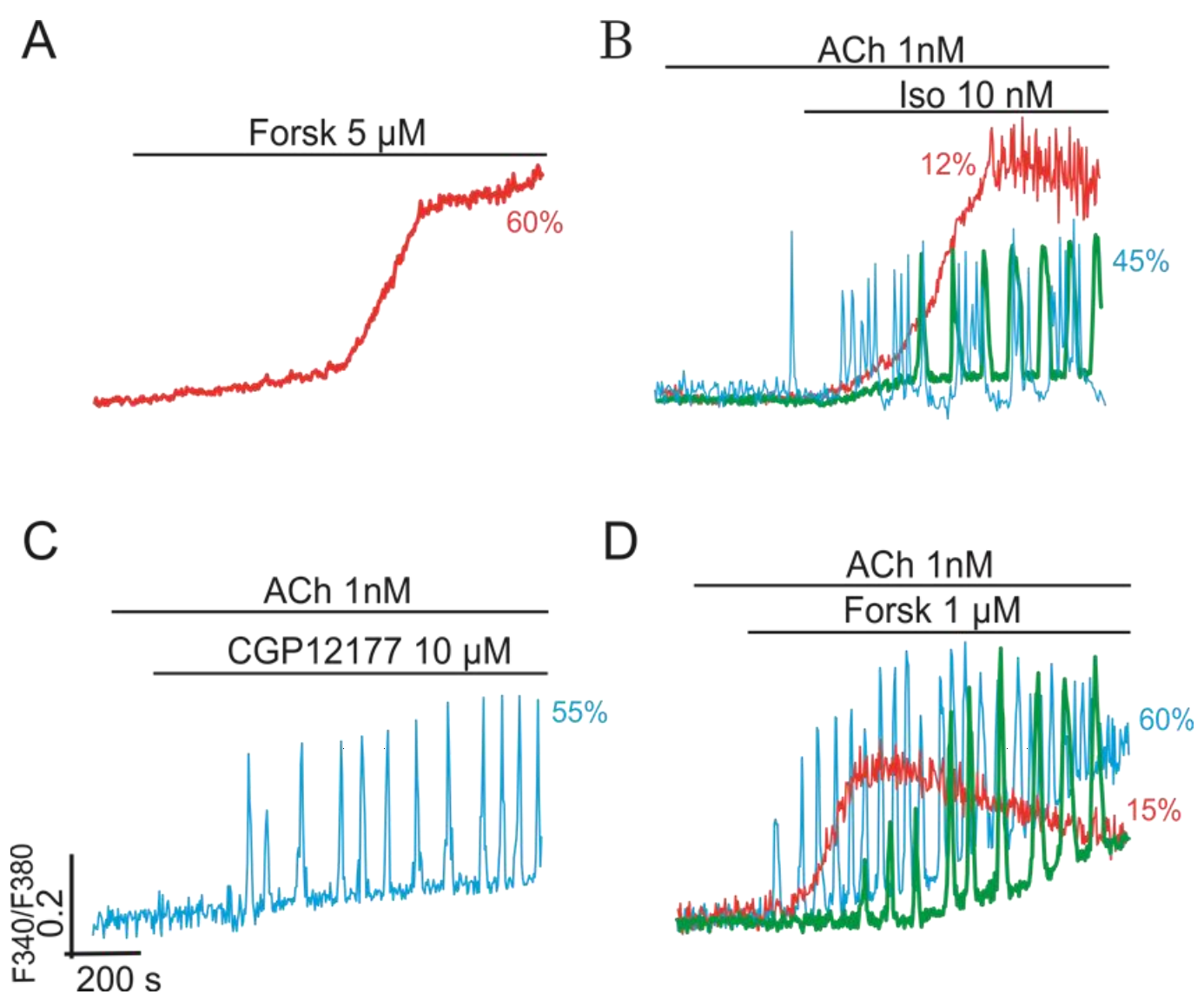
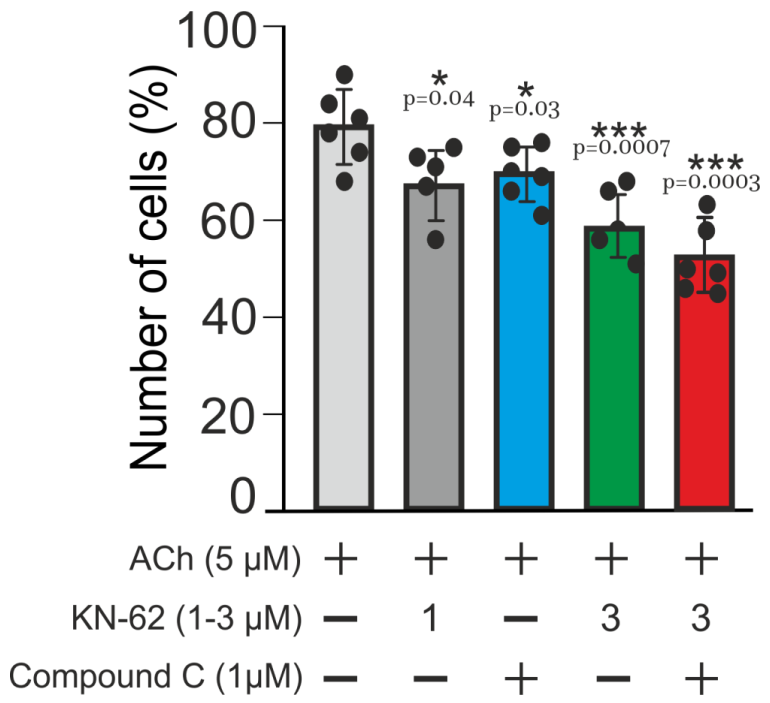
2.3. Multi-Loop Positive Feedback Control and Robustness of NOS-G
2.4. Impact of Obesity on Cultured Adipocytes
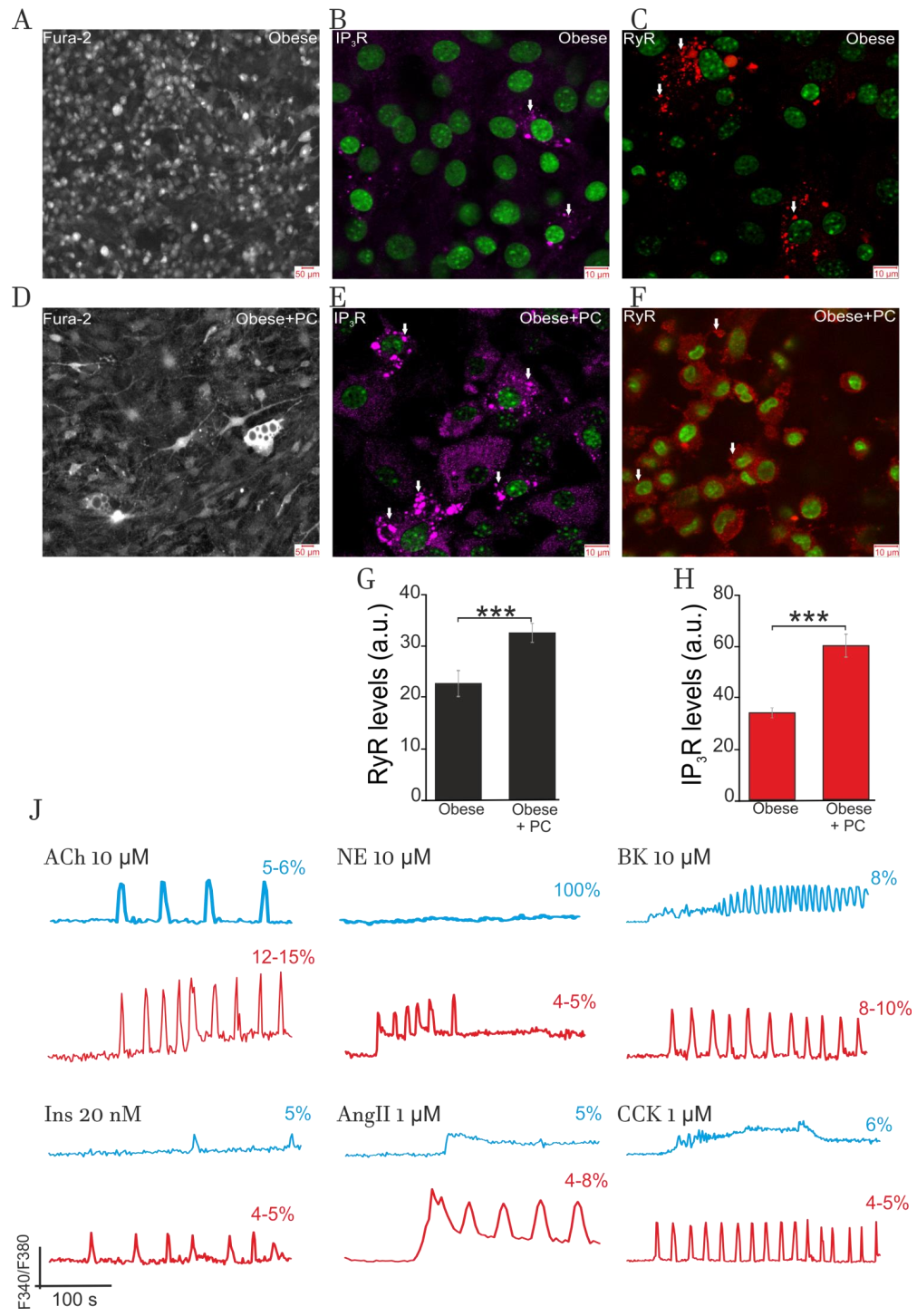
2.4.1. Effect of Obesity on Adipocytes Lipids Accumulation and Expression of IP3R and RyR
2.4.2. Impact of Obesity on Ca2+-Signaling
3. Discussion
3.1. Positive Feedback Loops Interplay
3.2. Extreme Sensitivity of NOS-G to Input Signal and G Proteins Interplay
3.3. Over-Activation of Input Axes and Multistability of NOS-G
3.4. Positive and Negative Feedbacks’ Crosstalk
3.5. Obesity-Induced General Hormonal Signaling Resistance of Adipocytes
4. Materials and Methods
4.1. Isolation and Cultivation of Preadipocytes
4.2. The Measurement of Cytosolic Calcium Concentration ([Ca2+]i)
4.3. Immunocytochemical Method
4.4. Animal Model of Obesity and Type 2 Diabetes
4.5. Metabolite and Blood Parameters Determinations
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
Abbreviations
| [Ca2+]i | intracellular free calcium concentration |
| PKB | protein kinase B |
| PI3Kα, γ | lipid kinases α, γ |
| eNOS | endothelial NO-synthase |
| AMPKα | AMP-activated protein kinase α |
| ACh | acetylcholine |
| NE | norepinephrine |
| BK | bradykinin |
| CCK | cholecystokinin |
| AngII | Angiotensin II |
| Ins | Insulin |
| Gβγ | G-protein βγ |
| Gαq | G-protein αq |
| α1,2-AR | α1,2-adrenoreceptors |
| β1-3-AR | β1-3-adrenoreceptors |
| eWAT | epididymal white adipose tissue |
| CICR | calcium- induced calcium release |
| NFL | negative feedback loop |
| PFL | positive feedback loop |
| m3-MR | m3-muscarinic receptors |
| ANP | atrial natriuretic peptide |
| NOS-G | eNOS- dependent rhythmic generator |
| PLC-G | PLC- dependent rhythmic generator |
| Gαs | G-protein αs |
| GPCR | G-protein coupled receptors |
| RTK | tyrosine kinase coupled receptors |
| T2D | type 2 diabetes |
| VP | vasopressin |
| ANP | atrial natriuretic peptide |
| ET-1 | endothelin-1 |
| PE | phenylephrine |
| SERCA | Ca2+-ATPase of endoplasmic reticulum |
| PMCA | Ca2+-ATPase of plasmalemma |
| AC | adenylate cyclase |
| PKA | protein kinase A |
| VEGF | vascular endothelial growth factor |
| CCK | cholecystokinin |
| BA | Bile acids |
| IP3R1,2 | inositol 1,4,5-trisphosphate receptors subtypes 1,2 |
| CaMKIIβ | Ca2+/calmodulin-dependent protein kinase IIβ |
| CaMKK | Ca2+/calmodulin-dependent kinase kinase |
| IP3 | inositol 1,4,5-trisphosphate |
| PLCβ,γ | phospholipase C β,γ |
| sGC | soluble guanylate cyclase |
| PKG1 | cGMP-dependent protein kinase 1 |
| CD38 | ectoenzyme ADP-ribosyl cyclase |
| PKB | protein kinase B |
| ARC | ADP-ribosyl cyclase |
| cADPr | cyclic ADP-ribose |
| TK | membrane tyrosine kinase |
| cSrc | cytosolic tyrosine kinase |
| RyR2,3 | ryanodine receptors subtypes 2,3 |
| RGS | Regulator of G protein signaling |
| IRAG | IP3R-associiated protein kinase G substrate |
References
- Thomas, A.P.; Bird, G.S.; Hajnóczky, G.; Robb-Gaspers, L.D.; Putney, J.W., Jr. Spatial and temporal aspects of cellular calcium signaling. FASEB J. 1996, 10, 1505–1517. [Google Scholar] [CrossRef] [PubMed]
- Larsen, A.Z.; Olsen, L.F.; Kummer, U. On the encoding and decoding of calcium signals in hepatocytes. Biophys. Chem. 2004, 107, 83–99. [Google Scholar] [CrossRef] [PubMed]
- Dupont, G.; Combettes, L. Fine tuning of cytosolic Ca2+ oscillations. F1000Research 2016, 5, 1000. [Google Scholar] [CrossRef] [PubMed]
- Samanta, K.; Parekh, A.B. Spatial Ca2+ profiling: Decrypting the universal cytosolic Ca2+ oscillation. J. Physiol. 2017, 595, 3053–3062. [Google Scholar] [CrossRef]
- Zhang, G.; Teggatz, E.G.; Zhang, A.Y.; Koeberl, M.J.; Yi, F.; Chen, L.; Li, P.-L. Cyclic ADP ribose-mediated Ca2+ signaling in mediating endothelial nitric oxide production in bovine coronary arteries. Am. J. Physiol. Heart. Circ. Physiol. 2006, 290, 1172–1181. [Google Scholar] [CrossRef]
- Gul, R.; Kim, S.-Y.; Park, K.-H.; Kim, B.-J.; Kim, S.-J.; Im, M.-J.; Kim, U.-H. A novel signaling pathway of ADP-ribosyl cyclase activation by angiotensin II in adult rat cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, 77–88. [Google Scholar] [CrossRef]
- Woods, N.M.; Cuthbertson, K.S.; Cobbold, P.H. Repetitive transient rises in cytoplasmic free calcium in hormone-stimulated hepatocytes. Nature 1986, 319, 600–602. [Google Scholar] [CrossRef]
- Rooney, T.A.; Joseph, S.K.; Queen, C.; Thomas, A.P. Cyclic GMP induces oscillatory calcium signals in rat hepatocytes. J. Biol. Chem. 1996, 271, 19817–19825. [Google Scholar] [CrossRef]
- Thorn, P.; Gerasimenko, O.; Petersen, O.H. Cyclic ADP-ribose regulation of ryanodine receptors involved in agonist evoked cytosolic Ca2+ oscillations in pancreatic acinar cells. EMBO J. 1994, 13, 2038–2043. [Google Scholar] [CrossRef]
- Prakash, Y.S.; Kannan, M.S.; Walseth, T.F.; Sieck, G.C. cADP ribose and [Ca2+]i regulation in rat cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, 1482–1489. [Google Scholar] [CrossRef]
- White, T.A.; Kannan, M.S.; Walseth, T.F. Intracellular calcium signaling through the cADPR pathway is agonist specific in porcine airway smooth muscle. FASEB J. 2003, 17, 482–484. [Google Scholar] [CrossRef]
- Kimura, C.; Oike, M.; Ohnaka, K.; Nose, Y.; Ito, Y. Constitutive nitric oxide production in bovine aortic and brain microvascular endothelial cells: A comparative study. J. Physiol. 2004, 554, 721–730. [Google Scholar] [CrossRef]
- Liu, H.N.; Ohya, S.; Wang, J.; Imaizumi, Y.; Nakayama, S. Involvement of ryanodine receptors in pacemaker Ca2+ oscillation in murine gastric ICC. Biochem. Biophys. Res. Commun. 2005, 328, 640–646. [Google Scholar] [CrossRef]
- Willmott, N.J.; Wong, K.; Strong, A.J. A fundamental role for the nitric oxide-G-kinase signaling pathway in mediating intercellular Ca2+ waves in glia. J. Neurosci. 2000, 20, 1767–1779. [Google Scholar] [CrossRef]
- Gaspers, L.D.; Pierobon, N.; Thomas, A.P. Intercellular calcium waves integrate hormonal control of glucose output in the intact liver. J. Physiol. 2019, 597, 2867–2885. [Google Scholar] [CrossRef]
- Decroly, O.; Goldbeter, A. Birhythmicity, chaos, and other patterns of temporal self-organization in a multiply regulated biochemical system. Proc. Natl. Acad. Sci. USA 1982, 79, 6917–6921. [Google Scholar] [CrossRef]
- Berridge, M.J.; Galione, A. Cytosolic calcium oscillators. FASEB J. 1988, 2, 3074–3082. [Google Scholar] [CrossRef]
- Westcott, E.B.; Goodwin, E.L.; Segal, S.S.; Jackson, W.F. Function and expression of ryanodine receptors and inositol 1,4,5-trisphosphate receptors in smooth muscle cells of murine feed arteries and arterioles. J. Physiol. 2012, 590, 1849–1869. [Google Scholar] [CrossRef]
- Mitsui, R.; Hashitani, H. Synchrony of spontaneous Ca2+ activity in microvascular mural cells. J. Smooth Muscle Res. 2020, 56, 1–18. [Google Scholar] [CrossRef]
- Mostafa, R.M.; Moustafa, Y.M.; Hamdy, H. Interstitial cells of Cajal, the Maestro in health and disease. World J. Gastroenterol. 2010, 16, 3239–3248. [Google Scholar] [CrossRef]
- Sanders, K.M.; Kito, Y.; Hwang, S.J.; Ward, S.M. Regulation of Gastrointestinal Smooth Muscle Function by Interstitial Cells. Physiology 2016, 31, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.; Seidler, B.; Kettenberger, A.; Sibaev, A.; Rohn, M.; Feil, R.; Allescher, H.-D.; Vanderwinden, J.-M.; Hofmann, F.; Schemann, M.; et al. Interstitial cells of Cajal integrate excitatory and inhibitory neurotransmission with intestinal slow-wave activity. Nat. Commun. 2013, 4, 1630. [Google Scholar] [CrossRef] [PubMed]
- Lang, D.A.; Matthews, D.R.; Burnett, M.; Turner, R.C. Brief, irregular oscillations of basal plasma insulin and glucose concentrations in diabetic man. Diabetes 1981, 30, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Hellman, B.; Salehi, A.; Grapengiesser, E.; Gylfe, E. Isolated mouse islets respond to glucose with an initial peak of glucagon release followed by pulses of insulin and somatostatin in antisynchrony with glucagon. Biochem. Biophys. Res. Commun. 2012, 417, 1219–1223. [Google Scholar] [CrossRef]
- Shimizu, K.; Ogura, H.; Matsumoto, N.; Ikeda, M.; Yamamoto, H.; Mori, M.; Morii, E.; Shimazu, T. Interstitial cells of Cajal are diminished in critically ill patients: Autopsy cases. Nutrition 2020, 70, 110591. [Google Scholar] [CrossRef]
- Rohrer, S.; Menge, B.A.; Grüber, L.; Deacon, C.F.; Schmidt, W.E.; Veldhuis, J.D.; Holst, J.J.; Meier, J.J. Impaired crosstalk between pulsatile insulin and glucagon secretion in prediabetic individuals. J. Clin. Endocrinol. Metab. 2012, 97, 791–795. [Google Scholar] [CrossRef][Green Version]
- Janah, L.; Kjeldsen, S.; Galsgaard, K.D.; Winther-Sørensen, M.; Stojanovska, E.; Pedersen, J.; Knop, F.K.; Holst, J.J.; Albrechtsen, N.J.W. Glucagon receptor signaling and glucagon resistance. Int. J. Mol. Sci. 2019, 20, 3314. [Google Scholar] [CrossRef]
- Rachdaoui, N. Insulin: The friend and the foe in the development of type 2 diabetes mellitus. Int. J. Mol. Sci. 2020, 21, 1770. [Google Scholar] [CrossRef]
- Andreeva, L.A.; Grishina, E.V.; Sergeev, A.I.; Lobanov, A.V.; Slastcheva, G.A.; Rykov, V.A.; Temyakov, A.V.; Dynnik, V.V. Emergence of acetylcholine resistance and loss of rhythmic activity associated with the development of hypertension, obesity, and Type 2 Diabetes. Biochem. Suppl. Ser. A Membr. Cell Biol. 2016, 10, 199–206. [Google Scholar] [CrossRef]
- Sanyal, A.; Naumann, J.; Hoffmann, L.S.; Chabowska-Kita, A.; Ehrlund, A.; Schlitzer, A.; Arner, P.; Blüher, M.; Pfeifer, A. Interplay between obesity-induced inflammation and cGMP signaling in white adipose tissue. Cell Rep. 2017, 18, 225–236. [Google Scholar] [CrossRef]
- Chen, J.-Y.; Ye, Z.-X.; Wang, X.-F.; Chang, J.; Yang, M.-W.; Zhong, H.-H.; Hong, F.-F.; Yang, S.-L. Nitric oxide bioavailability dysfunction involves in atherosclerosis. Biomed. Pharmacother. 2018, 97, 423–428. [Google Scholar] [CrossRef]
- Doronzo, G.; Viretto, M.; Russo, I.; Mattiello, L.; Di Martino, L.; Cavalot, F.; Anfossi, G.; Trovati, M. Nitric oxide activates PI3-K and MAPK signalling pathways in human and rat vascular smooth muscle cells: Influence of insulin resistance and oxidative stress. Atherosclerosis 2011, 216, 44–53. [Google Scholar] [CrossRef]
- Li, J.C.; Velagic, A.; Qin, C.X.; Li, M.; Leo, C.H.; Kemp-Harper, B.K.; Ritchie, R.H.; Woodman, O.L. Diabetes attenuates the contribution of endogenous nitric oxide but not nitroxyl to endothelium dependent relaxation of rat carotid arteries. Front. Pharmacol. 2021, 11, 585740. [Google Scholar] [CrossRef]
- Endo, M.; Tanaka, M.; Ogawa, Y. Calcium induced release of calcium from the sarcoplasmic reticulum of skinned skeletal muscle fibres. Nature 1970, 228, 34–36. [Google Scholar] [CrossRef]
- Ridgway, E.B.; Durham, A.C. Oscillations of calcium ion concentrations in Physarum polycephalum. J. Cell Biol. 1976, 69, 223–226. [Google Scholar] [CrossRef]
- Rooney, T.A.; Sass, E.J.; Thomas, A.P. Characterization of cytosolic calcium oscillations induced by phenylephrine and vasopressin in single fura-2-loaded hepatocytes. J. Biol. Chem. 1989, 264, 17131–17141. [Google Scholar] [CrossRef]
- De Young, G.W.; Keizer, J. A single-pool inositol 1,4,5-trisphosphate-receptor-based model for agonist-stimulated oscillations in Ca2+ concentration. Proc. Natl. Acad. Sci. USA 1992, 89, 9895–9899. [Google Scholar] [CrossRef]
- Borghans, J.M.; Dupont, G.; Goldbeter, A. Complex intracellular calcium oscillations. A theoretical exploration of possible mechanisms. Biophys. Chem. 1997, 66, 25–41. [Google Scholar] [CrossRef]
- Kummer, U.; Olsen, L.; Dixon, C.; Green, A.; Bornberg-Bauer, E.; Baier, G. Switching from simple to complex oscillations in calcium signaling. Biophys. J. 2000, 79, 1188–1195. [Google Scholar] [CrossRef]
- Thore, S.; Dyachok, O.; Gylfe, E.; Tengholm, A. Feedback activation of phospholipase C via intracellular mobilization and store-operated influx of Ca2+ in insulin-secreting beta-cells. J. Cell Sci. 2005, 118, 4463–4471. [Google Scholar] [CrossRef]
- Bartlett, P.J.; Cloete, I.; Sneyd, J.; Thomas, A.P. IP3-dependent Ca2+ oscillations switch into a dual oscillator mechanism in the presence of PLC-linked hormones. iScience 2020, 23, 101062. [Google Scholar] [CrossRef] [PubMed]
- Publicover, N.G.; Hammond, E.M.; Sanders, K.M. Amplification of nitric oxide signaling by interstitial cells isolated from canine colon. Proc. Natl. Acad. Sci. USA 1993, 90, 2087–2091. [Google Scholar] [CrossRef] [PubMed]
- Bredt, D.S.; Snyder, S.H. Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc. Natl. Acad. Sci. USA 1990, 87, 682–685. [Google Scholar] [CrossRef] [PubMed]
- Galione, A.; White, A.; Willmott, N.; Turner, M.; Potter, B.V.L.; Watson, S.P. cGMP mobilizes intracellular Ca2+ in sea urchin eggs by stimulating cyclic ADP-ribose synthesis. Nature 1993, 365, 456–459. [Google Scholar] [CrossRef] [PubMed]
- Willmott, N.; Sethi, J.K.; White, A.M.; Galione, A.; Walseth, T.F.; Lee, H.C. Nitric oxide-induced mobilization of intracellular calcium via the cyclic ADP-ribose signaling pathway. J. Biol. Chem. 1996, 271, 3699–3705. [Google Scholar] [CrossRef] [PubMed]
- Looms, D.K.; Tritsaris, K.; Nauntofte, B.; Dissing, S. Nitric oxide and cGMP activate Ca2+-release processes in rat parotid acinar cells. Biochem. J. 2001, 355, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Turovsky, E.A.; Turovskaya, M.V.; Dolgacheva, L.P.; Zinchenko, V.P.; Dynnik, V.V. Acetylcholine promotes Ca2+ and NO-oscillations in adipocytes implicating Ca2+→NO→cGMP→cADP-ribose→Ca2+ positive feedback loop--modulatory effects of norepinephrine and atrial natriuretic peptide. PLoS ONE 2013, 8, e63483. [Google Scholar] [CrossRef]
- Dedkova, E.N.; Ji, X.; Wang, Y.G.; Blatter, L.A.; Lipsius, S.L. Signaling mechanisms that mediate nitric oxide production induced by acetylcholine exposure and withdrawal in cat atrial myocytes. Circ. Res. 2003, 93, 1233–1240. [Google Scholar] [CrossRef]
- Liu, S.; Premont, R.T.; Kontos, C.D.; Huang, J.; Rockey, D.C. Endothelin-1 activates endothelial cell nitric-oxide synthase via heterotrimeric G-protein betagamma subunit signaling to protein jinase B/Akt. J. Biol. Chem. 2003, 278, 49929–49935. [Google Scholar] [CrossRef]
- Blanes, M.G.; Oubaha, M.; Rautureau, Y.; Gratton, J.P. Phosphorylation of tyrosine 801 of vascular endothelial growth factor receptor-2 is necessary for Akt-dependent endothelial nitric-oxide synthase activation and nitric oxide release from endothelial cells. J. Biol. Chem. 2007, 282, 10660–10669. [Google Scholar] [CrossRef]
- Symons, J.D.; McMillin, S.L.; Riehle, C.; Tanner, J.; Palionyte, M.; Hillas, E.; Jones, D.; Cooksey, R.C.; Birnbaum, M.J.; McClain, D.A.; et al. Contribution of insulin and Akt1 signaling to endothelial nitric oxide synthase in the regulation of endothelial function and blood pressure. Circ. Res. 2009, 104, 1085–1094. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, S.; Gil Wier, W.; Zhang, Q.; Jiang, H.; Li, Q.; Chen, S.; Tian, Z.; Li, Y.; Yu, X.; et al. Exercise improves the dilatation function of mesenteric arteries in postmyocardial infarction rats via a PI3K/Akt/eNOS pathway-mediated mechanism. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, 2097–2106. [Google Scholar] [CrossRef]
- Fischer, L.; Gukovskaya, A.S.; Penninger, J.M.; Mareninova, O.A.; Friess, H.; Gukovsky, I.; Pandol, S.J. Phosphatidylinositol 3-kinase facilitates bile acid-induced Ca2+ responses in pancreatic acinar cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, 875–886. [Google Scholar] [CrossRef]
- Turovsky, E.A.; Zinchenko, V.P.; Kaimachnikov, N.P. Attenuation of calmodulin regulation evokes Ca2+ oscillations: Evidence for the involvement of intracellular arachidonate-activated channels and connexons. Mol. Cell Biochem. 2019, 456, 191–204. [Google Scholar] [CrossRef]
- Dolgacheva, L.P.; Turovskaya, M.V.; Dynnik, V.V.; Zinchenko, V.P.; Goncharov, N.V.; Davletov, B.; Turovsky, E.A. Angiotensin II activates different calcium signaling pathways in adipocytes. Arch. Biochem. Biophys. 2016, 593, 38–49. [Google Scholar] [CrossRef]
- Dynnik, V.V.; Grishina, E.V.; Sirota, N.P.; Turovsky, E.A.; Djafarov, R.H.; Sergeev, A.I. Feedback control of second messengers signaling systems in white adipose tissue adipocytes in healthy state and its loss at adiposity. In Adipose Tissue; Szablewski, L., Ed.; IntechOpen: London, UK, 2018; pp. 227–250. [Google Scholar]
- Colyer, J. Phosphorylation states of phospholamban. Ann. N. Y. Acad. Sci. 1998, 853, 79–91. [Google Scholar] [CrossRef]
- Mattiazzi, A.; Mundiña-Weilenmann, C.; Guoxiang, C.; Vittone, L.; Kranias, E. Role of phospholamban phosphorylation on Thr17 in cardiac physiological and pathological conditions. Cardiovasc. Res. 2005, 68, 366–375. [Google Scholar] [CrossRef]
- Kasseckert, S.A.; Schäfer, C.; Kluger, A.; Gligorievski, D.; Tillmann, J.; Schlüter, K.-D.; Noll, T.; Sauer, H.; Piper, H.M.; Abdallah, Y. Stimulation of cGMP signalling protects coronary endothelium against reperfusion-induced intercellular gap formation. Cardiovasc. Res. 2009, 83, 381–387. [Google Scholar] [CrossRef][Green Version]
- Yoshida, Y.; Sun, H.T.; Cai, J.Q.; Imai, S. Cyclic GMP-dependent protein kinase stimulates the plasma membrane Ca2+ pump ATPase of vascular smooth muscle via phosphorylation of a 240-kDa protein. J. Biol. Chem. 1991, 266, 19819–19825. [Google Scholar] [CrossRef]
- DeMarco, S.J.; Strehler, E.E. Plasma membrane Ca2+-atpase isoforms 2b and 4b interact promiscuously and selectively with members of the membrane-associated guanylate kinase family of PDZ (PSD95/Dlg/ZO-1) domain-containing proteins. J. Biol. Chem. 2001, 276, 21594–21600. [Google Scholar] [CrossRef]
- Kruger, W.A.; Yun, C.C.; Monteith, G.R.; Poronnik, P. Muscarinic-induced recruitment of plasma membrane Ca2+-ATPase involves PSD-95/Dlg/Zo-1-mediated interactions. J. Biol. Chem. 2009, 284, 1820–1830. [Google Scholar] [CrossRef]
- Nalli, A.D.; Kumar, D.P.; Al-Shboul, O.; Mahavadi, S.; Kuemmerle, J.F.; Grider, J.R.; Murthy, K.S. Regulation of Gβγi-dependent PLC-β3 activity in smooth muscle: Inhibitory phosphorylation of PLC-β3 by PKA and PKG and stimulatory phosphorylation of Gαi-GTPase-activating protein RGS2 by PKG. Cell Biochem. Biophys. 2014, 70, 867–880. [Google Scholar] [CrossRef]
- Schlossmann, J.; Desch, M. IRAG and novel PKG targeting in the cardiovascular system. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, 672–682. [Google Scholar] [CrossRef]
- Fleming, I.; Fisslthaler, B.; Dimmeler, S.; Kemp, B.E.; Busse, R. Phosphorylation of Thr(495) regulates Ca2+/calmodulin-dependent endothelial nitric oxide synthase activity. Circ. Res. 2001, 88, 68–75. [Google Scholar] [CrossRef]
- Sessa, W.C. eNOS at a glance. J. Cell. Sci. 2004, 117, 2427–2429. [Google Scholar] [CrossRef]
- Oakhill, J.S.; Steel, R.; Chen, Z.-P.; Scott, J.W.; Ling, N.; Tam, S.; Kemp, B.E. AMPK is a direct adenylate charge-regulated protein kinase. Science 2011, 332, 1433–1435. [Google Scholar] [CrossRef]
- Lee, S.H.; Byun, J.S.; Kong, P.J.; Lee, H.J.; Kim, D.K.; Kim, H.S.; Sohn, J.-H.; Lee, J.J.; Lim, S.Y.; Chun, W.; et al. Inhibition of eNOS/sGC/PKG pathway decreases Akt phosphorylation induced by kainic acid in mouse hippocampus. Korean J. Physiol. Pharmacol. 2010, 14, 37–43. [Google Scholar] [CrossRef]
- Brusq, J.M.; Mayoux, E.; Guigui, L.; Kirilovsky, J. Effects of C-type natriuretic peptide on rat cardiac contractility. Br. J. Pharmacol. 1999, 128, 206–212. [Google Scholar] [CrossRef]
- Pierkes, M.; Gambaryan, S.; Bokník, P.; Lohmann, S.M.; Schmitz, W.; Potthast, R.; Holtwick, R.; Kuhn, M. Increased effects of C-type natriuretic peptide on cardiac ventricular contractility and relaxation in guanylyl cyclase A-deficient mice. Cardiovasc. Res. 2002, 53, 852–861. [Google Scholar] [CrossRef]
- Kleinert, M.; Clemmensen, C.; Hofmann, S.M.; Moore, M.C.; Renner, S.; Woods, S.C.; Huypens, P.; Beckers, J.; De Angelis, M.H.; Schürmann, A.; et al. Animal models of obesity and diabetes mellitus. Nat. Rev. Endocrinol. 2018, 14, 140–162. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agonists and Their Concentrations | |||||||
|---|---|---|---|---|---|---|---|
| ACh 1–5 µM | NE 1–5 µM | ANP 1–10 µM | CCK 3–20 nM | ANGII 0.2– 1 µM | BK 0.3–10 µM | Ins 3–20 nM | |
| Receptors and proteins | m3 Gβγ | α1 Gαq | NPR-A Gα | CCK-B Gαq, Gβγ | AT-1 Gαq, Gβγ | B2R Gαq, Gβγ | RTK TK, cSrc |
| PLC-G, % of cells with rhythmic activity | – | 30–40 | – | 25–40 | 25–40 | 30–40 | 20–30 |
| NOS-G, % of cells with rhythmic activity | 70–80 | – | 30–40 | 20–40 | 30–35 | 25–30 | 15–25 |
| Periods of Ca2+-oscillations (s) | 5–60 100–300 | 20–75 100–300 | 20–50 200–300 | 25–30 300–500 | 20–50 75–200 | 10–30 200–500 | 20–30 50–150 |
| Age (Month) | Weight (g) | Glucose (mM) | Insulin (ng/mL) | Cholest (mM) | TG (mM) | FFA (mM) | Blood Pressure (mmHg) |
| 2 | 24.4 ± 0.56 | 6.4 ± 0.15 | 0.51 ± 0.04 | 2.35 ± 0.09 | 1.48 ± 0.08 | 1.14 ± 0.11 | 121 ± 2.4 |
| 7 | 42.5 ± 0.72 | 8.01 ± 0.21 | 1.95 ± 0.11 | 3.5 ± 0.13 | 1.92 ± 0.15 | 1.82 ± 0.12 | 143 ± 1.53 |
| 10 | 56.4 ± 1.42 | 11.9 ± 0.41 | 3.39 ± 0.13 | 4.33 ± 0.18 | 2.62 ± 0.21 | 1.84 ± 0.16 | 177 ± 2.96 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turovsky, E.A.; Turovskaya, M.V.; Dynnik, V.V. Deregulation of Ca2+-Signaling Systems in White Adipocytes, Manifested as the Loss of Rhythmic Activity, Underlies the Development of Multiple Hormonal Resistance at Obesity and Type 2 Diabetes. Int. J. Mol. Sci. 2021, 22, 5109. https://doi.org/10.3390/ijms22105109
Turovsky EA, Turovskaya MV, Dynnik VV. Deregulation of Ca2+-Signaling Systems in White Adipocytes, Manifested as the Loss of Rhythmic Activity, Underlies the Development of Multiple Hormonal Resistance at Obesity and Type 2 Diabetes. International Journal of Molecular Sciences. 2021; 22(10):5109. https://doi.org/10.3390/ijms22105109
Chicago/Turabian StyleTurovsky, Egor A., Maria V. Turovskaya, and Vladimir V. Dynnik. 2021. "Deregulation of Ca2+-Signaling Systems in White Adipocytes, Manifested as the Loss of Rhythmic Activity, Underlies the Development of Multiple Hormonal Resistance at Obesity and Type 2 Diabetes" International Journal of Molecular Sciences 22, no. 10: 5109. https://doi.org/10.3390/ijms22105109
APA StyleTurovsky, E. A., Turovskaya, M. V., & Dynnik, V. V. (2021). Deregulation of Ca2+-Signaling Systems in White Adipocytes, Manifested as the Loss of Rhythmic Activity, Underlies the Development of Multiple Hormonal Resistance at Obesity and Type 2 Diabetes. International Journal of Molecular Sciences, 22(10), 5109. https://doi.org/10.3390/ijms22105109