
The Role of Inflammation in Breast and Prostate Cancer Metastasis to Bone

Abstract
1. Introduction
1.1. The Good and the Bad of Inflammation
1.2. Inflammation and Cancer
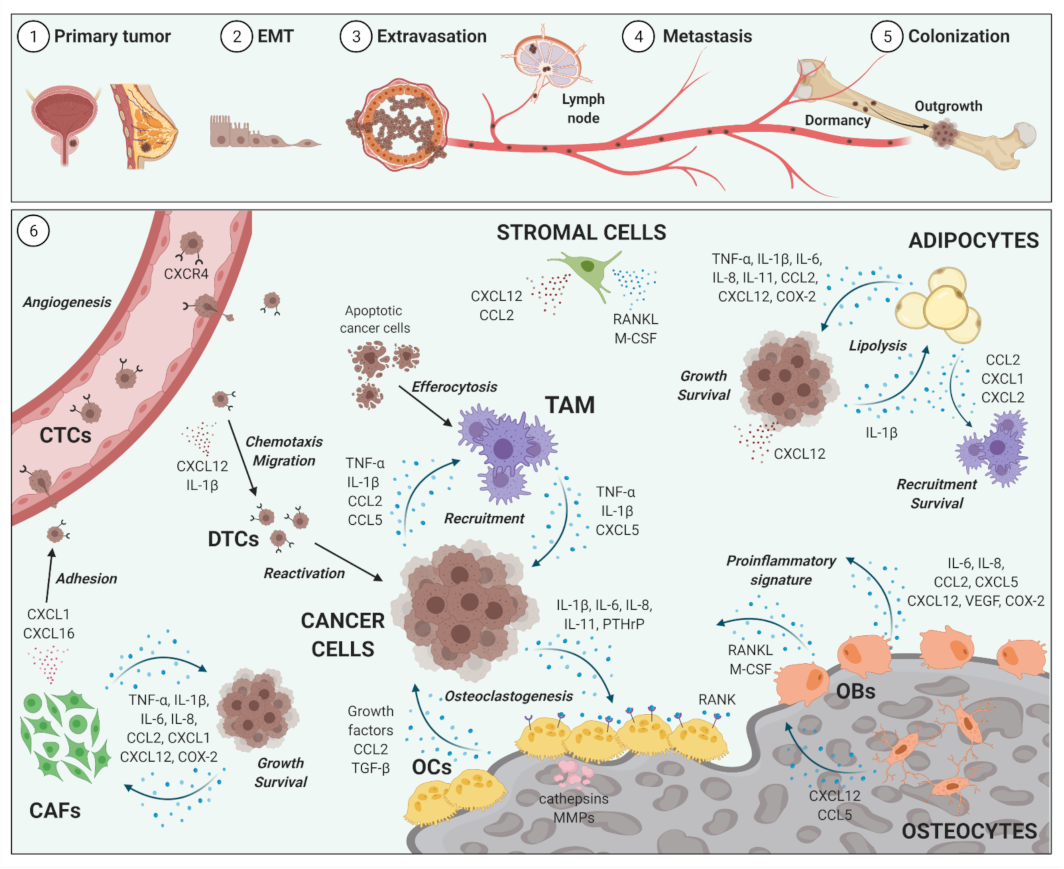
2. Impact of Inflammation on Cancer Metastases to Bone
2.1. Bone Metastases
2.2. Inflammatory Cellular and Soluble Mediators in Cancer Metastases to Bone Metastases
2.3. Selected Cellular Players in Inflammation and Bone-Metastatic Cancer
2.3.1. Osteoblasts and Osteoclasts
2.3.2. Tumor-Associated Macrophages
2.3.3. Bone Marrow Adipocytes
2.3.4. Cancer-Associated Fibroblasts
2.3.5. Selected Inflammatory Cytokines, Chemokines and Mediators
2.3.6. TNF-α
2.3.7. IL-1β and NF-κB Signaling
2.3.8. IL-6, CXCL-8/IL-8, IL-11
2.3.9. CXCL12/SDF-1
2.3.10. CCL2/MCP-1
2.3.11. CCL5/RANTES
2.3.12. Cyclooxygenase-2 and Prostaglandins

{kind=link}
| Mediator | Cellular Source | Mechanism of Action in Cancer Metastasis to Bone | Selected References |
|---|---|---|---|
| CCL2/MCP-1 | Adipocytes, ECs, OBs, OCs, TAM, stromal cells, tumor cells | Angiogenesis↑; COX-2↑; EMT↑; NF-κB activation↑; osteoclastogenesis↑; TAM recruitment and activation↑; tumor cell recruitment, growth, migration, metastasis↑; | [44,86,95,96,108,145,222,223,224,225,226,227,228,229,230,231,232,233,234,235,236,237,238,239,240,241,242] |
| CCL5 | Osteocytes, stromal cells, TAM, tumor cells | EMT↑; Osteoclastogenesis↑; TAM recruitment and activation ↑; tumor cell recruitment, growth, migration, metastasis↑ | [44,226,229,243,244,245,246,247,248] |
| COX-2/ prostaglandins | Adipocytes, OBs, TAM, tumor cells | CCL2↑; IL-8↑; IL-11↑; immunosuppression↑; osteoclastogenesis↑; prostaglandin E2↑; TAM recruitment and activation↑; tumor cell growth, migration, metastasis↑ | [59,108,113,249,250,251,252,253,254,255,256,257,258,259,260] |
| CXCL12/SDF1 | Adipocytes, CAFs, neutrophils, OBs, osteocytes; stromal cells, tumor cells | Angiogenesis↑; CAFs generation↑; chemotherapy resistance↑; dormancy↑; EMT↑; osteoclastogenesis↑; tumor cell recruitment, growth, migration, metastasis↑ | [78,88,89,147,193,194,195,199,200,201,202,203,204,205,206,207,208,209,210,211,212,213,214,215,216,217,218,219,220,221] |
| IL-1β/NF-κB | Adipocytes, CAFs, OBs, OCs, TAM, tumor cells | Adipocytic lipolysis↑; CAFs generation↑; CCL2↑; COX-2↑; dormancy↑; EMT↑; IL-6↑; IL-8↑; osteoclastogenesis↑; premetastatic niche formation↑; tumor cell recruitment, growth, migration, metastasis↑ | [44,81,113,120,140,148,149,150,151,152,153,154,155,156,157,158,159,160,161,162,163,164,165,166,167,168,169,170,171] |
| IL-6 | Adipocytes, CAFs, OBs; OCs, stromal cells, TAM, tumor cells | Angiogenesis↑; CAFs generation↑; CXCR4↑; chemotherapy resistance↑; COX-2↑; EMT↑; IL-8↑; IL-11↑; osteoblastogenesis↓; osteoclastogenesis↑; PTHrP↑; RANKL↑; tumor cell recruitment, growth, migration, metastasis↑ | [44,77,84,86,87,99,110, 121,132,138,144,163, 172,173,174,175,176,177,178,179,180,181,182,183,184,185,186,187,188,189,190,191,192,193,194,196,197,198] |
| IL-8/CXCL8 | Adipocytes, CAFs, ECs, OBs, TAM, tumor cells | Angiogenesis↑; CAFs generation↑; chemotherapy resistance↑; EMT↑; osteoclastogenesis↑; PTHrP↑; RANKL↑; tumor cell recruitment, growth, migration, metastasis↑ | [44,84,86,87,99,110,132,144,163,172,173,174,175,176,177,178,179,180,181,182,183,184,185,186,187,188,189,190,191,192,198] |
| IL-11 | Adipocytes, OBs, osteocytes; TAM; tumor cells | Chemotherapy resistance↑; osteoblastogenesis↓; OPG↓; osteoclastogenesis↑ | [77,84,86,87,89,144,179,180,181,182,183,184,185,186,187,195] |
| TNFα | Adipocytes, CAFs, ECs, neutrophils, TAM, tumor cells | Angiogenesis↑; chemotherapy resistance↑; dormancy↑; EMT↑; IL-6↑; osteoblastogenesis↓; osteoclastogenesis↑; OPG↓; TAM recruitment and activation↑; tumor cell recruitment, growth, migration, metastasis↑ | [9,33,34,35,44,78,99,110,141,142,143,144,145,146,147,159] |
3. Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Netea, M.G.; Balkwill, F.; Chonchol, M.; Cominelli, F.; Donath, M.Y.; Giamarellos-Bourboulis, E.J.; Golenbock, D.; Gresnigt, M.S.; Heneka, M.T.; Hoffman, H.M.; et al. A guiding map for inflammation. Nat. Immunol. 2017, 18, 826–831. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Savill, J. Resolution of inflammation: The beginning programs the end. Nat. Immunol. 2005, 6, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C.; Ahluwalia, N.; Albers, R.; Bosco, N.; Bourdet-Sicard, R.; Haller, D.; Holgate, S.T.; Jönsson, L.S.; Latulippe, M.E.; Marcos, A.; et al. A Consideration of Biomarkers to be Used for Evaluation of Inflammation in Human Nutritional Studies. Br. J. Nutr. 2013, 109, S1–S25. [Google Scholar] [CrossRef] [PubMed]
- Cronkite, D.A.; Strutt, T.M. The regulation of inflammation by innate and adaptive lymphocytes. J. Immunol. Res. 2018, 2018, 1467538. [Google Scholar] [CrossRef] [PubMed]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- Balkwill, F.; Charles, K.A.; Mantovani, A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell 2005, 7, 211–217. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R. A Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Bremnes, R.M.; Al-Shibli, K.; Donnem, T.; Sirera, R.; Al-Saad, S.; Andersen, S.; Stenvold, H.; Camps, C.; Busund, L.T. The role of tumor-infiltrating immune cells and chronic inflammation at the tumor site on cancer development, progression, and prognosis: Emphasis on non-small cell lung cancer. J. Thorac. Oncol. 2011, 6, 824–833. [Google Scholar] [CrossRef]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [CrossRef]
- Ponomarev, A.V.; Shubina, I.Z. Insights into mechanisms of tumor and immune system interaction: Association with wound healing. Front. Oncol. 2019, 9, 1115. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhao, B.; Wang, X. Tumor infiltrating immune cells (TIICs) as a biomarker for prognosis benefits in patients with osteosarcoma. BMC Cancer 2020, 20, 1022. [Google Scholar] [CrossRef]
- Ye, L.; Zhang, T.; Kang, Z.; Guo, G.; Sun, Y.; Lin, K.; Huang, Q.; Shi, X.; Ni, Z.; Ding, N.; et al. Tumor-infiltrating immune cells act as a marker for prognosis in colorectal cancer. Front. Immunol. 2019, 10, 2368. [Google Scholar] [CrossRef]
- Stanton, S.E.; Disis, M.L. Clinical significance of tumor-infiltrating lymphocytes in breast cancer. J. Immunother. Cancer 2016, 4, 59. [Google Scholar] [CrossRef] [PubMed]
- Pagès, F.; Galon, J.; Dieu-Nosjean, M.C.; Tartour, E.; Sautès-Fridman, C.; Fridman, W.H. Immune infiltration in human tumors: A prognostic factor that should not be ignored. Oncogene 2010, 29, 1093–1102. [Google Scholar] [CrossRef]
- Sousa, S.; Määttä, J. The role of tumour-associated macrophages in bone metastasis. J. Bone Oncol. 2016, 5, 135–138. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef]
- Graham, N.; Qian, B.Z. Mesenchymal stromal cells: Emerging roles in bone metastasis. Int. J. Mol. Sci. 2018, 19, 1121. [Google Scholar] [CrossRef]
- Lengyel, E.; Makowski, L.; DiGiovanni, J.; Kolonin, M.G. Cancer as a Matter of Fat: The Crosstalk between Adipose Tissue and Tumors. Trends Cancer 2018, 4, 374–384. [Google Scholar] [CrossRef]
- Raymaekers, K.; Stegen, S.; van Gastel, N.; Carmeliet, G. The vasculature: A vessel for bone metastasis. Bonekey Rep. 2015, 4, 742. [Google Scholar] [CrossRef][Green Version]
- Furesi, G.; Rauner, M.; Hofbauer, L.C. Emerging Players in Prostate Cancer–Bone Niche Communication. Trends Cancer 2020, 7, 112–121. [Google Scholar] [CrossRef]
- Bhome, R.; Bullock, M.D.; Al Saihati, H.A.; Goh, R.W.; Primrose, J.N.; Sayan, A.E.; Mirnezami, A.H. A top-down view of the tumor microenvironment: Structure, cells and signaling. Front. Cell Dev. Biol. 2015, 3, 33. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Vijayalekshmi, R.V.; Sung, B. Targeting inflammatory pathways for prevention and therapy of cancer: Short-term friend, long-term foe. Clin. Cancer Res. 2009, 15, 425–430. [Google Scholar] [CrossRef]
- Takahashi, H.; Ogata, H.; Nishigaki, R.; Broide, D.H.; Karin, M. Tobacco Smoke Promotes Lung Tumorigenesis by Triggering IKKβ- and JNK1-Dependent Inflammation. Cancer Cell 2010, 17, 89–97. [Google Scholar] [CrossRef]
- Park, E.J.; Lee, J.H.; Yu, G.Y.; He, G.; Ali, S.R.; Holzer, R.G.; Österreicher, C.H.; Takahashi, H.; Karin, M. Dietary and Genetic Obesity Promote Liver Inflammation and Tumorigenesis by Enhancing IL-6 and TNF Expression. Cell 2010, 140, 197–208. [Google Scholar] [CrossRef]
- Wroblewski, L.E.; Peek, R.M.; Wilson, K.T. Helicobacter pylori and gastric cancer: Factors that modulate disease risk. Clin. Microbiol. Rev. 2010, 23, 713–739. [Google Scholar] [CrossRef]
- Read, S.A.; Douglas, M.W. Virus induced inflammation and cancer development. Cancer Lett. 2014, 345, 174–181. [Google Scholar] [CrossRef]
- Roca, H.; McCauley, L.K. Inflammation and skeletal metastasis. Bonekey Rep. 2015, 4, 706. [Google Scholar] [CrossRef]
- Qiao, Y.; Yang, T.; Gan, Y.; Li, W.; Wang, C.; Gong, Y.; Lu, Z. Associations between aspirin use and the risk of cancers: A meta-analysis of observational studies. BMC Cancer 2018, 18, 288. [Google Scholar] [CrossRef]
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKβ links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004, 118, 285–296. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-B, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Montfort, A.; Colacios, C.; Levade, T.; Andrieu-Abadie, N.; Meyer, N.; Ségui, B. The TNF paradox in cancer progression and immunotherapy. Front. Immunol. 2019, 10, 1818. [Google Scholar] [CrossRef]
- Kumari, N.; Dwarakanath, B.S.; Das, A.; Bhatt, A.N. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumor Biol. 2016, 37, 11553–11572. [Google Scholar] [CrossRef]
- Kono, H.; Rock, K.L. How dying cells alert the immune system to danger. Nat. Rev. Immunol. 2008, 8, 279–289. [Google Scholar] [CrossRef]
- Hernandez, C.; Huebener, P.; Schwabe, R.F. Damage-associated molecular patterns in cancer: A double-edged sword. Oncogene 2016, 35, 5931–5941. [Google Scholar] [CrossRef]
- Lotfi, R.; Kaltenmeier, C.; Lotze, M.T.; Bergmann, C. Until Death Do Us Part: Necrosis and Oxidation Promote the Tumor Microenvironment. Transfus. Med. Hemotherapy 2016, 43, 120–132. [Google Scholar] [CrossRef]
- Sakurai, T.; He, G.; Matsuzawa, A.; Yu, G.Y.; Maeda, S.; Hardiman, G.; Karin, M. Hepatocyte Necrosis Induced by Oxidative Stress and IL-1α Release Mediate Carcinogen-Induced Compensatory Proliferation and Liver Tumorigenesis. Cancer Cell 2008, 14, 156–165. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Magbanua, M.J.M.; Yau, C.; Wolf, D.M.; Lee, J.S.; Chattopadhyay, A.; Scott, J.H.; Bowlby-Yoder, E.; Hwang, E.S.; Alvarado, M.; Ewing, C.A.; et al. Synchronous detection of circulating tumor cells in blood and disseminated tumor cells in bone marrow predicts adverse outcome in early breast cancer. Clin. Cancer Res. 2019, 25, 5388–5397. [Google Scholar] [CrossRef] [PubMed]
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.W.; Weinberg, R.A. Linking EMT programmes to normal and neoplastic epithelial stem cells. Nat. Rev. Cancer 2021, 21, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Carmona, M.; Lesage, J.; Cataldo, D.; Gilles, C. EMT and inflammation: Inseparable actors of cancer progression. Mol. Oncol. 2017, 11, 805–823. [Google Scholar] [CrossRef]
- Li, R.; Wen, A.; Lin, J. Pro-inflammatory cytokines in the formation of the pre-metastatic niche. Cancers 2020, 12, 3752. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Adekoya, T.O.; Richardson, R.M. Cytokines and chemokines as mediators of prostate cancer metastasis. Int. J. Mol. Sci. 2020, 21, 4449. [Google Scholar] [CrossRef]
- Jones, V.S.; Huang, R.Y.; Chen, L.P.; Chen, Z.S.; Fu, L.; Huang, R.P. Cytokines in cancer drug resistance: Cues to new therapeutic strategies. Biochim. Biophys. Acta Rev. Cancer 2016, 1865, 255–265. [Google Scholar] [CrossRef]
- Crusz, S.M.; Balkwill, F.R. Inflammation and cancer: Advances and new agents. Nat. Rev. Clin. Oncol. 2015, 12, 584–596. [Google Scholar] [CrossRef]
- Mundy, G.R. Metastasis to bone: Causes, consequences and therapeutic opportunities. Nat. Rev. Cancer 2002, 2, 584–593. [Google Scholar]
- Croucher, P.I.; McDonald, M.M.; Martin, T.J. Bone metastasis: The importance of the neighbourhood. Nat. Rev. Cancer 2016, 16, 373–386. [Google Scholar] [CrossRef]
- Kan, C.; Vargas, G.; Le Pape, F.; Clézardin, P. Cancer cell colonisation in the bone microenvironment. Int. J. Mol. Sci. 2016, 17, 1674. [Google Scholar] [CrossRef]
- Ortiz, A.; Lin, S.H. Osteolytic and osteoblastic bone metastases: Two extremes of the same spectrum? Recent Results Cancer Res. 2012, 192, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Florencio-Silva, R.; Sasso, G.R.D.S.; Sasso-Cerri, E.; Simões, M.J.; Cerri, P.S. Biology of Bone Tissue: Structure, Function, and Factors That Influence Bone Cells. Biomed Res. Int. 2015, 2015, 421746. [Google Scholar] [CrossRef]
- Sims, N.A.; Martin, T.J. Coupling the activities of bone formation and resorption: A multitude of signals within the basic multicellular unit. Bonekey Rep. 2014, 3, 481. [Google Scholar]
- Yasuda, H. Discovery of the RANKL/RANK/OPG system. J. Bone Miner. Metab. 2021, 39, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Xia, F.; Wei, Y.; Wei, X. Molecular mechanisms and clinical management of cancer bone metastasis. Bone Res. 2020, 8, 30. [Google Scholar] [CrossRef] [PubMed]
- Mundy, G.R. Mechanisms of bone metastasis. Cancer 1997, 80, 1546–1556. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Sosnoski, D.M.; Mastro, A.M. Breast cancer metastasis to the bone: Mechanisms of bone loss. Breast Cancer Res. 2010, 12, 215. [Google Scholar] [CrossRef]
- Hofbauer, L.C.; Rachner, T.D.; Coleman, R.E.; Jakob, F. Endocrine aspects of bone metastases. Lancet Diabetes Endocrinol. 2014, 2, 500–512. [Google Scholar] [CrossRef]
- Maurizi, A.; Rucci, N. The osteoclast in bone metastasis: Player and target. Cancers 2018, 10, 218. [Google Scholar] [CrossRef]
- Clezardin, P.; Teti, A. Bone metastasis: Pathogenesis and therapeutic implications. Clin. Exp. Metastasis 2007, 24, 599–608. [Google Scholar] [CrossRef] [PubMed]
- D’Oronzo, S.; Coleman, R.; Brown, J.; Silvestris, F. Metastatic bone disease: Pathogenesis and therapeutic options: Up-date on bone metastasis management. J. Bone Oncol. 2019, 15, 100205. [Google Scholar] [CrossRef]
- Coleman, R.E. Metastatic bone disease: Clinical features, pathophysiology and treatment strategies. Cancer Treat. Rev. 2001, 27, 165–176. [Google Scholar] [CrossRef]
- Paget, S. The distribution of secondary growths in cancer of the breast. Lancet 1889, 133, 571–573. [Google Scholar]
- Zarrer, J.; Haider, M.T.; Smit, D.J.; Taipaleenmäki, H. Pathological crosstalk between metastatic breast cancer cells and the bone microenvironment. Biomolecules 2020, 10, 337. [Google Scholar] [CrossRef]
- Weilbaecher, K.N.; Guise, T.A.; McCauley, L.K. Cancer to bone: A fatal attraction. Nat. Rev. Cancer 2011, 11, 411–425. [Google Scholar] [CrossRef]
- Padua, D.; Massagué, J. Roles of TGFβ in metastasis. Cell Res. 2009, 19, 89–102. [Google Scholar] [CrossRef]
- Breuksch, I.; Weinert, M.; Brenner, W. The role of extracellular calcium in bone metastasis. J. Bone Oncol. 2016, 5, 143–145. [Google Scholar] [CrossRef]
- Monteran, L.; Ershaid, N.; Sabah, I.; Fahoum, I.; Zait, Y.; Shani, O.; Cohen, N.; Eldar-Boock, A.; Satchi-Fainaro, R.; Erez, N. Bone metastasis is associated with acquisition of mesenchymal phenotype and immune suppression in a model of spontaneous breast cancer metastasis. Sci. Rep. 2020, 10, 13838. [Google Scholar] [CrossRef]
- Roy, L.D.; Ghosh, S.; Pathangey, L.B.; Tinder, T.L.; Gruber, H.E.; Mukherjee, P. Collagen induced arthritis increases secondary metastasis in MMTV-PyV MT mouse model of mammary cancer. BMC Cancer 2011, 11, 365. [Google Scholar] [CrossRef]
- Xiang, L.; Gilkes, D.M. The contribution of the immune system in bone metastasis pathogenesis. Int. J. Mol. Sci. 2019, 20, 999. [Google Scholar] [CrossRef]
- Buenrostro, D.; Mulcrone, P.L.; Owens, P.; Sterling, J.A. The Bone Microenvironment: A Fertile Soil for Tumor Growth. Curr. Osteoporos. Rep. 2016, 14, 151–158. [Google Scholar] [CrossRef]
- Waning, D.L.; Mohammad, K.S.; Reiken, S.; Xie, W.; Andersson, D.C.; John, S.; Chiechi, A.; Wright, L.E.; Umanskaya, A.; Niewolna, M.; et al. Excess TGF-β mediates muscle weakness associated with bone metastases in mice. Nat. Med. 2015, 21, 1262–1271. [Google Scholar] [CrossRef] [PubMed]
- Drabsch, Y.; ten Dijke, P. TGF-β Signaling in Breast Cancer Cell Invasion and Bone Metastasis. J. Mammary Gland Biol. Neoplasia 2011, 16, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Silvestris, F.; Lombardi, L.; De Matteo, M.; Bruno, A.; Dammacco, F. Myeloma bone disease: Pathogenetic mechanisms and clinical assessment. Leuk. Res. 2007, 31, 129–138. [Google Scholar] [CrossRef]
- Kudo, O.; Sabokbar, A.; Pocock, A.; Itonaga, I.; Fujikawa, Y.; Athanasou, N.A. Interleukin-6 and interleukin-11 support human osteoclast formation by a RANKL-independent mechanism. Bone 2003, 32, 1–7. [Google Scholar] [CrossRef]
- Coniglio, S.J. Role of tumor-derived chemokines in osteolytic bone metastasis. Front. Endocrinol. (Lausanne) 2018, 9, 313. [Google Scholar] [CrossRef] [PubMed]
- Rucci, N.; Millimaggi, D.; Mari, M.; Del Fattore, A.; Bologna, M.; Teti, A.; Angelucci, A.; Dolo, V. Receptor Activator of NF- B Ligand Enhances Breast Cancer-Induced Osteolytic Lesions through Upregulation of Extracellular Matrix Metalloproteinase Inducer/CD147. Cancer Res. 2010, 70, 6150–6160. [Google Scholar] [CrossRef]
- Jones, D.H.; Nakashima, T.; Sanchez, O.H.; Kozieradzki, I.; Komarova, S.V.; Sarosi, I.; Morony, S.; Rubin, E.; Sarao, R.; Hojilla, C.V.; et al. Regulation of cancer cell migration and bone metastasis by RANKL. Nature 2006, 440, 692–696. [Google Scholar]
- Tsuboi, M.; Kawakami, A.; Nakashima, T.; Matsuoka, N.; Urayama, S.; Kawabe, Y.; Fujiyama, K.; Kiriyama, T.; Aoyagi, T.; Maeda, K.; et al. Tumor necrosis factor-α and interleukin-1β increase the Fas-mediated apoptosis of human osteoblasts. J. Lab. Clin. Med. 1999, 134, 222–231. [Google Scholar] [CrossRef]
- Rachner, T.D.; Göbel, A.; Benad-Mehner, P.; Hofbauer, L.C.; Rauner, M. Dickkopf-1 as a mediator and novel target in malignant bone disease. Cancer Lett. 2014, 346, 172–177. [Google Scholar] [CrossRef]
- Delgado-Calle, J.; Anderson, J.; Cregor, M.D.; Hiasa, M.; Chirgwin, J.M.; Carlesso, N.; Yoneda, T.; Mohammad, K.S.; Plotkin, L.I.; Roodman, G.D.; et al. Bidirectional Notch Signaling and Osteocyte-Derived Factors in the Bone Marrow Microenvironment Promote Tumor Cell Proliferation and Bone Destruction in Multiple Myeloma. Cancer Res. 2016, 76, 1089–1100. [Google Scholar] [CrossRef] [PubMed]
- Kinder, M.; Chislock, E.; Bussard, K.M.; Shuman, L.; Mastro, A.M. Metastatic breast cancer induces an osteoblast inflammatory response. Exp. Cell Res. 2008, 314, 173–183. [Google Scholar] [CrossRef]
- Hsu, Y.L.; Hou, M.F.; Kuo, P.L.; Huang, Y.F.; Tsai, E.M. Breast tumor-associated osteoblast-derived CXCL5 increases cancer progression by ERK/MSK1/Elk-1/Snail signaling pathway. Oncogene 2013, 32, 4436–4447. [Google Scholar] [CrossRef]
- Bussard, K.M.; Venzon, D.J.; Mastro, A.M. Osteoblasts are a major source of inflammatory cytokines in the tumor microenvironment of bone metastatic breast cancer. J. Cell. Biochem. 2010, 111, 1138–1148. [Google Scholar] [CrossRef] [PubMed]
- Dhurjati, R.; Krishnan, V.; Shuman, L.A.; Mastro, A.M.; Vogler, E.A. Metastatic breast cancer cells colonize and degrade three-dimensional osteoblastic tissue in vitro. Clin. Exp. Metastasis 2008, 25, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Haider, M.-T.; Holen, I.; Dear, T.N.; Hunter, K.; Brown, H.K. Modifying the osteoblastic niche with zoledronic acid in vivo—Potential implications for breast cancer bone metastasis. Bone 2014, 66, 240–250. [Google Scholar] [CrossRef]
- Atkinson, E.G.; Delgado-Calle, J. The Emerging Role of Osteocytes in Cancer in Bone. JBMR Plus 2019, 3, e10186. [Google Scholar] [CrossRef]
- Zhou, J.; Tang, Z.; Gao, S.; Li, C.; Feng, Y.; Zhou, X. Tumor-Associated Macrophages: Recent Insights and Therapies. Front. Oncol. 2020, 10, 188. [Google Scholar] [CrossRef]
- Vasiliadou, I.; Holen, I. The role of macrophages in bone metastasis. J. Bone Oncol. 2013, 2, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Soki, F.N.; Cho, S.W.; Kim, Y.W.; Jones, J.D.; Park, S.I.; Koh, A.J.; Entezami, P.; Daignault-Newton, S.; Pienta, K.J.; Roca, H.; et al. Bone marrow macrophages support prostate cancer growth in bone. Oncotarget 2015, 6, 35782–35796. [Google Scholar] [CrossRef]
- Hiraoka, K.; Zenmyo, M.; Watari, K.; Iguchi, H.; Fotovati, A.; Kimura, Y.N.; Hosoi, F.; Shoda, T.; Nagata, K.; Osada, H.; et al. Inhibition of bone and muscle metastases of lung cancer cells by a decrease in the number of monocytes/macrophages. Cancer Sci. 2008, 99, 1595–1602. [Google Scholar] [CrossRef] [PubMed]
- Hsu, D.S.-S.; Wang, H.-J.; Tai, S.-K.; Chou, C.-H.; Hsieh, C.-H.; Chiu, P.-H.; Chen, N.-J.; Yang, M.-H. Acetylation of Snail Modulates the Cytokinome of Cancer Cells to Enhance the Recruitment of Macrophages. Cancer Cell 2014, 26, 534–548. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Kang, Y. Chemokine (C-C Motif) ligand 2 engages CCR2+ stromal cells of monocytic origin to promote breast cancer metastasis to lung and bone. J. Biol. Chem. 2009, 284, 29087–29096. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, K.; Sud, S.; McGregor, N.A.; Martinovski, G.; Rice, B.T.; Craig, M.J.; Varsos, Z.S.; Roca, H.; Pienta, K.J. The Chemokine CCL2 Increases Prostate Tumor Growth and Bone Metastasis through Macrophage and Osteoclast Recruitment. Neoplasia 2009, 11, 1235–1242. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, H.N.; Kim, K.O.; Jin, W.J.; Lee, S.; Kim, H.H.; Ha, H.; Lee, Z.H. CXCL10 promotes osteolytic bone metastasis by enhancing cancer outgrowth and osteoclastogenesis. Cancer Res. 2012, 72, 3175–3186. [Google Scholar] [CrossRef]
- Herroon, M.K.; Rajagurubandara, E.; Rudy, D.L.; Chalasani, A.; Hardaway, A.L.; Podgorski, I. Macrophage cathepsin K promotes prostate tumor progression in bone. Oncogene 2013, 32, 1580–1593. [Google Scholar] [CrossRef]
- Kim, S.W.; Kim, J.S.; Papadopoulos, J.; Choi, H.J.; He, J.; Maya, M.; Langley, R.R.; Fan, D.; Fidler, I.J.; Kim, S.J. Consistent interactions between tumor cell IL-6 and macrophage TNF-α enhance the growth of human prostate cancer cells in the bone of nude mouse. Int. Immunopharmacol. 2011, 11, 862–872. [Google Scholar] [CrossRef]
- Roca, H.; McCauley, L.K. Efferocytosis and prostate cancer skeletal metastasis: Implications for intervention. Oncoscience 2018, 5, 174–176. [Google Scholar] [CrossRef]
- Jones, J.D.; Sinder, B.P.; Paige, D.; Soki, F.N.; Koh, A.J.; Thiele, S.; Shiozawa, Y.; Hofbauer, L.C.; Daignault, S.; Roca, H.; et al. Trabectedin Reduces Skeletal Prostate Cancer Tumor Size in Association with Effects on M2 Macrophages and Efferocytosis. Neoplasia 2019, 21, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Roca, H.; Jones, J.D.; Purica, M.C.; Weidner, S.; Koh, A.J.; Kuo, R.; Wilkinson, J.E.; Wang, Y.; Daignault-Newton, S.; Pienta, K.J.; et al. Apoptosis-induced CXCL5 accelerates inflammation and growth of prostate tumor metastases in bone. J. Clin. Investig. 2018, 128, 248–266. [Google Scholar] [CrossRef] [PubMed]
- Sulciner, M.L.; Serhan, C.N.; Gilligan, M.M.; Mudge, D.K.; Chang, J.; Gartung, A.; Lehner, K.A.; Bielenberg, D.R.; Schmidt, B.; Dalli, J.; et al. Resolvins suppress tumor growth and enhance cancer therapy. J. Exp. Med. 2018, 215, 115–140. [Google Scholar] [CrossRef] [PubMed]
- Reilly, S.M.; Saltiel, A.R. Adapting to obesity with adipose tissue inflammation. Nat. Rev. Endocrinol. 2017, 13, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, Obesity, and Mortality from Cancer in a Prospectively Studied Cohort of U.S. Adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef]
- Freedland, S.J.; Aronson, W.J. Examining the relationship between obesity and prostate cancer. Rev. Urol. 2004, 6, 73–81. [Google Scholar]
- Jiralerspong, S.; Goodwin, P.J. Obesity and breast cancer prognosis: Evidence, challenges, and opportunities. J. Clin. Oncol. 2016, 34, 4203–4216. [Google Scholar] [CrossRef]
- Hardaway, A.L.; Herroon, M.K.; Rajagurubandara, E.; Podgorski, I. Bone marrow fat: Linking adipocyte-induced inflammation with skeletal metastases. Cancer Metastasis Rev. 2014, 33, 527–543. [Google Scholar] [CrossRef]
- Keto, C.J.; Aronson, W.J.; Terris, M.K.; Presti, J.C.; Kane, C.J.; Amling, C.L.; Freedland, S.J. Obesity is associated with castration-resistant disease and metastasis in men treated with androgen deprivation therapy after radical prostatectomy: Results from the SEARCH database. BJU Int. 2012, 110, 492–498. [Google Scholar] [CrossRef]
- Liu, C.; Zhao, Q.; Yu, X. Bone Marrow Adipocytes, Adipocytokines, and Breast Cancer Cells: Novel Implications in Bone Metastasis of Breast Cancer. Front. Oncol. 2020, 10, 561595. [Google Scholar] [CrossRef]
- Morris, E.V.; Edwards, C.M. The role of bone marrow adipocytes in bone metastasis. J. Bone Oncol. 2016, 5, 121–123. [Google Scholar] [CrossRef] [PubMed]
- Ambrosi, T.H.; Scialdone, A.; Graja, A.; Gohlke, S.; Jank, A.-M.; Bocian, C.; Woelk, L.; Fan, H.; Logan, D.W.; Schürmann, A.; et al. Adipocyte Accumulation in the Bone Marrow during Obesity and Aging Impairs Stem Cell-Based Hematopoietic and Bone Regeneration. Cell Stem Cell 2017, 20, 771–784.e6. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.; Koo, J.S. The role of adipokines and bone marrow adipocytes in breast cancer bone metastasis. Int. J. Mol. Sci. 2020, 21, 4967. [Google Scholar] [CrossRef]
- Nieman, K.M.; Romero, I.L.; Van Houten, B.; Lengyel, E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2013, 1831, 1533–1541. [Google Scholar] [CrossRef]
- Oh, S.R.; Sul, O.J.; Kim, Y.Y.; Kim, H.J.; Yu, R.; Suh, J.H.; Choi, H.S. Saturated fatty acids enhance osteoclast survival. J. Lipid Res. 2010, 51, 892–899. [Google Scholar] [CrossRef]
- Chen, J.-R.; Lazarenko, O.P.; Wu, X.; Tong, Y.; Blackburn, M.L.; Shankar, K.; Badger, T.M.; Ronis, M.J.J. Obesity Reduces Bone Density Associated with Activation of PPARγ and Suppression of Wnt/β-Catenin in Rapidly Growing Male Rats. PLoS ONE 2010, 5, e13704. [Google Scholar] [CrossRef]
- Wan, Y.; Chong, L.-W.; Evans, R.M. PPAR-γ regulates osteoclastogenesis in mice. Nat. Med. 2007, 13, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Halade, G.V.; El Jamali, A.; Williams, P.J.; Fajardo, R.J.; Fernandes, G. Obesity-mediated inflammatory microenvironment stimulates osteoclastogenesis and bone loss in mice. Exp. Gerontol. 2011, 46, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.D.; Hart, C.A.; Gazi, E.; Bagley, S.; Clarke, N.W. Promotion of prostatic metastatic migration towards human bone marrow stoma by Omega 6 and its inhibition by Omega 3 PUFAs. Br. J. Cancer 2006, 94, 842–853. [Google Scholar] [CrossRef] [PubMed]
- Templeton, Z.S.; Lie, W.-R.; Wang, W.; Rosenberg-Hasson, Y.; Alluri, R.V.; Tamaresis, J.S.; Bachmann, M.H.; Lee, K.; Maloney, W.J.; Contag, C.H.; et al. Breast Cancer Cell Colonization of the Human Bone Marrow Adipose Tissue Niche. Neoplasia 2015, 17, 849–861. [Google Scholar] [CrossRef]
- Chen, G.L.; Luo, Y.; Eriksson, D.; Meng, X.; Qian, C.; Bäuerle, T.; Chen, X.X.; Schett, G.; Bozec, A. High fat diet increases melanoma cell growth in the bone marrow by inducing osteopontin and interleukin 6. Oncotarget 2016, 7, 26653–26669. [Google Scholar] [CrossRef] [PubMed]
- Herroon, M.K.; Rajagurubandara, E.; Hardaway, A.L.; Powell, K.; Turchick, A.; Feldmann, D.; Podgorski, I. Bone marrow adipocytes promote tumor growth in bone via FABP4-dependent mechanisms. Oncotarget 2013, 4, 2108–2123. [Google Scholar] [CrossRef] [PubMed]
- Hardaway, A.L.; Herroon, M.K.; Rajagurubandara, E.; Podgorski, I. Marrow adipocyte-derived CXCL1 and CXCL2 contribute to osteolysis in metastatic prostate cancer. Clin. Exp. Metastasis 2015, 32, 353–368. [Google Scholar] [CrossRef] [PubMed]
- Gazi, E.; Gardner, P.; Lockyer, N.P.; Hart, C.A.; Brown, M.D.; Clarke, N.W. Direct evidence of lipid translocation between adipocytes and prostate cancer cells with imaging FTIR microspectroscopy. J. Lipid Res. 2007, 48, 1846–1856. [Google Scholar] [CrossRef]
- Tokuda, Y.; Satoh, Y.; Fujiyama, C.; Toda, S.; Sugihara, H.; Masaki, Z. Prostate cancer cell growth is modulated by adipocyte-cancer cell interaction. BJU Int. 2003, 91, 716–720. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhi, Z.; Wang, C.; Xing, H.; Song, G.; Yu, X.; Zhu, Y.; Wang, X.; Zhang, X.; Di, Y. Exogenous lipids promote the growth of breast cancer cells via CD36. Oncol. Rep. 2017, 38, 2105–2115. [Google Scholar] [CrossRef]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-Associated Adipocytes Exhibit an Activated Phenotype and Contribute to Breast Cancer Invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef]
- Fiori, M.E.; Di Franco, S.; Villanova, L.; Bianca, P.; Stassi, G.; De Maria, R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Mol. Cancer 2019, 18, 70. [Google Scholar] [CrossRef]
- Erez, N.; Truitt, M.; Olson, P.; Hanahan, D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-κB-Dependent Manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef]
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-associated fibroblasts: An emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 86. [Google Scholar] [CrossRef]
- Kim, H.M.; Jung, W.H.; Koo, J.S. Expression of cancer-associated fibroblast related proteins in metastatic breast cancer: An immunohistochemical analysis. J. Transl. Med. 2015, 13, 222. [Google Scholar] [CrossRef] [PubMed]
- Giannoni, E.; Bianchini, F.; Masieri, L.; Serni, S.; Torre, E.; Calorini, L.; Chiarugi, P. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res. 2010, 70, 6945–6956. [Google Scholar] [CrossRef] [PubMed]
- Prajapati, P.; Lambert, D.W. Cancer-associated fibroblasts—Not-so-innocent bystanders in metastasis to bone? J. Bone Oncol. 2016, 5, 128–131. [Google Scholar] [CrossRef]
- Zhang, X.H.F.; Jin, X.; Malladi, S.; Zou, Y.; Wen, Y.H.; Brogi, E.; Smid, M.; Foekens, J.A.; Massagué, J. Selection of bone metastasis seeds by mesenchymal signals in the primary tumor stroma. Cell 2013, 154, 1060–1073. [Google Scholar] [CrossRef]
- Li, X.; Sterling, J.A.; Fan, K.-H.; Vessella, R.L.; Shyr, Y.; Hayward, S.W.; Matrisian, L.M.; Bhowmick, N.A. Loss of TGF-β Responsiveness in Prostate Stromal Cells Alters Chemokine Levels and Facilitates the Development of Mixed Osteoblastic/Osteolytic Bone Lesions. Mol. Cancer Res. 2012, 10, 494–503. [Google Scholar] [CrossRef]
- Ershaid, N.; Sharon, Y.; Doron, H.; Raz, Y.; Shani, O.; Cohen, N.; Monteran, L.; Leider-Trejo, L.; Ben-Shmuel, A.; Yassin, M.; et al. NLRP3 inflammasome in fibroblasts links tissue damage with inflammation in breast cancer progression and metastasis. Nat. Commun. 2019, 10, 4375. [Google Scholar] [CrossRef] [PubMed]
- Sansone, P.; Berishaj, M.; Rajasekhar, V.K.; Ceccarelli, C.; Chang, Q.; Strillacci, A.; Savini, C.; Shapiro, L.; Bowman, R.L.; Mastroleo, C.; et al. Evolution of cancer stem-like cells in endocrine-resistant metastatic breast cancers is mediated by stromal microvesicles. Cancer Res. 2017, 77, 1927–1941. [Google Scholar] [CrossRef]
- Cheteh, E.H.; Sarne, V.; Ceder, S.; Bianchi, J.; Augsten, M.; Rundqvist, H.; Egevad, L.; Östman, A.; Wiman, K.G. Interleukin-6 derived from cancer-associated fibroblasts attenuates the p53 response to doxorubicin in prostate cancer cells. Cell Death Discov. 2020, 6, 42. [Google Scholar] [CrossRef]
- Ubellacker, J.M.; McAllister, S.S. The unresolved role of systemic factors in bone metastasis. J. Bone Oncol. 2016, 5, 96–99. [Google Scholar] [CrossRef]
- Salamanna, F.; Borsari, V.; Contartese, D.; Costa, V.; Giavaresi, G.; Fini, M. What is the role of interleukins in breast cancer bone metastases? A systematic review of preclinical and clinical evidence. Cancers 2019, 11, 2018. [Google Scholar] [CrossRef]
- Carswell, E.A.; Old, L.J.; Kassel, R.L.; Green, S.; Fiore, N.; Williamson, B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc. Natl. Acad. Sci. USA 1975, 72, 3666–3670. [Google Scholar] [CrossRef]
- Balkwill, F. TNF-α in promotion and progression of cancer. Cancer Metastasis Rev. 2006, 25, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Lu, X.; Shi, P.; Yang, G.; Zhou, Z.; Li, W.; Mao, X.; Jiang, D.; Chen, C. TNF-α increases breast cancer stem-like cells through up-regulating TAZ expression via the non-canonical NF-κB pathway. Sci. Rep. 2020, 10, 1804. [Google Scholar] [CrossRef] [PubMed]
- Michalaki, V.; Syrigos, K.; Charles, P.; Waxman, J. Serum levels of IL-6 and TNF-α correlate with clinicopathological features and patient survival in patients with prostate cancer. Br. J. Cancer 2004, 90, 2312–2316. [Google Scholar] [CrossRef]
- Neumark, E.; Sagi-Assif, O.; Shalmon, B.; Ben-Baruch, A.; Witz, I.P. Progression of mouse mammary tumors: MCP-1-TNF? cross-regulatory pathway and clonal expression of promalignancy and antimalignancy factors. Int. J. Cancer 2003, 106, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.F.; Huang, Y.; Han, Y.Y.; Lin, L.Y.; Sun, W.H.; Rabson, A.B.; Wang, Y.; Shi, Y.F. TNFα-activated mesenchymal stromal cells promote breast cancer metastasis by recruiting CXCR2+ neutrophils. Oncogene 2017, 36, 482–490. [Google Scholar] [CrossRef]
- Hamaguchi, T.; Wakabayashi, H.; Matsumine, A.; Sudo, A.; Uchida, A. TNF inhibitor suppresses bone metastasis in a breast cancer cell line. Biochem. Biophys. Res. Commun. 2011, 407, 525–530. [Google Scholar] [CrossRef]
- Rébé, C.; Ghiringhelli, F. Interleukin-1β and Cancer. Cancers 2020, 12, 1791. [Google Scholar] [CrossRef]
- Lopez-Castejon, G.; Brough, D. Understanding the mechanism of IL-1β secretion. Cytokine Growth Factor Rev. 2011, 22, 189–195. [Google Scholar] [CrossRef]
- Tulotta, C.; Ottewell, P. The role of IL-1B in breast cancer bone metastasis. Endocr. Relat. Cancer 2018, 25, R421–R434. [Google Scholar] [CrossRef]
- Liu, Q.; Russell, M.R.; Shahriari, K.; Jernigan, D.L.; Lioni, M.I.; Garcia, F.U.; Fatatis, A. Interleukin-1β promotes skeletal colonization and progression of metastatic prostate cancer cells with neuroendocrine features. Cancer Res. 2013, 73, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Nutter, F.; Holen, I.; Brown, H.K.; Cross, S.S.; Alyson Evans, C.; Walker, M.; Coleman, R.E.; Westbrook, J.A.; Selby, P.J.; Brown, J.E.; et al. Different molecular profiles are associated with breast cancer cell homing compared with colonisation of bone: Evidence using a novel bone-seeking cell line. Endocr. Relat. Cancer 2014, 21, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Holen, I.; Nutter, F.; Wilkinson, J.M.; Evans, C.A.; Avgoustou, P.; Ottewell, P.D. Human breast cancer bone metastasis in vitro and in vivo: A novel 3D model system for studies of tumour cell-bone cell interactions. Clin. Exp. Metastasis 2015, 32, 689–702. [Google Scholar] [CrossRef]
- Tulotta, C.; Lefley, D.V.; Freeman, K.; Gregory, W.M.; Hanby, A.M.; Heath, P.R.; Nutter, F.; Mark Wilkinson, J.; Spicer-Hadlington, A.R.; Liu, X.; et al. Endogenous production of IL1B by breast cancer cells drives metastasis and colonization of the bone microenvironment. Clin. Cancer Res. 2019, 25, 2769–2782. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.E.; Marshall, H.; Cameron, D.; Dodwell, D.; Burkinshaw, R.; Keane, M.; Gil, M.; Houston, S.J.; Grieve, R.J.; Barrett-Lee, P.J.; et al. Breast-cancer adjuvant therapy with zoledronic acid. N. Engl. J. Med. 2011, 365, 1396–1405. [Google Scholar] [CrossRef]
- Liu, F.; Ke, J.; Song, Y. Application of biomarkers for the prediction and diagnosis of bone metastasis in breast cancer. J. Breast Cancer 2020, 23, 588–598. [Google Scholar] [CrossRef]
- Holen, I.; Lefley, D.V.; Francis, S.E.; Rennicks, S.; Coleman, R.E.; Ottewell, P. IL-1 drives breast cancer growth and bone metastasis in vivo. Oncotarget 2016, 7, 75571–75584. [Google Scholar]
- Perez-Yepez, E.A.; Ayala-Sumuano, J.-T.; Lezama, R.; Meza, I. A novel β-catenin signaling pathway activated by IL-1β leads to the onset of epithelial–mesenchymal transition in breast cancer cells. Cancer Lett. 2014, 354, 164–171. [Google Scholar] [CrossRef]
- Sosnoski, D.M.; Norgard, R.J.; Grove, C.D.; Foster, S.J.; Mastro, A.M. Dormancy and growth of metastatic breast cancer cells in a bone-like microenvironment. Clin. Exp. Metastasis 2015, 32, 335–344. [Google Scholar] [CrossRef]
- Qu, X.; Tang, Y.; Hua, S. Immunological approaches towards cancer and inflammation: A cross talk. Front. Immunol. 2018, 9, 563. [Google Scholar] [CrossRef] [PubMed]
- Herroon, M.K.; Diedrich, J.D.; Rajagurubandara, E.; Martin, C.; Maddipati, K.R.; Kim, S.; Heath, E.I.; Granneman, J.; Podgorski, I. Prostate tumor cell-derived IL1β induces an inflammatory phenotype in bone marrow adipocytes and reduces sensitivity to docetaxel via lipolysis-dependent mechanisms. Mol. Cancer Res. 2019, 17, 2508–2521. [Google Scholar] [CrossRef] [PubMed]
- Eyre, R.; Alférez, D.G.; Santiago-Gómez, A.; Spence, K.; McConnell, J.C.; Hart, C.; Simões, B.M.; Lefley, D.; Tulotta, C.; Storer, J.; et al. Microenvironmental IL1β promotes breast cancer metastatic colonisation in the bone via activation of Wnt signalling. Nat. Commun. 2019, 10, 5016. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Wang, X.; Dong, Z.; Liu, J.; Zhang, S. Bone metastasis from breast cancer involves elevated IL-11 expression and the gp130/STAT3 pathway. Med. Oncol. 2013, 30, 634. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Park, B.K.; Zhang, H.; Zeng, Q.; Dai, J.; Keller, E.T.; Giordano, T.; Gu, K.; Shah, V.; Pei, L.; Zarbo, R.J.; et al. NF-κB in breast cancer cells promotes osteolytic bone metastasis by inducing osteoclastogenesis via GM-CSF. Nat. Med. 2007, 13, 62–69. [Google Scholar] [CrossRef]
- Jin, R.; Sterling, J.A.; Edwards, J.R.; DeGraff, D.J.; Lee, C.; Park, S.I.; Matusik, R.J. Activation of NF-kappa B Signaling Promotes Growth of Prostate Cancer Cells in Bone. PLoS ONE 2013, 8, e60983. [Google Scholar] [CrossRef]
- Ren, D.; Yang, Q.; Dai, Y.; Guo, W.; Du, H.; Song, L.; Peng, X. Oncogenic miR-210-3p promotes prostate cancer cell EMT and bone metastasis via NF-ΚB signaling pathway. Mol. Cancer 2017, 16, 117. [Google Scholar] [CrossRef]
- Bendinelli, P.; Matteucci, E.; Maroni, P.; Desiderio, M.A. NF-kappaB activation, dependent on acetylation/deacetylation, contributes to HIF-1 activity and migration of bone metastatic breast carcinoma cells. Mol. Cancer Res. 2009, 7, 1328–1341. [Google Scholar] [CrossRef] [PubMed]
- Wa, Q.; Huang, S.; Pan, J.; Tang, Y.; He, S.; Fu, X.; Peng, X.; Chen, X.; Yang, C.; Ren, D.; et al. miR-204-5p Represses Bone Metastasis via Inactivating NF-κB Signaling in Prostate Cancer. Mol. Ther. Nucleic Acids 2019, 18, 567–579. [Google Scholar] [CrossRef]
- Deng, J.; Liu, Y.; Lee, H.; Herrmann, A.; Zhang, W.; Zhang, C.; Shen, S.; Priceman, S.J.; Kujawski, M.; Pal, S.K.; et al. S1PR1-STAT3 Signaling Is Crucial for Myeloid Cell Colonization at Future Metastatic Sites. Cancer Cell 2012, 21, 642–654. [Google Scholar] [CrossRef]
- Lu, X.; Mu, E.; Wei, Y.; Riethdorf, S.; Yang, Q.; Yuan, M.; Yan, J.; Hua, Y.; Tiede, B.J.; Lu, X.; et al. VCAM-1 Promotes Osteolytic Expansion of Indolent Bone Micrometastasis of Breast Cancer by Engaging α4β1-Positive Osteoclast Progenitors. Cancer Cell 2011, 20, 701–714. [Google Scholar] [CrossRef] [PubMed]
- McCoy, E.M.; Hong, H.; Pruitt, H.C.; Feng, X. IL-11 produced by breast cancer cells augments osteoclastogenesis by sustaining the pool of osteoclast progenitor cells. BMC Cancer 2013, 13, 16. [Google Scholar] [CrossRef]
- Nakashima, J.; Tachibana, M.; Horiguchi, Y.; Oya, M.; Ohigashi, T.; Asakura, H.; Murai, M. Serum interleukin 6 as a prognostic factor in patients with prostate cancer. Clin. Cancer Res. 2000, 6, 2702–2706. [Google Scholar]
- Bachelot, T.; Ray-Coquard, I.; Menetrier-Caux, C.; Rastkha, M.; Duc, A.; Blay, J.-Y. Prognostic value of serum levels of interleukin 6 and of serum and plasma levels of vascular endothelial growth factor in hormone-refractory metastatic breast cancer patients. Br. J. Cancer 2003, 88, 1721–1726. [Google Scholar] [CrossRef] [PubMed]
- Benoy, I.H.; Salgado, R.; Van Dam, P.; Geboers, K.; Van Marck, E.; Scharpé, S.; Vermeulen, P.B.; Dirix, L.Y. Increased Serum Interleukin-8 in Patients with Early and Metastatic Breast Cancer Correlates with Early Dissemination and Survival. Clin. Cancer Res. 2004, 10, 7157–7162. [Google Scholar] [CrossRef] [PubMed]
- Jorcyk, C.; Tawara, K.; Jorcyk, C.L. Clinical significance of interleukin (IL)-6 in cancer metastasis to bone: Potential of anti-IL-6 therapies. Cancer Manag. Res. 2011, 3, 177–189. [Google Scholar] [CrossRef]
- Kim, M.-Y.; Oskarsson, T.; Acharyya, S.; Nguyen, D.X.; Zhang, X.H.-F.; Norton, L.; Massagué, J. Tumor Self-Seeding by Circulating Cancer Cells. Cell 2009, 139, 1315–1326. [Google Scholar] [CrossRef]
- Bendre, M.S.; Gaddy-Kurten, D.; Mon-Foote, T.; Akel, N.S.; Skinner, R.A.; Nicholas, R.W.; Suva, L.J. Expression of interleukin 8 and not parathyroid hormone-related protein by human breast cancer cells correlates with bone metastasis in vivo. Cancer Res 2002, 62, 5571–5579. [Google Scholar]
- Bendre, M.S.; Montague, D.C.; Peery, T.; Akel, N.S.; Gaddy, D.; Suva, L.J. Interleukin-8 stimulation of osteoclastogenesis and bone resorption is a mechanism for the increased osteolysis of metastatic bone disease. Bone 2003, 33, 28–37. [Google Scholar] [CrossRef]
- Bendre, M.S.; Margulies, A.G.; Walser, B.; Akel, N.S.; Bhattacharrya, S.; Skinner, R.A.; Swain, F.; Ramani, V.; Chirgwin, J.M.; Gaddy, D.; et al. Tumor-Derived Interleukin-8 Stimulates Osteolysis Independent of the Receptor Activator of Nuclear Factor- κ B Ligand Pathway Tumor-Derived Interleukin-8 Stimulates Osteolysis Independent of the Receptor Activator of Nuclear Factor-KB Ligand Pathway. Cancer Res. 2005, 65, 11001–11009. [Google Scholar] [CrossRef]
- Kamalakar, A.; Bendre, M.S.; Washam, C.L.; Fowler, T.W.; Carver, A.; Dilley, J.D.; Bracey, J.W.; Akel, N.S.; Margulies, A.G.; Skinner, R.A.; et al. Circulating interleukin-8 levels explain breast cancer osteolysis in mice and humans. Bone 2014, 61, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Adler, H.L.; McCurdy, M.A.; Kattan, M.W.; Timme, T.L.; Scardino, P.T.; Thompson, T.C. Elevated Levels of Circulating Interleukin-6 and Transforming Growth Factor-beta 1 in Patients with Metastatic Prostatic Carcinoma. J. Urol. 1999, 161, 182–187. [Google Scholar] [CrossRef]
- Tumminello, F.M.; Badalamenti, G.; Incorvaia, L.; Fulfaro, F.; D’Amico, C.; Leto, G. Serum interleukin-6 in patients with metastatic bone disease: Correlation with cystatin C. Med. Oncol. 2009, 26, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Noman, A.S.; Uddin, M.; Chowdhury, A.A.; Nayeem, M.J.; Raihan, Z.; Rashid, M.I.; Azad, A.K.; Rahman, M.L.; Barua, D.; Sultana, A.; et al. Serum sonic hedgehog (SHH) and interleukin-(IL-6) as dual prognostic biomarkers in progressive metastatic breast cancer. Sci. Rep. 2017, 7, 1796. [Google Scholar] [CrossRef]
- Liang, M.; Ma, Q.; Ding, N.; Luo, F.; Bai, Y.; Kang, F.; Gong, X.; Dong, R.; Dai, J.; Dai, Q.; et al. IL-11 is essential in promoting osteolysis in breast cancer bone metastasis via RANKL-independent activation of osteoclastogenesis. Cell Death Dis. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, S.; Diamond, E.J.; Mamkine, B.; Stone, N.N.; Stock, R.G. Serum Interleukin-8 is Elevated in Men with Prostate Cancer and Bone Metastases. Technol. Cancer Res. Treat. 2004, 3, 411. [Google Scholar] [CrossRef]
- Salgado, R.; Junius, S.; Benoy, I.; Van Dam, P.; Vermeulen, P.; Van Marck, E.; Huget, P.; Dirix, L.Y. Circulating interleukin-6 predicts survival in patients with metastatic breast cancer. Int. J. Cancer 2003, 103, 642–646. [Google Scholar] [CrossRef]
- Studebaker, A.W.; Storci, G.; Werbeck, J.L.; Sansone, P.; Sasser, A.K.; Tavolari, S.; Huang, T.; Chan, M.W.Y.; Marini, F.C.; Rosol, T.J.; et al. Fibroblasts Isolated from Common Sites of Breast Cancer Metastasis Enhance Cancer Cell Growth Rates and Invasiveness in an Interleukin-6–Dependent Manner. Cancer Res. 2008, 68, 9087–9095. [Google Scholar] [CrossRef]
- Gyamfi, J.; Lee, Y.-H.; Eom, M.; Choi, J. Interleukin-6/STAT3 signalling regulates adipocyte induced epithelial-mesenchymal transition in breast cancer cells. Sci. Rep. 2018, 8, 8859. [Google Scholar] [CrossRef]
- Deng, F.; Weng, Y.; Li, X.; Wang, T.; Fan, M.; Shi, Q. Overexpression of IL-8 promotes cell migration via PI3K-Akt signaling pathway and EMT in triple-negative breast cancer. Pathol. Res. Pract. 2020, 216, 152902. [Google Scholar] [CrossRef]
- Ara, T.; Declerck, Y. Interleukin-6 in bone metastasis and cancer progression. Eur. J. Cancer 2010, 46, 1223–1231. [Google Scholar] [CrossRef]
- Kim, S.J.; Uehara, H.; Karashima, T.; Mccarty, M.; Shih, N.; Fidler, I.J. Expression of Interleukin-8 Correlates with Angiogenesis, Tumorigenicity, and Metastasis of Human Prostate Cancer Cells Implanted Orthotopically in Nude Mice. Neoplasia 2001, 3, 33–42. [Google Scholar] [CrossRef]
- Masjedi, A.; Hashemi, V.; Hojjat-Farsangi, M.; Ghalamfarsa, G.; Azizi, G.; Yousefi, M.; Jadidi-Niaragh, F. The significant role of interleukin-6 and its signaling pathway in the immunopathogenesis and treatment of breast cancer. Biomed. Pharmacother. 2018, 108, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhang, J.; Dai, J.; Dehne, L.A.; Mizokami, A.; Yao, Z.; Keller, E.T. Osteoblasts induce prostate cancer proliferation and PSA expression through interleukin-6-mediated activation of the androgen receptor. Clin. Exp. Metastasis 2004, 21, 399–408. [Google Scholar] [CrossRef]
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordón-Cardo, C.; Guise, T.A.; Massagué, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef]
- Zheng, Y.; Chow, S.-O.; Boernert, K.; Basel, D.; Mikuscheva, A.; Kim, S.; Fong-Yee, C.; Trivedi, T.; Buttgereit, F.; Sutherland, R.L.; et al. Direct Crosstalk Between Cancer and Osteoblast Lineage Cells Fuels Metastatic Growth in Bone via Auto-Amplification of IL-6 and RANKL Signaling Pathways. J. Bone Miner. Res. 2014, 29, 1938–1949. [Google Scholar] [CrossRef]
- Wakabayashi, H.; Hamaguchi, T.; Nagao, N.; Kato, S.; Iino, T.; Nakamura, T.; Sudo, A. Interleukin-6 receptor inhibitor suppresses bone metastases in a breast cancer cell line. Breast Cancer 2018, 25, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Hartman, Z.C.; Poage, G.M.; Den Hollander, P.; Tsimelzon, A.; Hill, J.; Panupinthu, N.; Zhang, Y.; Mazumdar, A.; Hilsenbeck, S.G.; Mills, G.B.; et al. Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8. Cancer Res. 2013, 73, 3470–3480. [Google Scholar] [CrossRef]
- Zielińska, K.A.; Katanaev, V.L. The signaling duo CXCL12 and CXCR4: Chemokine fuel for breast cancer tumorigenesis. Cancers 2020, 12, 3071. [Google Scholar] [CrossRef]
- Jung, Y.; Kim, J.K.; Shiozawa, Y.; Wang, J.; Mishra, A.; Joseph, J.; Berry, J.E.; McGee, S.; Lee, E.; Sun, H.; et al. Recruitment of mesenchymal stem cells into prostate tumours promotes metastasis. Nat. Commun. 2013, 4, 1795. [Google Scholar] [CrossRef]
- Müller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef] [PubMed]
- García-Cuesta, E.M.; Santiago, C.A.; Vallejo-Díaz, J.; Juarranz, Y.; Rodríguez-Frade, J.M.; Mellado, M. The Role of the CXCL12/CXCR4/ACKR3 Axis in Autoimmune Diseases. Front. Endocrinol. (Lausanne) 2019, 10, 585. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Wu, T.; Lou, H.; Yu, X.; Taichman, R.S.; Lau, S.K.; Nie, S.; Umbreit, J.; Shim, H. Inhibition of breast cancer metastasis by selective synthetic polypeptide against CXCR4. Cancer Res. 2004, 64, 4302–4308. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.X.; Schneider, A.; Jung, Y.; Wang, J.; Dai, J.; Wang, J.; Cook, K.; Osman, N.I.; Koh-Paige, A.J.; Shim, H.; et al. Skeletal localization and neutralization of the SDF-1(CXCL12)/CXCR4 axis blocks prostate cancer metastasis and growth in osseous sites in vivo. J. Bone Miner. Res. 2005, 20, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Akashi, T.; Koizumi, K.; Tsuneyama, K.; Saiki, I.; Takano, Y.; Fuse, H. Chemokine receptor CXCR4 expression and prognosis in patients with metastatic prostate cancer. Cancer Sci. 2008, 99, 539–542. [Google Scholar] [CrossRef]
- Myers, T.J.; Longobardi, L.; Willcockson, H.; Temple, J.D.; Tagliafierro, L.; Ye, P.; Li, T.; Esposito, A.; Moats-Staats, B.M.; Spagnoli, A. BMP2 Regulation of CXCL12 Cellular, Temporal, and Spatial Expression Is Essential During Fracture Repair. J. Bone Miner. Res. 2015, 30, 2014–2027. [Google Scholar] [CrossRef]
- Yu, X.; Huang, Y.; Collin-Osdoby, P.; Osdoby, P. Stromal Cell-Derived Factor-1 (SDF-1) Recruits Osteoclast Precursors by Inducing Chemotaxis, Matrix Metalloproteinase-9 (MMP-9) Activity, and Collagen Transmigration. J. Bone Miner. Res. 2003, 18, 1404–1418. [Google Scholar] [CrossRef] [PubMed]
- Taichman, R.S.; Cooper, C.; Keller, E.T.; Pienta, K.J.; Taichman, N.S.; McCauley, L.K. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. 2002, 62, 1832–1837. [Google Scholar]
- Tulotta, C.; Stefanescu, C.; Chen, Q.; Torraca, V.; Meijer, A.H.; Snaar-Jagalska, B.E. CXCR4 signaling regulates metastatic onset by controlling neutrophil motility and response to malignant cells. Sci. Rep. 2019, 9, 2399. [Google Scholar] [CrossRef]
- Hung, C.-S.; Su, H.-Y.; Liang, H.-H.; Lai, C.-W.; Chang, Y.-C.; Ho, Y.-S.; Wu, C.-H.; Ho, J.-D.; Wei, P.-L.; Chang, Y.-J. High-level expression of CXCR4 in breast cancer is associated with early distant and bone metastases. Tumor Biol. 2014, 35, 1581–1588. [Google Scholar] [CrossRef]
- Darash-Yahana, M.; Pikarsky, E.; Abramovitch, R.; Zeira, E.; Pal, B.; Karplus, R.; Beider, K.; Avniel, S.; Kasem, S.; Galun, E.; et al. Role of high expression levels of CXCR4 in tumor growth, vascularization, and metastasis. FASEB J. 2004, 18, 1240–1242. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Diao, X.; Sun, J.; Chen, Z. Stromal cell-derived factor-1 and vascular endothelial growth factor as biomarkers for lymph node metastasis and poor cancer-specific survival in prostate cancer patients after radical prostatectomy. Urol. Oncol. Semin. Orig. Investig. 2013, 31, 312–317. [Google Scholar] [CrossRef]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal Fibroblasts Present in Invasive Human Breast Carcinomas Promote Tumor Growth and Angiogenesis through Elevated SDF-1/CXCL12 Secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Endo, M.; Yamamoto, Y.; Odagiri, H.; Kadomatsu, T.; Nakamura, T.; Tanoue, H.; Ito, H.; Yugami, M.; Miyata, K.; et al. ANGPTL2 increases bone metastasis of breast cancer cells through enhancing CXCR4 signaling. Sci. Rep. 2015, 5, 9170. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Pedersen, E.A.; Havens, A.M.; Jung, Y.; Mishra, A.; Joseph, J.; Kim, J.K.; Patel, L.R.; Ying, C.; Ziegler, A.M.; et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J. Clin. Investig. 2011, 121, 1298–1312. [Google Scholar] [CrossRef]
- Azab, A.K.; Runnels, J.M.; Pitsillides, C.; Moreau, A.-S.; Azab, F.; Leleu, X.; Jia, X.; Wright, R.; Ospina, B.; Carlson, A.L.; et al. CXCR4 inhibitor AMD3100 disrupts the interaction of multiple myeloma cells with the bone marrow microenvironment and enhances their sensitivity to therapy. Blood 2009, 113, 4341–4351. [Google Scholar] [CrossRef]
- Roy, L.D.; Sahraei, M.; Schettini, J.L.; Gruber, H.E.; Besmer, D.M.; Mukherjee, P. Systemic neutralization of IL-17A significantly reduces breast cancer associated metastasis in arthritic mice by reducing CXCL12/SDF-1 expression in the metastatic niches. BMC Cancer 2014, 14, 1–13. [Google Scholar] [CrossRef]
- Zannettino, A.C.W.; Farrugia, A.N.; Kortesidis, A.; Manavis, J.; To, L.B.; Martin, S.K.; Diamond, P.; Tamamura, H.; Lapidot, T.; Fujii, N.; et al. Elevated serum levels of stromal-derived factor-1α are associated with increase osteoclast activity and osteolytic bone disease in multiple myeloma patients. Cancer Res. 2005, 65, 1700–1709. [Google Scholar] [CrossRef]
- Conley-LaComb, M.K.; Semaan, L.; Singareddy, R.; Li, Y.; Heath, E.I.; Kim, S.; Cher, M.L.; Chinni, S.R. Pharmacological targeting of CXCL12/CXCR4 signaling in prostate cancer bone metastasis. Mol. Cancer 2016, 15, 68. [Google Scholar] [CrossRef]
- Zeng, H.; Wei, W.; Xu, X. Chemokine (C-X-C motif) receptor 4 RNA interference inhibits bone metastasis in breast cancer. Oncol. Lett. 2014, 8, 77–81. [Google Scholar] [CrossRef][Green Version]
- Wang, Q.; Diao, X.; Sun, J.; Chen, Z. Regulation of VEGF, MMP-9 and metastasis by CXCR4 in a prostate cancer cell line. Cell Biol. Int. 2011, 35, 897–904. [Google Scholar] [CrossRef]
- Li, X.; Loberg, R.; Liao, J.; Ying, C.; Snyder, L.A.; Pienta, K.J.; McCauley, L.K. A Destructive Cascade Mediated by CCL2 Facilitates Prostate Cancer Growth in Bone. Cancer Res. 2009, 69, 1685–1692. [Google Scholar] [CrossRef] [PubMed]
- Loberg, R.D.; Day, L.S.L.; Harwood, J.; Ying, C.; St. John, L.N.; Giles, R.; Neeley, C.K.; Pienta, K.J. CCL2 is a potent regulator of prostate cancer cell migration and proliferation. Neoplasia 2006, 8, 578–586. [Google Scholar] [CrossRef]
- Mulholland, B.S.; Forwood, M.R.; Morrison, N.A. Monocyte Chemoattractant Protein-1 (MCP-1/CCL2) Drives Activation of Bone Remodelling and Skeletal Metastasis. Curr. Osteoporos. Rep. 2019, 17, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Xiao, G.; Galson, D.L.; Nishio, Y.; Mizokami, A.; Keller, E.T.; Yao, Z.; Zhang, J. PTHrP-induced MCP-1 production by human bone marrow endothelial cells and osteoblasts promotes osteoclast differentiation and prostate cancer cell proliferation and invasionin vitro. Int. J. Cancer 2007, 121, 724–733. [Google Scholar] [CrossRef]
- Kim, M.S.; Day, C.J.; Morrison, N.A. MCP-1 Is Induced by Receptor Activator of Nuclear Factor-κB Ligand, Promotes Human Osteoclast Fusion, and Rescues Granulocyte Macrophage Colony-stimulating Factor Suppression of Osteoclast Formation. J. Biol. Chem. 2005, 280, 16163–16169. [Google Scholar] [CrossRef]
- Hopwood, B.; Tsykin, A.; Findlay, D.M.; Fazzalari, N.L. Gene expression profile of the bone microenvironment in human fragility fracture bone. Bone 2009, 44, 87–101. [Google Scholar] [CrossRef]
- Steiner, J.L.; Angela Murphy, E. Importance of chemokine (CC-motif) ligand 2 in breast cancer. Int. J. Biol. Markers 2012, 27, 179–185. [Google Scholar] [CrossRef]
- Soria, G.; Ben-Baruch, A. The inflammatory chemokines CCL2 and CCL5 in breast cancer. Cancer Lett. 2008, 267, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Craig, M.J.; Loberg, R.D. CCL2 (monocyte chemoattractant protein-1) in cancer bone metastases. Cancer Metastasis Rev. 2006, 25, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Roca, H.; Varsos, Z.S.; Sud, S.; Craig, M.J.; Ying, C.; Pienta, K.J. CCL2 and Interleukin-6 Promote Survival of Human CD11b+ Peripheral Blood Mononuclear Cells and Induce M2-type Macrophage Polarization. J. Biol. Chem. 2009, 284, 34342–34354. [Google Scholar] [CrossRef] [PubMed]
- Saji, H.; Koike, M.; Yamori, T.; Saji, S.; Seiki, M.; Matsushima, K.; Toi, M. Significant correlation of monocyte chemoattractant protein-1 expression with neovascularization and progression of breast carcinoma. Cancer 2001, 92, 1085–1091. [Google Scholar] [CrossRef]
- Ueno, T.; Toi, M.; Saji, H.; Muta, M.; Bando, H.; Kuroi, K.; Koike, M.; Inadera, H.; Matsushima, K. Significance of macrophage chemoattractant protein-1 in macrophage recruitment, angiogenesis, and survival in human breast cancer. Clin. Cancer Res. 2000, 6, 3282–3289. [Google Scholar] [PubMed]
- Tewari, B.N.; Singh Baghel, K.; Tripathi, C.; Dubey, P.; Bhatt, M.L.B.; Kumar, V.; Mati Goel, M.; Singh Negi, M.P.; Misra, S. A study on local expression of NF-κB, CCL2 and their involvement in intratumoral macrophage infiltration in breast cancer. Cell. Mol. Biol. (Noisy-le-Grand) 2016, 62, 116–125. [Google Scholar]
- Lu, Y.; Cai, Z.; Xiao, G.; Liu, Y.; Keller, E.T.; Yao, Z.; Zhang, J. CCR2 expression correlates with prostate cancer progression. J. Cell. Biochem. 2007, 101, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Chen, Q.; Corey, E.; Xie, W.; Fan, J.; Mizokami, A.; Zhang, J. Activation of MCP-1/CCR2 axis promotes prostate cancer growth in bone. Clin. Exp. Metastasis 2009, 26, 161–169. [Google Scholar] [CrossRef]
- Qian, B.-Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef]
- Hao, Q.; Vadgama, J.V.; Wang, P. CCL2/CCR2 signaling in cancer pathogenesis. Cell Commun. Signal 2020, 18. [Google Scholar] [CrossRef]
- Molloy, A.P.; Martin, F.T.; Dwyer, R.M.; Griffin, T.P.; Murphy, M.; Barry, F.P.; O’Brien, T.; Kerin, M.J. Mesenchymal stem cell secretion of chemokines during differentiation into osteoblasts, and their potential role in mediating interactions with breast cancer cells. Int. J. Cancer 2009, 124, 326–332. [Google Scholar] [CrossRef]
- Park, S.I.; Liao, J.; Berry, J.E.; Li, X.; Koh, A.J.; Michalski, M.E.; Eber, M.R.; Soki, F.N.; Sadler, D.; Sud, S.; et al. Cyclophosphamide Creates a Receptive Microenvironment for Prostate Cancer Skeletal Metastasis. Cancer Res. 2012, 72, 2522–2532. [Google Scholar] [CrossRef]
- Lim, S.Y.; Yuzhalin, A.E.; Gordon-Weeks, A.N.; Muschel, R.J. Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget 2016, 7, 28697–28710. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Miyazaki, H.; Furihata, M.; Sakai, H.; Konakahara, T.; Watanabe, M.; Okada, T. Chemokine CCL2/MCP-1 negatively regulates metastasis in a highly bone marrow-metastatic mouse breast cancer model. Clin. Exp. Metastasis 2009, 26, 817–828. [Google Scholar] [CrossRef]
- Luboshits, G.; Shina, S.; Kaplan, O.; Engelberg, S.; Nass, D.; Lifshitz-Mercer, B.; Chaitchik, S.; Keydar, I.; Ben-Baruch, A. Elevated expression of the CC chemokine regulated on activation, normal T cell expressed and secreted (RANTES) in advanced breast carcinoma. Cancer Res. 1999, 59, 4681–4687. [Google Scholar]
- Niwa, Y.; Akamatsu, H.; Niwa, H.; Sumi, H.; Ozaki, Y.; Abe, A. Correlation of tissue and plasma RANTES levels with disease course in patients with breast or cervical cancer. Clin. Cancer Res. 2001, 7, 285–289. [Google Scholar] [PubMed]
- Walens, A.; DiMarco, A.V.; Lupo, R.; Kroger, B.R.; Damrauer, J.S.; Alvarez, J.V. CCL5 promotes breast cancer recurrence through macrophage recruitment in residual tumors. eLife 2019, 8, e43653. [Google Scholar] [CrossRef]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Sottnik, J.L.; Dai, J.; Zhang, H.; Campbell, B.; Keller, E.T. Tumor-induced pressure in the bone microenvironment causes osteocytes to promote the growth of prostate cancer bone metastases. Cancer Res. 2015, 75, 2151–2158. [Google Scholar] [CrossRef]
- Huang, R.; Wang, S.; Wang, N.; Zheng, Y.; Zhou, J.; Yang, B.; Wang, X.; Zhang, J.; Guo, L.; Wang, S.; et al. CCL5 derived from tumor-associated macrophages promotes prostate cancer stem cells and metastasis via activating β-catenin/STAT3 signaling. Cell Death Dis. 2020, 11, 234. [Google Scholar] [CrossRef]
- Singh, B.; Lucci, A. Role of Cyclooxygenase-2 in Breast Cancer. J. Surg. Res. 2002, 108, 173–179. [Google Scholar] [CrossRef]
- Gasparini, G.; Longo, R.; Sarmiento, R.; Morabito, A. Inhibitors of cyclo-oxygenase 2: A new class of anticancer agents? Lancet Oncol. 2003, 4, 605–615. [Google Scholar] [CrossRef]
- Blackwell, K.A.; Raisz, L.G.; Pilbeam, C.C. Prostaglandins in bone: Bad cop, good cop? Trends Endocrinol. Metab. 2010, 21, 294–301. [Google Scholar] [CrossRef]
- Lucci, A.; Krishnamurthy, S.; Singh, B.; Bedrosian, I.; Meric-Bernstam, F.; Reuben, J.; Broglio, K.; Mosalpuria, K.; Lodhi, A.; Vincent, L.; et al. Cyclooxygenase-2 expression in primary breast cancers predicts dissemination of cancer cells to the bone marrow. Breast Cancer Res. Treat. 2009, 117, 61–68. [Google Scholar] [CrossRef][Green Version]
- Li, Z.; Schem, C.; Shi, Y.H.; Medina, D.; Zhang, M. Increased COX2 expression enhances tumor-induced osteoclastic lesions in breast cancer bone metastasis. Clin. Exp. Metastasis 2008, 25, 389–400. [Google Scholar] [CrossRef]
- Hiraga, T.; Myoui, A.; Choi, M.E.; Yoshikawa, H.; Yoneda, T. Stimulation of Cyclooxygenase-2 Expression by Bone-Derived Transforming Growth Factor-β Enhances Bone Metastases in Breast Cancer. Cancer Res. 2006, 66, 2067–2073. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ohshiba, T.; Miyaura, C.; Ito, A. Role of prostaglandin E produced by osteoblasts in osteolysis due to bone metastasis. Biochem. Biophys. Res. Commun. 2003, 300, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Valsecchi, M.E.; Pomerantz, S.C.; Jaslow, R.; Tester, W. Reduced risk of bone metastasis for patients with breast cancer who use cox-2 inhibitors. Clin. Breast Cancer 2009, 9, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Uehara, H.; Bando, Y.; Izumi, K. Soluble EP2 neutralizes prostaglandin E2-induced cell signaling and inhibits osteolytic tumor growth. Mol. Cancer Ther. 2008, 7, 2807–2816. [Google Scholar] [CrossRef] [PubMed]
- Takita, M.; Inada, M.; Maruyama, T.; Miyaura, C. Prostaglandin E receptor EP4 antagonist suppresses osteolysis due to bone metastasis of mouse malignant melanoma cells. FEBS Lett. 2007, 581, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.; Velez, R.; Romagosa, C.; Majem, B.; Pedrola, N.; Olivan, M.; Rigau, M.; Guiu, M.; Gomis, R.R.; Morote, J.; et al. Cyclooxygenase-2 inhibitor suppresses tumour progression of prostate cancer bone metastases in nude mice. BJU Int. 2014, 113, 164–177. [Google Scholar] [CrossRef]
- Singh, B.; Berry, J.A.; Shoher, A.; Ayers, G.D.; Wei, C.; Lucci, A. COX-2 involvement in breast cancer metastasis to bone. Oncogene 2007, 26, 3789–3796. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Göbel, A.; Dell’Endice, S.; Jaschke, N.; Pählig, S.; Shahid, A.; Hofbauer, L.C.; Rachner, T.D. The Role of Inflammation in Breast and Prostate Cancer Metastasis to Bone. Int. J. Mol. Sci. 2021, 22, 5078. https://doi.org/10.3390/ijms22105078
Göbel A, Dell’Endice S, Jaschke N, Pählig S, Shahid A, Hofbauer LC, Rachner TD. The Role of Inflammation in Breast and Prostate Cancer Metastasis to Bone. International Journal of Molecular Sciences. 2021; 22(10):5078. https://doi.org/10.3390/ijms22105078
Chicago/Turabian StyleGöbel, Andy, Stefania Dell’Endice, Nikolai Jaschke, Sophie Pählig, Amna Shahid, Lorenz C. Hofbauer, and Tilman D. Rachner. 2021. "The Role of Inflammation in Breast and Prostate Cancer Metastasis to Bone" International Journal of Molecular Sciences 22, no. 10: 5078. https://doi.org/10.3390/ijms22105078
APA StyleGöbel, A., Dell’Endice, S., Jaschke, N., Pählig, S., Shahid, A., Hofbauer, L. C., & Rachner, T. D. (2021). The Role of Inflammation in Breast and Prostate Cancer Metastasis to Bone. International Journal of Molecular Sciences, 22(10), 5078. https://doi.org/10.3390/ijms22105078