
Acceleration of TAA-Induced Liver Fibrosis by Stress Exposure Is Associated with Upregulation of Nerve Growth Factor and Glycopattern Deviations

 , , ,
, , ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Stress Exposure Accelerates TAA-Induced Fibrosis and Results in Increased Liver NGF Expression
2.2. Blocking of NGF Pathways Disrupt Deleterious Effect of Stress in Experimental TAA-Mediated Fibrosis
2.3. NGF Administration during Experimental TAA Model Boosts Liver Fibrosis
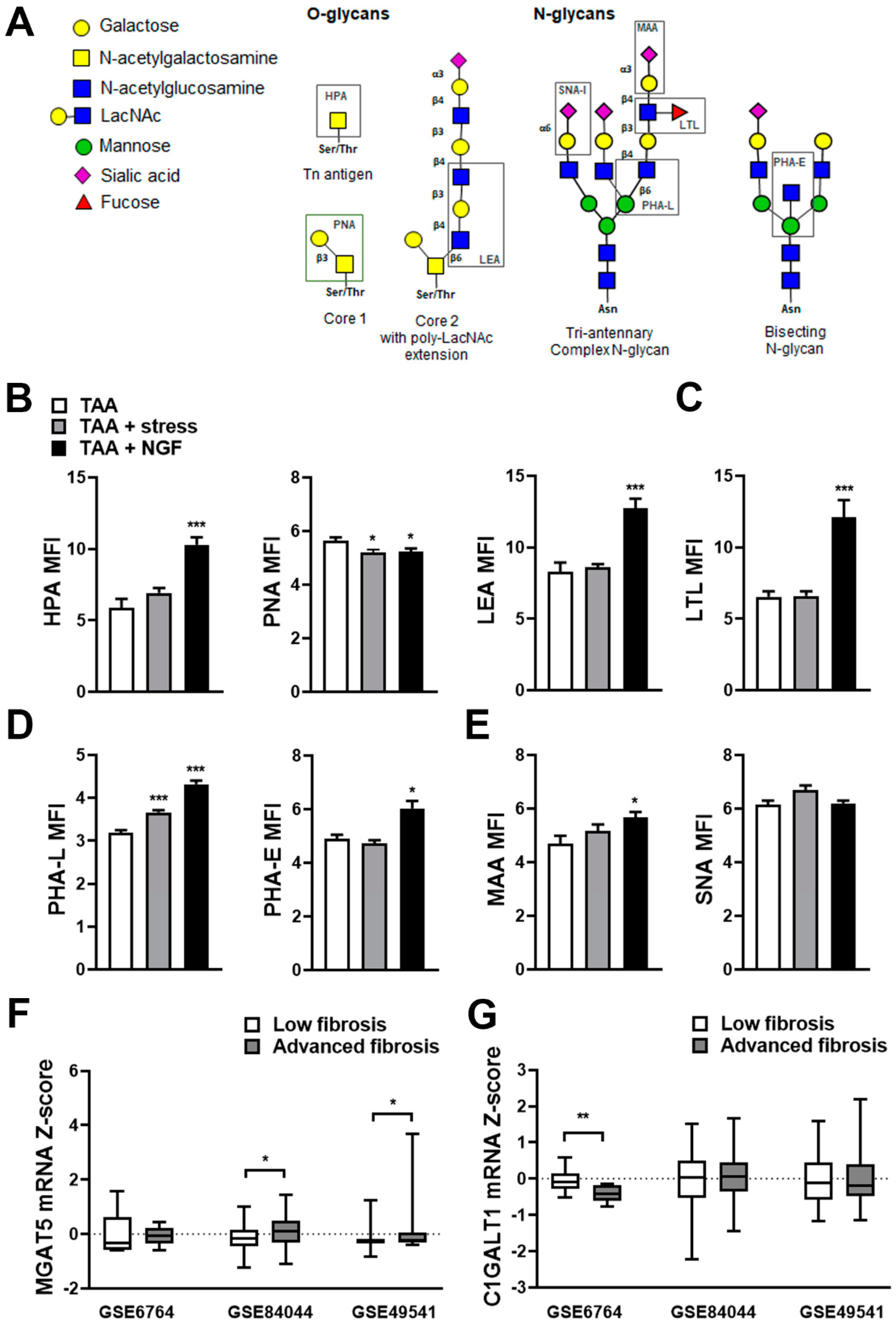
2.4. Altered Liver Glycosylation Accompanies the Progression of Stress-Induced Liver Fibrosis
2.5. Differential Expression of Glycosylation Enzymes Is Observed in Fibrosis Progression in Patients
3. Discussion
4. Materials and Methods
4.1. Liver Fibrosis Model and Animal Experiments
4.2. Histology and Liver Fibrosis Analysis
4.3. Liver Enzymes
4.4. Immunofluorescence
4.5. Terminal dUTP Nick-End Labeling (TUNEL) Staining
4.6. Glycophenotype Analysis
4.7. Bioinformatic Analysis
4.8. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef]
- Trautwein, C.; Friedman, S.L.; Schuppan, D.; Pinzani, M. Hepatic fibrosis: Concept to treatment. J. Hepatol. 2015, 62, 15–24. [Google Scholar] [CrossRef]
- Ringelhan, M.; Pfister, D.; O’Connor, T.; Pikarsky, E.; Heikenwalder, M. The immunology of hepatocellular carcinoma. Nat. Immunol. 2018, 19, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Koolhaas, J.M.; Bartolomucci, A.; Buwalda, B.; de Boer, S.F.; Flugge, G.; Korte, S.M.; Meerlo, P.; Murison, R.; Olivier, B.; Palanza, P.; et al. Stress revisited: A critical evaluation of the stress concept. Neurosci. Biobehav. Rev. 2011, 35, 1291–1301. [Google Scholar] [CrossRef] [PubMed]
- Hirose, S.; Hirayama, C.; Ikemi, Y. The influence of emotional stress on the liver blood flow. Kyushu J. Med. Sci. 1961, 12, 319–323. [Google Scholar] [PubMed]
- Fernandez, G.; Mena, M.P.; Arnau, A.; Sanchez, O.; Soley, M.; Ramirez, I. Immobilization stress induces c-Fos accumulation in liver. Cell Stress Chaperones 2000, 5, 306–312. [Google Scholar] [CrossRef]
- Chida, Y.; Sudo, N.; Sonoda, J.; Sogawa, H.; Kubo, C. Electric foot shock stress-induced exacerbation of alpha-galactosylceramide-triggered apoptosis in mouse liver. Hepatology 2004, 39, 1131–1140. [Google Scholar] [CrossRef]
- Zhu, Q.; Gu, L.; Wang, Y.; Jia, L.; Zhao, Z.; Peng, S.; Lei, L. The role of alpha-1 and alpha-2 adrenoceptors in restraint stress-induced liver injury in mice. PLoS ONE 2014, 9, e92125. [Google Scholar] [CrossRef] [PubMed]
- Fukudo, S.; Suzuki, J.; Tanaka, Y.; Iwahashi, S.; Nomura, T. Impact of stress on alcoholic liver injury; a histopathological study. J. Psychosom. Res. 1989, 33, 515–521. [Google Scholar] [CrossRef]
- Kunkel, E.J.; Kim, J.S.; Hann, H.W.; Oyesanmi, O.; Menefee, L.A.; Field, H.L.; Lartey, P.L.; Myers, R.E. Depression in Korean immigrants with hepatitis B and related liver diseases. Psychosomatics 2000, 41, 472–480. [Google Scholar] [CrossRef]
- Elwing, J.E.; Lustman, P.J.; Wang, H.L.; Clouse, R.E. Depression, anxiety, and nonalcoholic steatohepatitis. Psychosom. Med. 2006, 68, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Arck, P.C.; Knackstedt, M.K.; Blois, S.M. Current insights and future perspectives on neuro-endocrine-immune circuitry challenging pregnancy maintenance and fetal health. J. Reprod. Endokrinol. 2006, 3, 98–102. [Google Scholar]
- Welcome, M.O. Cellular mechanisms and molecular signaling pathways in stress-induced anxiety, depression, and blood-brain barrier inflammation and leakage. Inflammopharmacology 2020, 28, 643–665. [Google Scholar] [CrossRef]
- Foertsch, S.; Reber, S.O. The role of physical trauma in social stress-induced immune activation. Neurosci. Biobehav. Rev. 2020, 113, 169–178. [Google Scholar] [CrossRef]
- Tseilikman, V.; Kozochkin, D.; Synitsky, A.; Sibiriak, S.; Tseilikman, O.; Katashinsky, E.; Nikitina, A.; Vinogradov, D.; Simbirtsev, A. Does stress-induced release of interleukin-1 cause liver injury? Cell Mol. Neurobiol. 2012, 32, 1069–1078. [Google Scholar] [CrossRef]
- Cirulli, F.; Alleva, E. The NGF saga: From animal models of psychosocial stress to stress-related psychopathology. Front. Neuroendocrinol. 2009, 30, 379–395. [Google Scholar] [CrossRef]
- Tometten, M.; Blois, S.; Kuhlmei, A.; Stretz, A.; Klapp, B.F.; Arck, P.C. Nerve growth factor translates stress response and subsequent murine abortion via adhesion molecule-dependent pathways. Biol. Reprod. 2006, 74, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.J.; Reichardt, L.F. Trk receptors: Roles in neuronal signal transduction. Annu. Rev. Biochem. 2003, 72, 609–642. [Google Scholar] [CrossRef]
- Blomme, B.; Van Steenkiste, C.; Callewaert, N.; Van Vlierberghe, H. Alteration of protein glycosylation in liver diseases. J. Hepatol. 2009, 50, 592–603. [Google Scholar] [CrossRef]
- Dennis, J.W.; Granovsky, M.; Warren, C.E. Protein glycosylation in development and disease. Bioessays 1999, 21, 412–421. [Google Scholar] [CrossRef]
- Amado, M.; Almeida, R.; Schwientek, T.; Clausen, H. Identification and characterization of large galactosyltransferase gene families: Galactosyltransferases for all functions. Biochim. Biophys. Acta 1999, 1473, 35–53. [Google Scholar] [CrossRef]
- Joung, J.Y.; Cho, J.H.; Kim, Y.H.; Choi, S.H.; Son, C.G. A literature review for the mechanisms of stress-induced liver injury. Brain Behav. 2019, 9, 01235. [Google Scholar] [CrossRef]
- Skaper, S.D. Nerve growth factor: A neuroimmune crosstalk mediator for all seasons. Immunology 2017, 151, 1–15. [Google Scholar] [CrossRef]
- Cassiman, D.; Denef, C.; Desmet, V.J.; Roskams, T. Human and rat hepatic stellate cells express neurotrophins and neurotrophin receptors. Hepatology 2001, 33, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Antoni, M.H.; Lutgendorf, S.K.; Cole, S.W.; Dhabhar, F.S.; Sephton, S.E.; McDonald, P.G.; Stefanek, M.; Sood, A.K. The influence of bio-behavioural factors on tumour biology: Pathways and mechanisms. Nat. Rev. Cancer 2006, 6, 240–248. [Google Scholar] [CrossRef]
- Chida, Y.; Hamer, M.; Wardle, J.; Steptoe, A. Do stress-related psychosocial factors contribute to cancer incidence and survival? Nat. Clin. Pract. Oncol. 2008, 5, 466–475. [Google Scholar] [CrossRef]
- Antoni, M.H.; Dhabhar, F.S. The impact of psychosocial stress and stress management on immune responses in patients with cancer. Cancer 2019, 125, 1417–1431. [Google Scholar] [CrossRef]
- Krizanova, O.; Babula, P.; Pacak, K. Stress, catecholaminergic system and cancer. Stress 2016, 19, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Trim, N.; Morgan, S.; Evans, M.; Issa, R.; Fine, D.; Afford, S.; Wilkins, B.; Iredale, J. Hepatic stellate cells express the low affinity nerve growth factor receptor p75 and undergo apoptosis in response to nerve growth factor stimulation. Am. J. Pathol. 2000, 156, 1235–1243. [Google Scholar] [CrossRef]
- Pham, D.D.; Do, H.T.; Bruelle, C.; Kukkonen, J.P.; Eriksson, O.; Mogollon, I.; Korhonen, L.T.; Arumae, U.; Lindholm, D. p75 Neurotrophin Receptor Signaling Activates Sterol Regulatory Element-binding Protein-2 in Hepatocyte Cells via p38 Mitogen-activated Protein Kinase and Caspase-3. J. Biol. Chem. 2016, 291, 10747–10758. [Google Scholar] [CrossRef]
- Pham, D.D.; Bruelle, C.; Thi Do, H.; Pajanoja, C.; Jin, C.; Srinivasan, V.; Olkkonen, V.M.; Eriksson, O.; Jauhiainen, M.; Lalowski, M.; et al. Caspase-2 and p75 neurotrophin receptor (p75NTR) are involved in the regulation of SREBP and lipid genes in hepatocyte cells. Cell Death Dis. 2019, 10, 537. [Google Scholar] [CrossRef]
- Bonacchi, A.; Taddei, M.L.; Petrai, I.; Efsen, E.; Defranco, R.; Nosi, D.; Torcia, M.; Rosini, P.; Formigli, L.; Rombouts, K.; et al. Nuclear localization of TRK-A in liver cells. Histol. Histopathol. 2008, 23, 327–340. [Google Scholar]
- Amoras Eda, S.; Gomes, S.T.; Freitas, F.B.; Santana, B.B.; Ishak, G.; de Araujo, M.T.; Demachki, S.; da Silva Conde, S.R.; de Oliveira Guimaraes Ishak, M.; Ishak, R.; et al. NGF and P75NTR gene expression is associated with the hepatic fibrosis stage due to viral and non-viral causes. PLoS ONE 2015, 10, e0121754. [Google Scholar] [CrossRef]
- Passino, M.A.; Adams, R.A.; Sikorski, S.L.; Akassoglou, K. Regulation of hepatic stellate cell differentiation by the neurotrophin receptor p75NTR. Science 2007, 315, 1853–1856. [Google Scholar] [CrossRef] [PubMed]
- Oakley, F.; Trim, N.; Constandinou, C.M.; Ye, W.; Gray, A.M.; Frantz, G.; Hillan, K.; Kendall, T.; Benyon, R.C.; Mann, D.A.; et al. Hepatocytes express nerve growth factor during liver injury: Evidence for paracrine regulation of hepatic stellate cell apoptosis. Am. J. Pathol. 2003, 163, 1849–1858. [Google Scholar] [CrossRef]
- Asai, K.; Tamakawa, S.; Yamamoto, M.; Yoshie, M.; Tokusashi, Y.; Yaginuma, Y.; Kasai, S.; Ogawa, K. Activated hepatic stellate cells overexpress p75NTR after partial hepatectomy and undergo apoptosis on nerve growth factor stimulation. Liver Int. 2006, 26, 595–603. [Google Scholar] [CrossRef]
- Elsharkawy, A.M.; Oakley, F.; Mann, D.A. The role and regulation of hepatic stellate cell apoptosis in reversal of liver fibrosis. Apoptosis 2005, 10, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Helenius, A.; Aebi, M. Intracellular functions of N-linked glycans. Science 2001, 291, 2364–2369. [Google Scholar] [CrossRef]
- Gill, D.J.; Clausen, H.; Bard, F. Location, location, location: New insights into O-GalNAc protein glycosylation. Trends Cell Biol. 2011, 21, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Varki, A. Biological roles of glycans. Glycobiology 2017, 27, 3–49. [Google Scholar] [CrossRef]
- Chambers, W.; Thompson, S.; Skillen, A.W.; Record, C.O.; Turner, G.A. Abnormally fucosylated haptoglobin as a marker for alcoholic liver disease but not excessive alcohol consumption or non-alcoholic liver disease. Clin. Chim. Acta 1993, 219, 177–182. [Google Scholar] [CrossRef]
- Nakagawa, T.; Uozumi, N.; Nakano, M.; Mizuno-Horikawa, Y.; Okuyama, N.; Taguchi, T.; Gu, J.; Kondo, A.; Taniguchi, N.; Miyoshi, E. Fucosylation of N-glycans regulates the secretion of hepatic glycoproteins into bile ducts. J. Biol. Chem. 2006, 281, 29797–29806. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.; Pollacchi, A.; Hayes, P.; Therapondos, G.; Newsome, P.; Boyter, A.; Smith, K. A preliminary evaluation of the differences in the glycosylation of alpha-1-acid glycoprotein between individual liver diseases. Biomed. Chromatogr. 2002, 16, 365–372. [Google Scholar] [CrossRef]
- Lau, K.S.; Partridge, E.A.; Grigorian, A.; Silvescu, C.I.; Reinhold, V.N.; Demetriou, M.; Dennis, J.W. Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell 2007, 129, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Dennis, J.W.; Laferte, S. Oncodevelopmental expression of--GlcNAc beta 1-6Man alpha 1-6Man beta 1--branched asparagine-linked oligosaccharides in murine tissues and human breast carcinomas. Cancer Res. 1989, 49, 945–950. [Google Scholar] [PubMed]
- Partridge, E.A.; Le Roy, C.; Di Guglielmo, G.M.; Pawling, J.; Cheung, P.; Granovsky, M.; Nabi, I.R.; Wrana, J.L.; Dennis, J.W. Regulation of cytokine receptors by Golgi N-glycan processing and endocytosis. Science 2004, 306, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, S.Y.; Ooshima, A.; Yang, K.M.; Kang, J.M.; Kim, Y.W.; Kim, S.J. Glucosamine hydrochloride exerts a protective effect against unilateral ureteral obstruction-induced renal fibrosis by attenuating TGF-beta signaling. J. Mol. Med. 2013, 91, 1273–1284. [Google Scholar] [CrossRef]
- Blois, S.M.; Piccioni, F.; Freitag, N.; Tirado-Gonzalez, I.; Moschansky, P.; Lloyd, R.; Hensel-Wiegel, K.; Rose, M.; Garcia, M.G.; Alaniz, L.D.; et al. Dendritic cells regulate angiogenesis associated with liver fibrogenesis. Angiogenesis 2014, 17, 119–128. [Google Scholar] [CrossRef]
- Blois, S.M.; Joachim, R.; Kandil, J.; Margni, R.; Tometten, M.; Klapp, B.F.; Arck, P.C. Depletion of CD8+ cells abolishes the pregnancy protective effect of progesterone substitution with dydrogesterone in mice by altering the Th1/Th2 cytokine profile. J. Immunol. 2004, 172, 5893–5899. [Google Scholar] [CrossRef]
- Frank, P.; Barrientos, G.; Tirado-Gonzalez, I.; Cohen, M.; Moschansky, P.; Peters, E.M.; Klapp, B.F.; Rose, M.; Tometten, M.; Blois, S.M. Balanced levels of nerve growth factor are required for normal pregnancy progression. Reproduction 2014, 148, 179–189. [Google Scholar] [CrossRef]
- Ishak, K.; Baptista, A.; Bianchi, L.; Callea, F.; De Groote, J.; Gudat, F.; Denk, H.; Desmet, V.; Korb, G.; MacSween, R.N.; et al. Histological grading and staging of chronic hepatitis. J. Hepatol. 1995, 22, 696–699. [Google Scholar] [CrossRef]
- Fiore, E.J.; Bayo, J.M.; Garcia, M.G.; Malvicini, M.; Lloyd, R.; Piccioni, F.; Rizzo, M.; Peixoto, E.; Sola, M.B.; Atorrasagasti, C.; et al. Mesenchymal stromal cells engineered to produce IGF-I by recombinant adenovirus ameliorate liver fibrosis in mice. Stem Cells Dev. 2015, 24, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Joachim, R.A.; Handjiski, B.; Blois, S.M.; Hagen, E.; Paus, R.; Arck, P.C. Stress-induced neurogenic inflammation in murine skin skews dendritic cells towards maturation and migration: Key role of intercellular adhesion molecule-1/leukocyte function-associated antigen interactions. Am. J. Pathol. 2008, 173, 1379–1388. [Google Scholar] [CrossRef]
- Wurmbach, E.; Chen, Y.B.; Khitrov, G.; Zhang, W.; Roayaie, S.; Schwartz, M.; Fiel, I.; Thung, S.; Mazzaferro, V.; Bruix, J.; et al. Genome-wide molecular profiles of HCV-induced dysplasia and hepatocellular carcinoma. Hepatology 2007, 45, 938–947. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Gong, Q.; Zhang, J.; Chen, L.; Zhang, Z.; Lu, L.; Yu, D.; Han, Y.; Zhang, D.; Chen, P.; et al. Characterization of gene expression profiles in HBV-related liver fibrosis patients and identification of ITGBL1 as a key regulator of fibrogenesis. Sci. Rep. 2017, 7, 43446. [Google Scholar] [CrossRef] [PubMed]
- Moylan, C.A.; Pang, H.; Dellinger, A.; Suzuki, A.; Garrett, M.E.; Guy, C.D.; Murphy, S.K.; Ashley-Koch, A.E.; Choi, S.S.; Michelotti, G.A.; et al. Hepatic gene expression profiles differentiate presymptomatic patients with mild versus severe nonalcoholic fatty liver disease. Hepatology 2014, 59, 471–482. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atorrasagasti, C.; Piccioni, F.; Borowski, S.; Tirado-González, I.; Freitag, N.; Cantero, M.J.; Bayo, J.; Mazzolini, G.; Alaniz, L.D.; Blois, S.M.; et al. Acceleration of TAA-Induced Liver Fibrosis by Stress Exposure Is Associated with Upregulation of Nerve Growth Factor and Glycopattern Deviations. Int. J. Mol. Sci. 2021, 22, 5055. https://doi.org/10.3390/ijms22105055
Atorrasagasti C, Piccioni F, Borowski S, Tirado-González I, Freitag N, Cantero MJ, Bayo J, Mazzolini G, Alaniz LD, Blois SM, et al. Acceleration of TAA-Induced Liver Fibrosis by Stress Exposure Is Associated with Upregulation of Nerve Growth Factor and Glycopattern Deviations. International Journal of Molecular Sciences. 2021; 22(10):5055. https://doi.org/10.3390/ijms22105055
Chicago/Turabian StyleAtorrasagasti, Catalina, Flavia Piccioni, Sophia Borowski, Irene Tirado-González, Nancy Freitag, María José Cantero, Juan Bayo, Guillermo Mazzolini, Laura D. Alaniz, Sandra M. Blois, and et al. 2021. "Acceleration of TAA-Induced Liver Fibrosis by Stress Exposure Is Associated with Upregulation of Nerve Growth Factor and Glycopattern Deviations" International Journal of Molecular Sciences 22, no. 10: 5055. https://doi.org/10.3390/ijms22105055
APA StyleAtorrasagasti, C., Piccioni, F., Borowski, S., Tirado-González, I., Freitag, N., Cantero, M. J., Bayo, J., Mazzolini, G., Alaniz, L. D., Blois, S. M., & Garcia, M. G. (2021). Acceleration of TAA-Induced Liver Fibrosis by Stress Exposure Is Associated with Upregulation of Nerve Growth Factor and Glycopattern Deviations. International Journal of Molecular Sciences, 22(10), 5055. https://doi.org/10.3390/ijms22105055