Blood–Nerve Barrier (BNB) Pathology in Diabetic Peripheral Neuropathy and In Vitro Human BNB Model


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
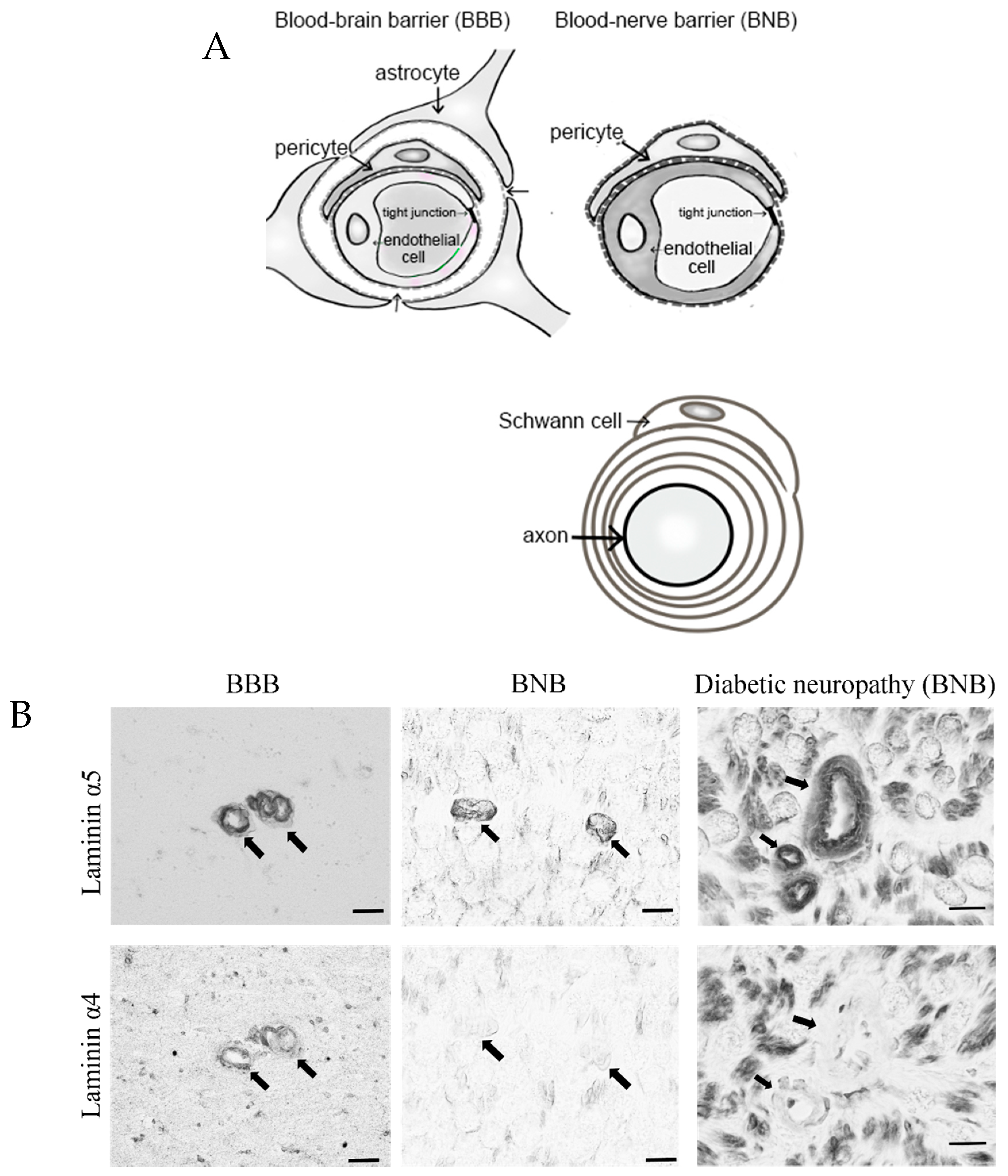
2. Anatomy of the BNB and Peripheral Nerves
2.1. Establishment of BNB-Forming Cell Lines and Functions of Pericytes
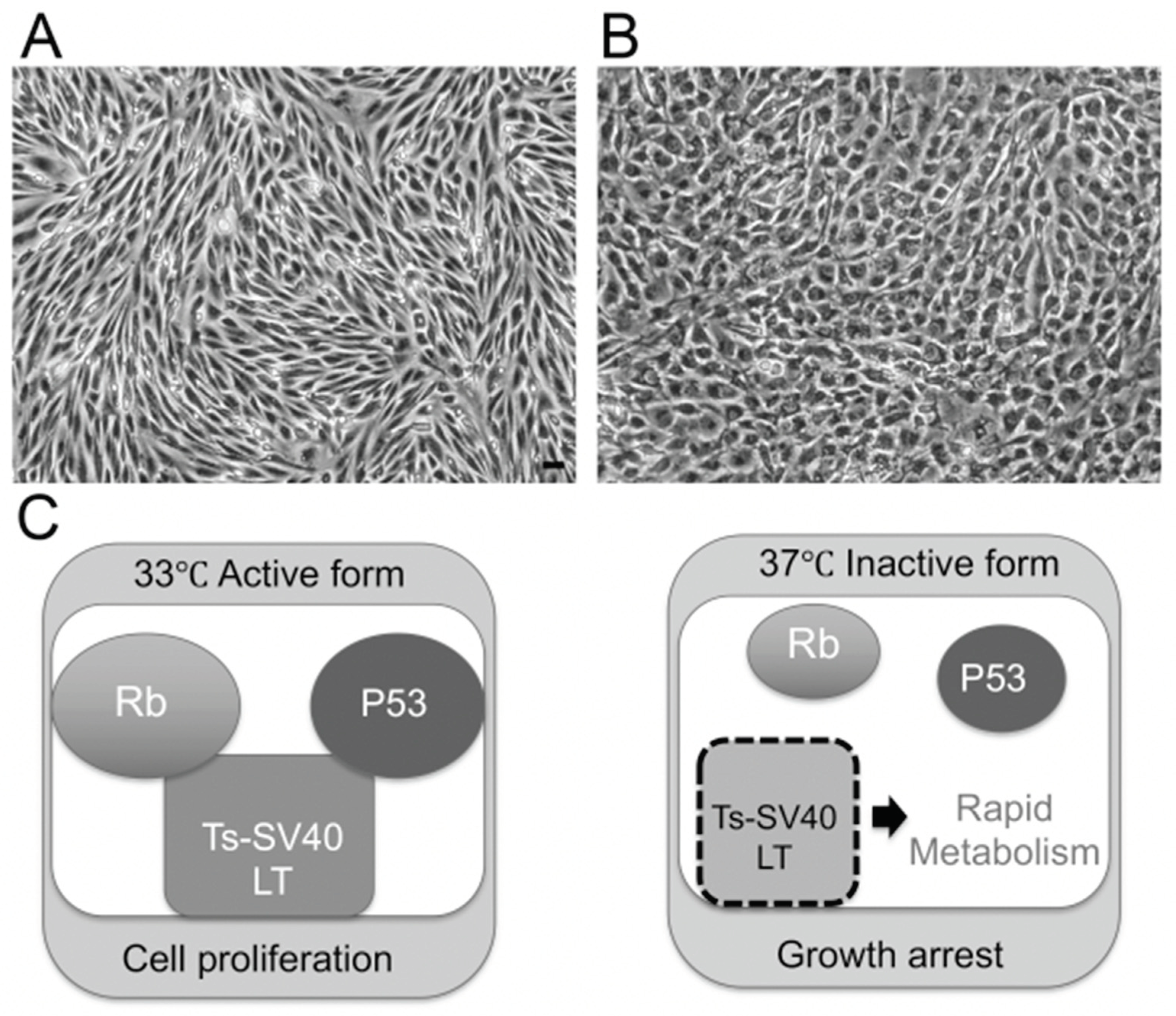
2.1.1. Establishment of BNB-Forming Cell Lines
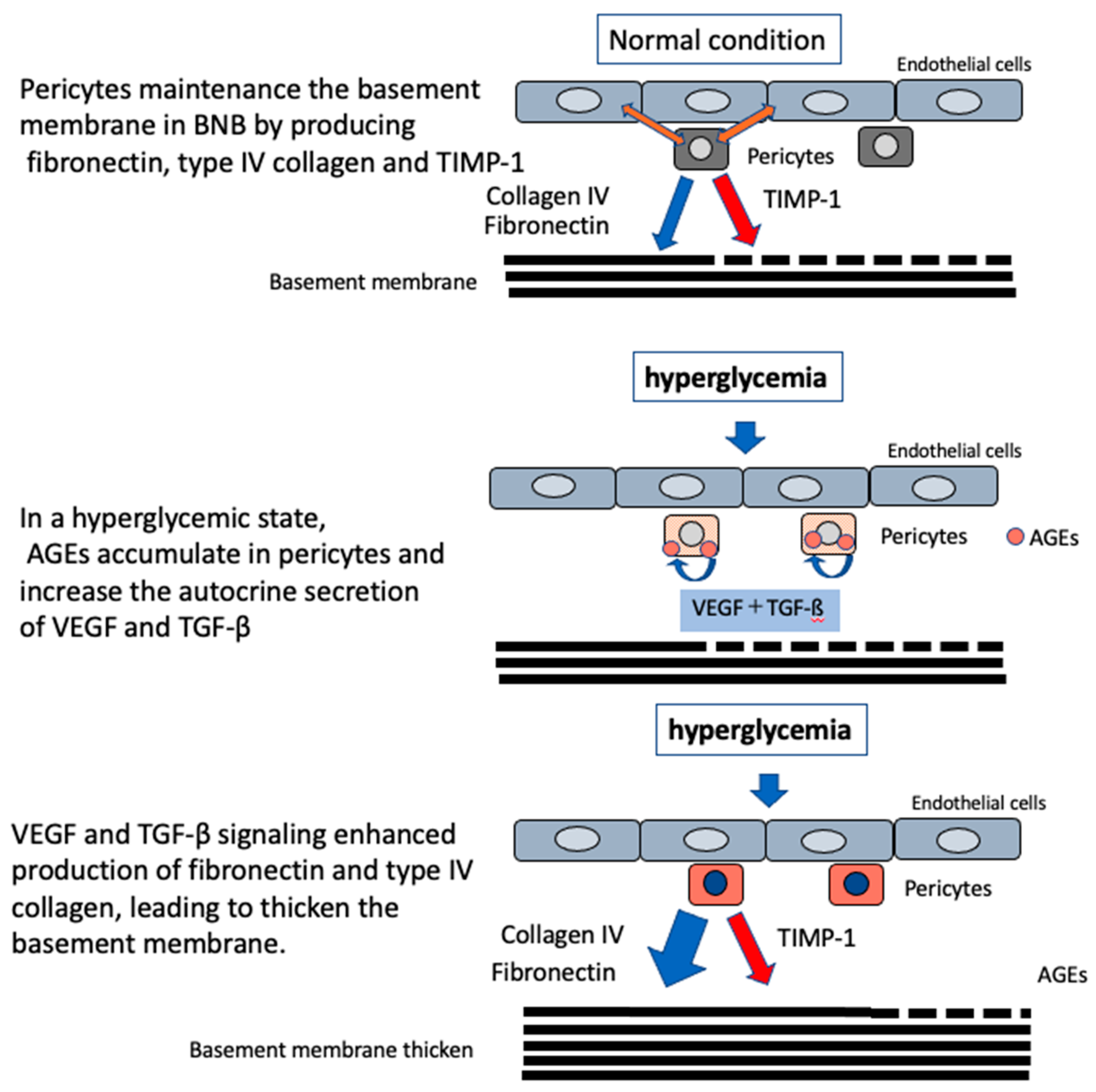
2.1.2. Function of Pericytes in the BNB
2.1.3. Construction of a New In Vitro BNB Model
3. BNB Disruption
3.1. Impaired Barrier Function Owing to Advanced Glycation End Products (AGEs)
3.2. Microvascular Basement Membrane Thickening in DPN
3.3. Induction of Local Inflammation
3.4. Disorders Due to Poor Circulation and Hypoxia
4. The Association between Disruption of Schwann Cells and BNB
5. Etiology of Conditions Other than Hyperglycemia
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AGEs | Advanced glycation end products |
| BBB | Blood–brain barrier |
| BNB | Blood–nerve barrier |
| DPN | Diabetic peripheral neuropathy |
| HIF-1 | Hypoxia inducible factor-1 |
| IgG | Immunoglobulin G |
| ROS | Reactive oxygen species |
| VEGFs | Vascular endothelial growth factors |
References
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. IDF Diabetes Atlas Committee. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef]
- Peltier, A.; Goutman, A.S.; Callaghan, B.C. Painful diabetic neuropathy. BMJ 2014, 348, 1799. [Google Scholar] [CrossRef]
- Harati, Y. Diabetic neuropathies: unanswered questions. Neurol. Clin. 2007, 25, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Ho, E.; Lam, K.; Chung, K.S. Contribution of polyol pathway to diabetes-induced oxidative stress. J. Am. Soc. Nephrol. 2003, 14, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Giri, B.; Dey, S.; Das, T.; Sarkar, M.; Banerjee, J.; Dash, S.K. Chronic hyperglycemia mediated physiological alteration and metabolic distortion leads to organ dysfunction infection cancer progression and other pathophysiological consequences: An update on glucose toxicity. Biomed. Pharmacother. 2018, 107, 306–328. [Google Scholar] [CrossRef] [PubMed]
- Kanda, T. Biology of the blood-nerve barrier and its alteration in immune mediated neuropathies. J. Neurol. Neurosurg. Psychiatry 2013, 84, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Abe, M.; Sano, Y.; Maeda, T.; Shimizu, F.; Kashiwamura, Y.; Haruki, H.; Saito, K.; Tasaki, A.; Kawai, M.; Terasaki, T.; et al. Establishment and characterization of humanperipheral nerve microvascular endothelialcell lines: A new in vitro blood-nerve barrier (BNB) model. Cell. Struct. Funct. 2012, 37, 89–100. [Google Scholar] [CrossRef]
- Poduslo, J.F.; Curran, G.L.; Berg, C.T. Macromolecular permeability across the blood-nerve and blood-brain barriers. Proc. Natl. Acad. Sci. USA 1994, 91, 5705–5709. [Google Scholar] [CrossRef]
- Ubogu, E.E. The molecular and biophysical characterization of the human blood-nerve barrier: Current concepts. J. Vasc. Res. 2013, 50, 289–303. [Google Scholar] [CrossRef]
- Cardoso, F.L.; Brites, D.; Brito, M.A. Looking at the blood-brain barrier: molecular anatomy and possible investigation approaches. Brain Res. Rev. 2010, 64, 328–363. [Google Scholar] [CrossRef]
- Takeshita, Y.; Omoto, M.; Fujikawa, S.; Kanda, T. Immunohistochemical analysis of laminin components in the blood–nerve barrier and blood–brain barrier. Clin. Exp. Neuroimmunol. 2017, 8, 49–53. [Google Scholar] [CrossRef][Green Version]
- Wu, C.; Ivans, F.; Anderson, P.; Hallmann, R.; Vestweber, D.; Nilsson, P.; Robenek, H.; Tryggvason, K.; Song, J.; Korpos, E.; et al. Endothelial basement membrane laminin alpha5 selectively inhibits T lymphocyte extravasation into the brain. Nat. Med. 2009, 15, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Kangwantas, K.; Pinteaux, E.; Penny, J. The extracellular matrix protein laminin-10 promotes blood-brain barrier repair after hypoxia and inflammation in vitro. J. Neuroinflamm. 2016, 13, 25. [Google Scholar] [CrossRef] [PubMed]
- Stierli, S.; Imperatore, V.; Lloyd, A.C. Schwann cell plasticity-roles in tissue homeostasis regeneration and disease. Glia 2019, 67, 2203–2215. [Google Scholar] [CrossRef]
- Sano, Y.; Shimizu, F.; Nakayama, H.; Abe, M.; Maeda, T.; Ohtsuki, S.; Terasaki, T.; Obinata, M.; Ueda, M.; Takahashi, R.; et al. Endothelial cells constituting blood-nerve barrier have highly specialized characteristics as barrier-forming cells. Cell Struct. Funct. 2007, 32, 139–147. [Google Scholar] [CrossRef]
- Shimizu, F.; Sano, Y.; Maeda, T.; Abe, M.; Nakayama, H.; Takahashi, R.; Ueda, M.; Ohtsuki, S.; Terasaki, T.; Obinata, M.; et al. Peripheral nerve pericytes originating fromthe blood-nerve barrier expresses tightjunctional molecules and transporters asbarrier-forming cells. J. Cell Physiol. 2008, 217, 388–399. [Google Scholar] [CrossRef]
- Shimizu, F.; Sano, Y.; Abe, M.; Maeda, T.; Ohtsui, S.; Terasaki, T.; Kanda, T. Peripheral nerve pericytes modify the blood-nerve barrier function and tight junctional molecules through the secretion of various soluble factors. J. Cell Physiol. 2011, 226, 255–266. [Google Scholar] [CrossRef]
- Yosef, N.; Xia, R.H.; Ubogu, E.E. Developmentand characterization of a novel human invitro blood-nerve barrier model usingprimary endoneurial endothelial cells. J. Neuropathol. Exp. Neurol. 2010, 69, 82–97. [Google Scholar] [CrossRef]
- Yosef, N.; Ubogu, E.E. An immortalized humanblood-nerve barrier endothelial cell line for invitro permeability studies. Cell Mol. Neurobiol. 2013, 33, 175–186. [Google Scholar] [CrossRef]
- Takeshita, Y.R.; Ansohoff, R.M. Inflammatorycell trafficking across the blood-brainbarrier: Chemokine regulation and in vitromodels. Immunol. Rev. 2012, 248, 228–239. [Google Scholar] [CrossRef]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at theblood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, P.; Johansson, B.R.; Levéen, P.; Betsholtz, C. Pericyte loss and microaneurysmformation in PDGF-B-deficient mice. Science 1997, 277, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the bloodbrain barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef]
- Shimizu, F.; Sano, Y.; Saito, K.; Abe, M.; Maeda, T.; Haruki, H.; Kanda, T. Pericyte-derived glial cellline-derived neurotrophic factor increase theexpression of claudin-5 in the blood-brainbarrier and the blood-nerve barrier. Neurochem. Res. 2012, 37, 401–409. [Google Scholar] [CrossRef]
- Rechthand, E.; Smith, Q.R.; Latker, C.H.; Rapoport, S.I. Altered blood-nerve barrier permeability to small molecules in experimental diabetes mellitus. J. Neuropathol. Exp. Neurol. 1987, 46, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Poduslo, J.F.; Curran, G.L.; Dyck, P.J. Increase in albumin IgG and IgM blood-nerve barrier indices in human diabetic neuropathy. Proc. Natl. Acad. Sci. USA 1988, 85, 4879–4883. [Google Scholar] [CrossRef] [PubMed]
- Mizisin, A.P.; Weerasuriya, A. Homeostatic regulation of the endoneurial microenvironment during development aging and in response to trauma disease and toxic insult. Acta Neuropathol. 2011, 121, 291–312. [Google Scholar] [CrossRef]
- Koschinsky, T.; He, C.J.; Mitsuhashi, T.; Bucala, R.; Liu, C.; Buenting, C.; Heitmann, H.; Vlassara, H. Orally absorbed reactive glycation products (glycotoxins): An environmental risk factorin diabetic nephropathy. Proc. Natl. Acad. Sci. USA 1997, 94, 6474–6479. [Google Scholar] [CrossRef]
- Shimizu, F.; Sano, Y.; Kanda, T. Advancedglycation end-products disrupt the bloodbrain barrier by stimulating the release ofTGF-β by pericytes and VEGF and MMP-2by endothelial cells in vitro. Neurobiol. Aging. 2013, 34, 1902–1912. [Google Scholar] [CrossRef]
- Wada, R.; Yagihashi, S. Role of advancedglycation end products and their receptors indevelopment of diabetic neuropathy. Ann. N. Y. Acad. Sci. 2005, 1043, 598–604. [Google Scholar] [CrossRef]
- Tsilibary, E.C. Microvascular basementmembranes in diabetes mellitus. J. Pathol. 2003, 200, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Canning, P.; Glenn, J.V.; Hsu, D.K.; Liu, F.T.; Gardiner, A.T.; Stitt, W.A. Inhibition of advanced glycation and absence ofgalectin-3 prevent blood-retinal barrierdysfunction during short-term diabetes. Exp. Diabetes Res. 2007, 2007, 51837. [Google Scholar] [CrossRef] [PubMed]
- Giannini, G.; Giannini, C.; Dyck, P.J. Basement membrane reduplication andpericyte degeneration precede developmentof diabetic polyneuropathy and are associatedwith its severity. Ann. Neurol. 1995, 37, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Vinik, A.I.; Mehrabyan, A. Diabeticneuropathies. Med. Clin. N. Am. 2004, 88, 947–999. [Google Scholar] [CrossRef]
- Richner, M.; Ferreira, N.; Dudele, A.; Jensen, S.T.; Vaegter, B.C.; Gonçalves, P.N. Functional and Structural Changes of the Blood-Nerve-Barrier in Diabetic Neuropathy. Front Neurosci. 2019, 12, 1038. [Google Scholar] [CrossRef]
- Shimizu, F.; Haruki, T.; Kanda, T. Advanced glycation end-products induce basement membrane hypertrophy in endoneurial microvessels and disrupt the blood-nerve barrier by stimulating the release of TGF-β and vascular endothelial growth factor (VEGF) by pericytes. Diabetologia 2011, 54, 1517–1526. [Google Scholar] [CrossRef]
- Rhee, S.Y.; Kim, S.Y. The Role of Advanced Glycation End Products in Diabetic Vascular Complications. Diabetes Metab. J. 2018, 42, 188–195. [Google Scholar]
- Talbot, S.; Chahmi, E.; Dias, J.P.; Couture, R. Key role for spinal dorsal horn microglial kinin B1 receptor in early diabetic pain neuropathy. J. Neuroinflamm. 2010, 7, 36. [Google Scholar] [CrossRef]
- O’Brien, P.D.; Hur, J.; Hayes, M.J.; Backus, C.; Sakowski, A.S.; Feldman, L.E. BTBR ob/ob mice as a novel diabetic neuropathy model: Neurological characterization and gene expression analyses. Neurobiol. Dis. 2015, 73, 348–355. [Google Scholar] [CrossRef]
- Hinder, L.M.; Murdock, B.J.; Park, M.; Bender, E.D.; O’Brien, P.D.; Rumora, E.A.; Hur, J.; Feldman, L.E. Transcriptional networks of progressive diabetic peripheral neuropathy in the db/db mouse model of type 2 diabetes: An inflammatory story. Exp. Neurol. 2018, 305, 33–43. [Google Scholar] [CrossRef]
- Nguyen, D.V.; Shaw, C.L.; Grant, B.M. Inflammation in the pathogenesis of microvascular complications in diabetes. Front. Endocrinol. 2012, 3, 170. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Meng, Q.; Ji, J.; Lou, X.; Zhang, L. Toll-like receptor 4 and tumor necrosis factor-alpha as diagnostic biomarkers for diabetic peripheral neuropathy. Neurosci. Lett. 2015, 585, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Kan, H.; Hsieh, J.-H.; Chien, H.-F.; Lin, Y.-H.; Yeh, T.-Y.; Chao, C.-C.; Hsieh, S.-T. CD40-mediated HIF-1α expression underlying microangiopathy in diabetic nerve pathology. Dis. Model. Mech. 2018, 11, dmm033647. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, P.N.; Jager, E.S.; Richner, M.; Murray, S.S.; Mohseni, S.; Jensen, S.T.; Vaegter, B.C. Schwann cell p75 neurotrophin receptor modulates small fiber degeneration in diabetic neuropathy. Glia 2020, 68, 2725–2743. [Google Scholar] [CrossRef] [PubMed]
- Dyck, P.J.; Karnes, L.J.; Okazaki, H.; Lais, A.; Engelstad, J. The spatial distribution of fiber loss in diabetic polyneuropathy suggests ischemia. Ann. Neurol. 1986, 19, 440–449. [Google Scholar]
- Tuck, R.R.; Schmelzer, J.D.; Low, A.P. Endoneurial blood flow and oxygen tension in the sciatic nerves of rats with experimental diabetic neuropathy. Brain 1984, 107, 935–950. [Google Scholar] [CrossRef] [PubMed]
- Newrick, P.G.; Wilson, J.A.; Jakubowski, J.; Boulton, J.A.; Ward, D.J. Sural nerve oxygen tension in diabetes. Br. Med. J. 1986, 293, 1053–1054. [Google Scholar] [CrossRef]
- Cameron, N.E.; Cotter, A.M.; Dines, C.K.; Maxfield, K.E.; Carey, F.; Mirrless, J.D. Aldose reductase inhibition nerve perfusion oxygenation and function in streptozotocin-diabetic rats: Dose-response considerations and independence from a myo-inositol mechanism. Diabetologia 1994, 37, 651–663. [Google Scholar] [CrossRef]
- Ibrahim, S.; Harris, D.N.; Radatz, M.; Seimi, F.; Rajbhandari, S.; Brady, L.; Jakubowski, J.; Ward, D.J. A new minimally invasive technique to show nerve ischaemia in diabetic neuropathy. Diabetologia 1999, 42, 737–742. [Google Scholar] [CrossRef]
- Lim, T.; Shi, X.; Johnson, M.J.; Rone, B.M.; Antel, P.J.; David, S.; Zhang, J. Peripheral nerve injury induces persistent vascular dysfunction and endoneurial hypoxia contributing to the genesis of neuropathic pain. J. Neurosci. 2015, 35, 3346–3359. [Google Scholar] [CrossRef]
- Xiong, M.; Elson, G.; Legarda, D.; Leibovich, J.S. Production of vascular endothelial growth factor by murine macrophages: Regulation by hypoxia lactate and the inducible nitric oxide synthase pathway. Am. J. Pathol. 1998, 153, 587–598. [Google Scholar] [CrossRef]
- Lokmic, Z.; Musyoka, J.; Hewitson, D.T.; Darby, A.I. Hypoxia and hypoxia signaling in tissue repair and fibrosis. Int. Rev. Cell Mol. Biol. 2012, 296, 139–185. [Google Scholar] [PubMed]
- Park, K.K.; Liu, K.; Hu, Y.; Kanter, L.J.; He, Z. PTEN/mTOR and axon regeneration. Exp. Neurol. 2010, 223, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Singh, V.; Krishnan, A.; Koshy, K.; Martinez, A.J.; Cheng, C.; Almquist, C.; Zochodne, W.D. Regeneration of diabetic axons is enhanced by selective knockdown of the PTEN gene. Brain 2014, 137, 1051–1067. [Google Scholar] [CrossRef] [PubMed]
- Yuan, G.; Khan, A.S.; Luo, W.; Nanduri, J.; Semenza, L.G.; Prabhakar, R.N. Hypoxia-inducible factor 1 mediates increased expression of NADPH oxidase-2 in response to intermittent hypoxia. J. Cell Physiol. 2011, 226, 2925–2933. [Google Scholar] [CrossRef]
- Gonçalves, N.P.; Vægter, B.C.; Andersen, H.; Østergaard, L.; Calcutt, A.N.; Jensen, S.T. Schwann cell interactions with axons and microvessels in diabetic neuropathy. Nat. Rev. Neurol. 2017, 13, 135–147. [Google Scholar] [CrossRef]
- Pun, P.B.; Lu, J.; Moochhala, S. Involvement of ROS in BBB dysfunction. Free Radic. Res. 2009, 43, 348–364. [Google Scholar] [CrossRef]
- Ng, T.F.; Lee, K.F.; Song, T.Z.; Calcutt, A.N.; Lee, Y.A.; Chung, S.; Chung, S.K. Effects of sorbitol dehydrogenase deficiency on nerve conduction in experimental diabetic mice. Diabetes 1998, 47, 961–966. [Google Scholar] [CrossRef]
- Song, Z.; Fu, W.T.D.; Chan, Y.-S.; Leung, S.; Chung, M.S.S.; Chung, K.S. Transgenic mice overexpressing aldose reductase in Schwann cells show more severe nerve conduction velocity deficit and oxidative stress under hyperglycemic stress. Mol. Cell. Neurosci. 2003, 23, 638–647. [Google Scholar] [CrossRef]
- Uehara, K.; Yamagishi, S.; Otsuki, S.; Chin, S.; Yagihashi, S. Effects of polyol pathway hyperactivity on protein kinase C activity nociceptive peptide expression and neuronal structure in dorsal root ganglia in diabetic mice. Diabetes 2004, 53, 3239–3247. [Google Scholar] [CrossRef]
- Viader, A.; Sasaki, Y.; Kim, S.; Strickland, A.; Workman, S.C.; Yang, K.; Gross, W.R.; Milbrandt, J. Aberrant Schwann cell lipid metabolism linked to mitochondrial deficits leads to axon degeneration and neuropathy. Neuron 2013, 77, 886–898. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Lv, Q.; Chen, X.; Zou, J.; Liu, Z.; Shi, Y. CD8(+) T cell-mediated cytotoxicity toward Schwann cells promotes diabetic peripheral neuropathy. Cell. Physiol. Biochem. 2013, 32, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Tesfaye, S.; Stevens, K.L.; Stephenson, M.J.; Fuller, H.J.; Plater, M.; Ionescu-Tirgoviste, C.; Nuber, A.; Pozza, G.; Ward, D.J. Prevalence of diabetic peripheral neuropathy and its relation to glycaemic control and potential risk factors: The EURODIAB IDDM Complications Study. Diabetologia 1996, 39, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- Tesfaye, S.; Chaturvedi, N.; Eaton, M.E.S.; Ward, D.J.; Manes, C.; Ionescu-Tirgoviste, C.; Witte, R.D.; Fuller, H.J. EURODIAB Prospective Complications Study Group. Vascular risk factors and diabetic neuropathy. N. Engl. J. Med. 2005, 352, 341–350. [Google Scholar] [CrossRef] [PubMed]
- The Diabetes Control and Complications Trial Research Group. N. Engl. J. Med. 1993, 329, 977–986.
- Linn, T.; Ortac, K.; Laube, H.; Federlin, K. Intensive therapy in adult insulin-dependent diabetes mellitus is associated with improved insulin sensitivity and reserve: a randomized controlled prospective study over 5 years in newly diagnosed patients. Metabolism 1996, 45, 1508–1513. [Google Scholar] [CrossRef]
- Duckworth, W.; Abraira, C.; Moritz, T.; Reda, D.; Emanuele, N.; Reaven, D.P.; Zieve, J.F.; Marks, J.; Davis, N.S.; Hayward, R.; et al. Glucose control and vascular complications in veterans with type 2 diabetes. N. Engl. J. Med. 2009, 360, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Ismail-Beigi, F.; Craven, T.; Banerji, A.M.; Basile, J.; Calles, J.; Cohen, M.R.; Cuddihy, R.; Cushman, C.W.; Genuth, S.; Grimm, H.R.; et al. Effect of intensive treatment of hyperglycaemia on microvascular outcomes in type 2 diabetes: An analysis of the ACCORD randomised trial. Lancet 2010, 376, 419–430. [Google Scholar] [CrossRef]
- Callaghan, B.C.; Cheng, T.H.; Stables, L.C.; Smith, L.A.; Feldman, L.E. Diabetic neuropathy: Clinical manifestations and current treatments. Lancet Neurol. 2012, 11, 521–534. [Google Scholar] [CrossRef]
- Ang, L.; Jaiswal, M.; Martin, C.; Pop-Busui, R. Glucose control and diabetic neuropathy: Lessons from recent large clinical trials. Curr. Diabetes Rep. 2014, 14, 528. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takeshita, Y.; Sato, R.; Kanda, T. Blood–Nerve Barrier (BNB) Pathology in Diabetic Peripheral Neuropathy and In Vitro Human BNB Model. Int. J. Mol. Sci. 2021, 22, 62. https://doi.org/10.3390/ijms22010062
Takeshita Y, Sato R, Kanda T. Blood–Nerve Barrier (BNB) Pathology in Diabetic Peripheral Neuropathy and In Vitro Human BNB Model. International Journal of Molecular Sciences. 2021; 22(1):62. https://doi.org/10.3390/ijms22010062
Chicago/Turabian StyleTakeshita, Yukio, Ryota Sato, and Takashi Kanda. 2021. "Blood–Nerve Barrier (BNB) Pathology in Diabetic Peripheral Neuropathy and In Vitro Human BNB Model" International Journal of Molecular Sciences 22, no. 1: 62. https://doi.org/10.3390/ijms22010062
APA StyleTakeshita, Y., Sato, R., & Kanda, T. (2021). Blood–Nerve Barrier (BNB) Pathology in Diabetic Peripheral Neuropathy and In Vitro Human BNB Model. International Journal of Molecular Sciences, 22(1), 62. https://doi.org/10.3390/ijms22010062