Depletion of Mitochondrial Components from Extracellular Vesicles Secreted from Astrocytes in a Mouse Model of Fragile X Syndrome

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
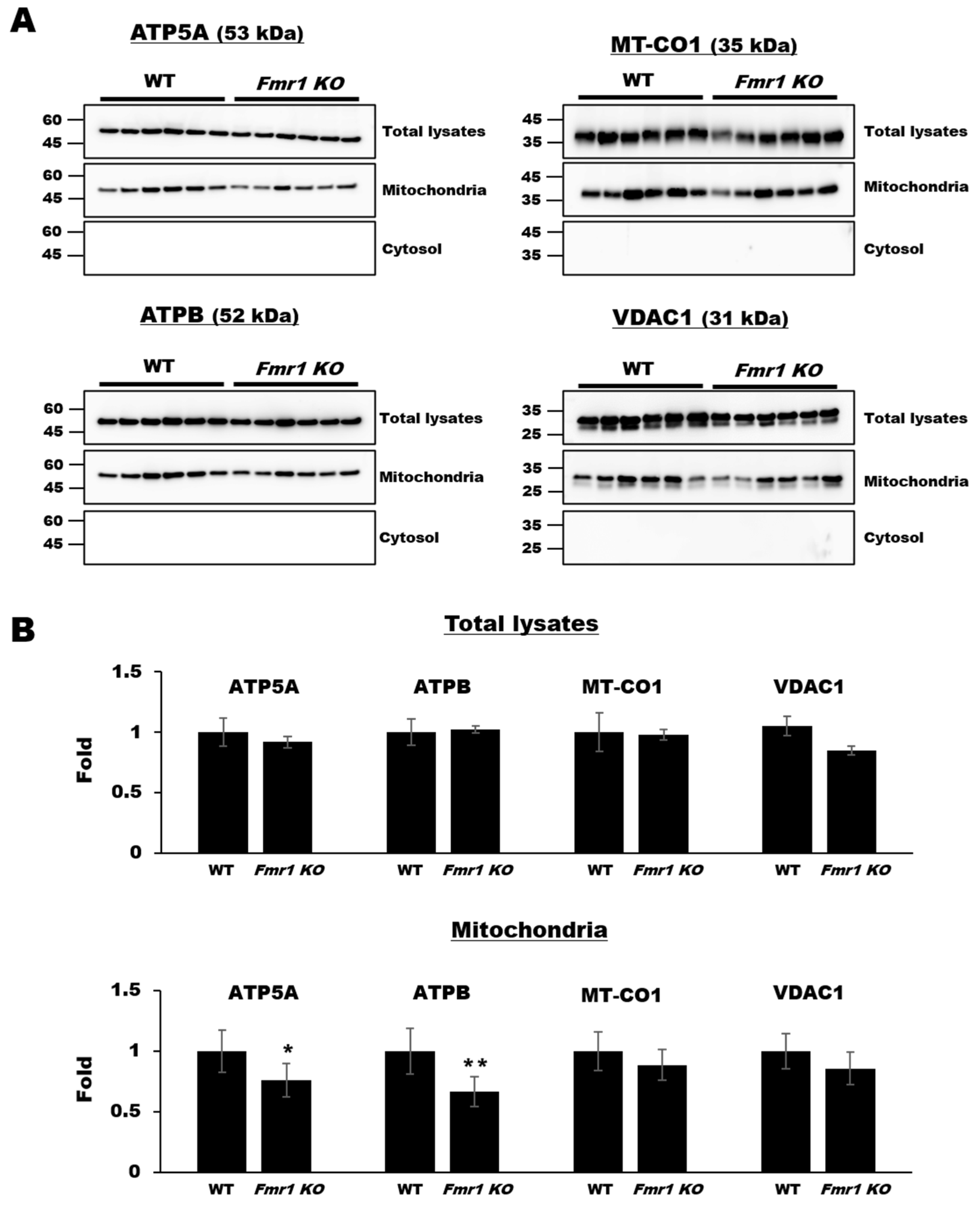
2.1. Decreased Expression of Mitochondrial Components in Mitochondrial Fractions but Not Cortical Lysates from Fmr1 KO Mice
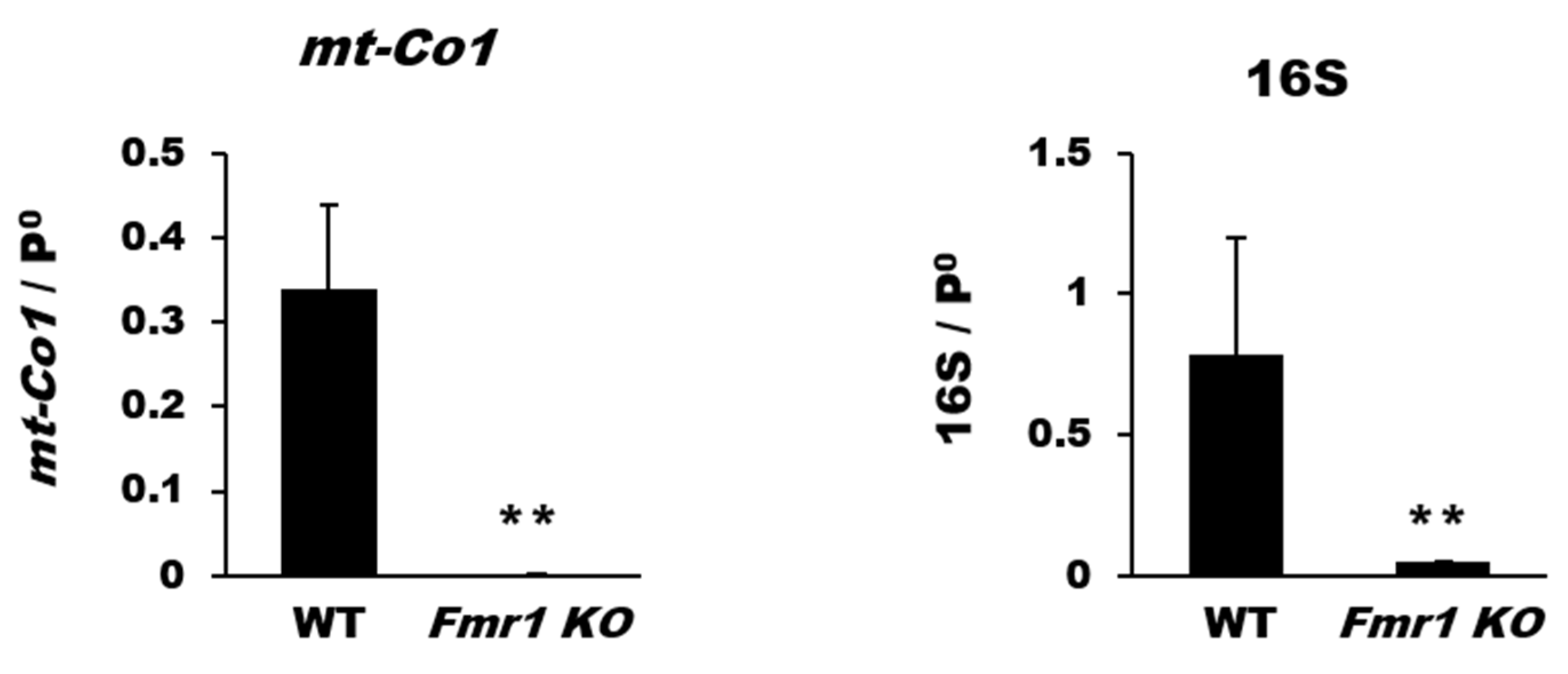
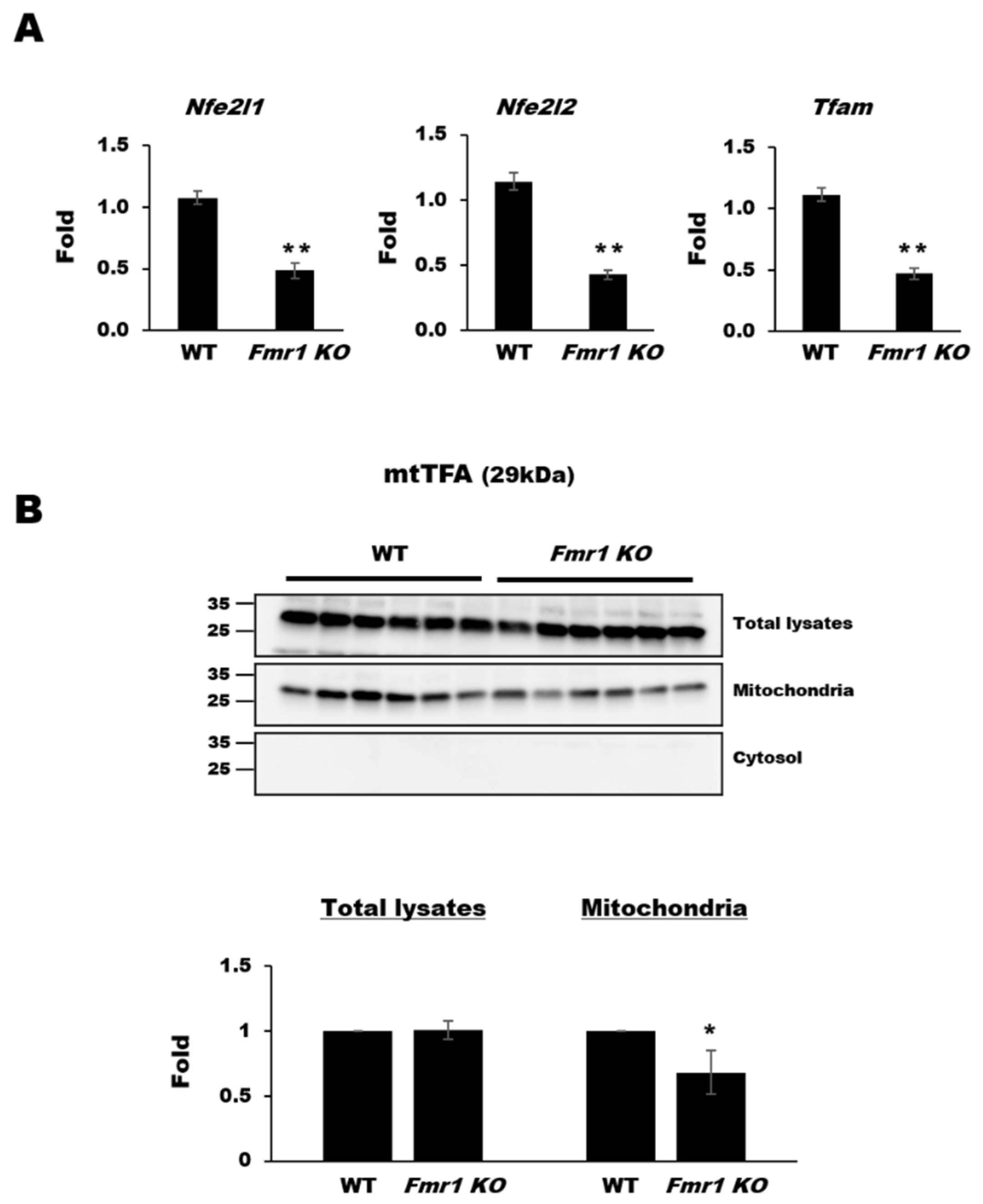
2.2. Reduction in Mitochondrial Biogenesis and Transcriptional Activity in Cortices of Fmr1 KO Mice
2.3. Reduced Levels of Mitochondrial Proteins in Mitochondrial Fractions from Astrocytes of Fmr1 KO Mice
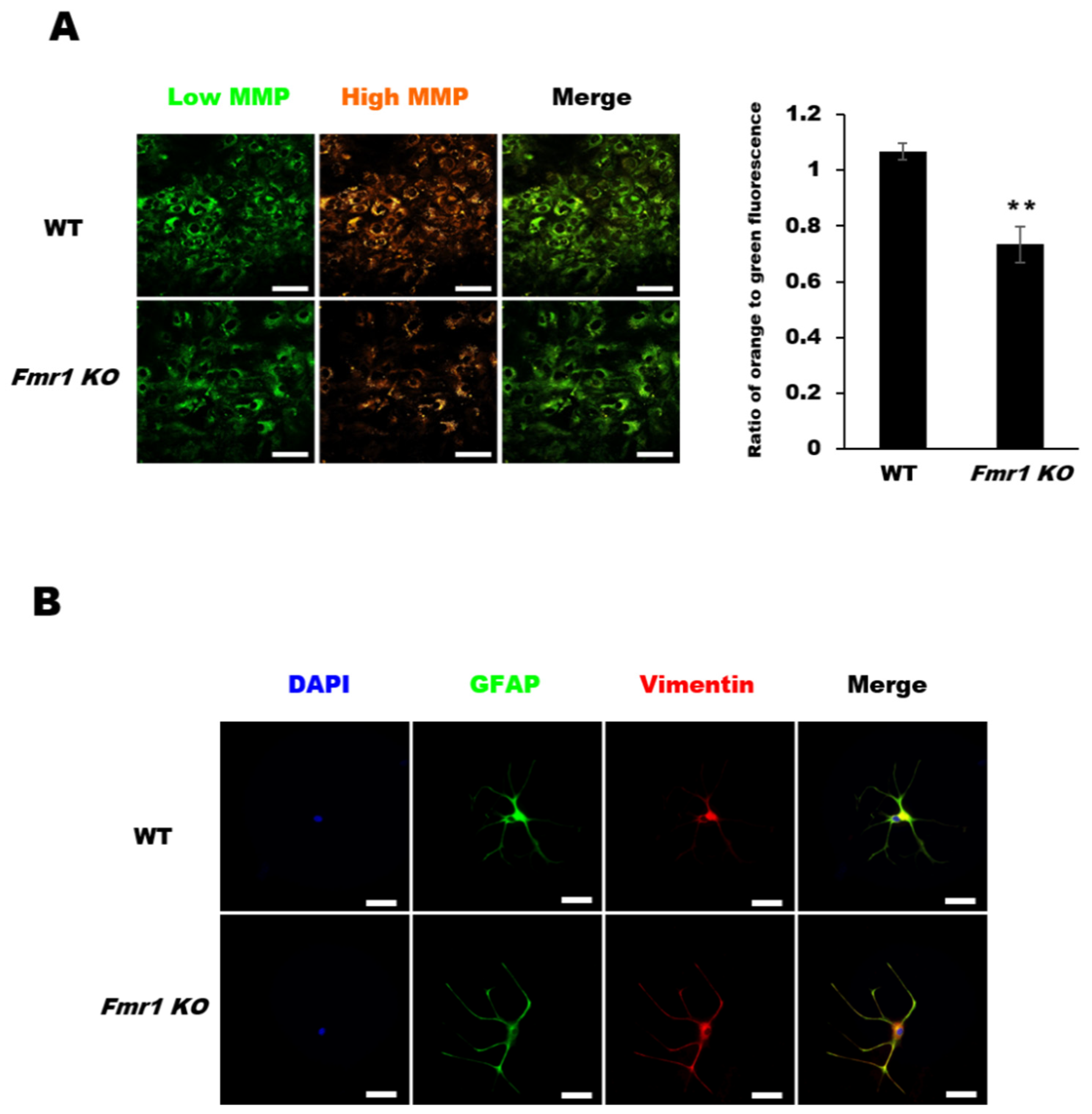
2.4. Decreased Mitochondrial Membrane Potential (MMP) in Astrocytes from Fmr1 KO Mice
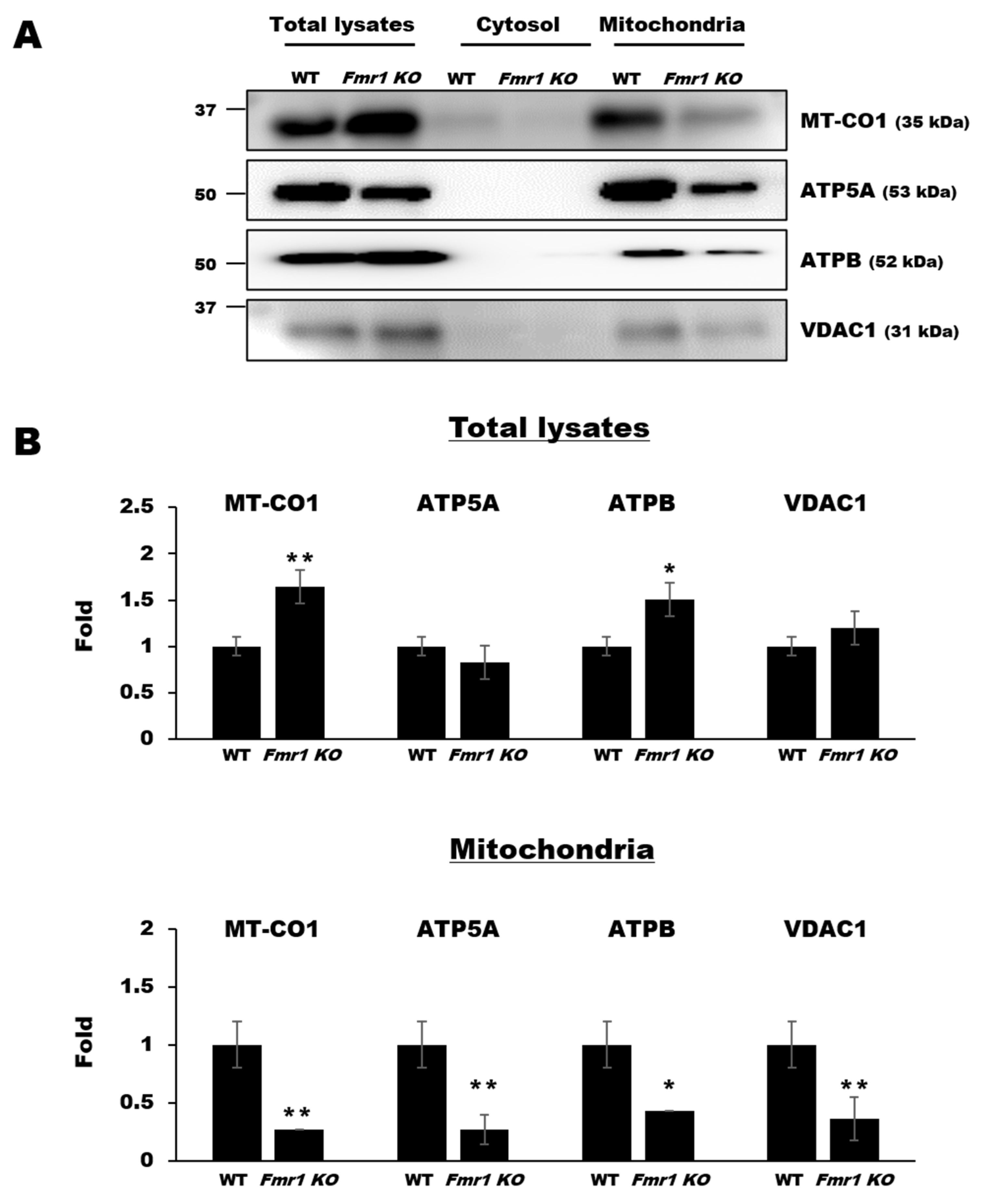
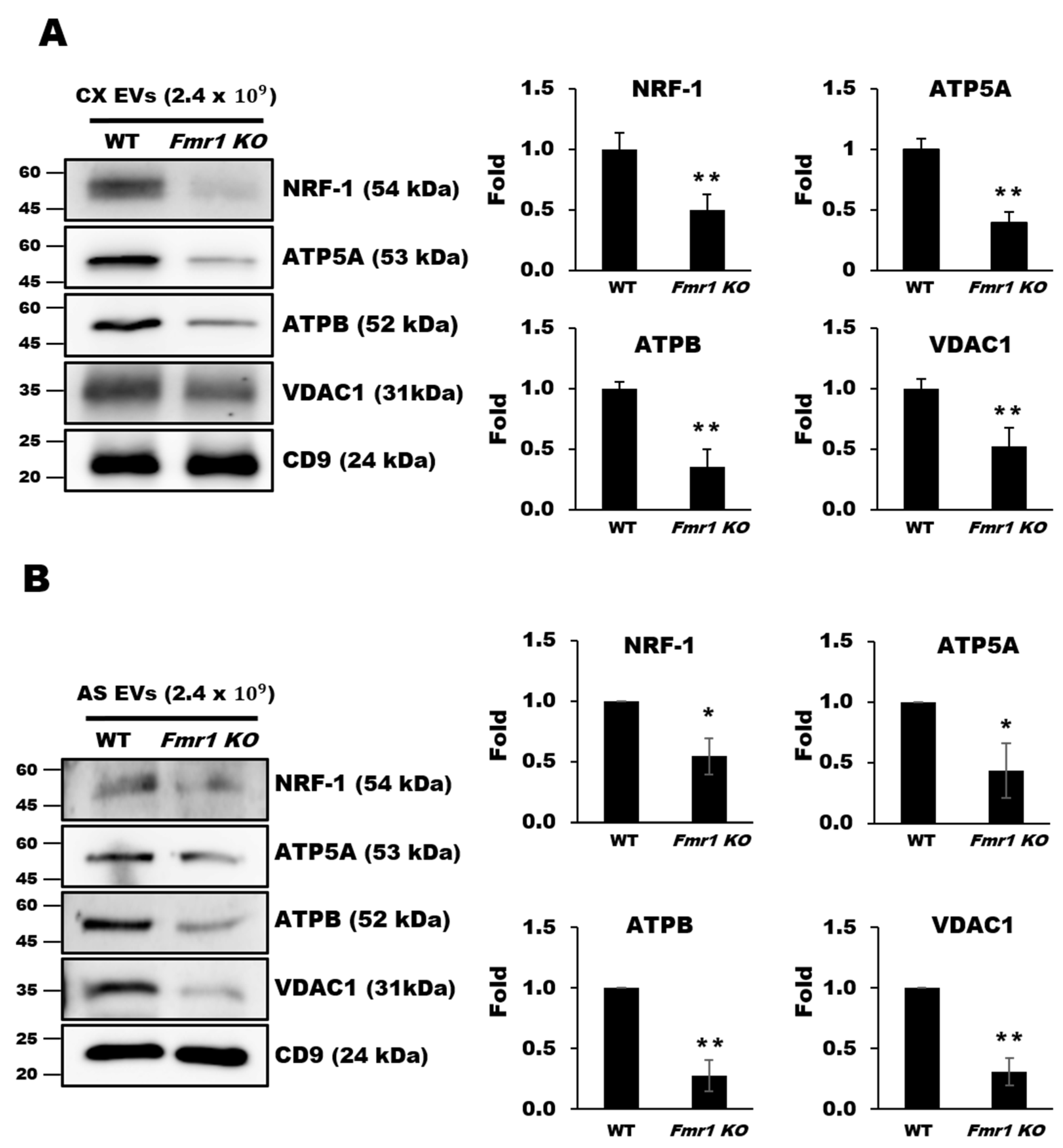
2.5. Mitochondrial Components are Depleted in EVs Secreted by Cortices and Astrocytes of Fmr1 KO Mice
3. Discussion
4. Materials and Methods
4.1. Animal Maintenance
4.2. Primary Astrocyte Cultures
4.3. qRT-PCR Analysis
4.4. Mitochondria Fractionation
4.5. EVs Isolation and Purification
4.6. Nanoparticle Tracking Analysis (NTA)
4.7. Transmission Electron Microscopy (TEM)
4.8. Western Blot Analysis
4.9. Measurement of MMP
4.10. Immunocytochemistry
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ALS | Amyotrophic lateral sclerosis |
| ASD | Autism spectrum disorders |
| ATP5A | ATP synthase alpha-subunit |
| ATPB | ATP synthase beta-subunit |
| CD9 | Cluster of Differentiation 9 |
| CD81 | Cluster of Differentiation 81 |
| COX | Cytochrome c oxidase subunit |
| ΔΔCT | Delta cycle threshold |
| DIV | Days in vitro |
| EVs | Extracellular vesicles |
| FMRP | Fragile X mental retardation protein |
| Fmr1 KO | Fmr1 knockout (Fmr1-/y) |
| FXS | Fragile X syndrome |
| GFAP | Glial fibrillary acidic protein |
| JC-1 | Tetraethyl benzimidazolyl carbocyanine iodide |
| LC/MS | Liquid chromatography-mass spectrometry |
| MMP | Mitochondrial membrane potential |
| MT-CO1 | COX subunit I encoded by mitochondrial DNA |
| mtTFA | Mitochondrial transcription factor A |
| NTA | Nanoparticle tracking analysis |
| NRF-1 | Nuclear factor, erythroid derived 2, like 1 |
| NRF-2 | Nuclear factor, erythroid derived 2, like 2 |
| P2 | Postnatal days 2 |
| PBS | Phosphate buffered saline |
| PD | Parkinson’s disease |
| PINK1 | PTEN-induced kinase 1 |
| RPLP0 | Nuclear gene ribosomal protein large p0 |
| TEM | Transmission electron microscopy |
| VDAC1 | Voltage-dependent anion-selective channel 1 |
| VIF | Vimentin intermediate filament |
| WT | Wild-type (Fmr1+/y) |
References
- Saldarriaga, W.; Tassone, F.; Gonzalez-Teshima, L.Y.; Forero-Forero, J.V.; Ayala-Zapata, S.; Hagerman, R. Fragile X syndrome. Colomb. Med. 2014, 45, 190–198. [Google Scholar] [CrossRef]
- Brown, V.; Jin, P.; Ceman, S.; Darnell, J.C.; O’Donnell, W.T.; Tenenbaum, S.A.; Jin, X.; Feng, Y.; Wilkinson, K.D.; Keene, J.D.; et al. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell 2001, 107, 477–487. [Google Scholar] [CrossRef]
- Schon, E.A.; Przedborski, S. Mitochondria: The next (neurode)generation. Neuron 2011, 70, 1033–1053. [Google Scholar] [CrossRef] [PubMed]
- Qin, M.; Kang, J.; Smith, C.B. Increased rates of cerebral glucose metabolism in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. USA 2002, 99, 15758–15763. [Google Scholar] [CrossRef] [PubMed]
- Davidovic, L.; Navratil, V.; Bonaccorso, C.M.; Catania, M.V.; Bardoni, B.; Dumas, M.E. A metabolomic and systems biology perspective on the brain of the fragile X syndrome mouse model. Genome Res. 2011, 21, 2190–2202. [Google Scholar] [CrossRef] [PubMed]
- Lima-Cabello, E.; Garcia-Guirado, F.; Calvo-Medina, R.; el Bekay, R.; Perez-Costillas, L.; Quintero-Navarro, C.; Sanchez-Salido, L.; de Diego-Otero, Y. An Abnormal Nitric Oxide Metabolism Contributes to Brain Oxidative Stress in the Mouse Model for the Fragile X Syndrome, a Possible Role in Intellectual Disability. Oxidative Med. Cell. Longev. 2016, 2016, 8548910. [Google Scholar] [CrossRef] [PubMed]
- el Bekay, R.; Romero-Zerbo, Y.; Decara, J.; Sanchez-Salido, L.; Del Arco-Herrera, I.; Rodriguez-de Fonseca, F.; de Diego-Otero, Y. Enhanced markers of oxidative stress, altered antioxidants and NADPH-oxidase activation in brains from Fragile X mental retardation 1-deficient mice, a pathological model for Fragile X syndrome. Eur. J. Neurosci. 2007, 26, 3169–3180. [Google Scholar] [CrossRef]
- Lumaban, J.G.; Nelson, D.L. The Fragile X proteins Fmrp and Fxr2p cooperate to regulate glucose metabolism in mice. Hum. Mol. Genet. 2015, 24, 2175–2184. [Google Scholar] [CrossRef]
- Shen, M.; Wang, F.; Li, M.; Sah, N.; Stockton, M.E.; Tidei, J.J.; Gao, Y.; Korabelnikov, T.; Kannan, S.; Vevea, J.D.; et al. Reduced mitochondrial fusion and Huntingtin levels contribute to impaired dendritic maturation and behavioral deficits in Fmr1-mutant mice. Nat. Neurosci. 2019, 22, 386–400. [Google Scholar] [CrossRef]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef]
- Dong, L.F.; Kovarova, J.; Bajzikova, M.; Bezawork-Geleta, A.; Svec, D.; Endaya, B.; Sachaphibulkij, K.; Coelho, A.R.; Sebkova, N.; Ruzickova, A.; et al. Horizontal transfer of whole mitochondria restores tumorigenic potential in mitochondrial DNA-deficient cancer cells. Elife 2017, 6, e22187. [Google Scholar] [CrossRef] [PubMed]
- Morrison, T.J.; Jackson, M.V.; Cunningham, E.K.; Kissenpfennig, A.; McAuley, D.F.; O’Kane, C.M.; Krasnodembskaya, A.D. Mesenchymal Stromal Cells Modulate Macrophages in Clinically Relevant Lung Injury Models by Extracellular Vesicle Mitochondrial Transfer. Am. J. Respir. Crit. Care Med. 2017, 196, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- Soto-Heredero, G.; Baixauli, F.; Mittelbrunn, M. Interorganelle Communication between Mitochondria and the Endolysosomal System. Front. Cell Dev. Biol. 2017, 5, 95. [Google Scholar] [CrossRef] [PubMed]
- Phinney, D.G.; Di Giuseppe, M.; Njah, J.; Sala, E.; Shiva, S.; St Croix, C.M.; Stolz, D.B.; Watkins, S.C.; Di, Y.P.; Leikauf, G.D.; et al. Mesenchymal stem cells use extracellular vesicles to outsource mitophagy and shuttle microRNAs. Nat. Commun. 2015, 6, 8472. [Google Scholar] [CrossRef]
- Jang, S.C.; Crescitelli, R.; Cvjetkovic, A.; Belgrano, V.; Olofsson Bagge, R.; Sundfeldt, K.; Ochiya, T.; Kalluri, R.; Lotvall, J. Mitochondrial protein enriched extracellular vesicles discovered in human melanoma tissues can be detected in patient plasma. J. Extracell. Vesicles 2019, 8, 1635420. [Google Scholar] [CrossRef]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell. Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef]
- Liu, S.; Hossinger, A.; Gobbels, S.; Vorberg, I.M. Prions on the run: How extracellular vesicles serve as delivery vehicles for self-templating protein aggregates. Prion 2017, 11, 98–112. [Google Scholar] [CrossRef]
- Eitan, E.; Hutchison, E.R.; Marosi, K.; Comotto, J.; Mustapic, M.; Nigam, S.M.; Suire, C.; Maharana, C.; Jicha, G.A.; Liu, D.; et al. Extracellular Vesicle-Associated Aβ Mediates Trans-Neuronal Bioenergetic and Ca2+-Handling Deficits in Alzheimer’s Disease Models. NPJ Aging Mech. Dis. 2016, 2, 16019. [Google Scholar] [CrossRef]
- Polanco, J.C.; Scicluna, B.J.; Hill, A.F.; Gotz, J. Extracellular Vesicles Isolated from the Brains of rTg4510 Mice Seed Tau Protein Aggregation in a Threshold-dependent Manner. J. Biol. Chem. 2016, 291, 12445–12466. [Google Scholar] [CrossRef]
- Gosselin, R.D.; Meylan, P.; Decosterd, I. Extracellular microvesicles from astrocytes contain functional glutamate transporters: Regulation by protein kinase C and cell activation. Front. Cell. Neurosci. 2013, 10, 251. [Google Scholar] [CrossRef]
- Wang, S.; Cesca, F.; Loers, G.; Schweizer, M.; Buck, F.; Benfenati, F.; Schachner, M.; Kleene, R. Synapsin I is an oligomannose-carrying glycoprotein, acts as an oligomannose-binding lectin, and promotes neurite outgrowth and neuronal survival when released via glia-derived exosomes. J. Neurosci. 2011, 31, 7275–7290. [Google Scholar] [CrossRef] [PubMed]
- Ignatenko, O.; Chilov, D.; Paetau, I.; de Miguel, E.; Jackson, C.B.; Capin, G.; Paetau, A.; Terzioglu, M.; Euro, L.; Suomalainen, A. Loss of mtDNA activates astrocytes and leads to spongiotic encephalopathy. Nat. Commun. 2018, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Brennan, K.; Martin, K.; FitzGerald, S.P.; O’Sullivan, J.; Wu, Y.; Blanco, A.; Richardson, C.; Mc Gee, M.M. A comparison of methods for the isolation and separation of extracellular vesicles from protein and lipid particles in human serum. Sci. Rep. 2020, 10, 1039. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Wang, T.; Wan, H.; Han, L.; Qin, X.; Zhang, Y.; Wang, J.; Yu, C.; Berton, F.; Francesconi, W.; et al. Fmr1 deficiency promotes age-dependent alterations in the cortical synaptic proteome. Proc. Natl. Acad. Sci. USA 2015, 112, E4697–E4706. [Google Scholar] [CrossRef]
- D’Antoni, S.; de Bari, L.; Valenti, D.; Borro, M.; Bonaccorso, C.M.; Simmaco, M.; Vacca, R.A.; Catania, M.V. Aberrant mitochondrial bioenergetics in the cerebral cortex of the Fmr1 knockout mouse model of fragile X syndrome. Biol. Chem. 2020, 401, 497–503. [Google Scholar] [CrossRef]
- Bechara, E.G.; Didiot, M.C.; Melko, M.; Davidovic, L.; Bensaid, M.; Martin, P.; Castets, M.; Pognonec, P.; Khandjian, E.W.; Moine, H.; et al. A novel function for fragile X mental retardation protein in translational activation. PLoS Biol. 2009, 7, e16. [Google Scholar] [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Javadov, S.; Grimm, M.; Margreiter, R.; Ausserlechner, M.J.; Hagenbuchner, J. Crosstalk between Mitochondria and Cytoskeleton in Cardiac Cells. Cells 2020, 9, 222. [Google Scholar] [CrossRef]
- Chernoivanenko, I.S.; Matveeva, E.A.; Gelfand, V.I.; Goldman, R.D.; Minin, A.A. Mitochondrial membrane potential is regulated by vimentin intermediate filaments. FASEB J. 2015, 29, 820–827. [Google Scholar] [CrossRef]
- Upadhya, R.; Zingg, W.; Shetty, S.; Shetty, A.K. Astrocyte-derived extracellular vesicles: Neuroreparative properties and role in the pathogenesis of neurodegenerative disorders. J. Control. Release 2020, 323, 225–239. [Google Scholar] [CrossRef]
- Moidunny, S.; Vinet, J.; Wesseling, J.; Bijzet, J.; Shieh, C.H.; van Ijzendoorn, S.C.; Bezzi, P.; Boddeke, H.W.; Biber, K. Adenosine A2B receptor-mediated leukemia inhibitory factor release from astrocytes protects cortical neurons against excitotoxicity. J. Neuroinflamm. 2012, 9, 198. [Google Scholar] [CrossRef] [PubMed]
- Schiera, G.; Di Liegro, C.M.; Di Liegro, I. Extracellular membrane vesicles as vehicles for brain cell-to-cell interactions in physiological as well as pathological conditions. Biomed. Res. Int. 2015, 2015, 152926. [Google Scholar] [CrossRef] [PubMed]
- Silverman, J.M.; Christy, D.; Shyu, C.C.; Moon, K.M.; Fernando, S.; Gidden, Z.; Cowan, C.M.; Ban, Y.; Stacey, R.G.; Grad, L.I.; et al. CNS-derived extracellular vesicles from superoxide dismutase 1 (SOD1)G93A ALS mice originate from astrocytes and neurons and carry misfolded SOD1. J. Biol. Chem. 2019, 294, 3744–3759. [Google Scholar] [CrossRef] [PubMed]
- Bylicky, M.A.; Mueller, G.P.; Day, R.M. Mechanisms of Endogenous Neuroprotective Effects of Astrocytes in Brain Injury. Oxid. Med. Cell. Longev. 2018, 2018, 6501031. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Shao, A.; Yao, Y.; Tu, S.; Deng, Y.; Zhang, J. Dual roles of astrocytes in plasticity and reconstruction after traumatic brain injury. Cell Commun. Signal. 2020, 18, 62. [Google Scholar] [CrossRef]
- Chen, W.; Zheng, P.; Hong, T.; Wang, Y.; Liu, N.; He, B.; Zou, S.; Ren, D.; Duan, J.; Zhao, L.; et al. Astrocytes-derived exosomes induce neuronal recovery after traumatic brain injury via delivering gap junction alpha 1-20 k. J. Tissue Eng. Regen. Med. 2020, 14, 412–423. [Google Scholar] [CrossRef]
- Araki, T.; Ikegaya, Y.; Koyama, R. The effects of microglia- and astrocyte-derived factors on neurogenesis in health and disease. Eur. J. Neurosci. 2020. [Google Scholar] [CrossRef]
- Song, H.; Stevens, C.F.; Gage, F.H. Astroglia induce neurogenesis from adult neural stem cells. Nature 2002, 417, 39–44. [Google Scholar] [CrossRef]
- Sourial, M.; Doering, L.C. Astrocyte-Secreted Factors Selectively Alter Neural Stem and Progenitor Cell Proliferation in the Fragile X Mouse. Front. Cell. Neurosci. 2016, 10, 126. [Google Scholar] [CrossRef]
- Barres, B.A. The mystery and magic of glia: A perspective on their roles in health and disease. Neuron 2008, 60, 430–440. [Google Scholar] [CrossRef]
- Naskar, S.; Chattarji, S. Stress Elicits Contrasting Effects on the Structure and Number of Astrocytes in the Amygdala versus Hippocampus. eNeuro 2019, 6. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.; Kim, J.; Jeong, H.K.; Kim, B.; Jou, I.; Park, S.M.; Chen, L.; Kang, U.J.; Zhuang, X.; Joe, E.H. PINK1 deficiency attenuates astrocyte proliferation through mitochondrial dysfunction, reduced AKT and increased p38 MAPK activation, and downregulation of EGFR. Glia 2013, 61, 800–812. [Google Scholar] [CrossRef] [PubMed]
- Lee, F.H.F.; Lai, T.K.Y.; Su, P.; Liu, F. Altered cortical Cytoarchitecture in the Fmr1 knockout mouse. Mol. Brain 2019, 12, 56. [Google Scholar] [CrossRef] [PubMed]
- Pacey, L.K.; Guan, S.; Tharmalingam, S.; Thomsen, C.; Hampson, D.R. Persistent astrocyte activation in the fragile X mouse cerebellum. Brain Behav. 2015, 5, e00400. [Google Scholar] [CrossRef]
- Falchi, A.M.; Sogos, V.; Saba, F.; Piras, M.; Congiu, T.; Piludu, M. Astrocytes shed large membrane vesicles that contain mitochondria, lipid droplets and ATP. Histochem. Cell. Biol. 2013, 139, 221–231. [Google Scholar] [CrossRef]
- Torralba, D.; Baixauli, F.; Sanchez-Madrid, F. Mitochondria Know No Boundaries: Mechanisms and Functions of Intercellular Mitochondrial Transfer. Front. Cell. Dev. Biol. 2016, 4, 107. [Google Scholar] [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Coelho-Junior, H.J.; Bossola, M.; Landi, F.; Bernabei, R.; Bucci, C.; Marzetti, E. Generation and Release of Mitochondrial-Derived Vesicles in Health, Aging and Disease. J. Clin. Med. 2020, 9, 1440. [Google Scholar] [CrossRef]
- Yanez-Mo, M.; Siljander, P.R.; Andreu, Z.; Zavec, A.B.; Borras, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef]
- Abels, E.R.; Breakefield, X.O. Introduction to Extracellular Vesicles: Biogenesis, RNA Cargo Selection, Content, Release, and Uptake. Cell. Mol. Neurobiol. 2016, 36, 301–312. [Google Scholar] [CrossRef]
- Quiros, P.M.; Goyal, A.; Jha, P.; Auwerx, J. Analysis of mtDNA/nDNA Ratio in Mice. Curr. Protoc. Mouse Biol. 2017, 7, 47–54. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ha, B.G.; Heo, J.-Y.; Jang, Y.-J.; Park, T.-S.; Choi, J.-Y.; Jang, W.Y.; Jeong, S.-J. Depletion of Mitochondrial Components from Extracellular Vesicles Secreted from Astrocytes in a Mouse Model of Fragile X Syndrome. Int. J. Mol. Sci. 2021, 22, 410. https://doi.org/10.3390/ijms22010410
Ha BG, Heo J-Y, Jang Y-J, Park T-S, Choi J-Y, Jang WY, Jeong S-J. Depletion of Mitochondrial Components from Extracellular Vesicles Secreted from Astrocytes in a Mouse Model of Fragile X Syndrome. International Journal of Molecular Sciences. 2021; 22(1):410. https://doi.org/10.3390/ijms22010410
Chicago/Turabian StyleHa, Byung Geun, Jung-Yoon Heo, Yu-Jin Jang, Tae-Shin Park, Ju-Yeon Choi, Woo Young Jang, and Sung-Jin Jeong. 2021. "Depletion of Mitochondrial Components from Extracellular Vesicles Secreted from Astrocytes in a Mouse Model of Fragile X Syndrome" International Journal of Molecular Sciences 22, no. 1: 410. https://doi.org/10.3390/ijms22010410
APA StyleHa, B. G., Heo, J.-Y., Jang, Y.-J., Park, T.-S., Choi, J.-Y., Jang, W. Y., & Jeong, S.-J. (2021). Depletion of Mitochondrial Components from Extracellular Vesicles Secreted from Astrocytes in a Mouse Model of Fragile X Syndrome. International Journal of Molecular Sciences, 22(1), 410. https://doi.org/10.3390/ijms22010410