Clinical Impact of Inherited and Acquired Genetic Variants in Mastocytosis

 ,
,  , , , , , , and
, , , , , , and 
Abstract
1. Introduction
2. Studies of the Common Germline Genetic Variants in Patients with Mastocytosis
2.1. IL4R and IL13 Variants
2.2. IL31 Variants
2.3. IL6 and IL6R Variants
2.4. TLR2 Gene Variants
3. mRNA Expression Studies in Patients with Mastocytosis
4. Missense MRGPRX2, ADGRE2, and PLCG2 Genes Variants and Mast Cell Activation
5. Hereditary Alpha-Tryptasemia in Clonal MC Disease
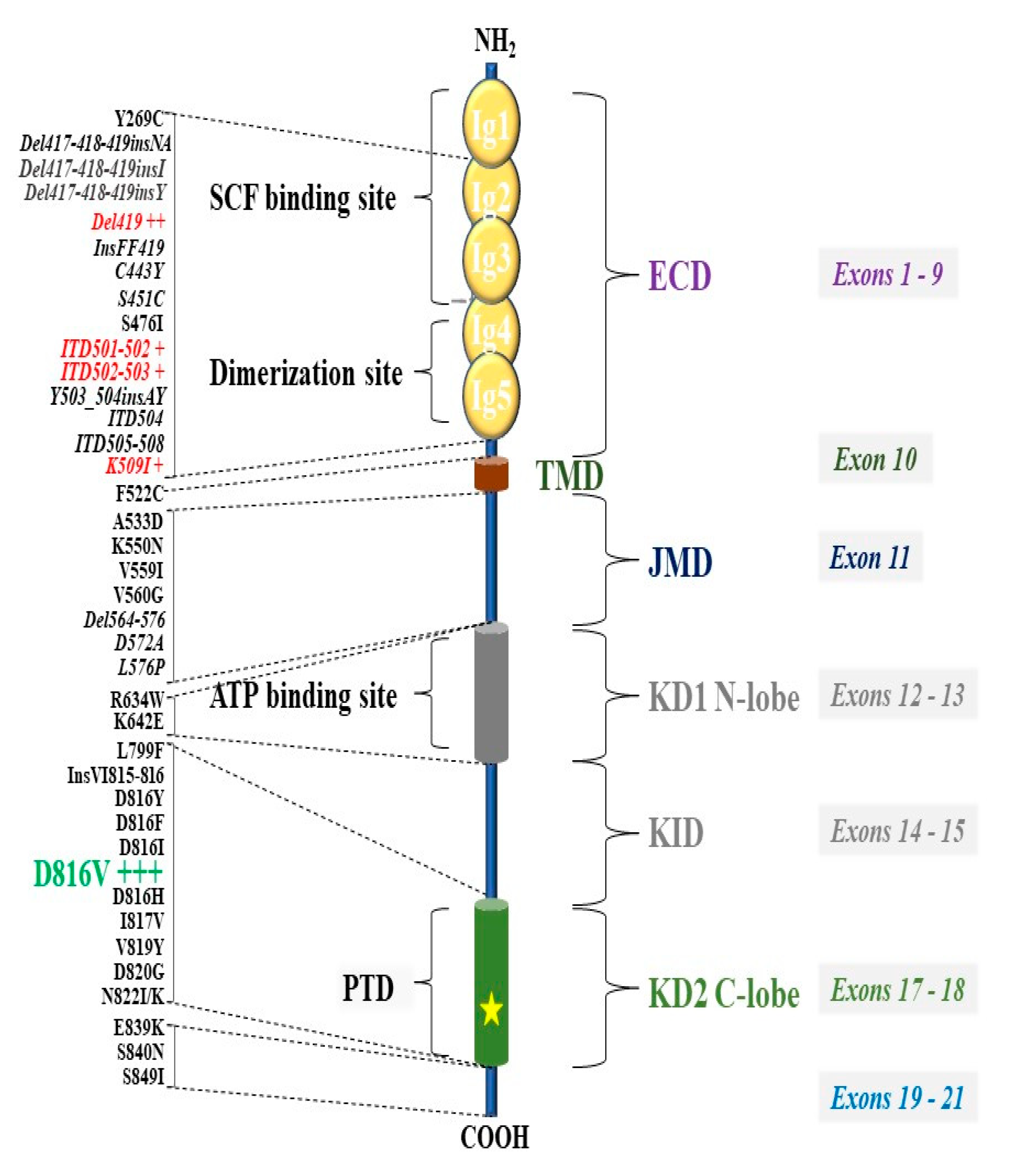
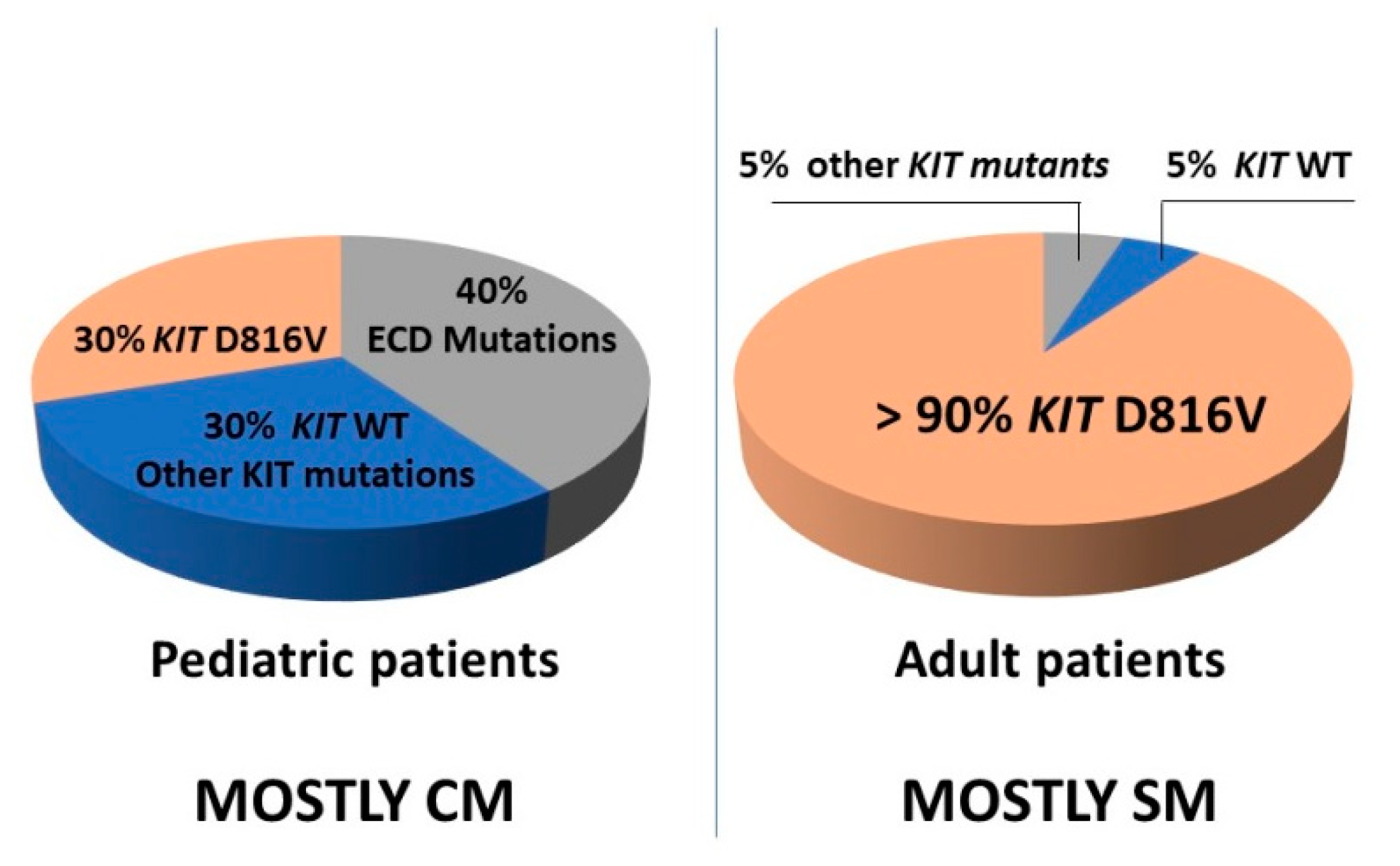
6. KIT Variants and Their Role in the Pathogenesis of Mastocytosis
7. Role of Additional Genetic Variants (Mutations) as Drivers of Advanced SM
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADGRE2 | Adhesion G protein–coupled receptor E2 |
| ASM | Aggressive Systemic mastocytosis |
| ASXL1 | ASXL Transcriptional Regulator 1 |
| BST | basal serum tryptase |
| CBL | Cbl Proto-Oncogene |
| CM | Cutaneous mastocytosis |
| DNMT3A | DNA Methyltransferase 3 Alpha |
| ECD | Extracellular domain of KIT |
| EZH2 | Enhancer Of Zeste 2 Polycomb Repressive Complex 2 Subunit |
| HSC | hematopoietic stem cells |
| HαT | hereditary alpha-tryptasemia |
| HVA | Hymenoptera venom allergy |
| ISM | Indolent systemic mastocytosis |
| JMD: | Juxtamembrane domain of KIT |
| KID | Kinase insert domain of KIT |
| KIT | KIT Proto-Oncogene, Receptor Tyrosine Kinase |
| MCL | Mast cell leukemia |
| MC | Mast cells |
| PTD | phosphotransferase domain of KIT |
| SM | systemic mastocytosis |
| SM-AHN | Systemic mastocytosis with associated hematologic neoplasm |
| MCS | Mast cell sarcoma |
| TPSAB1 | Genes encoding α- and β-tryptases |
| MCAS | MC activation syndrome |
| MRGPRX2 | Mas-related G protein-coupled receptor |
| NRAS | NRAS Proto-Oncogene, GTPase |
| PLCG2 | Phospholipase Cγ2 |
| RUNX1 | RUNX Family Transcription Factor 1 |
| SCF | Stem cell factor |
| SF3B1 | Splicing Factor 3b Subunit 1 |
| SRSF2 | Serine And Arginine Rich Splicing Factor 2 |
| TET2 | Tet Methylcytosine Dioxygenase 2 |
| TLR | Toll-like receptor |
| U2AF1 | U2 Small Nuclear RNA Auxiliary Factor 1 |
| WDSM | well-differentiated systemic mastocytosis |
References
- Födinger, M.; Fritsch, G.; Winkler, K.; Emminger, W.; Mitterbauer, G.; Gadner, H.; Valent, P.; Mannhalter, C. Origin of human mast cells: Development from transplanted hematopoietic stem cells after allogeneic bone marrow transplantation. Blood 1994, 84, 2954–2959. [Google Scholar] [CrossRef] [PubMed]
- Eisenwort, G.; Sadovnik, I.; Schwaab, J.; Jawhar, M.; Keller, A.; Stefanzl, G.; Berger, D.; Blatt, K.; Hoermann, G.; Bilban, M.; et al. Identification of a leukemia-initiating stem cell in human mast cell leukemia. Leukemia 2019, 33, 2673–2684. [Google Scholar] [CrossRef] [PubMed]
- Schwaab, J.; Schnittger, S.; Sotlar, K.; Walz, C.; Fabarius, A.; Pfirrmann, M.; Kohlmann, A.; Grossmann, V.; Meggendorfer, M.; Horny, H.-P.; et al. Comprehensive mutational profiling in advanced systemic mastocytosis. Blood 2013, 122, 2460–2466. [Google Scholar] [CrossRef] [PubMed]
- Jawhar, M.; Schwaab, J.; Schnittger, S.; Sotlar, K.; Horny, H.P.; Metzgeroth, G.; Müller, N.; Schneider, S.; Naumann, N.; Walz, C.; et al. Molecular profiling of myeloid progenitor cells in multi-mutated advanced systemic mastocytosis identifies KIT D816V as a distinct and late event. Leukemia 2015, 29, 1115–1122. [Google Scholar] [CrossRef]
- Yun, H.D.; Antony, M.L.; Linden, M.A.; Noble-Orcutt, K.E.; Eckfeldt, C.; Ustun, C.; Nelson, A.C.; Sachs, Z. Evolution of clonal dynamics and differential response to targeted therapy in a case of systemic mastocytosis with associated myelodysplastic syndrome. Leuk. Res. 2020, 95, 106404. [Google Scholar] [CrossRef]
- Valent, P.; Horny, H.-P.; Escribano, L.; Longley, B.; Li, C.Y.; Schwartz, L.B.; Marone, G.; Nuñez, R.; Akin, C.; Sotlar, K.; et al. Diagnostic criteria and classification of mastocytosis: A consensus proposal. Leuk. Res. 2001, 25, 603–625. [Google Scholar] [CrossRef]
- Valent, P.; Sperr, W.R.; Schwartz, L.B.; Horny, H.-P. Diagnosis and classification of mast cell proliferative disorders: Delineation from immunologic diseases and non–mast cell hematopoietic neoplasms. J. Allergy Clin. Immunol. 2004, 114, 3–11. [Google Scholar] [CrossRef]
- Valent, P.; Akin, C.; Escribano, L.; Födinger, M.; Hartmann, K.; Brockow, K.; Castells, M.; Sperr, W.R.; Kluin-Nelemans, H.C.; Hamdy, N.A.T.; et al. Standards and standardization in mastocytosis: Consensus Statements on Diagnostics, Treatment Recommendations and Response Criteria. Eur. J. Clin. Investig. 2007, 37, 435–453. [Google Scholar] [CrossRef]
- Horny, H.-P.; Sotlar, K.; Valent, P. Mastocytosis: State of the Art. Pathobiology 2007, 74, 121–132. [Google Scholar] [CrossRef]
- Horny, H.P.; Akin, C.; Arber, D.; Peterson, L.A.; Tefferi, A.; Metcalfe, D.D.; Bennett, J.M.; Bain, B.; Escribano, L.; Valent, P. Mastocytosis. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Arber, D.A., Hasserjian, R.P., Le Beau, M.M., et al., Eds.; IARC Press: Lyon, France, 2017; Volume 3, pp. 62–69. [Google Scholar]
- Valent, P.; Akin, C.; Metcalfe, D.D. Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood 2017, 129, 1420–1427. [Google Scholar] [CrossRef]
- Georgin-Lavialle, S.; Lhermitte, L.; Dubreuil, P.; Chandesris, M.-O.; Hermine, O.; Damaj, G. Mast cell leukemia. Blood 2013, 121, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Ustun, C.; Arock, M.; Kluin-Nelemans, H.C.; Reiter, A.; Sperr, W.R.; George, T.; Horny, H.-P.; Hartmann, K.; Sotlar, K.; Damaj, G.; et al. Advanced systemic mastocytosis: From molecular and genetic progress to clinical practice. Haematologica 2016, 101, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Arock, M.; Sotlar, K.; Akin, C.; Broesby-Olsen, S.; Hoermann, G.; Escribano, L.; Kristensen, T.K.; Kluin-Nelemans, H.C.; Hermine, O.; Dubreuil, P.; et al. KIT mutation analysis in mast cell neoplasms: Recommendations of the European Competence Network on Mastocytosis. Leukemia 2015, 29, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Montero, A.G.; Jara-Acevedo, M.; Teodosio, C.; Sánchez-Muñoz, L.; Núñez, R.; Prados, A.; Aldanondo, I.; Domínguez, M.; Botana, L.M.; Sánchez-Jiménez, F.; et al. KIT mutation in mast cells and other bone marrow hematopoietic cell lineages in systemic mast cell disorders: A prospective study of the Spanish Network on Mastocytosis (REMA) in a series of 113 patients. Blood 2006, 108, 2366–2372. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-González, J.I.; Álvarez-Twose, I.; Jara-Acevedo, M.; Henriques, A.; Viñas, E.; Prieto, C.; Sánchez-Muñoz, L.; Caldas, C.; Mayado, A.; Matito, A.; et al. Frequency and prognostic impact of KIT and other genetic variants in indolent systemic mastocytosis. Blood 2019, 134, 456–468. [Google Scholar] [CrossRef]
- Teodosio, C.; García-Montero, A.C.; Jara-Acevedo, M.; Álvarez-Twose, I.; Sánchez-Muñoz, L.; Almeida, J.; Morgado, J.M.; Matito, A.; Escribano, L.; Orfao, A. An immature immunophenotype of bone marrow mast cells predicts for multilineage D816V KIT mutation in systemic mastocytosis. Leukemia 2011, 26, 951–958. [Google Scholar] [CrossRef]
- Kristensen, T.K.; Broesby-Olsen, S.; Vestergaard, H.; Bindslev-Jensen, C.; Moller, M.B.; on behalf of the Mastocytosis Centre Odense University Hospital (MastOUH). Circulating KITD816V mutation-positive non-mast cells in peripheral blood are characteristic of indolent systemic mastocytosis. Eur. J. Haematol. 2012, 89, 42–46. [Google Scholar] [CrossRef]
- Hartmann, K.; Escribano, L.; Grattan, C.; Brockow, K.; Carter, M.C.; Alvarez-Twose, I.; Matito, A.; Broesby-Olsen, S.; Siebenhaar, F.; Lange, M.; et al. Cutaneous manifestations in patients with mastocytosis: Consensus report of the European Competence Network on Mastocytosis; the American Academy of Allergy, Asthma & Immunology; and the European Academy of Allergology and Clinical Immunology. J. Allergy Clin. Immunol. 2016, 137, 35–45. [Google Scholar] [CrossRef]
- Matito, A.; Azaña, J.M.; Torrelo, A.; Álvarez-Twose, I. Cutaneous Mastocytosis in Adults and Children. Immunol. Allergy Clin. N. Am. 2018, 38, 351–363. [Google Scholar] [CrossRef]
- Lange, M.; Nedoszytko, B.; Górska, A.; Zawrocki, A.; Sobjanek, M.; Kozlowski, D. Mastocytosis in children and adults: Clinical disease heterogeneity. Arch. Med. Sci. 2012, 8, 533–541. [Google Scholar] [CrossRef]
- Lange, M.; Niedoszytko, M.; Renke, J.; Gleń, J.; Nedoszytko, B. Clinical aspects of paediatric mastocytosis: A review of 101 cases. J. Eur. Acad. Dermatol. Venereol. 2011, 27, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Akin, C.; Arock, M.; Brockow, K.; Butterfield, J.H.; Carter, M.C.; Castells, M.; Escribano, L.; Hartmann, K.; Lieberman, P.; et al. Definitions, Criteria and Global Classification of Mast Cell Disorders with Special Reference to Mast Cell Activation Syndromes: A Consensus Proposal. Int. Arch. Allergy Immunol. 2011, 157, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Valent, P. Mast cell activation syndromes: Definition and classification. Allergy 2013, 68, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Akin, C.; Bonadonna, P.; Hartmann, K.; Brockow, K.; Niedoszytko, M.; Nedoszytko, B.; Siebenhaar, F.; Sperr, W.R.; Elberink, J.N.O.; et al. Proposed Diagnostic Algorithm for Patients with Suspected Mast Cell Activation Syndrome. J. Allergy Clin. Immunol. Pract. 2019, 7, 1125–1133.e1. [Google Scholar] [CrossRef]
- Daley, T.; Metcalfe, D.D.; Akin, C. Association of the Q576R polymorphism in the interleukin-4 receptor α chain with indolent mastocytosis limited to the skin. Blood 2001, 98, 880–882. [Google Scholar] [CrossRef]
- Nedoszytko, B.; Niedoszytko, M.; Lange, M.; Van Doormaal, J.; Gleń, J.; Zabłotna, M.; Renke, J.; Vales, A.; Buljubasic, F.; Jassem, E.; et al. Interleukin-13promoter gene polymorphism -1112C/T is associated with the systemic form of mastocytosis. Allergy 2009, 64, 287–294. [Google Scholar] [CrossRef]
- Lange, M.; Gleń, J.; Zabłotna, M.; Nedoszytko, B.; Sokołowska-Wojdyło, M.; Rębała, K.; Ługowska-Umer, H.; Niedoszytko, M.; Górska, A.; Sikorska, M.; et al. Interleukin-31 Polymorphisms and Serum IL-31 Level in Patients with Mastocytosis: Correlation with Clinical Presen-tation and Pruritus. Acta Derm. Venereol. 2017, 97, 47–53. [Google Scholar] [CrossRef]
- Rausz, E.; Szilágyi, Á.; Nedoszytko, B.; Lange, M.; Niedoszytko, M.; Lautner-Csorba, O.; Falus, A.; Aladzsity, I.; Kókai, M.; Valent, P.; et al. Comparative analysis of IL6 and IL6 receptor gene polymorphisms in mastocytosis. Br. J. Haematol. 2013, 160, 216–219. [Google Scholar] [CrossRef]
- Nedoszytko, B.; Lange, M.; Renke, J.; Niedoszytko, M.; Zabłotna, M.; Gleń, J.; Nowicki, R. The Possible Role of Gene Variant Coding Nonfunctional Toll-Like Receptor 2 in the Pathogenesis of Mastocytosis. Int. Arch. Allergy Immunol. 2018, 177, 80–86. [Google Scholar] [CrossRef]
- Pulendran, B.; Artis, D. New Paradigms in Type 2 Immunity. Science 2012, 337, 431–435. [Google Scholar] [CrossRef]
- Seyfizadeh, N.; Seyfizadeh, N.; Gharibi, T.; Babaloo, Z. Interleukin-13 as an important cytokine: A review on its roles in some human diseases. Acta Microbiol. Immunol. Hung. 2015, 62, 341–378. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Laporte, S.L.; Juo, Z.S.; Vaclavikova, J.; Colf, L.A.; Qi, X.; Heller, N.M.; Keegan, A.D.; Garcia, K.C. Molecular and Structural Basis of Cytokine Receptor Pleiotropy in the Interleukin-4/13 System. Cell 2008, 132, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Kaur, D.; Hollins, F.; Woodman, L.; Yang, W.; Monk, P.; May, R.; Bradding, P.; Brightling, C.E. Mast cells express IL-13R alpha 1: IL-13 promotes human lung mast cell proliferation and Fc epsilon RI expression. Allergy 2006, 61, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- McLeod, J.J.; Baker, B.; Ryan, J.J. Mast cell production and response to IL-4 and IL-13. Cytokine 2015, 75, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Toru, H.; Pawankar, R.; Ra, C.; Yata, J.; Nakahata, T. Human mast cells produce IL-13 by high-affinity IgE receptor cross-linking: Enhanced IL-13 production by IL-4–primed human mast cells. J. Allergy Clin. Immunol. 1998, 102, 491–502. [Google Scholar] [CrossRef]
- Potaczek, D.P.; Kabesch, M. Current concepts of IgE regulation and impact of genetic determinants. Clin. Exp. Allergy 2012, 42, 852–871. [Google Scholar] [CrossRef]
- Cornelissen, C.G.; Lüscher-Firzlaff, J.; Baron, J.M.; Lüscher, B. Signaling by IL-31 and functional consequences. Eur. J. Cell Biol. 2012, 91, 552–566. [Google Scholar] [CrossRef]
- Rabenhorst, A.; Hartmann, K. Interleukin-31: A Novel Diagnostic Marker of Allergic Diseases. Curr. Allergy Asthma Rep. 2014, 14, 1–7. [Google Scholar] [CrossRef]
- Bilsborough, J.; Leung, D.Y.; Maurer, M.; Howell, M.; Boguniewicz, M.; Yao, L.; Storey, H.; LeCiel, C.; Harder, B.; Gross, J.A. IL-31 is associated with cutaneous lymphocyte antigen–positive skin homing T cells in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2006, 117, 418–425. [Google Scholar] [CrossRef]
- Ishii, T.; Wang, J.; Zhang, W.; Mascarenhas, J.; Hoffman, R.; Dai, Y.; Wisch, N.; Xu, M. Pivotal role of mast cells in pruritogenesis in patients with myeloproliferative disorders. Blood 2009, 113, 5942–5950. [Google Scholar] [CrossRef]
- Niyonsaba, F.; Ushio, H.; Hara, M.; Yokoi, H.; Tominaga, M.; Takamori, K.; Kajiwara, N.; Saito, H.; Nagaoka, I.; Ogawa, H.; et al. Antimicrobial Peptides Human β-Defensins and Cathelicidin LL-37 Induce the Secretion of a Pruritogenic Cytokine IL-31 by Human Mast Cells. J. Immunol. 2010, 184, 3526–3534. [Google Scholar] [CrossRef] [PubMed]
- Raap, U.; Wichmann, K.; Bruder, M.; Ständer, S.; Wedi, B.; Kapp, A.; Werfel, T. Correlation of IL-31 serum levels with severity of atopic dermatitis. J. Allergy Clin. Immunol. 2008, 122, 421–423. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, K.; Wagner, N.; Rabenhorst, A.; Pflanz, L.; Leja, S.; Förster, A.; Gehring, M.; Kapp, A.; Raap, U. Serum IL-31 levels are increased in a subset of patients with mastocytosis and correlate with disease severity in adult patients. J. Allergy Clin. Immunol. 2013, 132, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Schulz, F.; Marenholz, I.; Fölster-Holst, R.; Chen, C.; Sternjak, A.; Baumgrass, R.; Esparza-Gordillo, J.; Gruber, C.; Nickel, R.; Schreiber, S.; et al. A common haplotype of the IL-31 gene influencing gene expression is associated with nonatopic eczema. J. Allergy Clin. Immunol. 2007, 120, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Sokołowska-Wojdyło, M.; Gleń, J.; Zabłotna, M.; Rębała, K.; Sikorska, M.; Florek, A.; Trzeciak, M.; Barańska-Rybak, W.; Malek, M.; Nedoszytko, B. Association of distinct IL-31 polymorphisms with pruritus and severity of atopic dermatitis. J. Eur. Acad. Dermatol. Venereol. 2013, 27, 662–664. [Google Scholar] [CrossRef] [PubMed]
- Sokołowska-Wojdyło, M.; Gleń, J.; Zabłotna, M.; Rębała, K.; Trzeciak, M.; Sikorska, M.; Ruckemann-Dziurdzińska, K.; Msc, B.N.; Florek, A.; Nowicki, R. The frequencies of haplotypes defined by three polymorphisms of theIL-31gene: −1066, −2057, and IVS2+12 in Polish patients with atopic dermatitis. Int. J. Dermatol. 2014, 54, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Brockow, K.; Akin, C.; Huber, M.; Scott, L.M.; Schwartz, L.B.; Metcalfe, D.D. Levels of mast-cell growth factors in plasma and in suction skin blister fluid in adults with mastocytosis: Correlation with dermal mast-cell numbers and mast-cell tryptase. J. Allergy Clin. Immunol. 2002, 109, 82–88. [Google Scholar] [CrossRef]
- Brockow, K.; Akin, C.; Huber, M.; Metcalfe, D.D. IL-6 levels predict disease variant and extent of organ involvement in patients with mastocytosis. Clin. Immunol. 2005, 115, 216–223. [Google Scholar] [CrossRef]
- Mayado, A.; Teodosio, C.; Garcia-Montero, A.C.; Matito, A.; Rodriguez-Caballero, A.; Morgado, J.M.; Muñiz, C.; Jara-Acevedo, M.; Álvarez-Twose, I.; Sanchez-Muñoz, L.; et al. Increased IL6 plasma levels in indolent systemic mastocytosis patients are associated with high risk of disease progression. Leukemia 2016, 30, 124–130. [Google Scholar] [CrossRef]
- Theoharides, T.C.; Boucher, W.; Spear, K. Serum interleukin-6 reflects disease severity and osteoporosis in mastocytosis patients. Int. Arch. Allergy Immunol. 2002, 128, 344–350. [Google Scholar] [CrossRef]
- Lange, M.; Renke, J.; Gleń, J.; Niedoszytko, M.; Nedoszytko, B. Serum tryptase, interleukin 6 and SCORMA Index as disease severity parameters in childhood mastocytosis. Postępy Dermatol. Alergol. 2010, 27, 238. [Google Scholar]
- Tobío, A.; Bandara, G.; Morris, D.A.; Kim, D.K.; O’Connell, M.P.; Komarow, H.D.; Carter, M.C.; Smrz, D.; Metcalfe, D.D.; Olivera, A. Oncogenic D816V-KIT signaling in mast cells causes persistent IL-6 production. Haematologica 2020, 105, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Valent, P. KIT D816V and the cytokine storm in mastocytosis: Production and role of interleukin-6. Haematologica 2020, 105, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Sandig, H.; Bulfone-Paus, S. TLR signaling in mast cells: Common and unique features. Front. Immunol. 2012, 3, 185. [Google Scholar] [CrossRef]
- Olivera, A.; Beaven, M.A.; Metcalfe, D.D. Mast cells signal their importance in health and disease. J. Allergy Clin. Immunol. 2018, 142, 381–393. [Google Scholar] [CrossRef]
- Lin, M.; Yu, B.; Zhang, W.; Zhang, W.; Xiao, Z.; Mao, Z.; Lai, Y.; Lin, D.; Ma, Q.; Pan, E.; et al. Toll-like receptor 2-mediated MAPKs and NF-κB activation requires the GNAO1- dependent pathway in human mast cells. Integr. Biol. 2016, 12, 968–975. [Google Scholar]
- Salpietro, C.; Rigoli, L.; Del Giudice, M.; Cuppari, C.; Di Bella, C.; Salpietro, A.; Maiello, N.; La Rosa, M.; Marseglia, G.L.; Leonardi, S.; et al. TLR2 and TLR4 gene polymorphisms and atopic dermatitis in Italian children: A multicenter study. Int. J. Immunopathol. Pharmacol. 2011, 24, 33–40. [Google Scholar] [CrossRef]
- Niebuhr, M.; Langnickel, J.; Draing, C.; Renz, H.; Kapp, A.; Werfel, T. Dysregulation of toll-like receptor-2 (TLR-2)-induced effects in monocytes from patients with atopic dermatitis: Impact of the TLR-2 R753Q polymorphism. Allergy 2008, 63, 728–734. [Google Scholar] [CrossRef]
- Mrabet-Dahbi, S.; Dalpke, A.H.; Niebuhr, M.; Frey, M.; Draing, C.; Brand, S.; Heeg, K.; Werfel, T.; Renz, H. The Toll-like receptor 2 R753Q mutation modifies cytokine production and Toll-like receptor expression in atopic dermatitis. J. Allergy Clin. Immunol. 2008, 121, 1013–1019. [Google Scholar] [CrossRef]
- Zabłotna, M.; Sobjanek, M.; Purzycka-Bohdan, D.; Szczerkowska-Dobosz, A.; Nedoszytko, B.; Nowicki, R.J. The significance of Toll-like receptor (TLR) 2 and 9 gene polymorphisms in psoriasis. Adv. Dermatol. Allergol. 2017, 34, 85–86. [Google Scholar] [CrossRef]
- D’Ambrosio, C.; Akin, C.; Wu, Y.; Magnusson, M.K.; Metcalfe, D.D. Gene expression analysis in mastocytosis reveals a highly consistent profile with candidate molecular markers. J. Allergy Clin. Immunol. 2003, 112, 1162–1170. [Google Scholar] [CrossRef] [PubMed]
- Teodosio, C.; García-Montero, A.C.; Jara-Acevedo, M.; Sánchez-Muñoz, L.; Pedreira, C.E.; Álvarez-Twose, I.; Matarraz, S.; Morgado, J.M.; Bárcena, P.; Matito, A.; et al. Gene expression profile of highly purified bone marrow mast cells in systemic mastocytosis. J. Allergy Clin. Immunol. 2013, 131, 1213–1224.e4. [Google Scholar] [CrossRef]
- Niedoszytko, M.; Elberink, J.N.G.O.; Bruinenberg, M.; Nedoszytko, B.; De Monchy, J.G.R.; Meerman, G.J.T.; Weersma, R.K.; Mulder, A.B.; Jassem, E.; Van Doormaal, J.J. Gene expression profile, pathways, and transcriptional system regulation in indolent systemic mastocytosis. Allergy 2010, 66, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Niedoszytko, M.; Bruinenberg, M.; Van Doormaal, J.J.; De Monchy, J.G.R.; Nedoszytko, B.; Koppelman, G.H.; Nawijn, M.C.; Wijmenga, C.; Jassem, E.; Elberink, J.N.G.O. Gene expression analysis predicts insect venom anaphylaxis in indolent systemic mastocytosis. Allergy 2010, 66, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Górska, A.; Gruchala-Niedoszytko, M.; Niedoszytko, M.; Maciejewska, A.; Chełmińska, M.; Skrzypski, M.; Wasąg, B.; Kaczkan, M.; Lange, M.; Nedoszytko, B.; et al. The Role of TRAF4 and B3GAT1 Gene Expression in the Food Hypersensitivity and Insect Venom Allergy in Mastocytosis. Arch. Immunol. Ther. Exp. 2016, 64, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Bulfone-Paus, S.; Nilsson, G.; Draber, P.; Blank, U.; Levi-Schaffer, F. Positive and Negative Signals in Mast Cell Activation. Trends Immunol. 2017, 38, 657–667. [Google Scholar] [CrossRef] [PubMed]
- McNeil, B.D.; Pundir, P.P.; Meeker, S.; Han, L.; Undem, B.J.; Kulka, M.; Dong, X. Identification of a mast-cell-specific receptor crucial for pseudo-allergic drug reactions. Nat. Cell Biol. 2015, 519, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, H.; Gupta, K.; Ali, H. Roles of Mas-related G protein-coupled receptor X2 on mast cell-mediated host defense, pseudoallergic drug reactions, and chronic inflammatory diseases. J. Allergy Clin. Immunol. 2016, 138, 700–710. [Google Scholar] [CrossRef]
- Tatemoto, K.; Nozaki, Y.; Tsuda, R.; Konno, S.; Tomura, K.; Furuno, M.; Ogasawara, H.; Edamura, K.; Takagi, H.; Iwamura, H.; et al. Immunoglobulin E-independent activation of mast cell is mediated by Mrg receptors. Biochem. Biophys. Res. Commun. 2006, 349, 1322–1328. [Google Scholar] [CrossRef]
- Alkanfari, I.; Gupta, K.; Jahan, T.; Ali, H. Naturally Occurring Missense MRGPRX2 Variants Display Loss of Function Phenotype for Mast Cell Degranulation in Response to Substance P, Hemokinin-1, Human β-Defensin-3, and Icatibant. J. Immunol. 2018, 201, 343–349. [Google Scholar] [CrossRef]
- Na Ayudhya, C.C.; Roy, S.; Alkanfari, I.; Ganguly, A.; Ali, H. Identification of Gain and Loss of Function Missense Variants in MRGPRX2’s Transmembrane and Intracellular Domains for Mast Cell Activation by Substance P. Int. J. Mol. Sci. 2019, 20, 5247. [Google Scholar] [CrossRef] [PubMed]
- Tatemoto, K.; Nozaki, Y.; Tsuda, R.; Kaneko, S.; Tomura, K.; Furuno, M.; Ogasawara, H.; Edamura, K.; Takagi, H.; Iwamura, H.; et al. Endogenous protein and enzyme fragments induce immunoglobulin E-independent activation of mast cells via a G protein-coupled receptor, MRGPRX2. Scand. J. Immunol. 2018, 87, e12655. [Google Scholar] [CrossRef] [PubMed]
- Giavina-Bianchi, P.; Gonçalves, D.G.; Zanandréa, A.; De Castro, R.B.; Garro, L.S.; Kalil, J.; Castells, M. Anaphylaxis to quinolones in mastocytosis: Hypothesis on the mechanism. J. Allergy Clin. Immunol. Pract. 2019, 7, 2089–2090. [Google Scholar] [CrossRef] [PubMed]
- Weiler, C.R. Mastocytosis, Quinolones, MRGPRX2, and Anaphylaxis. J. Allergy Clin. Immunol. Pract. 2019, 7, 2091–2092. [Google Scholar] [CrossRef]
- Boyden, S.E.; Desai, A.; Cruse, G.; Young, M.L.; Bolan, H.C.; Scott, L.M.; Eisch, A.R.; Long, R.D.; Lee, C.-C.R.; Satorius, C.L.; et al. Vibratory Urticaria Associated with a Missense Variant in ADGRE2. N. Engl. J. Med. 2016, 374, 656–663. [Google Scholar] [CrossRef]
- Ombrello, M.J.; Remmers, E.F.; Sun, G.; Freeman, A.F.; Datta, S.; Torabi-Parizi, P.; Subramanian, N.; Bunney, T.D.; Baxendale, R.W.; Martins, M.S.; et al. Cold Urticaria, Immunodeficiency, and Autoimmunity Related toPLCG2Deletions. N. Engl. J. Med. 2012, 366, 330–338. [Google Scholar] [CrossRef]
- Gandhi, C.; Healy, C.; Wanderer, A.A.; Hoffman, H.M. Familial atypical cold urticaria: Description of a new hereditary disease. J. Allergy Clin. Immunol. 2009, 124, 1245–1250. [Google Scholar] [CrossRef]
- Aderibigbe, O.M.; Priel, D.L.; Lee, C.R.; Ombrello, M.J.; Prajapati, V.H.; Liang, M.G.; Lyons, J.J.; Kuhns, D.B.; Cowen, E.W.; Milner, J.D. Distinct Cutaneous Manifestations and Cold-Induced Leukocyte Activation Associated With PLCG2 Mutations. JAMA Dermatol. 2015, 151, 627–634. [Google Scholar] [CrossRef]
- Lyons, J.J.; Sun, G.; Stone, K.D.; Nelson, C.M.; Wisch, L.B.; O’Brien, M.; Jones, N.; Lindsley, A.; Komarow, H.D.; Bai, Y.; et al. Mendelian inheritance of elevated serum tryptase associated with atopy and connective tissue abnormalities. J. Allergy Clin. Immunol. 2014, 133, 1471–1474. [Google Scholar] [CrossRef]
- Lyons, J.J.; Yu, X.; Hughes, J.D.; Le, Q.T.; Jamil, A.; Bai, Y.; Ho, N.; Zhao, M.; Liu, Y.; O’Connell, M.P.; et al. Elevated basal serum tryptase identifies a multisystem disorder associated with increased TPSAB1 copy number. Nat. Genet. 2016, 48, 1564–1569. [Google Scholar] [CrossRef]
- Lyons, J.J. Hereditary Alpha Tryptasemia: Genotyping and Associated Clinical Features. Immunol. Allergy Clin. N. Am. 2018, 38, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Greiner, G.; Sprinzl, B.; Górska, A.; Ratzinger, F.; Gurbisz, M.; Witzeneder, N.; Schmetterer, K.G.; Gisslinger, B.; Uyanik, G.; Hadzijusufovic, E.; et al. Hereditary alpha tryptasemia is a valid genetic biomarker for severe mediator-related symptoms in mastocytosis. Blood 2020. [Google Scholar] [CrossRef] [PubMed]
- Lyons, J.J.; Chovanec, J.; O’Connell, M.P.; Liu, Y.; Šelb, J.; Zanotti, R.; Bai, Y.; Kim, J.; DiMaggio, T.; Schwartz, L.B.; et al. Heritable risk for severe anaphylaxis associated with increased α-tryptase-encoding germline copy number at TPSAB1. J. Allergy Clin. Immunol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.C.; Wilcock, A.; Bonin, H.; Beaman, G.; Myers, B.; Grattan, C.; Briggs, T.A.; Arkwright, P.D. Hereditary Alpha-Tryptasemia: UK Prevalence and Variability in Disease Expression. J. Allergy Clin. Immunol. Pract. 2020, 8, 3549–3556. [Google Scholar] [CrossRef]
- Le, Q.T.; Lyons, J.J.; Naranjo, A.N.; Olivera, A.; Lazarus, R.A.; Metcalfe, D.D.; Milner, J.D.; Schwartz, L.B. Impact of naturally forming human alpha/beta-tryptase heterotetramers in the pathogenesis of hereditary alpha-tryptasemia. J. Exp. Med. 2019, 216, 2348–2361. [Google Scholar] [CrossRef]
- Sabato, V.; Chovanec, J.; Faber, M.A.; Milner, J.D.; Ebo, D.; Lyons, J.J. First Identification of an Inherited TPSAB1 Quintuplication in a Patient with Clonal Mast Cell Disease. J. Clin. Immunol. 2018, 38, 457–459. [Google Scholar] [CrossRef]
- Lennartsson, J.; Rönnstrand, L. Stem Cell Factor Receptor/c-Kit: From Basic Science to Clinical Implications. Physiol. Rev. 2012, 92, 1619–1649. [Google Scholar] [CrossRef]
- Cruse, G.; Metcalfe, D.D.; Olivera, A. Functional deregulation of KIT: Link to mast cell proliferative diseases and other neoplasms. Immunol. Allergy Clin. N. Am. 2014, 34, 219–237. [Google Scholar] [CrossRef]
- Bodemer, C.; Hermine, O.; Palmérini, F.; Yang, Y.; Grandpeix-Guyodo, C.; Leventhal, P.S.; Hadj-Rabia, S.; Nasca, L.; Georgin-Lavialle, S.; Cohen-Akenine, A.; et al. Pediatric Mastocytosis Is a Clonal Disease Associated with D816V and Other Activating c-KIT Mutations. J. Investig. Dermatol. 2010, 130, 804–815. [Google Scholar] [CrossRef]
- Orfao, A.; Montero, A.G.; Sánchez-Muñoz, L.; Escribano, L. Recent advances in the understanding of mastocytosis: The role of KIT mutations. Br. J. Haematol. 2007, 138, 12–30. [Google Scholar] [CrossRef]
- Bibi, S.; Langenfeld, F.; Jeanningros, S.; Brenet, F.; Soucie, E.; Hermine, O.; Damaj, G.; Dubreuil, P.; Arock, M. Molecular defects in mastocytosis: KIT and beyond KIT. Immunol. Allergy Clin. N. Am. 2014, 34, 239–262. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Twose, I.; Jara-Acevedo, M.; Morgado, J.M.; Garcia-Montero, A.; Sanchez-Munoz, L.; Teodosio, C.; Matito, A.; Mayado, A.; Caldas, C.; Mollejo, M.; et al. Clinical, immunophenotypic, and molecular characteristics of well-differentiated systemic mastocytosis. J. Allergy Clin. Immunol. 2016, 137, 168–178.e1. [Google Scholar] [CrossRef] [PubMed]
- Grootens, J.; Ungerstedt, J.S.; Ekoff, M.; Ronnberg, E.; Klimkowska, M.; Amini, R.M.; Arock, M.; Soderlund, S.; Mattsson, M.; Nilsson, G.; et al. Single-cell analysis reveals the KIT D816V mutation in haematopoietic stem and progenitor cells in systemic mastocytosis. EBioMedicine 2019, 43, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A. Systemic mastocytosis in adults: 2017 update on diagnosis, risk stratification and management. Am. J. Hematol. 2016, 91, 1146–1159. [Google Scholar] [CrossRef]
- Lasho, T.; Finke, C.; Zblewski, D.; Hanson, C.A.; Ketterling, R.P.; Butterfield, J.H.; Tefferi, A.; Pardanani, A. Concurrent activating KIT mutations in systemic mastocytosis. Br. J. Haematol. 2016, 173, 153–156. [Google Scholar] [CrossRef]
- de Beauchene, I.C.; Allain, A.; Panel, N.; Laine, E.; Trouve, A.; Dubreuil, P.; Tchertanov, L. Hotspot mutations in KIT receptor differentially modulate its allosterically coupled conformational dynamics: Impact on activation and drug sensitivity. PLoS Comput. Biol. 2014, 10, e1003749. [Google Scholar]
- Laine, E.; de Beauchene, I.C.; Perahia, D.; Auclair, C.; Tchertanov, L. Mutation D816V alters the internal structure and dynamics of c-KIT receptor cytoplasmic region: Implications for dimerization and activation mechanisms. PLoS Comput. Biol. 2011, 7, e1002068. [Google Scholar] [CrossRef]
- Piao, X.; Paulson, R.; van der Geer, P.; Pawson, T.; Bernstein, A. Oncogenic mutation in the Kit receptor tyrosine kinase alters substrate specificity and induces degradation of the protein tyrosine phosphatase SHP-1. Proc. Natl. Acad. Sci. USA 1996, 93, 14665–14669. [Google Scholar] [CrossRef]
- Bibi, S.; Arslanhan, M.D.; Langenfeld, F.; Jeanningros, S.; Cerny-Reiterer, S.; Hadzijusufovic, E.; Tchertanov, L.; Moriggl, R.; Valent, P.; Arock, M. Co-operating STAT5 and AKT signaling pathways in chronic myeloid leukemia and mastocytosis: Possible new targets of therapy. Haematologica 2014, 99, 417–429. [Google Scholar] [CrossRef]
- Harir, N.; Boudot, C.; Friedbichler, K.; Sonneck, K.; Kondo, R.; Martin-Lannerée, S.; Kenner, L.; Kerenyi, M.; Yahiaoui, S.; Gouilleux-Gruart, V.; et al. Oncogenic Kit controls neoplastic mast cell growth through a Stat5/PI3-kinase signaling cascade. Blood 2008, 112, 2463–2473. [Google Scholar] [CrossRef]
- Baumgartner, C.; Cerny-Reiterer, S.; Sonneck, K.; Mayerhofer, M.; Gleixner, K.V.; Fritz, R.; Kerenyi, M.; Boudot, C.; Gouilleux, F.; Kornfeld, J.W.; et al. Expression of activated STAT5 in neoplastic mast cells in systemic mastocytosis: Subcellular distribution and role of the transforming oncoprotein KIT D816V. Am. J. Pathol. 2009, 175, 2416–2429. [Google Scholar] [CrossRef] [PubMed]
- Peter, B.; Bibi, S.; Eisenwort, G.; Wingelhofer, B.; Berger, D.; Stefanzl, G.; Blatt, K.; Herrmann, H.; Hadzijusufovic, E.; Hoermann, G.; et al. Drug-induced inhibition of phosphorylation of STAT5 overrides drug resistance in neoplastic mast cells. Leukemia 2018, 32, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Voisset, E.; Lopez, S.; Dubreuil, P.; De Sepulveda, P. The tyrosine kinase FES is an essential effector of KITD816V proliferation signal. Blood 2007, 110, 2593–2599. [Google Scholar] [CrossRef] [PubMed]
- Smrz, D.; Kim, M.S.; Zhang, S.; Mock, B.A.; Smrzova, S.; DuBois, W.; Simakova, O.; Maric, I.; Wilson, T.M.; Metcalfe, D.D.; et al. mTORC1 and mTORC2 differentially regulate homeostasis of neoplastic and non-neoplastic human mast cells. Blood 2011, 118, 6803–6813. [Google Scholar] [CrossRef] [PubMed]
- Gabillot-Carre, M.; Lepelletier, Y.; Humbert, M.; de Sepuvelda, P.; Hamouda, N.B.; Zappulla, J.P.; Liblau, R.; Ribadeau-Dumas, A.; Machavoine, F.; Letard, S.; et al. Rapamycin inhibits growth and survival of D816V-mutated c-kit mast cells. Blood 2006, 108, 1065–1072. [Google Scholar] [CrossRef] [PubMed]
- Gleixner, K.V.; Mayerhofer, M.; Cerny-Reiterer, S.; Hormann, G.; Rix, U.; Bennett, K.L.; Hadzijusufovic, E.; Meyer, R.A.; Pickl, W.F.; Gotlib, J.; et al. KIT-D816V-independent oncogenic signaling in neoplastic cells in systemic mastocytosis: Role of Lyn and Btk activation and disruption by dasatinib and bosutinib. Blood 2011, 118, 1885–1898. [Google Scholar] [CrossRef]
- Hoermann, G.; Cerny-Reiterer, S.; Perne, A.; Klauser, M.; Hoetzenecker, K.; Klein, K.; Mullauer, L.; Groger, M.; Nijman, S.M.; Klepetko, W.; et al. Identification of oncostatin M as a STAT5-dependent mediator of bone marrow remodeling in KIT D816V-positive systemic mastocytosis. Am. J. Pathol. 2011, 178, 2344–2356. [Google Scholar] [CrossRef]
- Lee, Y.N.; Brandal, S.; Noel, P.; Wentzel, E.; Mendell, J.T.; McDevitt, M.A.; Kapur, R.; Carter, M.; Metcalfe, D.D.; Takemoto, C.M. KIT signaling regulates MITF expression through miRNAs in normal and malignant mast cell proliferation. Blood 2011, 117, 3629–3640. [Google Scholar] [CrossRef]
- Aichberger, K.J.; Mayerhofer, M.; Gleixner, K.V.; Krauth, M.T.; Gruze, A.; Pickl, W.F.; Wacheck, V.; Selzer, E.; Mullauer, L.; Agis, H.; et al. Identification of MCL1 as a novel target in neoplastic mast cells in systemic mastocytosis: Inhibition of mast cell survival by MCL1 antisense oligonucleotides and synergism with PKC412. Blood 2007, 109, 3031–3041. [Google Scholar] [CrossRef]
- Jawhar, M.; Schwaab, J.; Schnittger, S.; Meggendorfer, M.; Pfirrmann, M.; Sotlar, K.; Horny, H.P.; Metzgeroth, G.; Kluger, S.; Naumann, N. Additional mutations in SRSF2, ASXL1 and/or RUNX1 identify ahigh-risk group of patients with KIT D816V+ advanced systemic mastocytosis. Leukemia 2016, 30, 136. [Google Scholar] [CrossRef]
- Jawhar, M.; Schwaab, J.; Hausmann, D.; Clemens, J.; Naumann, N.; Henzler, T.; Horny, H.P.; Sotlar, K.; Schoenberg, S.O.; Cross, N.C.P. Splenomegaly, elevated alkaline phosphatase and mutations in the SRSF2/ASXL1/RUNX1 gene panel are strong adverse prognostic markers in patients with systemic mastocytosis. Leukemia 2016, 30, 2342–2350. [Google Scholar] [CrossRef] [PubMed]
- Damaj, M.J.G.; Chandesris, O.; Hanssens, K.; Soucie, E.; Canioni, D.; Kolb, B.; Durieu, I.; Gyan, E.; Livideanu, C.; Chèze, S.; et al. ASXL1 but not TET2 mutations adversely impact overall survival of patients suffering systemic mastocytosis with associated clonal hematologic non-mast-cell diseases. PLoS ONE 2014, 9, e85362. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.; Lasho, T.L.; Finke, C.; Zblewski, D.; Abdelrahman, R.; Wassie, E.; Gangat, N.; Hanson, C.A.; Ketterling, R.P.; Tefferi, A. ASXL1 and CBL mutations are independently predictive of inferior survival in advanced systemic mastocytosis. Br. J. Haematol. 2015, 175, 534–536. [Google Scholar] [CrossRef] [PubMed]
- Jawhar, M.; Schwaab, J.; Álvarez-Twose, I.; Shoumariyeh, K.; Naumann, N.; Lübke, J.; Perkins, C.; Muñoz-González, J.I.; Meggendorfer, M.; Kennedy, V.; et al. MARS: Mutation-Adjusted Risk Score for Advanced Systemic Mastocytosis. J. Clin. Oncol. 2019, 37, 2846–2856. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-González, J.I.; Jara-Acevedo, M.; Alvarez-Twose, I.; Merker, J.D.; Teodosio, C.; Hou, Y.; Henriques, A.; Roskin, K.M.; Sanchez-Muñoz, L.; Tsai, A.G.; et al. Impact of somatic and germline mutations on the outcome of systemic mastocytosis. Blood Adv. 2018, 2, 2814–2828. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.; Shah, S.; Mannelli, F.; Elala, Y.C.; Guglielmelli, P.; Lasho, T.L.; Patnaik, M.M.; Gangat, N.; Ketterling, R.P.; Reichard, K.K.; et al. Mayo alliance prognostic system for mastocytosis: Clinical and hybrid clinical-molecular models. Blood Adv. 2018, 2, 2964–2972. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Cutaneous mastocytosis (CM) |
| maculopapular cutaneous mastocytosis (MPCM) = urticaria pigmentosa (UP) |
| diffuse cutaneous mastocytosis (DCM) |
| Mastocytoma of skin (cutaneous mastocytoma) |
| Systemic mastocytosis (SM) |
| Indolent systemic mastocytosis (ISM) |
| Smoldering systemic mastocytosis (SSM) |
| Systemic mastocytosis with associated hematologic neoplasm (SM-AHN) |
| Aggressive Systemic mastocytosis (ASM) |
| Mast cell leukemia (MCL) |
| Mast cell sarcoma (MCS) |
| Gene | Polymorhism | Effect on Gene/Protein Function | Effects on Risk of Mastocytosis and Prognosis | References |
|---|---|---|---|---|
| IL4R | p.Q576R (rs 1801275) | gain-of-function | Association p.Q576R of IL4R gene with CM and more favorable prognosis | [26] |
| IL13 | −1112C > T (rs 18 00925) | −1112T high transcription variant | Presence of −1112T variant increase the risk of SM and correlate with increased tryptase level | [27] |
| Gene | Polymorhism | Effect on Gene/Protein Function | Effects on Risk of Mastocytosis and Prognosis |
|---|---|---|---|
| IL31 | −1066G > A (rs 11608363) | Not known | Not increase mastocytosis risk |
| −2057G > A (rs 6489188) | Not known | −2057AA genotype increase the risk of mastocytosis in adult patients with CM and SM | |
| IVS2 + 12A > G | Noncoding intronic variant | AA and AG genotypes increased the risk of mastocytosis in adults and children with CM |
| Gene | Polymorhism | Effect on Gene/Protein Function | Effects on Risk of Mastocytosis and Prognosis | References |
|---|---|---|---|---|
| IL6 | −174G > C (rs 1800795) | −174G high transcription variant | No association with mastocytosis | [29] |
| IL6R | p.D358A (rs 9192284) | Misense variant | homozygous AA carriers of the missense variant had lower risk for mastocytosis than those with the AC or CC genotypes | [29] |
| Gene | Polymorphism | Effect on Gene/Protein Function | Effects on Risk of Mastocytosis and Prognosis | References |
|---|---|---|---|---|
| TLR2 | p.R753Q (rs5743708) | 753Q variant encode non-functional receptor | Presence of 753Q variant of TLR-2 gene increased the risk of systemic mastocytosis | [30] |
| TLR4 | 896A > G (rs496790) | intronic variant | Not increase mastocytosis risk | [30] |
| TLR9 | −1237C > T (rs5743836) | CC lower transcription rate | No association with mastocytosis | [30] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nedoszytko, B.; Arock, M.; Lyons, J.J.; Bachelot, G.; Schwartz, L.B.; Reiter, A.; Jawhar, M.; Schwaab, J.; Lange, M.; Greiner, G.; et al. Clinical Impact of Inherited and Acquired Genetic Variants in Mastocytosis. Int. J. Mol. Sci. 2021, 22, 411. https://doi.org/10.3390/ijms22010411
Nedoszytko B, Arock M, Lyons JJ, Bachelot G, Schwartz LB, Reiter A, Jawhar M, Schwaab J, Lange M, Greiner G, et al. Clinical Impact of Inherited and Acquired Genetic Variants in Mastocytosis. International Journal of Molecular Sciences. 2021; 22(1):411. https://doi.org/10.3390/ijms22010411
Chicago/Turabian StyleNedoszytko, Boguslaw, Michel Arock, Jonathan J. Lyons, Guillaume Bachelot, Lawrence B. Schwartz, Andreas Reiter, Mohamad Jawhar, Juliana Schwaab, Magdalena Lange, Georg Greiner, and et al. 2021. "Clinical Impact of Inherited and Acquired Genetic Variants in Mastocytosis" International Journal of Molecular Sciences 22, no. 1: 411. https://doi.org/10.3390/ijms22010411
APA StyleNedoszytko, B., Arock, M., Lyons, J. J., Bachelot, G., Schwartz, L. B., Reiter, A., Jawhar, M., Schwaab, J., Lange, M., Greiner, G., Hoermann, G., Niedoszytko, M., Metcalfe, D. D., & Valent, P. (2021). Clinical Impact of Inherited and Acquired Genetic Variants in Mastocytosis. International Journal of Molecular Sciences, 22(1), 411. https://doi.org/10.3390/ijms22010411