Neuropsychiatric Disorders Due to Limbic Encephalitis: Immunologic Aspect



Abstract
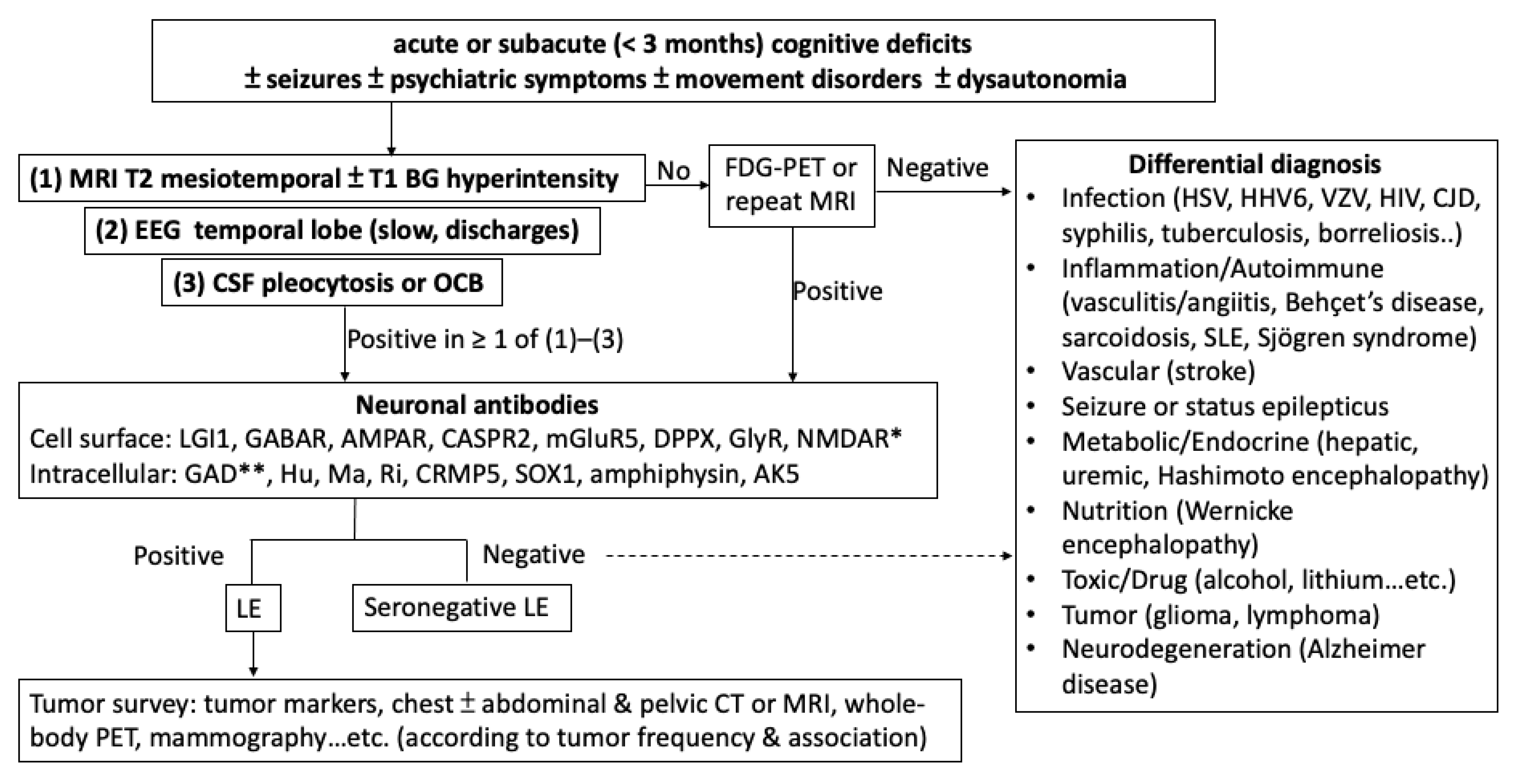
1. Introduction
2. Antibodies
3. Immunopathology
4. Inflammatory Mediators
5. Genetic Factors
6. Cancers
7. Infection
8. Immune Checkpoint Inhibitors
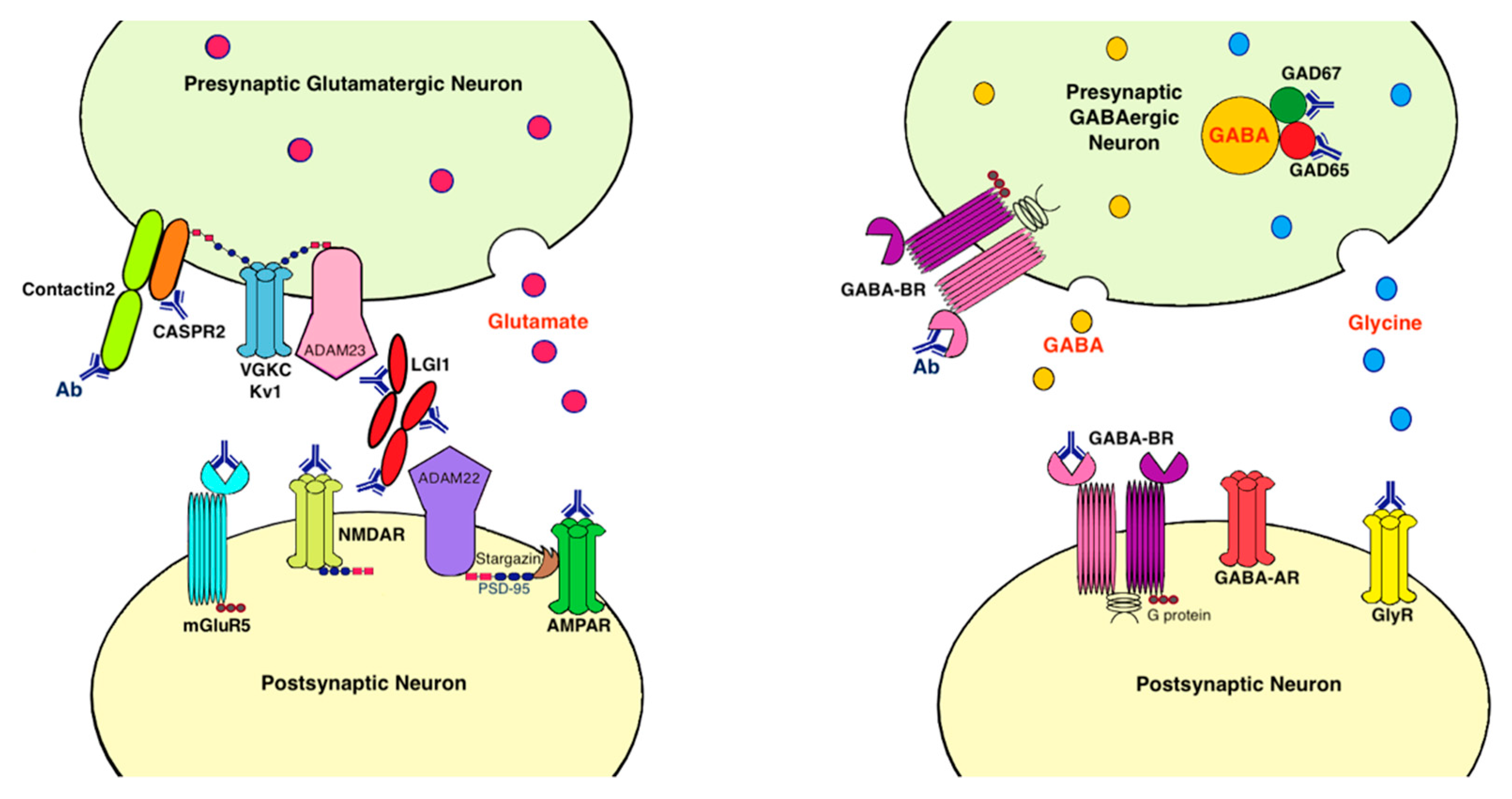
9. Antigens Associated with LE
9.1. Voltage-Gated Potassium Channel Complex (VGKC)
9.2. Leucine-Rich, Glioma Inactivated 1 Protein (LGI1)
9.3. Contactin-Associated Protein-Like 2 (CASPR2)
9.4. AMPA Receptor
9.5. Metabotropic Glutamate Receptor 5 (mGluR5)
9.6. Dipeptidyl Peptidase-Like Protein 6 (DPP6)/DPPX
9.7. GABA-B Receptor
9.8. Glutamic Acid Decarboxylase (GAD)
10. Intracellular Antibodies
11. Treatment
11.1. First-Line Treatment
11.2. Second-Line Treatment
11.3. Maintenance Therapy
12. Prognosis
13. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AE | autoimmune encephalitis |
| AK5 | adenylate kinase 5 |
| AMPAR | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor |
| BBB | blood-brain barrier |
| BG | basal ganglia |
| CASPR2 | contactin-associated protein-like 2 |
| CJD | Creutzfeldt-Jakob disease |
| CNS | central nervous system |
| CRMP5 | collapsin response-mediator protein-5 |
| CSF | cerebrospinal fluid |
| DPPX/DPP6 | dipeptidyl peptidase-like protein 6 |
| EEG | electroencephalography |
| FDG-PET | fluorodeoxyglucose-positron emission tomography |
| FLAIR | fluid-attenuated inversion recovery |
| GABA-BR | γ-aminobutyric acid B receptor |
| GAD | glutamic acid decarboxylase |
| GlyR | glycine receptor |
| HIV | human immunodeficiency virus |
| HSV | herpes simplex virus |
| HHV6 | human herpesvirus 6 |
| ICI | immune checkpoint inhibitor |
| IL | interleukin |
| INF-γ | interferon-γ |
| LE | limbic encephalitis |
| LGI1 | leucine-rich, glioma inactivated 1 |
| mGluR5 | metabotropic glutamate receptor subtype 5 |
| MRI | magnetic resonance imaging |
| NK | natural killer |
| NMDAR | N-methyl-D-aspartate receptor |
| OCB | oligoclonal band |
| PNS | paraneoplastic syndrome |
| SLE | systemic lupus erythematosus |
| SOX1 | sex-determining region Y (SRY)-box 1 |
| SPS | stiff-person syndrome |
| VZV | varicella zoster virus |
References
- Graus, F.; Titulaer, M.J.; Balu, R.; Benseler, S.; Bien, C.G.; Cellucci, T.; Cortese, I.; Dale, R.C.; Gelfand, J.M.; Geschwind, M.; et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016, 15, 391–404. [Google Scholar] [CrossRef]
- Linnoila, J.J.; Binnicker, M.J.; Majed, M.; Klein, C.J.; McKeon, A. CSF herpes virus and autoantibody profiles in the evaluation of encephalitis. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e245. [Google Scholar] [CrossRef]
- Graus, F.; Dalmau, J. Paraneoplastic neurological syndromes in the era of immune-checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2019, 16, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Budhram, A.; Leung, A.; Nicolle, M.W.; Burneo, J.G. Diagnosing autoimmune limbic encephalitis. CMAJ 2019, 191, E529–E534. [Google Scholar] [CrossRef] [PubMed]
- Pape, H.C.; Pare, D. Plastic synaptic networks of the amygdala for the acquisition, expression, and extinction of conditioned fear. Physiol. Rev. 2010, 90, 419–463. [Google Scholar] [CrossRef]
- Aroniadou-Anderjaska, V.; Fritsch, B.; Qashu, F.; Braga, M.F. Pathology and pathophysiology of the amygdala in epileptogenesis and epilepsy. Epilepsy Res. 2008, 78, 102–116. [Google Scholar] [CrossRef]
- Urbach, H.; Soeder, B.M.; Jeub, M.; Klockgether, T.; Meyer, B.; Bien, C.G. Serial MRI of limbic encephalitis. Neuroradiology 2006, 48, 380–386. [Google Scholar] [CrossRef]
- Gadoth, A.; Pittock, S.J.; Dubey, D.; McKeon, A.; Britton, J.W.; Schmeling, J.E.; Smith, A.; Kotsenas, A.L.; Watson, R.E.; Lachance, D.H.; et al. Expanded phenotypes and outcomes among 256 LGI1/CASPR2-IgG-positive patients. Ann. Neurol. 2017, 82, 79–92. [Google Scholar] [CrossRef]
- Liu, X.; Shan, W.; Zhao, X.; Ren, J.; Ren, G.; Chen, C.; Shi, W.; Lv, R.; Li, Z.; Liu, Y.; et al. The Clinical Value of (18) F-FDG-PET in Autoimmune Encephalitis Associated With LGI1 Antibody. Front. Neurol. 2020, 11, 418. [Google Scholar] [CrossRef]
- Boyko, M.; Au, K.L.K.; Casault, C.; de Robles, P.; Pfeffer, G. Systematic review of the clinical spectrum of CASPR2 antibody syndrome. J. Neurol. 2020, 267, 1137–1146. [Google Scholar] [CrossRef]
- Graus, F.; Escudero, D.; Oleaga, L.; Bruna, J.; Villarejo-Galende, A.; Ballabriga, J.; Barcelo, M.I.; Gilo, F.; Popkirov, S.; Stourac, P.; et al. Syndrome and outcome of antibody-negative limbic encephalitis. Eur. J. Neurol. 2018, 25, 1011–1016. [Google Scholar] [CrossRef] [PubMed]
- Giannoccaro, M.P.; Menassa, D.A.; Jacobson, L.; Coutinho, E.; Prota, G.; Lang, B.; Leite, M.I.; Cerundolo, V.; Liguori, R.; Vincent, A. Behaviour and neuropathology in mice injected with human contactin-associated protein 2 antibodies. Brain 2019, 142, 2000–2012. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.; Bi, M.; Murchison, A.G.; Makuch, M.; Bien, C.G.; Chu, K.; Farooque, P.; Gelfand, J.M.; Geschwind, M.D.; Hirsch, L.J.; et al. The importance of early immunotherapy in patients with faciobrachial dystonic seizures. Brain 2018, 141, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Joubert, B.; Saint-Martin, M.; Noraz, N.; Picard, G.; Rogemond, V.; Ducray, F.; Desestret, V.; Psimaras, D.; Delattre, J.Y.; Antoine, J.C.; et al. Characterization of a Subtype of Autoimmune Encephalitis with Anti-Contactin-Associated Protein-like 2 Antibodies in the Cerebrospinal Fluid, Prominent Limbic Symptoms, and Seizures. JAMA Neurol. 2016, 73, 1115–1124. [Google Scholar] [CrossRef]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG subclasses and allotypes: From structure to effector functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef]
- Zuliani, L.; Nosadini, M.; Gastaldi, M.; Spatola, M.; Iorio, R.; Zoccarato, M.; Mariotto, S.; De Gaspari, P.; Perini, F.; Ferrari, S.; et al. Management of antibody-mediated autoimmune encephalitis in adults and children: Literature review and consensus-based practical recommendations. Neurol. Sci. 2019, 40, 2017–2030. [Google Scholar] [CrossRef]
- Bien, C.G.; Vincent, A.; Barnett, M.H.; Becker, A.J.; Blumcke, I.; Graus, F.; Jellinger, K.A.; Reuss, D.E.; Ribalta, T.; Schlegel, J.; et al. Immunopathology of autoantibody-associated encephalitides: Clues for pathogenesis. Brain 2012, 135, 1622–1638. [Google Scholar] [CrossRef]
- Bauer, J.; Bien, C.G. Neuropathology of autoimmune encephalitides. Handb. Clin. Neurol. 2016, 133, 107–120. [Google Scholar] [CrossRef]
- van Sonderen, A.; Petit-Pedrol, M.; Dalmau, J.; Titulaer, M.J. The value of LGI1, Caspr2 and voltage-gated potassium channel antibodies in encephalitis. Nat. Rev. Neurol. 2017, 13, 290–301. [Google Scholar] [CrossRef]
- Alexopoulos, H.; Dalakas, M.C. The immunobiology of autoimmune encephalitides. J. Autoimmun. 2019, 104, 102339. [Google Scholar] [CrossRef]
- Lancaster, E.; Dalmau, J. Neuronal autoantigens—Pathogenesis, associated disorders and antibody testing. Nat. Rev. Neurol. 2012, 8, 380–390. [Google Scholar] [CrossRef] [PubMed]
- van Coevorden-Hameete, M.H.; de Graaff, E.; Titulaer, M.J.; Hoogenraad, C.C.; Sillevis Smitt, P.A. Molecular and cellular mechanisms underlying anti-neuronal antibody mediated disorders of the central nervous system. Autoimmun. Rev. 2014, 13, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Irani, S.R.; Gelfand, J.M.; Al-Diwani, A.; Vincent, A. Cell-surface central nervous system autoantibodies: Clinical relevance and emerging paradigms. Ann. Neurol. 2014, 76, 168–184. [Google Scholar] [CrossRef] [PubMed]
- Pittock, S.J.; Kryzer, T.J.; Lennon, V.A. Paraneoplastic antibodies coexist and predict cancer, not neurological syndrome. Ann. Neurol. 2004, 56, 715–719. [Google Scholar] [CrossRef] [PubMed]
- Dalmau, J.; Geis, C.; Graus, F. Autoantibodies to Synaptic Receptors and Neuronal Cell Surface Proteins in Autoimmune Diseases of the Central Nervous System. Physiol. Rev. 2017, 97, 839–887. [Google Scholar] [CrossRef] [PubMed]
- Ghimire, P.K.U.; Gajurel, B.P.; Karn, R.; Rajbhandari, R.; Paudel, S.; Gautam, N.; Ojha, R. Anti-LGI1, anti-GABABR, and Anti-CASPR2 encephalitides in Asia: A systematic review. Brain Behav. 2020, 10, e01793. [Google Scholar] [CrossRef]
- Joubert, B.; Kerschen, P.; Zekeridou, A.; Desestret, V.; Rogemond, V.; Chaffois, M.O.; Ducray, F.; Larrue, V.; Daubail, B.; Idbaih, A.; et al. Clinical Spectrum of Encephalitis Associated with Antibodies Against the alpha-Amino-3-Hydroxy-5-Methyl-4-Isoxazolepropionic Acid Receptor: Case Series and Review of the Literature. JAMA Neurol. 2015, 72, 1163–1169. [Google Scholar] [CrossRef]
- Fukuda, T.G.; do Rosario, M.S.; Branco, R.C.C.; Fukuda, J.S.; de Souza, E.S.R.A.; Oliveira-Filho, J.; de Jesus, P.A.P. Multiple paraneoplastic antibodies (anti-SOX1, anti-Hu, and anti-Amphiphysin) detected in a patient with limbic encephalitis and small cell lung cancer. Neurol. India 2017, 65, 1127–1128. [Google Scholar] [CrossRef]
- Jia, Y.; Wang, J.; Xue, L.; Hou, Y. Limbic encephalitis associated with AMPA receptor and CRMP5 antibodies: A case report and literature review. Brain Behav. 2020, 10, e01528. [Google Scholar] [CrossRef]
- Yoneda, M. Hashimoto’s Encephalopathy and Autoantibodies. Brain Nerve 2018, 70, 305–314. [Google Scholar] [CrossRef]
- Pilli, D.; Zou, A.; Tea, F.; Dale, R.C.; Brilot, F. Expanding Role of T Cells in Human Autoimmune Diseases of the Central Nervous System. Front. Immunol. 2017, 8, 652. [Google Scholar] [CrossRef] [PubMed]
- Melzer, N.; Meuth, S.G.; Wiendl, H. CD8+ T cells and neuronal damage: Direct and collateral mechanisms of cytotoxicity and impaired electrical excitability. FASEB J. 2009, 23, 3659–3673. [Google Scholar] [CrossRef] [PubMed]
- Ehling, P.; Melzer, N.; Budde, T.; Meuth, S.G. CD8(+) T Cell-Mediated Neuronal Dysfunction and Degeneration in Limbic Encephalitis. Front. Neurol. 2015, 6, 163. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, N.J.; Sutton, V.R.; Sedelies, K.A.; Ciccone, A.; Jenkins, M.; Turner, S.J.; Bird, P.I.; Trapani, J.A. Cytotoxic T lymphocyte-induced killing in the absence of granzymes A and B is unique and distinct from both apoptosis and perforin-dependent lysis. J. Cell Biol. 2006, 173, 133–144. [Google Scholar] [CrossRef]
- Kreutzfeldt, M.; Bergthaler, A.; Fernandez, M.; Bruck, W.; Steinbach, K.; Vorm, M.; Coras, R.; Blumcke, I.; Bonilla, W.V.; Fleige, A.; et al. Neuroprotective intervention by interferon-gamma blockade prevents CD8+ T cell-mediated dendrite and synapse loss. J. Exp. Med. 2013, 210, 2087–2103. [Google Scholar] [CrossRef]
- Chung, W.S.; Welsh, C.A.; Barres, B.A.; Stevens, B. Do glia drive synaptic and cognitive impairment in disease? Nat. Neurosci. 2015, 18, 1539–1545. [Google Scholar] [CrossRef]
- Wesselingh, R.; Butzkueven, H.; Buzzard, K.; Tarlinton, D.; O’Brien, T.J.; Monif, M. Innate Immunity in the Central Nervous System: A Missing Piece of the Autoimmune Encephalitis Puzzle? Front. Immunol. 2019, 10, 2066. [Google Scholar] [CrossRef]
- Khan, N.L.; Jeffree, M.A.; Good, C.; Macleod, W.; Al-Sarraj, S. Histopathology of VGKC antibody-associated limbic encephalitis. Neurology 2009, 72, 1703–1705. [Google Scholar] [CrossRef]
- Amhaoul, H.; Hamaide, J.; Bertoglio, D.; Reichel, S.N.; Verhaeghe, J.; Geerts, E.; Van Dam, D.; De Deyn, P.P.; Kumar-Singh, S.; Katsifis, A.; et al. Brain inflammation in a chronic epilepsy model: Evolving pattern of the translocator protein during epileptogenesis. Neurobiol. Dis. 2015, 82, 526–539. [Google Scholar] [CrossRef]
- Yang, M.T.; Lin, Y.C.; Ho, W.H.; Liu, C.L.; Lee, W.T. Everolimus is better than rapamycin in attenuating neuroinflammation in kainic acid-induced seizures. J. Neuroinflamm. 2017, 14, 15. [Google Scholar] [CrossRef]
- Tomczak, A.; Su, E.; Tugizova, M.; Carlson, A.M.; Kipp, L.B.; Feng, H.; Han, M.H. A case of GFAP-astroglial autoimmunity presenting with reversible parkinsonism. Mult. Scler. Relat. Disord. 2019, 39, 101900. [Google Scholar] [CrossRef] [PubMed]
- Ismail, F.S.; Faustmann, P.M. Astrocytes and their potential role in anti-NMDA receptor encephalitis. Med. Hypotheses 2020, 139, 109612. [Google Scholar] [CrossRef] [PubMed]
- Habbas, S.; Santello, M.; Becker, D.; Stubbe, H.; Zappia, G.; Liaudet, N.; Klaus, F.R.; Kollias, G.; Fontana, A.; Pryce, C.R.; et al. Neuroinflammatory TNFalpha Impairs Memory via Astrocyte Signaling. Cell 2015, 163, 1730–1741. [Google Scholar] [CrossRef] [PubMed]
- Hebert, J.; Gros, P.; Lapointe, S.; Amtashar, F.S.; Steriade, C.; Maurice, C.; Wennberg, R.A.; Day, G.S.; Tang-Wai, D.F. Searching for autoimmune encephalitis: Beware of normal CSF. J. Neuroimmunol. 2020, 345, 577285. [Google Scholar] [CrossRef]
- Hansen, N.; Schwing, K.; Onder, D.; Widman, G.; Leelaarporn, P.; Prusseit, I.; Surges, R.; Melzer, N.; Gross, C.; Becker, A.J.; et al. Low CSF CD4/CD8+ T-cell proportions are associated with blood-CSF barrier dysfunction in limbic encephalitis. Epilepsy Behav. 2020, 102, 106682. [Google Scholar] [CrossRef]
- Lin, Y.T.; Yang, X.; Lv, J.W.; Liu, X.W.; Wang, S.J. CXCL13 Is A Biomarker of Anti-Leucine-Rich Glioma-Inactivated Protein 1 Encephalitis Patients. Neuropsychiatr. Dis. Treat. 2019, 15, 2909–2915. [Google Scholar] [CrossRef]
- Roberts, W.K.; Blachere, N.E.; Frank, M.O.; Dousmanis, A.; Ransohoff, R.M.; Darnell, R.B. A destructive feedback loop mediated by CXCL10 in central nervous system inflammatory disease. Ann. Neurol. 2015, 78, 619–629. [Google Scholar] [CrossRef]
- Kortvelyessy, P.; Goihl, A.; Guttek, K.; Schraven, B.; Pruss, H.; Reinhold, D. Serum and CSF cytokine levels mirror different neuroimmunological mechanisms in patients with LGI1 and Caspr2 encephalitis. Cytokine 2020, 135, 155226. [Google Scholar] [CrossRef]
- Michael, B.D.; Griffiths, M.J.; Granerod, J.; Brown, D.; Davies, N.W.; Borrow, R.; Solomon, T. Characteristic Cytokine and Chemokine Profiles in Encephalitis of Infectious, Immune-Mediated, and Unknown Aetiology. PLoS ONE 2016, 11, e0146288. [Google Scholar] [CrossRef]
- Jiang, J.X.; Fewings, N.; Dervish, S.; Fois, A.F.; Duma, S.R.; Silsby, M.; Bandodkar, S.; Ramanathan, S.; Bleasel, A.; John, B.; et al. Novel Surrogate Markers of CNS Inflammation in CSF in the Diagnosis of Autoimmune Encephalitis. Front. Neurol. 2019, 10, 1390. [Google Scholar] [CrossRef]
- Spolski, R.; Leonard, W.J. Interleukin-21: A double-edged sword with therapeutic potential. Nat. Rev. Drug Discov. 2014, 13, 379–395. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Guo, S.; Hibbert, J.M.; Jain, V.; Singh, N.; Wilson, N.O.; Stiles, J.K. CXCL10/IP-10 in infectious diseases pathogenesis and potential therapeutic implications. Cytokine Growth Factor Rev. 2011, 22, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.J.; Lee, S.T.; Moon, J.; Sunwoo, J.S.; Byun, J.I.; Lim, J.A.; Shin, Y.W.; Jun, J.S.; Lee, H.S.; Lee, W.J.; et al. Anti-LGI1 encephalitis is associated with unique HLA subtypes. Ann. Neurol. 2017, 81, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Binks, S.; Varley, J.; Lee, W.; Makuch, M.; Elliott, K.; Gelfand, J.M.; Jacob, S.; Leite, M.I.; Maddison, P.; Chen, M.; et al. Distinct HLA associations of LGI1 and CASPR2-antibody diseases. Brain 2018, 141, 2263–2271. [Google Scholar] [CrossRef] [PubMed]
- Muniz-Castrillo, S.; Ambati, A.; Dubois, V.; Vogrig, A.; Joubert, B.; Rogemond, V.; Picard, G.; Lin, L.; Fabien, N.; Mignot, E.; et al. Primary DQ effect in the association between HLA and neurological syndromes with anti-GAD65 antibodies. J. Neurol. 2020, 267, 1906–1911. [Google Scholar] [CrossRef] [PubMed]
- Shojima, Y.; Nishioka, K.; Watanabe, M.; Jo, T.; Tanaka, K.; Takashima, H.; Noda, K.; Okuma, Y.; Urabe, T.; Yokoyama, K.; et al. Clinical Characterization of Definite Autoimmune Limbic Encephalitis: A 30-case Series. Intern. Med. 2019, 58, 3369–3378. [Google Scholar] [CrossRef]
- Dalmau, J.; Lancaster, E.; Martinez-Hernandez, E.; Rosenfeld, M.R.; Balice-Gordon, R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol. 2011, 10, 63–74. [Google Scholar] [CrossRef]
- Small, M.; Treilleux, I.; Couillault, C.; Pissaloux, D.; Picard, G.; Paindavoine, S.; Attignon, V.; Wang, Q.; Rogemond, V.; Lay, S.; et al. Genetic alterations and tumor immune attack in Yo paraneoplastic cerebellar degeneration. Acta Neuropathol. 2018, 135, 569–579. [Google Scholar] [CrossRef]
- Melzer, N.; Meuth, S.G.; Wiendl, H. Paraneoplastic and non-paraneoplastic autoimmunity to neurons in the central nervous system. J. Neurol. 2013, 260, 1215–1233. [Google Scholar] [CrossRef]
- Vogrig, A.; Gigli, G.L.; Segatti, S.; Corazza, E.; Marini, A.; Bernardini, A.; Valent, F.; Fabris, M.; Curcio, F.; Brigo, F.; et al. Epidemiology of paraneoplastic neurological syndromes: A population-based study. J. Neurol. 2020, 267, 26–35. [Google Scholar] [CrossRef]
- Gultekin, S.H.; Rosenfeld, M.R.; Voltz, R.; Eichen, J.; Posner, J.B.; Dalmau, J. Paraneoplastic limbic encephalitis: Neurological symptoms, immunological findings and tumour association in 50 patients. Brain 2000, 123 Pt 7, 1481–1494. [Google Scholar] [CrossRef]
- Erkmen, C.P.; Fadul, C.E.; Dalmau, J.; Erkmen, K. Thymoma-associated paraneoplastic encephalitis (TAPE): Diagnosis and treatment of a potentially fatal condition. J. Thorac. Cardiovasc. Surg. 2011, 141, e17–e20. [Google Scholar] [CrossRef] [PubMed]
- van Coevorden-Hameete, M.H.; de Bruijn, M.; de Graaff, E.; Bastiaansen, D.; Schreurs, M.W.J.; Demmers, J.A.A.; Ramberger, M.; Hulsenboom, E.S.P.; Nagtzaam, M.M.P.; Boukhrissi, S.; et al. The expanded clinical spectrum of anti-GABABR encephalitis and added value of KCTD16 autoantibodies. Brain 2019, 142, 1631–1643. [Google Scholar] [CrossRef] [PubMed]
- Irani, S.R.; Alexander, S.; Waters, P.; Kleopa, K.A.; Pettingill, P.; Zuliani, L.; Peles, E.; Buckley, C.; Lang, B.; Vincent, A. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan’s syndrome and acquired neuromyotonia. Brain 2010, 133, 2734–2748. [Google Scholar] [CrossRef] [PubMed]
- Irani, S.R.; Pettingill, P.; Kleopa, K.A.; Schiza, N.; Waters, P.; Mazia, C.; Zuliani, L.; Watanabe, O.; Lang, B.; Buckley, C.; et al. Morvan syndrome: Clinical and serological observations in 29 cases. Ann. Neurol. 2012, 72, 241–255. [Google Scholar] [CrossRef]
- Dalmau, J.; Rosenfeld, M.R. Paraneoplastic syndromes of the CNS. Lancet Neurol. 2008, 7, 327–340. [Google Scholar] [CrossRef]
- Armangue, T.; Spatola, M.; Vlagea, A.; Mattozzi, S.; Carceles-Cordon, M.; Martinez-Heras, E.; Llufriu, S.; Muchart, J.; Erro, M.E.; Abraira, L.; et al. Frequency, symptoms, risk factors, and outcomes of autoimmune encephalitis after herpes simplex encephalitis: A prospective observational study and retrospective analysis. Lancet Neurol. 2018, 17, 760–772. [Google Scholar] [CrossRef]
- Mohammad, S.S.; Sinclair, K.; Pillai, S.; Merheb, V.; Aumann, T.D.; Gill, D.; Dale, R.C.; Brilot, F. Herpes simplex encephalitis relapse with chorea is associated with autoantibodies to N-Methyl-D-aspartate receptor or dopamine-2 receptor. Mov. Disord. 2014, 29, 117–122. [Google Scholar] [CrossRef]
- Peters, J.; Wesley, S.F. Case of concurrent herpes simplex and autoimmune encephalitis. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7. [Google Scholar] [CrossRef]
- Hoftberger, R.; Armangue, T.; Leypoldt, F.; Graus, F.; Dalmau, J. Clinical Neuropathology practice guide 4-2013: Post-herpes simplex encephalitis: N-methyl-Daspartate receptor antibodies are part of the problem. Clin. Neuropathol. 2013, 32, 251–254. [Google Scholar] [CrossRef]
- Seeley, W.W.; Marty, F.M.; Holmes, T.M.; Upchurch, K.; Soiffer, R.J.; Antin, J.H.; Baden, L.R.; Bromfield, E.B. Post-transplant acute limbic encephalitis: Clinical features and relationship to HHV6. Neurology 2007, 69, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Dubey, D.; David, W.S.; Amato, A.A.; Reynolds, K.L.; Clement, N.F.; Chute, D.F.; Cohen, J.V.; Lawrence, D.P.; Mooradian, M.J.; Sullivan, R.J.; et al. Varied phenotypes and management of immune checkpoint inhibitor-associated neuropathies. Neurology 2019, 93, e1093–e1103. [Google Scholar] [CrossRef] [PubMed]
- Vogrig, A.; Muniz-Castrillo, S.; Joubert, B.; Picard, G.; Rogemond, V.; Marchal, C.; Chiappa, A.M.; Chanson, E.; Skowron, F.; Leblanc, A.; et al. Central nervous system complications associated with immune checkpoint inhibitors. J. Neurol. Neurosurg. Psychiatry 2020, 91, 772–778. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, K.P.; Romero, R.S.; Gonzalez, G.; Dix, J.E.; Lowy, I.; Fury, M. Anti-Hu-Associated Autoimmune Limbic Encephalitis in a Patient with PD-1 Inhibitor-Responsive Myxoid Chondrosarcoma. Oncologist 2018, 23, 118–120. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.; Jaffer, M.; Verma, N.; Mokhtari, S.; Ramsakal, A.; Peguero, E. Immune checkpoint inhibitor induced anti-glutamic acid decarboxylase 65 (Anti-GAD 65) limbic encephalitis responsive to intravenous immunoglobulin and plasma exchange. J. Neurol. 2020, 267, 1023–1025. [Google Scholar] [CrossRef]
- Vogrig, A.; Fouret, M.; Joubert, B.; Picard, G.; Rogemond, V.; Pinto, A.L.; Muniz-Castrillo, S.; Roger, M.; Raimbourg, J.; Dayen, C.; et al. Increased frequency of anti-Ma2 encephalitis associated with immune checkpoint inhibitors. Neurol. Neuroimmunol. Neuroinflamm. 2019, 6. [Google Scholar] [CrossRef]
- Brown, M.P.; Hissaria, P.; Hsieh, A.H.; Kneebone, C.; Vallat, W. Autoimmune limbic encephalitis with anti-contactin-associated protein-like 2 antibody secondary to pembrolizumab therapy. J. Neuroimmunol. 2017, 305, 16–18. [Google Scholar] [CrossRef]
- Hacohen, Y.; Singh, R.; Rossi, M.; Lang, B.; Hemingway, C.; Lim, M.; Vincent, A. Clinical relevance of voltage-gated potassium channel-complex antibodies in children. Neurology 2015, 85, 967–975. [Google Scholar] [CrossRef]
- Lang, B.; Makuch, M.; Moloney, T.; Dettmann, I.; Mindorf, S.; Probst, C.; Stoecker, W.; Buckley, C.; Newton, C.R.; Leite, M.I.; et al. Intracellular and non-neuronal targets of voltage-gated potassium channel complex antibodies. J. Neurol. Neurosurg. Psychiatry 2017, 88, 353–361. [Google Scholar] [CrossRef]
- Lilleker, J.B.; Jones, M.S.; Mohanraj, R. VGKC complex antibodies in epilepsy: Diagnostic yield and therapeutic implications. Seizure 2013, 22, 776–779. [Google Scholar] [CrossRef] [PubMed]
- van Sonderen, A.; Schreurs, M.W.; Wirtz, P.W.; Sillevis Smitt, P.A.; Titulaer, M.J. From VGKC to LGI1 and Caspr2 encephalitis: The evolution of a disease entity over time. Autoimmun. Rev. 2016, 15, 970–974. [Google Scholar] [CrossRef] [PubMed]
- van Sonderen, A.; Schreurs, M.W.; de Bruijn, M.A.; Boukhrissi, S.; Nagtzaam, M.M.; Hulsenboom, E.S.; Enting, R.H.; Thijs, R.D.; Wirtz, P.W.; Sillevis Smitt, P.A.; et al. The relevance of VGKC positivity in the absence of LGI1 and Caspr2 antibodies. Neurology 2016, 86, 1692–1699. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.J.; Lennon, V.A.; Aston, P.A.; McKeon, A.; O’Toole, O.; Quek, A.; Pittock, S.J. Insights from LGI1 and CASPR2 potassium channel complex autoantibody subtyping. JAMA Neurol. 2013, 70, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Michael, S.; Waters, P.; Irani, S.R. Stop testing for autoantibodies to the VGKC-complex: Only request LGI1 and CASPR2. Pr. Neurol. 2020, 20, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Fukata, Y.; Yokoi, N.; Miyazaki, Y.; Fukata, M. The LGI1-ADAM22 protein complex in synaptic transmission and synaptic disorders. Neurosci. Res. 2017, 116, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Ohkawa, T.; Satake, S.; Yokoi, N.; Miyazaki, Y.; Ohshita, T.; Sobue, G.; Takashima, H.; Watanabe, O.; Fukata, Y.; Fukata, M. Identification and characterization of GABA(A) receptor autoantibodies in autoimmune encephalitis. J. Neurosci. 2014, 34, 8151–8163. [Google Scholar] [CrossRef]
- Boillot, M.; Huneau, C.; Marsan, E.; Lehongre, K.; Navarro, V.; Ishida, S.; Dufresnois, B.; Ozkaynak, E.; Garrigue, J.; Miles, R.; et al. Glutamatergic neuron-targeted loss of LGI1 epilepsy gene results in seizures. Brain 2014, 137, 2984–2996. [Google Scholar] [CrossRef]
- Ohkawa, T.; Fukata, Y.; Yamasaki, M.; Miyazaki, T.; Yokoi, N.; Takashima, H.; Watanabe, M.; Watanabe, O.; Fukata, M. Autoantibodies to epilepsy-related LGI1 in limbic encephalitis neutralize LGI1-ADAM22 interaction and reduce synaptic AMPA receptors. J. Neurosci. 2013, 33, 18161–18174. [Google Scholar] [CrossRef]
- Petit-Pedrol, M.; Sell, J.; Planaguma, J.; Mannara, F.; Radosevic, M.; Haselmann, H.; Ceanga, M.; Sabater, L.; Spatola, M.; Soto, D.; et al. LGI1 antibodies alter Kv1.1 and AMPA receptors changing synaptic excitability, plasticity and memory. Brain 2018, 141, 3144–3159. [Google Scholar] [CrossRef]
- Irani, S.R.; Michell, A.W.; Lang, B.; Pettingill, P.; Waters, P.; Johnson, M.R.; Schott, J.M.; Armstrong, R.J.; Zagami, A.S.; Bleasel, A.; et al. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann. Neurol. 2011, 69, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Malter, M.P.; Frisch, C.; Schoene-Bake, J.C.; Helmstaedter, C.; Wandinger, K.P.; Stoecker, W.; Urbach, H.; Surges, R.; Elger, C.E.; Vincent, A.V.; et al. Outcome of limbic encephalitis with VGKC-complex antibodies: Relation to antigenic specificity. J. Neurol. 2014, 261, 1695–1705. [Google Scholar] [CrossRef] [PubMed]
- Bastiaansen, A.E.M.; van Sonderen, A.; Titulaer, M.J. Autoimmune encephalitis with anti-leucine-rich glioma-inactivated 1 or anti-contactin-associated protein-like 2 antibodies (formerly called voltage-gated potassium channel-complex antibodies). Curr. Opin. Neurol. 2017, 30, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Saint-Martin, M.; Joubert, B.; Pellier-Monnin, V.; Pascual, O.; Noraz, N.; Honnorat, J. Contactin-associated protein-like 2, a protein of the neurexin family involved in several human diseases. Eur. J. Neurosci. 2018, 48, 1906–1923. [Google Scholar] [CrossRef]
- Ogawa, Y.; Oses-Prieto, J.; Kim, M.Y.; Horresh, I.; Peles, E.; Burlingame, A.L.; Trimmer, J.S.; Meijer, D.; Rasband, M.N. ADAM22, a Kv1 channel-interacting protein, recruits membrane-associated guanylate kinases to juxtaparanodes of myelinated axons. J. Neurosci. 2010, 30, 1038–1048. [Google Scholar] [CrossRef]
- Patterson, K.R.; Dalmau, J.; Lancaster, E. Mechanisms of Caspr2 antibodies in autoimmune encephalitis and neuromyotonia. Ann. Neurol. 2018, 83, 40–51. [Google Scholar] [CrossRef]
- Dawes, J.M.; Weir, G.A.; Middleton, S.J.; Patel, R.; Chisholm, K.I.; Pettingill, P.; Peck, L.J.; Sheridan, J.; Shakir, A.; Jacobson, L.; et al. Immune or Genetic-Mediated Disruption of CASPR2 Causes Pain Hypersensitivity Due to Enhanced Primary Afferent Excitability. Neuron 2018, 97, 806–822.e10. [Google Scholar] [CrossRef]
- Klein, C.J.; Lennon, V.A.; Aston, P.A.; McKeon, A.; Pittock, S.J. Chronic pain as a manifestation of potassium channel-complex autoimmunity. Neurology 2012, 79, 1136–1144. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef]
- Sprengel, R. Role of AMPA receptors in synaptic plasticity. Cell Tissue Res. 2006, 326, 447–455. [Google Scholar] [CrossRef]
- Lai, M.; Hughes, E.G.; Peng, X.; Zhou, L.; Gleichman, A.J.; Shu, H.; Mata, S.; Kremens, D.; Vitaliani, R.; Geschwind, M.D.; et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann. Neurol. 2009, 65, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Hughes, E.G.; Moscato, E.H.; Parsons, T.D.; Dalmau, J.; Balice-Gordon, R.J. Cellular plasticity induced by anti-alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor encephalitis antibodies. Ann. Neurol. 2015, 77, 381–398. [Google Scholar] [CrossRef] [PubMed]
- Laurido-Soto, O.; Brier, M.R.; Simon, L.E.; McCullough, A.; Bucelli, R.C.; Day, G.S. Patient characteristics and outcome associations in AMPA receptor encephalitis. J. Neurol. 2019, 266, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, F.; Bockaert, J.; Collingridge, G.L.; Conn, P.J.; Ferraguti, F.; Schoepp, D.D.; Wroblewski, J.T.; Pin, J.P. Metabotropic glutamate receptors: From the workbench to the bedside. Neuropharmacology 2011, 60, 1017–1041. [Google Scholar] [CrossRef] [PubMed]
- Spatola, M.; Sabater, L.; Planaguma, J.; Martinez-Hernandez, E.; Armangue, T.; Pruss, H.; Iizuka, T.; Caparo Oblitas, R.L.; Antoine, J.C.; Li, R.; et al. Encephalitis with mGluR5 antibodies: Symptoms and antibody effects. Neurology 2018, 90, e1964–e1972. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, E.; Martinez-Hernandez, E.; Titulaer, M.J.; Boulos, M.; Weaver, S.; Antoine, J.C.; Liebers, E.; Kornblum, C.; Bien, C.G.; Honnorat, J.; et al. Antibodies to metabotropic glutamate receptor 5 in the Ophelia syndrome. Neurology 2011, 77, 1698–1701. [Google Scholar] [CrossRef]
- Boronat, A.; Gelfand, J.M.; Gresa-Arribas, N.; Jeong, H.Y.; Walsh, M.; Roberts, K.; Martinez-Hernandez, E.; Rosenfeld, M.R.; Balice-Gordon, R.; Graus, F.; et al. Encephalitis and antibodies to dipeptidyl-peptidase-like protein-6, a subunit of Kv4.2 potassium channels. Ann. Neurol. 2013, 73, 120–128. [Google Scholar] [CrossRef]
- Nadal, M.S.; Ozaita, A.; Amarillo, Y.; Vega-Saenz de Miera, E.; Ma, Y.; Mo, W.; Goldberg, E.M.; Misumi, Y.; Ikehara, Y.; Neubert, T.A.; et al. The CD26-related dipeptidyl aminopeptidase-like protein DPPX is a critical component of neuronal A-type K+ channels. Neuron 2003, 37, 449–461. [Google Scholar] [CrossRef]
- Clark, B.D.; Kwon, E.; Maffie, J.; Jeong, H.Y.; Nadal, M.; Strop, P.; Rudy, B. DPP6 Localization in Brain Supports Function as a Kv4 Channel Associated Protein. Front. Mol. Neurosci. 2008, 1, 8. [Google Scholar] [CrossRef]
- Tobin, W.O.; Lennon, V.A.; Komorowski, L.; Probst, C.; Clardy, S.L.; Aksamit, A.J.; Appendino, J.P.; Lucchinetti, C.F.; Matsumoto, J.Y.; Pittock, S.J.; et al. DPPX potassium channel antibody: Frequency, clinical accompaniments, and outcomes in 20 patients. Neurology 2014, 83, 1797–1803. [Google Scholar] [CrossRef]
- Hara, M.; Arino, H.; Petit-Pedrol, M.; Sabater, L.; Titulaer, M.J.; Martinez-Hernandez, E.; Schreurs, M.W.; Rosenfeld, M.R.; Graus, F.; Dalmau, J. DPPX antibody-associated encephalitis: Main syndrome and antibody effects. Neurology 2017, 88, 1340–1348. [Google Scholar] [CrossRef] [PubMed]
- Linnoila, J.J.; Rosenfeld, M.R.; Dalmau, J. Neuronal surface antibody-mediated autoimmune encephalitis. Semin. Neurol. 2014, 34, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Emson, P.C. GABA(B) receptors: Structure and function. Prog. Brain Res. 2007, 160, 43–57. [Google Scholar] [CrossRef]
- Bettler, B.; Kaupmann, K.; Mosbacher, J.; Gassmann, M. Molecular structure and physiological functions of GABA(B) receptors. Physiol. Rev. 2004, 84, 835–867. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, E.; Lai, M.; Peng, X.; Hughes, E.; Constantinescu, R.; Raizer, J.; Friedman, D.; Skeen, M.B.; Grisold, W.; Kimura, A.; et al. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: Case series and characterisation of the antigen. Lancet Neurol. 2010, 9, 67–76. [Google Scholar] [CrossRef]
- Hoftberger, R.; Titulaer, M.J.; Sabater, L.; Dome, B.; Rozsas, A.; Hegedus, B.; Hoda, M.A.; Laszlo, V.; Ankersmit, H.J.; Harms, L.; et al. Encephalitis and GABAB receptor antibodies: Novel findings in a new case series of 20 patients. Neurology 2013, 81, 1500–1506. [Google Scholar] [CrossRef]
- Jeffery, O.J.; Lennon, V.A.; Pittock, S.J.; Gregory, J.K.; Britton, J.W.; McKeon, A. GABAB receptor autoantibody frequency in service serologic evaluation. Neurology 2013, 81, 882–887. [Google Scholar] [CrossRef]
- Dogan Onugoren, M.; Deuretzbacher, D.; Haensch, C.A.; Hagedorn, H.J.; Halve, S.; Isenmann, S.; Kramme, C.; Lohner, H.; Melzer, N.; Monotti, R.; et al. Limbic encephalitis due to GABAB and AMPA receptor antibodies: A case series. J. Neurol. Neurosurg. Psychiatry 2015, 86, 965–972. [Google Scholar] [CrossRef]
- Arino, H.; Hoftberger, R.; Gresa-Arribas, N.; Martinez-Hernandez, E.; Armangue, T.; Kruer, M.C.; Arpa, J.; Domingo, J.; Rojc, B.; Bataller, L.; et al. Paraneoplastic Neurological Syndromes and Glutamic Acid Decarboxylase Antibodies. JAMA Neurol. 2015, 72, 874–881. [Google Scholar] [CrossRef]
- Zhang, X.; Lang, Y.; Sun, L.; Zhang, W.; Lin, W.; Cui, L. Clinical characteristics and prognostic analysis of anti-gamma-aminobutyric acid-B (GABA-B) receptor encephalitis in Northeast China. BMC Neurol. 2020, 20, 1. [Google Scholar] [CrossRef]
- Blinder, T.; Lewerenz, J. Cerebrospinal Fluid Findings in Patients With Autoimmune Encephalitis—A Systematic Analysis. Front. Neurol. 2019, 10, 804. [Google Scholar] [CrossRef] [PubMed]
- Moser, A.; Hanssen, H.; Wandinger, K.P. Excessively increased CSF glutamate levels in GABAB-receptor antibody associated encephalitis: A case report. J. Neurol. Sci. 2018, 388, 10–11. [Google Scholar] [CrossRef] [PubMed]
- Golombeck, K.S.; Bonte, K.; Monig, C.; van Loo, K.M.; Hartwig, M.; Schwindt, W.; Widman, G.; Lindenau, M.; Becker, A.J.; Glatzel, M.; et al. Evidence of a pathogenic role for CD8(+) T cells in anti-GABAB receptor limbic encephalitis. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e232. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Bu, H.; He, J.; Zhao, Z.; Han, W.; Gao, R.; Li, X.; Li, Q.; Guo, X.; Zou, Y. The gamma-aminobutyric acid-B receptor (GABAB) encephalitis: Clinical manifestations and response to immunotherapy. Int. J. Neurosci. 2018, 128, 627–633. [Google Scholar] [CrossRef]
- Solimena, M.; Folli, F.; Aparisi, R.; Pozza, G.; De Camilli, P. Autoantibodies to GABA-ergic neurons and pancreatic beta cells in stiff-man syndrome. N. Engl. J. Med. 1990, 322, 1555–1560. [Google Scholar] [CrossRef] [PubMed]
- Ali, F.; Rowley, M.; Jayakrishnan, B.; Teuber, S.; Gershwin, M.E.; Mackay, I.R. Stiff-person syndrome (SPS) and anti-GAD-related CNS degenerations: Protean additions to the autoimmune central neuropathies. J. Autoimmun. 2011, 37, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Fenalti, G.; Buckle, A.M. Structural biology of the GAD autoantigen. Autoimmun. Rev. 2010, 9, 148–152. [Google Scholar] [CrossRef]
- Dalakas, M.C. Progress and stiff challenges in understanding the role of GAD-antibodies in stiff-person syndrome. Exp. Neurol. 2013, 247, 303–307. [Google Scholar] [CrossRef]
- Walikonis, J.E.; Lennon, V.A. Radioimmunoassay for glutamic acid decarboxylase (GAD65) autoantibodies as a diagnostic aid for stiff-man syndrome and a correlate of susceptibility to type 1 diabetes mellitus. Mayo Clin. Proc. 1998, 73, 1161–1166. [Google Scholar] [CrossRef]
- Munoz-Lopetegi, A.; de Bruijn, M.; Boukhrissi, S.; Bastiaansen, A.E.M.; Nagtzaam, M.M.P.; Hulsenboom, E.S.P.; Boon, A.J.W.; Neuteboom, R.F.; de Vries, J.M.; Sillevis Smitt, P.A.E.; et al. Neurologic syndromes related to anti-GAD65: Clinical and serologic response to treatment. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7. [Google Scholar] [CrossRef]
- Raju, R.; Foote, J.; Banga, J.P.; Hall, T.R.; Padoa, C.J.; Dalakas, M.C.; Ortqvist, E.; Hampe, C.S. Analysis of GAD65 autoantibodies in Stiff-Person syndrome patients. J. Immunol. 2005, 175, 7755–7762. [Google Scholar] [CrossRef]
- Vianello, M.; Giometto, B.; Vassanelli, S.; Canato, M.; Betterle, C.; Mucignat, C. Peculiar labeling of cultured hippocampal neurons by different sera harboring anti-glutamic acid decarboxylase autoantibodies (GAD-Ab). Exp. Neurol. 2006, 202, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Burton, A.R.; Baquet, Z.; Eisenbarth, G.S.; Tisch, R.; Smeyne, R.; Workman, C.J.; Vignali, D.A. Central nervous system destruction mediated by glutamic acid decarboxylase-specific CD4+ T cells. J. Immunol. 2010, 184, 4863–4870. [Google Scholar] [CrossRef] [PubMed]
- Cheramy, M.; Hampe, C.S.; Ludvigsson, J.; Casas, R. Characteristics of in-vitro phenotypes of glutamic acid decarboxylase 65 autoantibodies in high-titre individuals. Clin. Exp. Immunol. 2013, 171, 247–254. [Google Scholar] [CrossRef]
- Malter, M.P.; Helmstaedter, C.; Urbach, H.; Vincent, A.; Bien, C.G. Antibodies to glutamic acid decarboxylase define a form of limbic encephalitis. Ann. Neurol. 2010, 67, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.; Alexopoulos, H.; Pettingill, P.; McMenamin, M.; Deacon, R.; Erdelyi, F.; Szabo, G.; Buckley, C.J.; Vincent, A. Immunization against GAD induces antibody binding to GAD-independent antigens and brainstem GABAergic neuronal loss. PLoS ONE 2013, 8, e72921. [Google Scholar] [CrossRef]
- Haberlandt, E.; Bast, T.; Ebner, A.; Holthausen, H.; Kluger, G.; Kravljanac, R.; Kroll-Seger, J.; Kurlemann, G.; Makowski, C.; Rostasy, K.; et al. Limbic encephalitis in children and adolescents. Arch. Dis. Child. 2011, 96, 186–191. [Google Scholar] [CrossRef]
- Baizabal-Carvallo, J.F. The neurological syndromes associated with glutamic acid decarboxylase antibodies. J. Autoimmun. 2019, 101, 35–47. [Google Scholar] [CrossRef]
- Gagnon, M.M.; Savard, M. Limbic Encephalitis Associated With GAD65 Antibodies: Brief Review of the Relevant literature. Can. J. Neurol. Sci. 2016, 43, 486–493. [Google Scholar] [CrossRef]
- Hansen, N.; Widman, G.; Witt, J.A.; Wagner, J.; Becker, A.J.; Elger, C.E.; Helmstaedter, C. Seizure control and cognitive improvement via immunotherapy in late onset epilepsy patients with paraneoplastic versus GAD65 autoantibody-associated limbic encephalitis. Epilepsy Behav. 2016, 65, 18–24. [Google Scholar] [CrossRef]
- Liu, B.; Zhou, Y.; Meng, L.; Skinner, H. A Survival Case of Super-refractory Status Epilepticus due to Glutamic Acid Decarboxylase Antibodies-associated Limbic Encephalitis. Cureus 2018, 10, e3125. [Google Scholar] [CrossRef] [PubMed]
- Boronat, A.; Sabater, L.; Saiz, A.; Dalmau, J.; Graus, F. GABA(B) receptor antibodies in limbic encephalitis and anti-GAD-associated neurologic disorders. Neurology 2011, 76, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Ringelstein, M.; Haas, J.; Serysheva, I.I.; Komorowski, L.; Fechner, K.; Wandinger, K.P.; Albrecht, P.; Hefter, H.; Moser, A.; et al. Inositol 1,4,5-trisphosphate receptor type 1 autoantibodies in paraneoplastic and non-paraneoplastic peripheral neuropathy. J. Neuroinflamm. 2016, 13, 278. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Berger, B.; Bischler, P.; Dersch, R.; Hottenrott, T.; Rauer, S.; Stich, O. “Non-classical” paraneoplastic neurological syndromes associated with well-characterized antineuronal antibodies as compared to “classical” syndromes—More frequent than expected. J. Neurol. Sci. 2015, 352, 58–61. [Google Scholar] [CrossRef]
- Sun, X.; Tan, J.; Sun, H.; Liu, Y.; Guan, W.; Jia, J.; Wang, Z. Anti-SOX1 Antibodies in Paraneoplastic Neurological Syndrome. J. Clin. Neurol. 2020, 16, 530–546. [Google Scholar] [CrossRef]
- Yamamoto, T.; Tsuji, S. Anti-Ma2-associated encephalitis and paraneoplastic limbic encephalitis. Brain Nerve 2010, 62, 838–851. [Google Scholar]
- Shen, K.; Xu, Y.; Guan, H.; Zhong, W.; Chen, M.; Zhao, J.; Li, L.; Wang, M. Paraneoplastic limbic encephalitis associated with lung cancer. Sci. Rep. 2018, 8, 6792. [Google Scholar] [CrossRef]
- Ortega Suero, G.; Sola-Valls, N.; Escudero, D.; Saiz, A.; Graus, F. Anti-Ma and anti-Ma2-associated paraneoplastic neurological syndromes. Neurologia 2018, 33, 18–27. [Google Scholar] [CrossRef]
- Kohler, W.; Ehrlich, S.; Dohmen, C.; Haubitz, M.; Hoffmann, F.; Schmidt, S.; Klingel, R.; Kraft, A.; Neumann-Haefelin, T.; Topka, H.; et al. Tryptophan immunoadsorption for the treatment of autoimmune encephalitis. Eur. J. Neurol. 2015, 22, 203–206. [Google Scholar] [CrossRef]
- Heine, J.; Ly, L.T.; Lieker, I.; Slowinski, T.; Finke, C.; Pruss, H.; Harms, L. Immunoadsorption or plasma exchange in the treatment of autoimmune encephalitis: A pilot study. J. Neurol. 2016, 263, 2395–2402. [Google Scholar] [CrossRef]
- Dogan Onugoren, M.; Golombeck, K.S.; Bien, C.; Abu-Tair, M.; Brand, M.; Bulla-Hellwig, M.; Lohmann, H.; Munstermann, D.; Pavenstadt, H.; Tholking, G.; et al. Immunoadsorption therapy in autoimmune encephalitides. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e207. [Google Scholar] [CrossRef] [PubMed]
- Rossling, R.; Pruss, H. Apheresis in Autoimmune Encephalitis and Autoimmune Dementia. J. Clin. Med. 2020, 9, 2683. [Google Scholar] [CrossRef] [PubMed]
- Dalakas, M.C. Neurological complications of immune checkpoint inhibitors: What happens when you ‘take the brakes off’ the immune system. Ther. Adv. Neurol. Disord. 2018, 11, 1756286418799864. [Google Scholar] [CrossRef]
- Barbagallo, M.; Vitaliti, G.; Pavone, P.; Romano, C.; Lubrano, R.; Falsaperla, R. Pediatric Autoimmune Encephalitis. J. Pediatr. Neurosci. 2017, 12, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Shapiro-Shelef, M.; Calame, K. Regulation of plasma-cell development. Nat. Rev. Immunol. 2005, 5, 230–242. [Google Scholar] [CrossRef]
- Kosmidis, M.L.; Dalakas, M.C. Practical considerations on the use of rituximab in autoimmune neurological disorders. Ther. Adv. Neurol. Disord. 2010, 3, 93–105. [Google Scholar] [CrossRef]
- Hoftberger, R.; van Sonderen, A.; Leypoldt, F.; Houghton, D.; Geschwind, M.; Gelfand, J.; Paredes, M.; Sabater, L.; Saiz, A.; Titulaer, M.J.; et al. Encephalitis and AMPA receptor antibodies: Novel findings in a case series of 22 patients. Neurology 2015, 84, 2403–2412. [Google Scholar] [CrossRef]
- Brummaier, T.; Pohanka, E.; Studnicka-Benke, A.; Pieringer, H. Using cyclophosphamide in inflammatory rheumatic diseases. Eur. J. Intern. Med. 2013, 24, 590–596. [Google Scholar] [CrossRef]
- Kanter, I.C.; Huttner, H.B.; Staykov, D.; Biermann, T.; Struffert, T.; Kerling, F.; Hilz, M.J.; Schellinger, P.D.; Schwab, S.; Bardutzky, J. Cyclophosphamide for anti-GAD antibody-positive refractory status epilepticus. Epilepsia 2008, 49, 914–920. [Google Scholar] [CrossRef]
- Gea-Banacloche, J.C. Rituximab-associated infections. Semin. Hematol. 2010, 47, 187–198. [Google Scholar] [CrossRef]
- Mihara, M.; Kasutani, K.; Okazaki, M.; Nakamura, A.; Kawai, S.; Sugimoto, M.; Matsumoto, Y.; Ohsugi, Y. Tocilizumab inhibits signal transduction mediated by both mIL-6R and sIL-6R, but not by the receptors of other members of IL-6 cytokine family. Int. Immunopharmacol. 2005, 5, 1731–1740. [Google Scholar] [CrossRef]
- Chavele, K.M.; Merry, E.; Ehrenstein, M.R. Cutting edge: Circulating plasmablasts induce the differentiation of human T follicular helper cells via IL-6 production. J. Immunol. 2015, 194, 2482–2485. [Google Scholar] [CrossRef]
- Lee, W.J.; Lee, S.T.; Moon, J.; Sunwoo, J.S.; Byun, J.I.; Lim, J.A.; Kim, T.J.; Shin, Y.W.; Lee, K.J.; Jun, J.S.; et al. Tocilizumab in Autoimmune Encephalitis Refractory to Rituximab: An Institutional Cohort Study. Neurotherapeutics 2016, 13, 824–832. [Google Scholar] [CrossRef]
- Benucci, M.; Tramacere, L.; Infantino, M.; Manfredi, M.; Grossi, V.; Damiani, A.; Gobbi, F.L.; Piccininni, M.; Zaccara, G.; Cincotta, M. Efficacy of Tocilizumab in Limbic Encephalitis with Anti-CASPR2 Antibodies. Case Rep. Neurol. Med. 2020, 2020, 5697670. [Google Scholar] [CrossRef]
- Randell, R.L.; Adams, A.V.; Van Mater, H. Tocilizumab in Refractory Autoimmune Encephalitis: A Series of Pediatric Cases. Pediatr. Neurol. 2018, 86, 66–68. [Google Scholar] [CrossRef]
- Scheibe, F.; Ostendorf, L.; Reincke, S.M.; Pruss, H.; von Brunneck, A.C.; Kohnlein, M.; Alexander, T.; Meisel, C.; Meisel, A. Daratumumab treatment for therapy-refractory anti-CASPR2 encephalitis. J. Neurol. 2020, 267, 317–323. [Google Scholar] [CrossRef]
- Nosadini, M.; Mohammad, S.S.; Toldo, I.; Sartori, S.; Dale, R.C. Mycophenolate mofetil, azathioprine and methotrexate usage in paediatric anti-NMDAR encephalitis: A systematic literature review. Eur. J. Paediatr. Neurol. 2019, 23, 7–18. [Google Scholar] [CrossRef]
- Dale, R.C.; Gorman, M.P.; Lim, M. Autoimmune encephalitis in children: Clinical phenomenology, therapeutics, and emerging challenges. Curr. Opin. Neurol. 2017, 30, 334–344. [Google Scholar] [CrossRef]
- Dutra, L.A.; Abrantes, F.; Toso, F.F.; Pedroso, J.L.; Barsottini, O.G.P.; Hoftberger, R. Autoimmune encephalitis: A review of diagnosis and treatment. Arq. Neuropsiquiatr. 2018, 76, 41–49. [Google Scholar] [CrossRef]
- Argyropoulos, G.P.D.; Moore, L.; Loane, C.; Roca-Fernandez, A.; Lage-Martinez, C.; Gurau, O.; Irani, S.R.; Zeman, A.; Butler, C.R. Pathologic tearfulness after limbic encephalitis: A novel disorder and its neural basis. Neurology 2020, 94, e1320–e1335. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Symptoms | Tumor Association | Image | Prognosis | |
|---|---|---|---|---|
| Cell-surface | ||||
| LGI1 | LE, seizure including FBDS, hyponatremia, paroxysmal dizziness spells, pain, dysautonomia | <10% SCLC, thymoma | Normal or nonspecific changes; T2-mesiotemporal hyperintensity; T1-bright basal ganglia in FBDS | Almost fair response to immunotherapy but high relapse rate (15–35%) |
| CASPR2 | LE, Morvan syndrome, neuromyotonia, seizure, neuropathic pain, dysautonomia | 10–40% (44% for LGI1 and CASPR2 dual seropositivity) thymoma | Normal or nonspecific changes; T2 mesiotemporal hyperintensity | Favorable response to immunotherapy but high relapse rate (35%) |
| AMPAR | LE, confusion, psychiatric symptoms, seizure | 50–70% lung, breast, thymoma | T2 mesiotemporal hyperintensity | Favorable response to immunotherapy but common relapse |
| GABA-BR | LE, seizure, dysautonomia, movement disorder, rapidly progressive dementia | 50–60% SCLC | T2 mesiotemporal hyperintensity | Frequent co-expression with other Abs; poor prognosis with concurrent tumor or convulsive SE; relapse rate (20%) |
| mGluR5 | LE, Ophelia syndrome seizures, movement disorders | 50% Hodgkin lymphoma | Limbic and extra-limbic (thalamus, pons, frontal or parieto-occipital cortex, cerebellum) | Complete or partial recovery to immunotherapy |
| DPPX | LE, BE, diarrhea, CNS hyperexcitability, PERM, dysautonomia | <10–30% B cell tumor | Normal or nonspecific T2/FLAIR white matter abnormalities | Chronic and second-line immunotherapy frequently required, relapse rate (23%) |
| Synaptic | ||||
| GAD | LE, SPS, PERM, seizure, CA, oculomotor dysfunction, diabetes | <15% (higher with coexisting Abs, esp. GABA-BR Ab) lung, thymoma | T2 mesiotemporal hyperintensity | 70% had partial improvement with immunotherapy |
| Intracellular | ||||
| Ma | LE, BE, diencephalic encephalitis, seizure, CS | 90% testicular, lung and pleural | Nonspecific | Favorable in anti-Ma2 but poorer in anti-Ma |
| Hu | LE, CS, BE, dysautonomia, sensory neuropathy | >90% SCLC | Nonspecific | Poor |
| Ri | LE, CS, BE, OMS, movement disorders | >90% woman-breast, man-lung and bladder | Nonspecific | Common co-expression with other Abs |
| CRMP5 | LE, encephalomyelitis, CS, SPS | >90% SCLC, thymoma | Normal or T2 mesiotemporal hyperintensity | Poor |
| SOX1 | LE, LEMS, CS, neuropathy, LEMS | 30–60% SCLC | Normal or T2 mesiotemporal hyperintensity | Common co-expression with other Abs; poor response to treatment |
| AK5 | LE | No data | Hippocampal atrophy | Poor response to treatment |
| Dose & Duration | Mechanism | Note | |
|---|---|---|---|
| First Line | |||
| Methylprednisolone | Adult: 1 g daily Children: 30 mg/kg/day (max. 1 g) for 3–5 days | Inhibits NF-κB → anti-inflammation | 30 mg/kg/day (max. 1 g) once monthly for 3–6 months |
| IVIg | 2 g/kg in 2 or 5 days | Neutralizes Abs and cytokines, decreases B cells, inhibits complement activation, modulates regulatory T cells | 1 g/kg monthly for 3 months or longer Should not be given immediately prior to plasmapheresis |
| Plasmapheresis (PE or IA) | 5–7 exchanges in 10–14 days | Remove Ab | |
| Second Line | |||
| Rituximab | 375 mg/m2 weekly for 4 weeks, or 750 mg/m2 (max. 1000 mg/dose) for two doses 2 weeks apart | Anti-CD20 Ab → depletion of B cells and plasmablasts | |
| Cyclophosphamide | 750–1000 mg/m2 (max. 1000–1500 mg/dose) monthly for 3–6 months | Akylating agents inhibiting DNA synthesis → suppress B and T cells | Cause infertility in repeated doses |
| Tocilizumab | 8–12 mg/kg (max. 800 mg) monthly for 6 months | Anti-IL6 receptor Ab → inhibits B and T cells | |
| Daratumumab | 16 mg/kg weekly in cycle 1–8, every two weeks in cycle 9–13 | anti-CD38 Ab → depletion of plasma cells | One case of anti-CASPR2 encephalitis |
| Bortezomib | 1.3 mg/m2 on day 1, 4, 8, and 11 of a 21-day cycle, total 3 cycles | proteasome inhibitor → depletion of plasma cells | Clinical trial (NCT03993262) |
| Maintenance therapy | |||
| Prednisolone | 1–2 mg/kg/day once daily or divided for 4 weeks; tapered over several weeks to months | Inhibits NF-κB → anti-inflammation | |
| Mycophenolate mofetil (MMF) | Initial 300 mg/m2/day, target 600 mg/m2/day, 1–1.5 g/day, twice daily | Inhibits purine nucleotides → inhibits B and T cells | |
| Azathioprine (AZA) | 1–2.5 mg/kg/day (max.150 mg/day), once daily | Inhibits purine synthesis → inhibits B and T cells | |
| Methotrexate (MTX) | Oral: 10 mg/m2 weekly Intrathecal: 10 mg weekly for 4 weeks | Inhibits NF-κB → anti-inflammation | |
| Symptoms | Antibodies |
| Seizure | GABA-BR (~100%), LGI1 (80–100%), GAD (60–100%), CASPR2 (75%), NMDAR*(70%), AMPAR (33%) FBDS in LGI1 (33–67%) |
| Paroxysmal dizzy spells | LGI1 (14%) |
| Cramps or neuropathic pain | CASPR2, LGI1 |
| Movement disorders | NMDAR *, CRMP5, Hu, GlyR, GABA-BR, DPPX, CASPR2, Ri |
| Dysautonomia | NMDAR *, LGI1, CASPR2, GABA-BR, DPPX, GlyR |
| GI symptoms (diarrhea) | DPPX |
| Laboratory findings | Antibodies |
| Hyponatremia | GLI1 (~50%), CASPR2 (~25%) |
| Hyperglycemia | GAD |
| CSF pleocytosis + OCB | GABA-BR, GAD, mGluR5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kao, Y.-C.; Lin, M.-I.; Weng, W.-C.; Lee, W.-T. Neuropsychiatric Disorders Due to Limbic Encephalitis: Immunologic Aspect. Int. J. Mol. Sci. 2021, 22, 389. https://doi.org/10.3390/ijms22010389
Kao Y-C, Lin M-I, Weng W-C, Lee W-T. Neuropsychiatric Disorders Due to Limbic Encephalitis: Immunologic Aspect. International Journal of Molecular Sciences. 2021; 22(1):389. https://doi.org/10.3390/ijms22010389
Chicago/Turabian StyleKao, Yu-Chia, Ming-I Lin, Wen-Chin Weng, and Wang-Tso Lee. 2021. "Neuropsychiatric Disorders Due to Limbic Encephalitis: Immunologic Aspect" International Journal of Molecular Sciences 22, no. 1: 389. https://doi.org/10.3390/ijms22010389
APA StyleKao, Y.-C., Lin, M.-I., Weng, W.-C., & Lee, W.-T. (2021). Neuropsychiatric Disorders Due to Limbic Encephalitis: Immunologic Aspect. International Journal of Molecular Sciences, 22(1), 389. https://doi.org/10.3390/ijms22010389