Restoration of Sarcoplasmic Reticulum Ca2+ ATPase (SERCA) Activity Prevents Age-Related Muscle Atrophy and Weakness in Mice


 ,
,
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
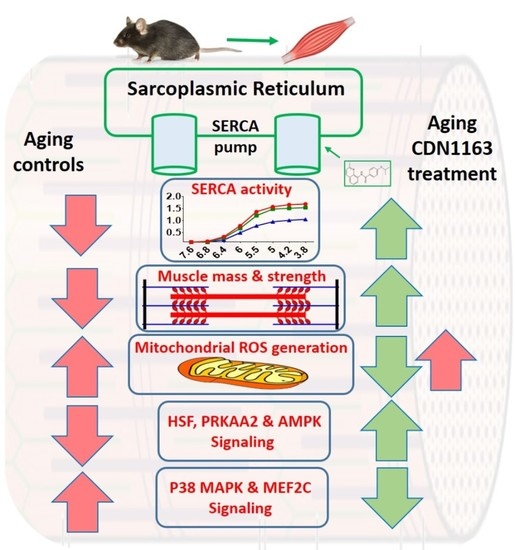
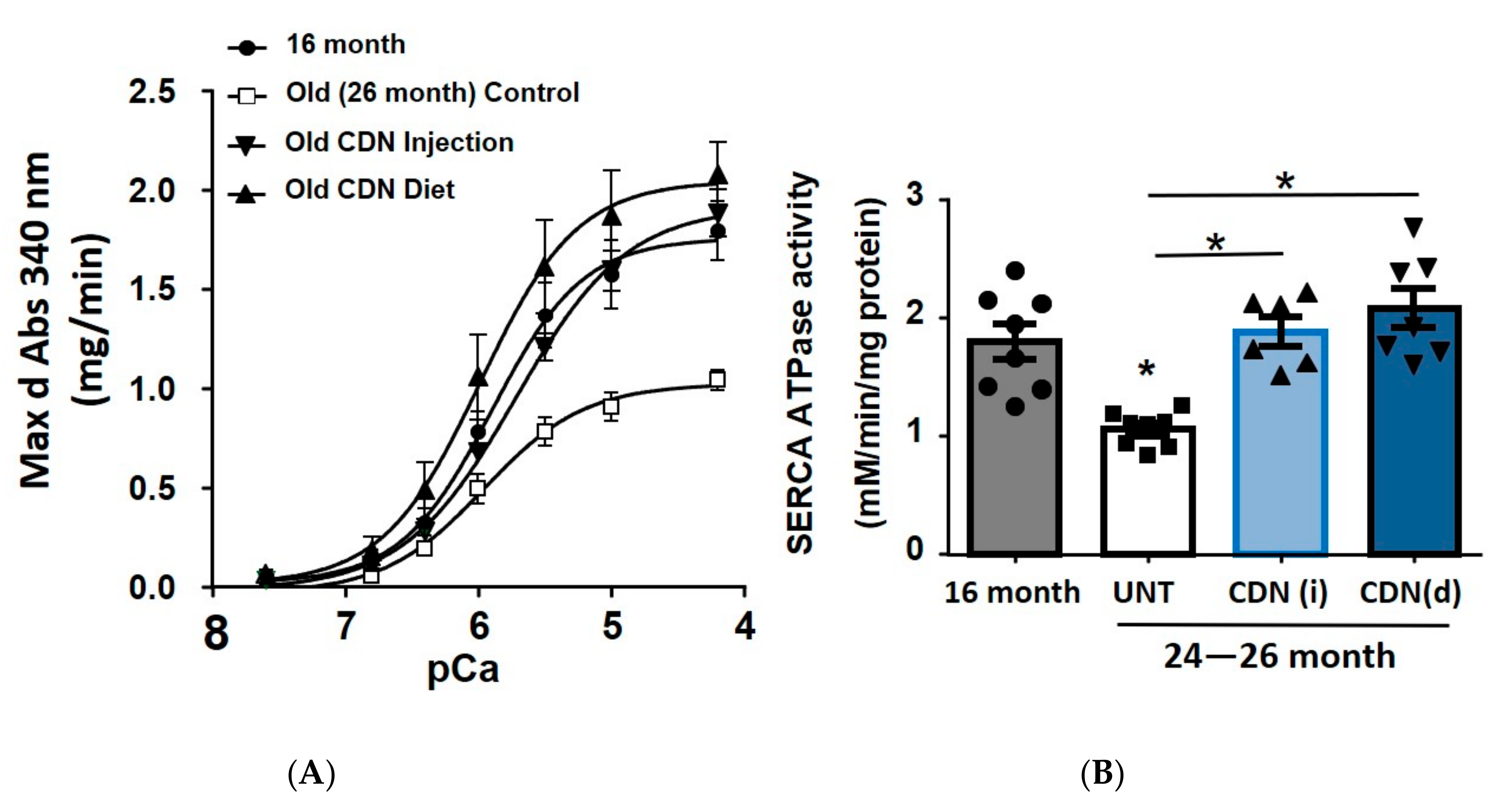
2.1. CDN1163 Prevents Decline in SERCA ATPase Activity in the Gastrocnemius Muscle of Aging Mice
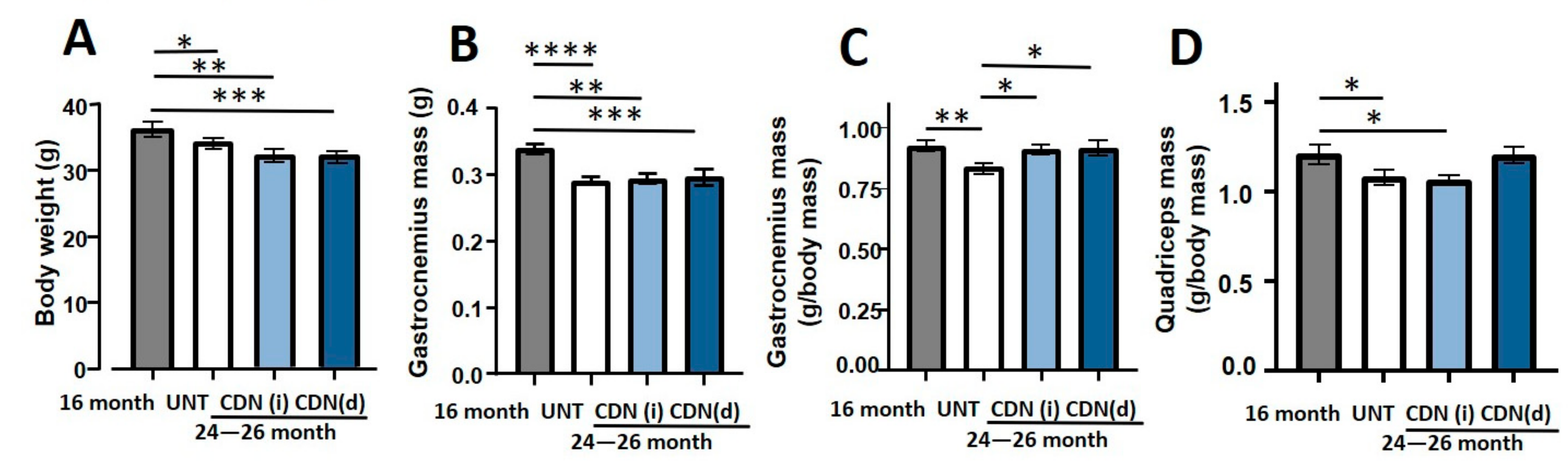
2.2. CDN1163 Prevents Muscles Atrophy in the Aging Mice
2.3. CDN1163 Prevents Contractile Dysfunction of the Extensor Digitorum Longus (EDL) Muscle in the Aging Mice
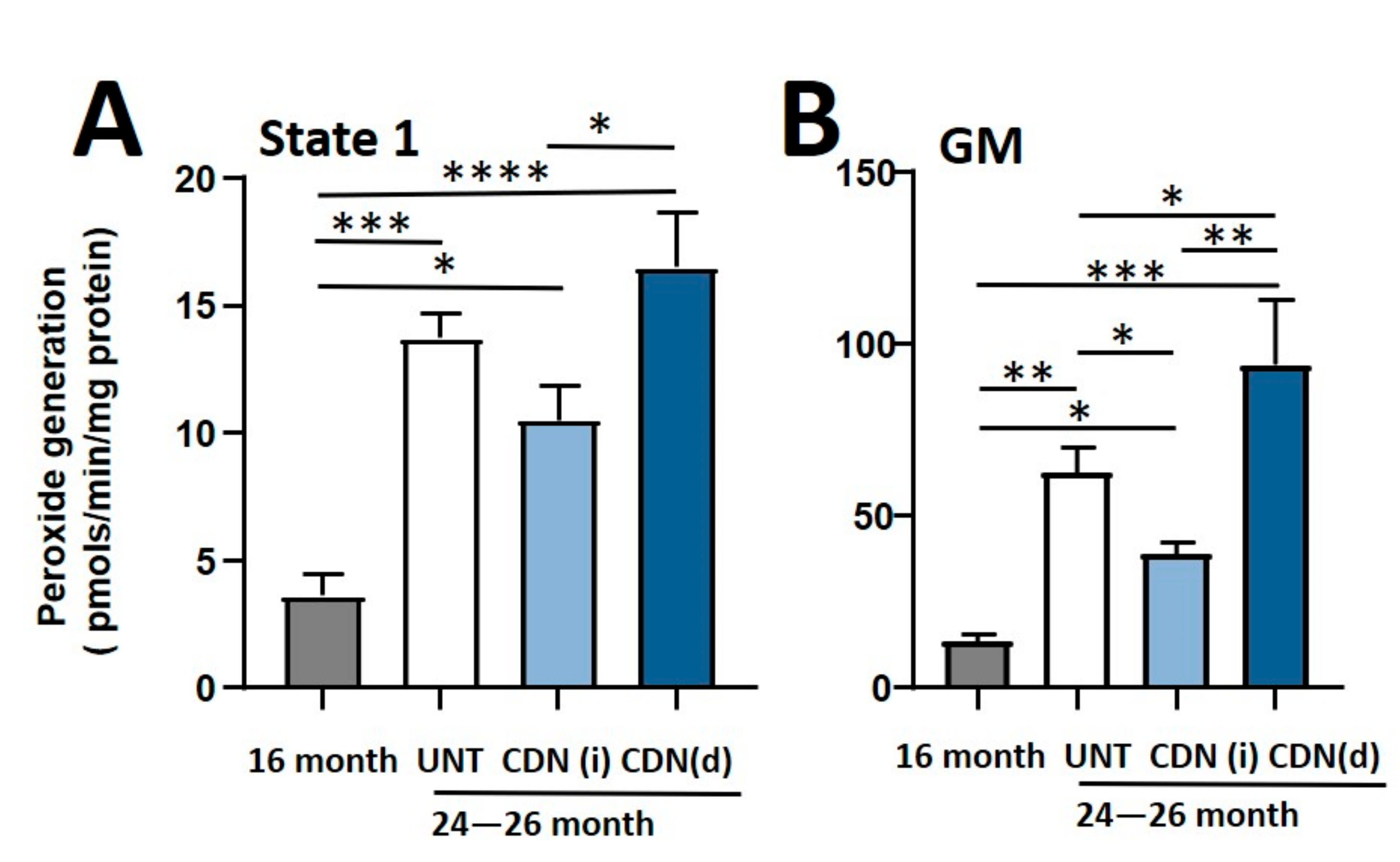
2.4. CDN1163 Attenuates Mitochondrial Dysfunction in Sarcopenia
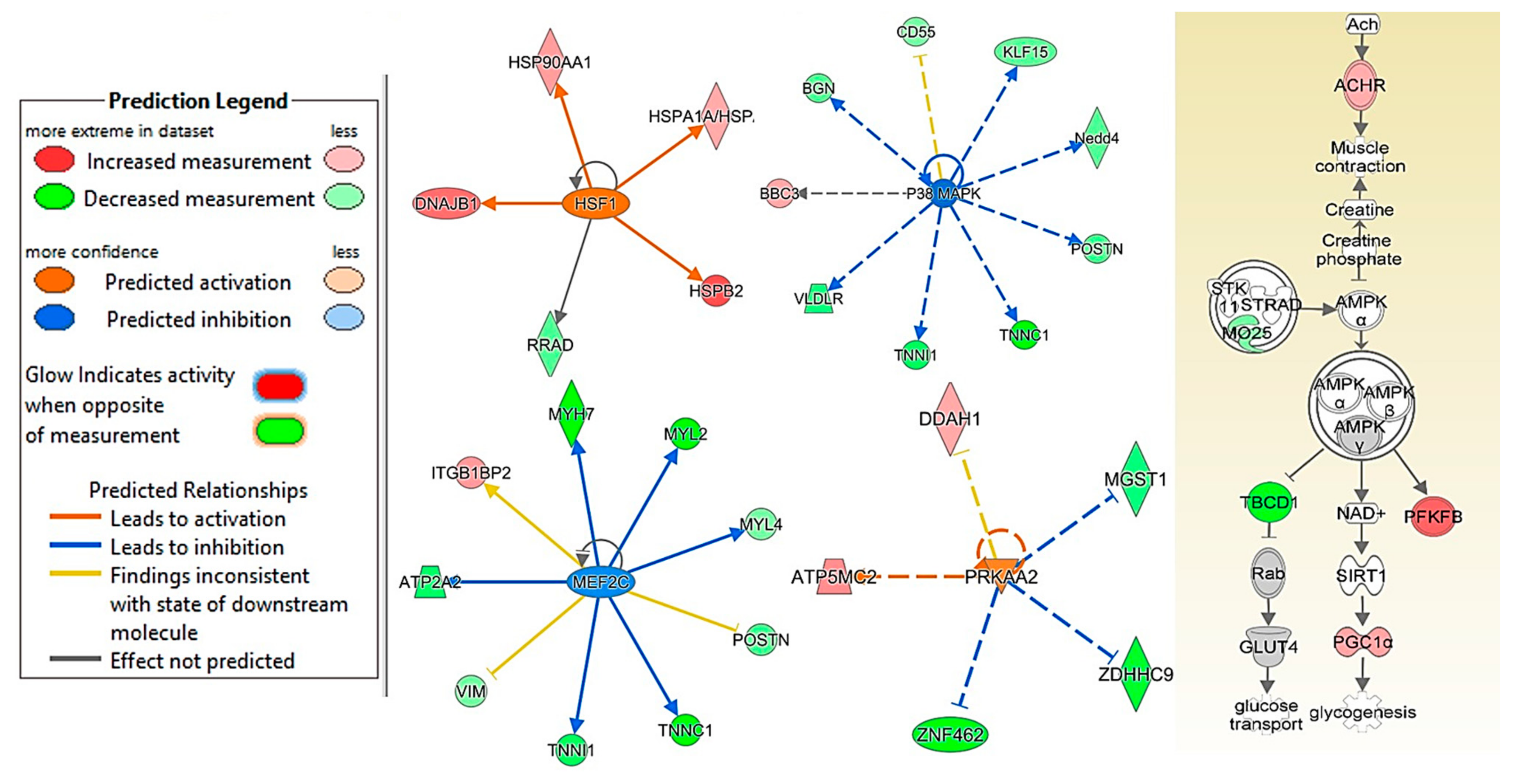
2.5. CDN1163 Treatment via Diet and Injection Prevents Age-Related Alterations in HSF1, P38 MAPK, MEF2C, PRKAA2 and APMK Pathways
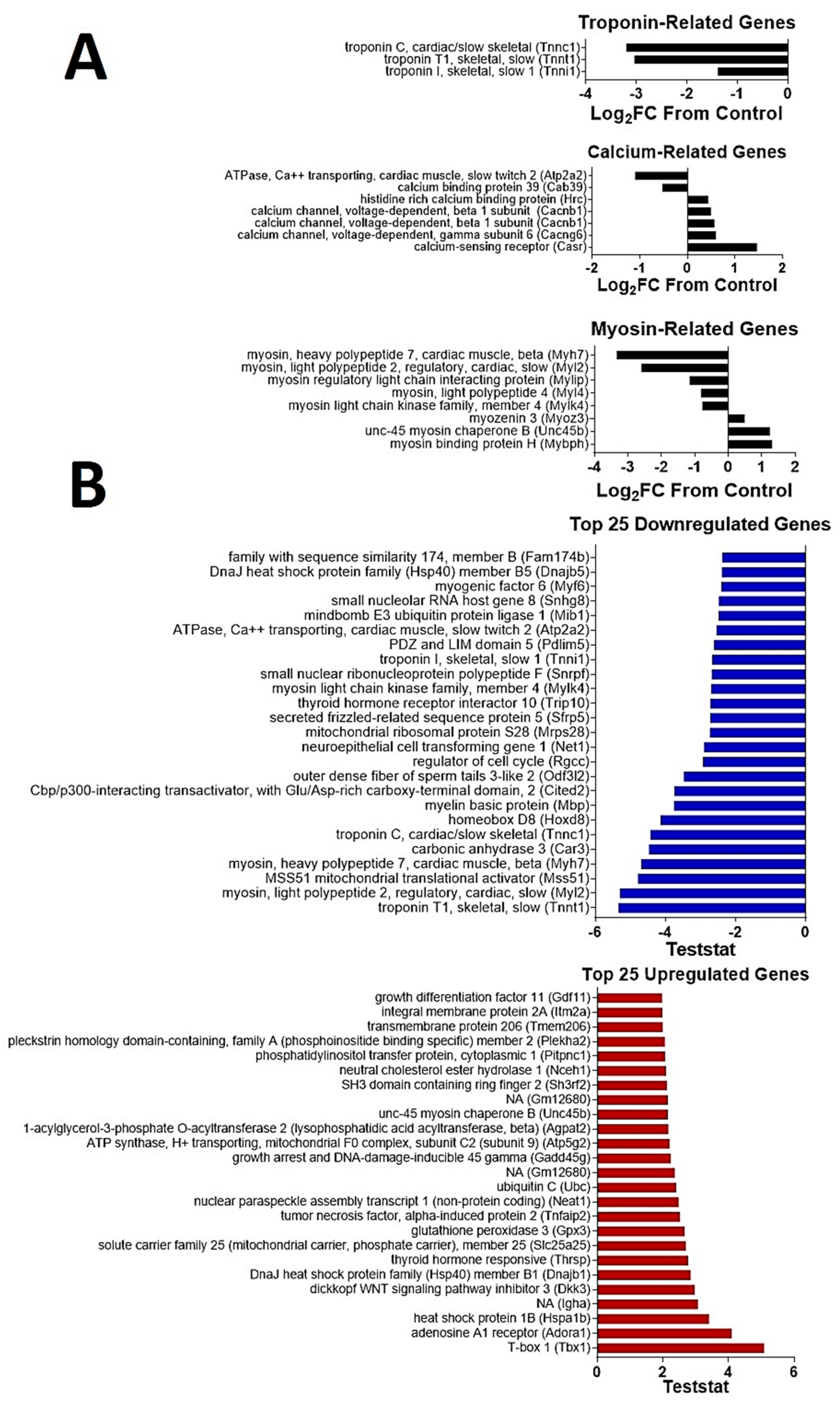
2.6. CDN1163 Reverses Selected Changes in Skeletal Muscle Gene Signature with A Wging
3. Discussion
4. Material and Methods
4.1. Animals and Study Design
4.2. SERCA Assay
4.3. Muscle Contractile Function
4.4. Mitochondrial Hydroperoxides Generation
4.5. Transcriptome Sequencing Data Generation
4.6. Differential Gene Expression Analysis
4.7. Ingenuity Upstream Regulator and Pathway Analysis
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Lang, T.; Streeper, T.; Cawthon, P.; Baldwin, K.; Taaffe, D.R.; Harris, T.B. Sarcopenia: Etiology, clinical consequences, intervention, and assessment. Osteoporos. Int. 2010, 21, 543–559. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Yakabe, M.; Akishita, M. Age-related sarcopenia and its pathophysiological bases. Inflamm. Regen. 2016, 36, 17. [Google Scholar] [CrossRef] [PubMed]
- Qaisar, R.; Bhaskaran, S.; Premkumar, P.; Ranjit, R.; Natarajan, K.S.; Ahn, B.; Riddle, K.; Claflin, D.R.; Richardson, A.; Brooks, S.V.; et al. Oxidative stress-induced dysregulation of excitation-contraction coupling contributes to muscle weakness. J. Cachexia Sarcopenia Muscle 2018, 9, 1003–1017. [Google Scholar] [CrossRef] [PubMed]
- Qaisar, R.; Bhaskaran, S.; Ranjit, R.; Sataranatarajan, K.; Premkumar, P.; Huseman, K.; Van Remmen, H. Restoration of SERCA ATPase prevents oxidative stress-related muscle atrophy and weakness. Redox Biol. 2019, 20, 68–74. [Google Scholar] [CrossRef]
- Schertzer, J.D.; Plant, D.R.; Ryall, J.G.; Beitzel, F.; Stupka, N.; Lynch, G.S. Beta2-agonist administration increases sarcoplasmic reticulum Ca2+-ATPase activity in aged rat skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E526–E533. [Google Scholar] [CrossRef][Green Version]
- Thomas, M.M.; Vigna, C.; Betik, A.C.; Tupling, A.R.; Hepple, R.T. Initiating treadmill training in late middle age offers modest adaptations in Ca2+ handling but enhances oxidative damage in senescent rat skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1269–R1278. [Google Scholar] [CrossRef]
- Fodor, J.; Al-Gaadi, D.; Czirjak, T.; Olah, T.; Dienes, B.; Csernoch, L.; Szentesi, P. Improved Calcium Homeostasis and Force by Selenium Treatment and Training in Aged Mouse Skeletal Muscle. Sci. Rep. 2020, 10, 1707. [Google Scholar] [CrossRef]
- Qaisar, R.; Karim, A.; Elmoselhi, A.B. Muscle unloading: A comparison between spaceflight and ground-based models. Acta Physiol. 2020, 228, e13431. [Google Scholar] [CrossRef]
- Narici, M.V.; Maffulli, N. Sarcopenia: Characteristics, mechanisms and functional significance. Br. Med. Bull. 2010, 95, 139–159. [Google Scholar] [CrossRef]
- Andersson, D.C.; Betzenhauser, M.J.; Reiken, S.; Meli, A.C.; Umanskaya, A.; Xie, W.; Shiomi, T.; Zalk, R.; Lacampagne, A.; Marks, A.R. Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab. 2011, 14, 196–207. [Google Scholar] [CrossRef]
- Delrio-Lorenzo, A.; Rojo-Ruiz, J.; Alonso, M.T.; Garcia-Sancho, J. Sarcoplasmic reticulum Ca(2+) decreases with age and correlates with the decline in muscle function in Drosophila. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef] [PubMed]
- Goonasekera, S.A.; Lam, C.K.; Millay, D.P.; Sargent, M.A.; Hajjar, R.J.; Kranias, E.G.; Molkentin, J.D. Mitigation of muscular dystrophy in mice by SERCA overexpression in skeletal muscle. J. Clin. Investig. 2011, 121, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Voit, A.; Patel, V.; Pachon, R.; Shah, V.; Bakhutma, M.; Kohlbrenner, E.; McArdle, J.J.; Dell’Italia, L.J.; Mendell, J.R.; Xie, L.H.; et al. Reducing sarcolipin expression mitigates Duchenne muscular dystrophy and associated cardiomyopathy in mice. Nat. Commun. 2017, 8, 1068. [Google Scholar] [CrossRef] [PubMed]
- Tadini-Buoninsegni, F.; Smeazzetto, S.; Gualdani, R.; Moncelli, M.R. Drug Interactions With the Ca(2+)-ATPase From Sarco(Endo)Plasmic Reticulum (SERCA). Front. Mol. Biosci. 2018, 5, 36. [Google Scholar] [CrossRef]
- Dahl, R. A new target for Parkinson’s disease: Small molecule SERCA activator CDN1163 ameliorates dyskinesia in 6-OHDA-lesioned rats. Bioorg. Med. Chem. 2017, 25, 53–57. [Google Scholar] [CrossRef]
- Kang, S.; Dahl, R.; Hsieh, W.; Shin, A.; Zsebo, K.M.; Buettner, C.; Hajjar, R.J.; Lebeche, D. Small Molecular Allosteric Activator of the Sarco/Endoplasmic Reticulum Ca2+-ATPase (SERCA) Attenuates Diabetes and Metabolic Disorders. J. Biol. Chem. 2016, 291, 5185–5198. [Google Scholar] [CrossRef]
- Krajnak, K.; Dahl, R. A new target for Alzheimer’s disease: A small molecule SERCA activator is neuroprotective in vitro and improves memory and cognition in APP/PS1 mice. Bioorg. Med. Chem. Lett. 2018, 28, 1591–1594. [Google Scholar] [CrossRef]
- Eisner, V.; Csordas, G.; Hajnoczky, G. Interactions between sarco-endoplasmic reticulum and mitochondria in cardiac and skeletal muscle-pivotal roles in Ca(2)(+) and reactive oxygen species signaling. J. Cell Sci. 2013, 126, 2965–2978. [Google Scholar] [CrossRef]
- Jang, Y.C.; Lustgarten, M.S.; Liu, Y.; Muller, F.L.; Bhattacharya, A.; Liang, H.; Salmon, A.B.; Brooks, S.V.; Larkin, L.; Hayworth, C.R. Increased superoxide in vivo accelerates age-associated muscle atrophy through mitochondrial dysfunction and neuromuscular junction degeneration. FASEB J. 2010, 24, 1376–1390. [Google Scholar] [CrossRef]
- Muller, F.L.; Song, W.; Jang, Y.C.; Liu, Y.; Sabia, M.; Richardson, A.; Van Remmen, H. Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R1159–R1168. [Google Scholar] [CrossRef]
- Cristea, A.; Qaisar, R.; Edlund, P.K.; Lindblad, J.; Bengtsson, E.; Larsson, L. Effects of aging and gender on the spatial organization of nuclei in single human skeletal muscle cells. Aging Cell 2010, 9, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Lin, I.H.; Chang, J.L.; Hua, K.; Huang, W.C.; Hsu, M.T.; Chen, Y.F. Skeletal muscle in aged mice reveals extensive transformation of muscle gene expression. BMC Genet. 2018, 19, 55. [Google Scholar] [CrossRef] [PubMed]
- Fajardo, V.A.; Rietze, B.A.; Chambers, P.J.; Bellissimo, C.; Bombardier, E.; Quadrilatero, J.; Tupling, A.R. Effects of sarcolipin deletion on skeletal muscle adaptive responses to functional overload and unload. Am. J. Physiol. Cell Physiol. 2017, 313, C154–C161. [Google Scholar] [CrossRef] [PubMed]
- Periasamy, M.; Kalyanasundaram, A. SERCA pump isoforms: Their role in calcium transport and disease. Muscle Nerve 2007, 35, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Dufresne, S.S.; Dumont, N.A.; Boulanger-Piette, A.; Fajardo, V.A.; Gamu, D.; Kake-Guena, S.A.; David, R.O.; Bouchard, P.; Lavergne, E.; Penninger, J.M.; et al. Muscle RANK is a key regulator of Ca2+ storage, SERCA activity, and function of fast-twitch skeletal muscles. Am. J. Physiol. Cell Physiol. 2016, 310, C663–C672. [Google Scholar] [CrossRef] [PubMed]
- Ferrington, D.A.; Krainev, A.G.; Bigelow, D.J. Altered turnover of calcium regulatory proteins of the sarcoplasmic reticulum in aged skeletal muscle. J. Biol. Chem. 1998, 273, 5885–5891. [Google Scholar] [CrossRef]
- Vallejo-Illarramendi, A.; Toral-Ojeda, I.; Aldanondo, G.; Lopez de Munain, A. Dysregulation of calcium homeostasis in muscular dystrophies. Expert Rev. Mol. Med. 2014, 16, e16. [Google Scholar] [CrossRef]
- Mazala, D.A.; Pratt, S.J.P.; Chen, D.; Molkentin, J.D.; Lovering, R.M.; Chin, E.R. SERCA1 overexpression minimizes skeletal muscle damage in dystrophic mouse models. Am. J. Physiol. Cell Physiol. 2015, 308, C699–C709. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Muller, F.L.; Liu, Y.; Sabia, M.; Liang, H.; Song, W.; Jang, Y.C.; Ran, Q.; Van Remmen, H. Denervation induces cytosolic phospholipase A2-mediated fatty acid hydroperoxide generation by muscle mitochondria. J. Biol. Chem. 2009, 284, 46–55. [Google Scholar] [CrossRef]
- Zhou, J.; Dhakal, K.; Yi, J. Mitochondrial Ca(2+) uptake in skeletal muscle health and disease. Sci. China Life Sci. 2016, 59, 770–776. [Google Scholar] [CrossRef]
- Waldeck-Weiermair, M.; Deak, A.T.; Groschner, L.N.; Alam, M.R.; Jean-Quartier, C.; Malli, R.; Graier, W.F. Molecularly distinct routes of mitochondrial Ca2+ uptake are activated depending on the activity of the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA). J. Biol. Chem. 2013, 288, 15367–15379. [Google Scholar] [CrossRef] [PubMed]
- Alway, S.E.; Mohamed, J.S.; Myers, M.J. Mitochondria Initiate and Regulate Sarcopenia. Exerc. Sport Sci. Rev. 2017, 45, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Liu, Y.; Sugiura, Y.; Allen, P.D.; Gregg, R.G.; Lin, W. Neuromuscular synaptic patterning requires the function of skeletal muscle dihydropyridine receptors. Nat. Neurosci. 2011, 14, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Adjeitey, C.N.; Mailloux, R.J.; Dekemp, R.A.; Harper, M.E. Mitochondrial uncoupling in skeletal muscle by UCP1 augments energy expenditure and glutathione content while mitigating ROS production. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E405–E415. [Google Scholar] [CrossRef]
- Singh, R.; Moreno, P.; Hajjar, R.J.; Lebeche, D. A role for calcium in resistin transcriptional activation in diabetic hearts. Sci. Rep. 2018, 8, 15633. [Google Scholar] [CrossRef]
- Yuan, L.; Wang, H.; Liu, Q.; Wang, Z.; Zhang, M.; Zhao, Y.; Liang, K.; Chen, L.; Xu, T.; Xu, P. Etoposide-induced protein 2.4 functions as a regulator of the calcium ATPase and protects pancreatic beta-cell survival. J. Biol. Chem. 2018, 293, 10128–10140. [Google Scholar] [CrossRef]
- Hall, D.T.; Griss, T.; Ma, J.F.; Sanchez, B.J.; Sadek, J.; Tremblay, A.M.K.; Mubaid, S.; Omer, A.; Ford, R.J.; Bedard, N.; et al. The AMPK agonist 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR), but not metformin, prevents inflammation-associated cachectic muscle wasting. EMBO Mol. Med. 2018, 10. [Google Scholar] [CrossRef]
- Koya, T.; Nishizawa, S.; Ohno, Y.; Goto, A.; Ikuta, A.; Suzuki, M.; Ohira, T.; Egawa, T.; Nakai, A.; Sugiura, T.; et al. Heat shock transcription factor 1-deficiency attenuates overloading-associated hypertrophy of mouse soleus muscle. PLoS ONE 2013, 8, e77788. [Google Scholar] [CrossRef]
- Perez, F.P.; Moinuddin, S.S.; ul ain Shamim, Q.; Joseph, D.J.; Morisaki, J.; Zhou, X. Longevity pathways: HSF1 and FoxO pathways, a new therapeutic target to prevent age-related diseases. Curr. Aging Sci. 2012, 5, 87–95. [Google Scholar] [CrossRef]
- Bernet, J.D.; Doles, J.D.; Hall, J.K.; Kelly Tanaka, K.; Carter, T.A.; Olwin, B.B. p38 MAPK signaling underlies a cell-autonomous loss of stem cell self-renewal in skeletal muscle of aged mice. Nat. Med. 2014, 20, 265–271. [Google Scholar] [CrossRef]
- Ingemann, L.; Kirkegaard, T. Lysosomal storage diseases and the heat shock response: Convergences and therapeutic opportunities. J. Lipid Res. 2014, 55, 2198–2210. [Google Scholar] [CrossRef] [PubMed]
- Kefaloyianni, E.; Gaitanaki, C.; Beis, I. ERK1/2 and p38-MAPK signalling pathways, through MSK1, are involved in NF-kappaB transactivation during oxidative stress in skeletal myoblasts. Cell Signal. 2006, 18, 2238–2251. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Dyar, K.A.; Calabria, E. Skeletal muscle mass is controlled by the MRF4-MEF2 axis. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 164–167. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Na, R.; Gu, M.; Salmon, A.B.; Liu, Y.; Liang, H.; Qi, W.; Van Remmen, H.; Richardson, A.; Ran, Q. Reduction of mitochondrial H2O2 by overexpressing peroxiredoxin 3 improves glucose tolerance in mice. Aging Cell 2008, 7, 866–878. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qaisar, R.; Pharaoh, G.; Bhaskaran, S.; Xu, H.; Ranjit, R.; Bian, J.; Ahn, B.; Georgescu, C.; Wren, J.D.; Van Remmen, H. Restoration of Sarcoplasmic Reticulum Ca2+ ATPase (SERCA) Activity Prevents Age-Related Muscle Atrophy and Weakness in Mice. Int. J. Mol. Sci. 2021, 22, 37. https://doi.org/10.3390/ijms22010037
Qaisar R, Pharaoh G, Bhaskaran S, Xu H, Ranjit R, Bian J, Ahn B, Georgescu C, Wren JD, Van Remmen H. Restoration of Sarcoplasmic Reticulum Ca2+ ATPase (SERCA) Activity Prevents Age-Related Muscle Atrophy and Weakness in Mice. International Journal of Molecular Sciences. 2021; 22(1):37. https://doi.org/10.3390/ijms22010037
Chicago/Turabian StyleQaisar, Rizwan, Gavin Pharaoh, Shylesh Bhaskaran, Hongyang Xu, Rojina Ranjit, Jan Bian, Bumsoo Ahn, Constantin Georgescu, Jonathan D. Wren, and Holly Van Remmen. 2021. "Restoration of Sarcoplasmic Reticulum Ca2+ ATPase (SERCA) Activity Prevents Age-Related Muscle Atrophy and Weakness in Mice" International Journal of Molecular Sciences 22, no. 1: 37. https://doi.org/10.3390/ijms22010037
APA StyleQaisar, R., Pharaoh, G., Bhaskaran, S., Xu, H., Ranjit, R., Bian, J., Ahn, B., Georgescu, C., Wren, J. D., & Van Remmen, H. (2021). Restoration of Sarcoplasmic Reticulum Ca2+ ATPase (SERCA) Activity Prevents Age-Related Muscle Atrophy and Weakness in Mice. International Journal of Molecular Sciences, 22(1), 37. https://doi.org/10.3390/ijms22010037