The Proteomic Landscape of Resting and Activated CD4+ T Cells Reveal Insights into Cell Differentiation and Function

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
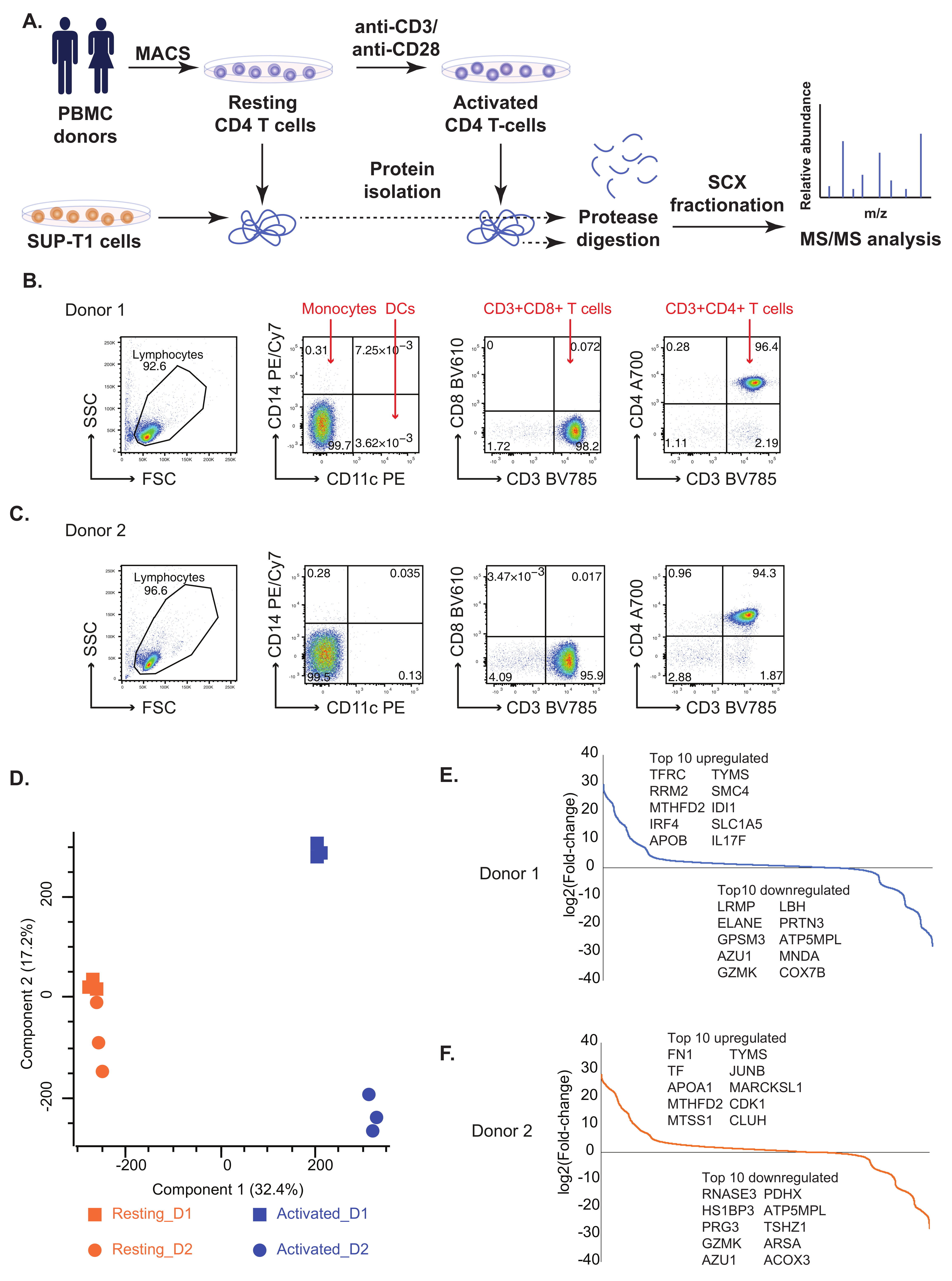
2.1. Comparative Proteomic Analysis of Resting and Activated Primary CD4+ T Cells and SUP-T1 T Lymphoblastic Cell Line
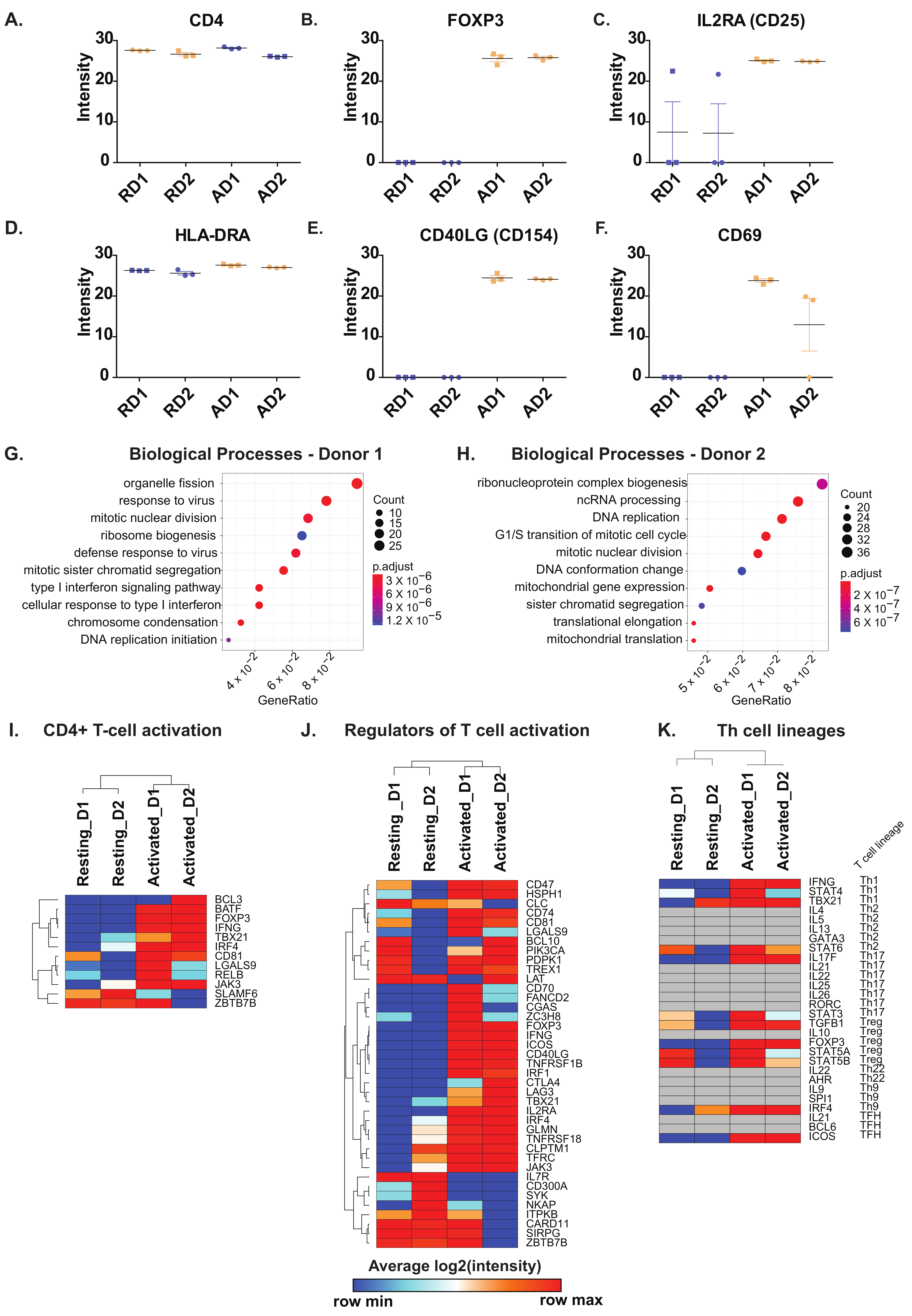
2.2. Known and Novel Molecular Markers of Resting and Activated CD4+ T Cells
2.3. Activation of CD4+ T Cells Influences Processes and Signaling Pathways
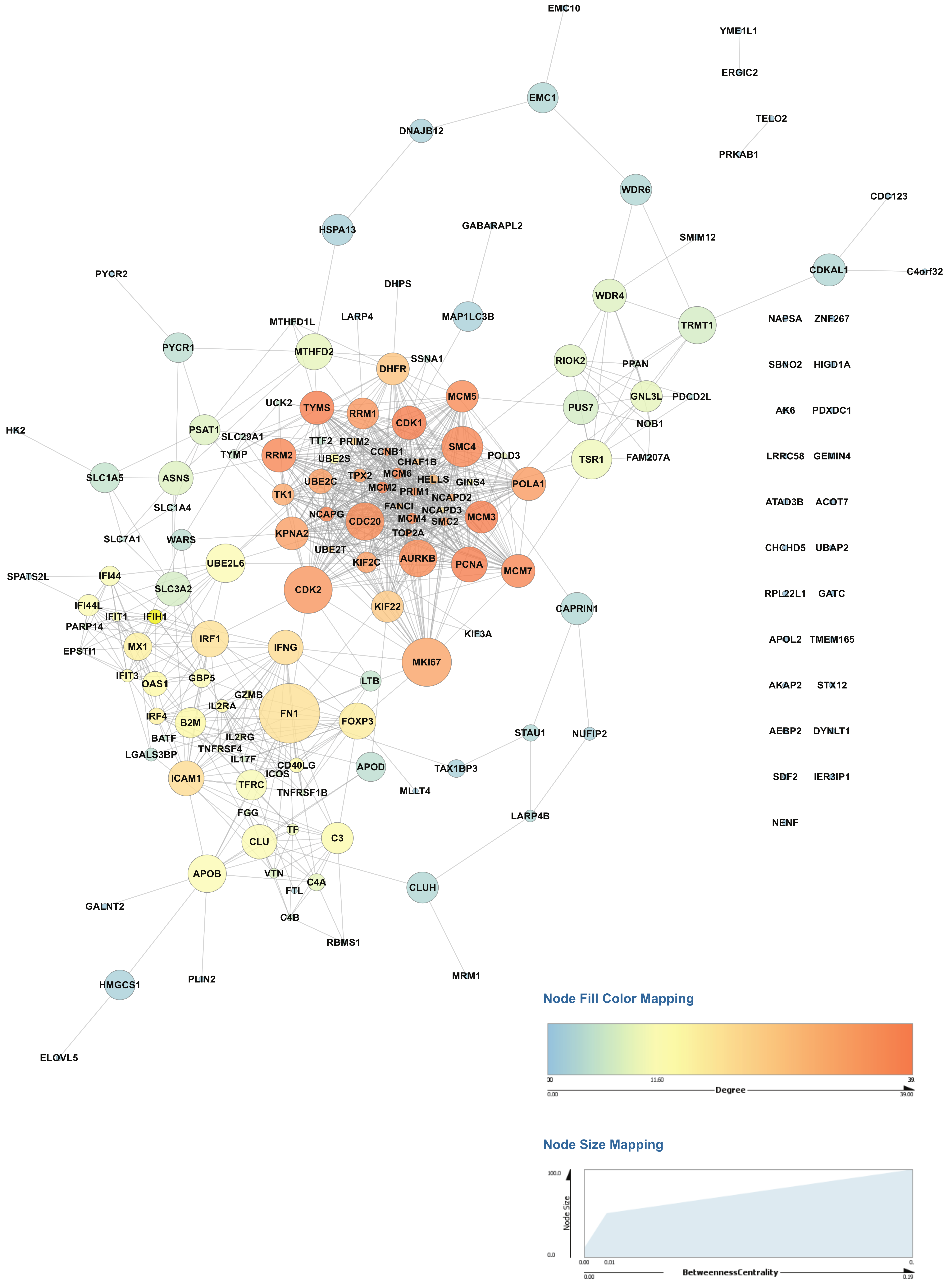
2.4. Activation of CD4+ T Cells Influences Protein Signaling Networks
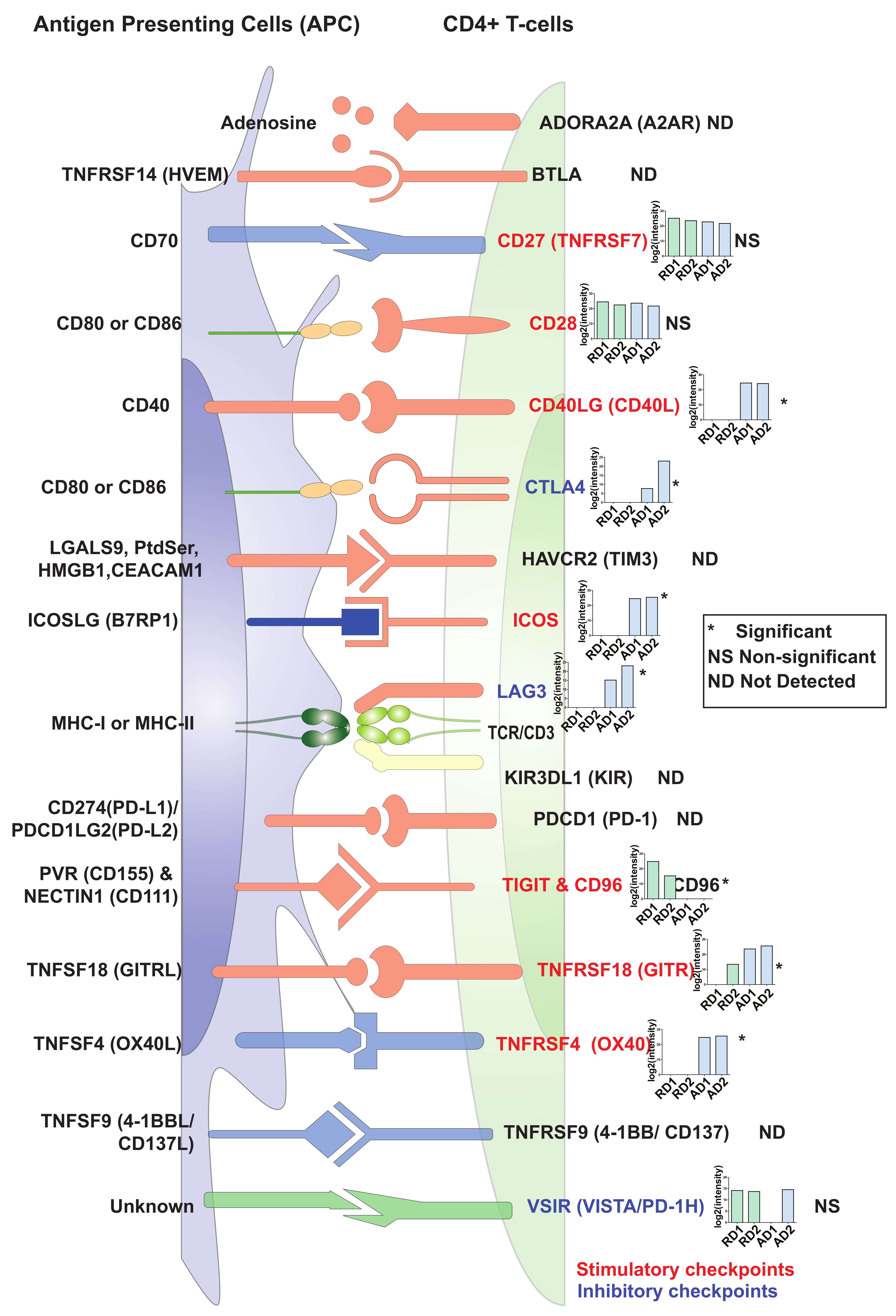
2.5. Changes in Expression Levels of Immune Checkpoint Proteins in Activated/Resting CD4+ T Cells
2.6. Comparison of Primary T Cell Data with Proteomes of Human Lymphoblastic T Cell Lines and Published Primary Human and Mouse CD4+ T Cell Datasets
3. Discussion
4. Materials and Methods
4.1. Cells
4.2. CD4+ T Cell Activation
4.3. Sample Preparation of CD4+ T Cell for Proteomics
4.4. Tandem Mass Spectrometry (MS/MS) Analysis
4.5. Bioinformatics Analysis of Mass Spectrometry Data
4.6. Isolation of SUP-T1 Cell Proteome and MS/MS Analysis
4.7. Comparison with Published Datasets
4.8. Data Availability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AA | Amino Acid |
| FDR | False Discovery Rate |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| MACS | Magnetic-Activated Cell Sorting |
| NCBI | National Center for Biotechnology Information |
| PBMC | Peripheral Blood Mononuclear Cell |
| PSM | Peptide Spectrum Match |
| SDS | Sodium Dodecyl Sulphate |
| TCR | T Cell Receptor |
| TEABC | Triethyl Ammonium Bicarbonate |
| Th cells | T Helper Cells |
| TPCK | L-(tosylamido-2-phenyl) ethyl chloromethyl ketone |
References
- Luckheeram, R.V.; Zhou, R.; Verma, A.D.; Xia, B. CD4+ T Cells: Differentiation and Functions. Clin. Dev. Immunol. 2012, 2012, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Famili, F.; Wiekmeijer, A.-S.; Staal, F.J.T. The development of T cells from stem cells in mice and humans. Future Sci. OA 2017, 3, FSO186. [Google Scholar] [CrossRef] [PubMed]
- Mondino, A.; Khoruts, A.; Jenkins, M.K. The anatomy of T-cell activation and tolerance. Proc. Natl. Acad. Sci. USA 1996, 93, 2245–2252. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.R.; Byeon, Y.; Kim, D.; Park, S.G. Recent insights of T cell receptor-mediated signaling pathways for T cell activation and development. Exp. Mol. Med. 2020, 52, 750–761. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T.R.; Cherwinski, H.; Bond, M.W.; Giedlin, M.A.; Coffman, R.L. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 1986, 136, 2348–2357. [Google Scholar] [PubMed]
- Geginat, J.; Paroni, M.; Maglie, S.; Alfen, J.S.; Kastirr, I.; Gruarin, P.; De Simone, M.; Pagani, M.; Abrignani, S. Plasticity of Human CD4 T Cell Subsets. Front. Immunol. 2014, 5, 630. [Google Scholar] [CrossRef]
- Raphael, I.; Nalawade, S.; Eagar, T.N.; Forsthuber, T.G. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 2015, 74, 5–17. [Google Scholar] [CrossRef]
- Christie, D.; Zhu, J. Transcriptional Regulatory Networks for CD4 T Cell Differentiation. Curr. Top. Microbiol. Immunol. 2014, 381, 125–172. [Google Scholar] [CrossRef]
- Oestreich, K.J.; Weinmann, A.S. Master regulators or lineage-specifying? Changing views on CD4+ T cell transcription factors. Nat. Rev. Immunol. 2012, 12, 799–804. [Google Scholar] [CrossRef]
- Berger, E.A.; Doms, R.W.; Fenyö, E.-M.; Korber, B.T.M.; Littman, D.R.; Moore, J.P.; Sattentau, Q.J.; Schuitemaker, H.; Sodroski, J.; Weiss, R.A. A new classification for HIV-1. Nat. Cell Biol. 1998, 391, 240. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniam, M.; Pandhare, J.; Dash, C. Immune Control of HIV. JoLS J. Life Sci. 2019, 1, 4–37. [Google Scholar] [CrossRef]
- Okoye, A.A.; Picker, L.J. CD4+ T-cell depletion in HIV infection: Mechanisms of immunological failure. Immunol. Rev. 2013, 254, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Geldmacher, C.; Koup, R.A. Pathogen-specific T cell depletion and reactivation of opportunistic pathogens in HIV infection. Trends Immunol. 2012, 33, 207–214. [Google Scholar] [CrossRef]
- Hirahara, K.; Nakayama, T. CD4+ T-cell subsets in inflammatory diseases: Beyond the Th1/Th2 paradigm. Int. Immunol. 2016, 28, 163–171. [Google Scholar] [CrossRef]
- Chemin, K.; Gerstner, C.; Malmström, V. Effector Functions of CD4+ T Cells at the Site of Local Autoimmune Inflammation—Lessons From Rheumatoid Arthritis. Front. Immunol. 2019, 10, 353. [Google Scholar] [CrossRef]
- Zhao, Y.; Lin, L.; Li, J.; Xiao, Z.G.; Chen, B.; Wan, L.; Li, M.; Wu, X.; Cho, C.H.; Shen, J. CD4+ T cells in obesity and obesity-associated diseases. Cell. Immunol. 2018, 332, 1–6. [Google Scholar] [CrossRef]
- Antony, P.A.; Piccirillo, C.A.; Akpinarli, A.; Finkelstein, S.E.; Speiss, P.J.; Surman, D.R.; Palmer, D.C.; Chan, C.-C.; Klebanoff, C.A.; Overwijk, W.W.; et al. CD8+T Cell Immunity Against a Tumor/Self-Antigen Is Augmented by CD4+ T Helper Cells and Hindered by Naturally Occurring T Regulatory Cells. J. Immunol. 2005, 174, 2591–2601. [Google Scholar] [CrossRef]
- Williams, M.A.; Tyznik, A.J.; Bevan, M.J. Interleukin-2 signals during priming are required for secondary expansion of CD8+ memory T cells. Nat. Cell Biol. 2006, 441, 890–893. [Google Scholar] [CrossRef]
- Janssen, E.M.; Lemmens, E.E.; Wolfe, T.L.; Christen, U.; Von Herrath, M.G.; Schoenberger, S.P. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nat. Cell Biol. 2003, 421, 852–856. [Google Scholar] [CrossRef]
- Laidlaw, B.J.; Craft, J.E.; Kaech, S.M. The multifaceted role of CD4+ T cells in CD8+ T cell memory. Nat. Rev. Immunol. 2016, 16, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.M.; Wilson, N.S.; Waithman, J.; Villadangos, J.A.; Carbone, F.R.; Heath, W.R.; Belz, G.T. Cognate CD4+ T cell licensing of dendritic cells in CD8+ T cell immunity. Nat. Immunol. 2004, 5, 1143–1148. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, A.; Badr, M.E.S.G.; Miyauchi, K.; Ishihara, C.; Onishi, R.; Guo, Z.; Sasaki, Y.; Ike, H.; Takumi, A.; Tsuji, N.M.; et al. CRTAM determines the CD4+ cytotoxic T lymphocyte lineage. J. Exp. Med. 2016, 213, 123–138. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Akpinarli, A.; Maris, C.; Hipkiss, E.L.; Lane, M.; Kwon, E.-K.M.; Muranski, P.; Restifo, N.P.; Antony, P.A. Naive tumor-specific CD4+ T cells differentiated in vivo eradicate established melanoma. J. Exp. Med. 2010, 207, 651–667. [Google Scholar] [CrossRef]
- Quezada, S.A.; Simpson, T.R.; Peggs, K.S.; Merghoub, T.; Vider, J.; Fan, X.; Blasberg, R.; Yagita, H.; Muranski, P.; Antony, P.A.; et al. Tumor-reactive CD4+ T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J. Exp. Med. 2010, 207, 637–650. [Google Scholar] [CrossRef]
- Matsuzaki, J.; Tsuji, T.; Luescher, I.F.; Shiku, H.; Mineno, J.; Okamoto, S.; Old, L.J.; Shrikant, P.; Gnjatic, S.; Odunsi, K. Direct tumor recognition by a human CD4+ T-cell subset potently mediates tumor growth inhibition and orchestrates anti-tumor immune responses. Sci. Rep. 2015, 5, 14896. [Google Scholar] [CrossRef]
- Reed, C.M.; Cresce, N.D.; Mauldin, I.S.; Slingluff, C.L.; Olson, W.C. Vaccination with Melanoma Helper Peptides Induces Antibody Responses Associated with Improved Overall Survival. Clin. Cancer Res. 2015, 21, 3879–3887. [Google Scholar] [CrossRef]
- Gnjatic, S.; Atanackovic, D.; Jäger, E.; Matsuo, M.; Selvakumar, A.; Altorki, N.K.; Maki, R.G.; Dupont, B.; Ritter, G.; Chen, Y.-T.; et al. Survey of naturally occurring CD4+ T cell responses against NY-ESO-1 in cancer patients: Correlation with antibody responses. Proc. Natl. Acad. Sci. USA 2003, 100, 8862–8867. [Google Scholar] [CrossRef]
- Marchingo, J.M.; Cantrell, D.A. The active inner life of naive T cells. Nat. Immunol. 2020, 21, 827–828. [Google Scholar] [CrossRef]
- Gerner, M.C.; Niederstaetter, L.; Ziegler, L.; Bileck, A.; Slany, A.; Janker, L.; Schmidt, R.L.; Gerner, C.; Del Favero, G.; Schmetterer, K.G. Proteome Analysis Reveals Distinct Mitochondrial Functions Linked to Interferon Response Patterns in Activated CD4+ and CD8+ T Cells. Front. Pharmacol. 2019, 10, 727. [Google Scholar] [CrossRef]
- Tan, H.; Yang, K.; Li, Y.; Shaw, T.I.; Wang, Y.; Blanco, D.B.; Wang, X.; Cho, J.-H.; Wang, H.; Rankin, S.; et al. Integrative Proteomics and Phosphoproteomics Profiling Reveals Dynamic Signaling Networks and Bioenergetics Pathways Underlying T Cell Activation. Immunity 2017, 46, 488–503. [Google Scholar] [CrossRef] [PubMed]
- Duguet, F.; Locard-Paulet, M.; Marcellin, M.; Chaoui, K.; Bernard, I.; Andreoletti, O.; Lesourne, R.; Burlet-Schiltz, O.; De Peredo, A.G.; Saoudi, A. Proteomic Analysis of Regulatory T Cells Reveals the Importance of Themis1 in the Control of Their Suppressive Function. Mol. Cell. Proteom. 2017, 16, 1416–1432. [Google Scholar] [CrossRef] [PubMed]
- Kubach, J.; Lutter, P.; Bopp, T.; Stoll, S.; Becker, C.; Huter, E.; Richter, C.; Weingarten, P.; Warger, T.; Knop, J.; et al. Human CD4+CD25+ regulatory T cells: Proteome analysis identifies galectin-10 as a novel marker essential for their anergy and suppressive function. Blood 2007, 110, 1550–1558. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, I.; Nousiainen, K.; Bhosale, S.D.; Starskaia, I.; Moulder, R.; Rokka, A.; Cheng, F.; Mohanasundaram, P.; Eriksson, J.E.; Goodlett, D.R.; et al. Quantitative proteomic characterization and comparison of T helper 17 and induced regulatory T cells. PLoS Biol. 2018, 16, e2004194. [Google Scholar] [CrossRef] [PubMed]
- Wolf, T.; Jin, W.; Zoppi, G.; Vogel, I.A.; Akhmedov, M.; Bleck, C.K.E.; Beltraminelli, T.; Rieckmann, J.C.; Ramirez, N.J.; Benevento, M.; et al. Dynamics in protein translation sustaining T cell preparedness. Nat. Immunol. 2020, 21, 927–937. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Hwang, S.; Rezaul, K.; Lu, L.J.; Mayya, V.; Gerstein, M.; Eng, J.K.; Lundgren, D.H.; Han, D.K. Global Survey of Human T Leukemic Cells by Integrating Proteomics and Transcriptomics Profiling. Mol. Cell. Proteom. 2007, 6, 1343–1353. [Google Scholar] [CrossRef]
- Marin-Acevedo, J.A.; Dholaria, B.; Soyano, A.E.; Knutson, K.L.; Chumsri, S.; Lou, Y. Next generation of immune checkpoint therapy in cancer: New developments and challenges. J. Hematol. Oncol. 2018, 11, 1–20. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Darvin, P.; Toor, S.M.; Nair, V.S.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef]
- Zahavi, D.; Weiner, L.M. Targeting Multiple Receptors to Increase Checkpoint Blockade Efficacy. Int. J. Mol. Sci. 2019, 20, 158. [Google Scholar] [CrossRef]
- Howden, A.J.; Hukelmann, J.L.; Brenes, A.; Spinelli, L.; Sinclair, L.V.; Lamond, A.; Cantrell, D.A. Quantitative analysis of T cell proteomes and environmental sensors during T cell differentiation. Nat. Immunol. 2019, 20, 1542–1554. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, C.J.; Getnet, D.; Kim, M.; Manda, S.S.; Kumar, P.; Huang, T.-C.; Pinto, S.M.; Nirujogi, R.S.; Iwasaki, M.; Shaw, P.G.; et al. A multi-omic analysis of human naïve CD4+ T cells. BMC Syst. Biol. 2015, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Rieckmann, J.C.; Geiger, R.; Hornburg, D.; Wolf, T.; Kveler, K.; Jarrossay, D.; Sallusto, F.; Shen-Orr, S.S.; Lanzavecchia, A.; Mann, M.; et al. Social network architecture of human immune cells unveiled by quantitative proteomics. Nat. Immunol. 2017, 18, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Naldi, A.; Carneiro, J.; Chaouiya, C.; Thieffry, D. Diversity and Plasticity of Th Cell Types Predicted from Regulatory Network Modelling. PLoS Comput. Biol. 2010, 6, e1000912. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.; Liu, G.; Yin, J.; Tan, B.; Wu, G.; Bazer, F.W.; Peng, Y.; Yin, Y. Amino-acid transporters in T-cell activation and differentiation. Cell Death Dis. 2017, 8, e2655. [Google Scholar] [CrossRef] [PubMed]
- Levring, T.B.; Hansen, A.K.; Nielsen, B.L.; Kongsbak, M.; Von Essen, M.R.; Woetmann, A.; Ødum, N.; Bonefeld, C.M.; Geisler, C. Activated human CD4+ T cells express transporters for both cysteine and cystine. Sci. Rep. 2012, 2, 266. [Google Scholar] [CrossRef]
- Werner, A.; Amann, E.; Schnitzius, V.; Habermeier, A.; Luckner-Minden, C.; Leuchtner, N.; Rupp, J.; Closs, E.I.; Munder, M. Induced arginine transport via cationic amino acid transporter-1 is necessary for human T-cell proliferation. Eur. J. Immunol. 2015, 46, 92–103. [Google Scholar] [CrossRef]
- Kelly, A.P.; Finlay, D.K.; Hinton, H.J.; Clarke, R.G.; Fiorini, E.; Radtke, F.; Cantrell, D.A. Notch-induced T cell development requires phosphoinositide-dependent kinase 1. EMBO J. 2007, 26, 3441–3450. [Google Scholar] [CrossRef]
- Cantor, J.M.; Slepak, M.; Ege, N.; Chang, J.T.; Ginsberg, M.H. Loss of T Cell CD98 H Chain Specifically Ablates T Cell Clonal Expansion and Protects from Autoimmunity. J. Immunol. 2011, 187, 851–860. [Google Scholar] [CrossRef]
- Ron-Harel, N.; Santos, D.; Ghergurovich, J.M.; Sage, P.T.; Reddy, A.; Lovitch, S.B.; Dephoure, N.; Satterstrom, F.K.; Sheffer, M.; Spinelli, J.B.; et al. Mitochondrial Biogenesis and Proteome Remodeling Promote One-Carbon Metabolism for T Cell Activation. Cell Metab. 2016, 24, 104–117. [Google Scholar] [CrossRef]
- Ron-Harel, N.; Notarangelo, G.; Ghergurovich, J.M.; Paulo, J.A.; Sage, P.T.; Santos, D.; Satterstrom, F.K.; Gygi, S.P.; Rabinowitz, J.D.; Sharpe, A.H.; et al. Defective respiration and one-carbon metabolism contribute to impaired naïve T cell activation in aged mice. Proc. Natl. Acad. Sci. USA 2018, 115, 13347–13352. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, Y.; Verhaaf, B.; Van Gastel-Mol, E.J.; Wolvers-Tettero, I.L.M.; De Vos, J.; MacLeod, R.A.F.; Noordzij, J.G.; Dik, W.A.; Van Dongen, J.; Langerak, A.W. Human T-cell lines with well-defined T-cell receptor gene rearrangements as controls for the BIOMED-2 multiplex polymerase chain reaction tubes. Leukemia 2006, 21, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Burger, R.; Hansen-Hagge, T.E.; Drexler, H.G.; Gramatzki, M. Heterogeneity of T-acute lymphoblastic leukemia (T-ALL) cell lines: Suggestion for classification by immunophenotype and T-cell receptor studies. Leuk. Res. 1999, 23, 19–27. [Google Scholar] [CrossRef]
- Bird, J.J.; Brown, D.R.; Mullen, A.C.; Moskowitz, N.H.; Mahowald, M.A.; Sider, J.R.; Gajewski, T.F.; Wang, C.-R.; Reiner, S.L. Helper T Cell Differentiation Is Controlled by the Cell Cycle. Immunity 1998, 9, 229–237. [Google Scholar] [CrossRef]
- Chapman, N.M.; Chi, H. Hallmarks of T-cell Exit from Quiescence. Cancer Immunol. Res. 2018, 6, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.H.; Kim, A.-R.; Park, N.-H.; Park, J.W.; Han, A.I.-S. DRG2 Regulates G2/M Progression via the Cyclin B1-Cdk1 Complex. Mol. Cells 2016, 39, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Jackman, M.; Lindon, C.; Nigg, E.A.; Pines, J. Active cyclin B1–Cdk1 first appears on centrosomes in prophase. Nat. Cell Biol. 2003, 5, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Wells, A.D.; Morawski, P.A. New roles for cyclin-dependent kinases in T cell biology: Linking cell division and differentiation. Nat. Rev. Immunol. 2014, 14, 261–270. [Google Scholar] [CrossRef]
- Chunder, N.; Wang, L.; Chen, C.; Hancock, W.W.; Wells, A.D. Cyclin-Dependent Kinase 2 Controls Peripheral Immune Tolerance. J. Immunol. 2012, 189, 5659–5666. [Google Scholar] [CrossRef]
- Song, J.; Salek-Ardakani, S.; So, T.; Croft, M. The kinases aurora B and mTOR regulate the G1–S cell cycle progression of T lymphocytes. Nat. Immunol. 2006, 8, 64–73. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed]
- Read, R.; Fenton, T.; Gomez, G.G.; Wykosky, J.; Vandenberg, S.R.; Babic, I.; Iwanami, A.; Yang, H.; Cavenee, W.K.; Mischel, P.S.; et al. A Kinome-Wide RNAi Screen in Drosophila Glia Reveals That the RIO Kinases Mediate Cell Proliferation and Survival through TORC2-Akt Signaling in Glioblastoma. PLoS Genet. 2013, 9, e1003253. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Li, C.; Jin, L.; Xing, J.; Sha, Z.; Zhang, T.; Ji, D.; Yu, R.; Gao, S. RIOK2 is negatively regulated by miR-4744 and promotes glioma cell migration/invasion through epithelial-mesenchymal transition. J. Cell. Mol. Med. 2020, 24, 4494–4509. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-Cerca, S.; Sagar, V.; Schäfer, T.; Diop, M.; Wesseling, A.-M.; Lu, H.; Chai, E.; Hurt, E.; LaRonde-LeBlanc, N. ATPase-dependent role of the atypical kinase Rio2 on the evolving pre-40S ribosomal subunit. Nat. Struct. Mol. Biol. 2012, 19, 1316–1323. [Google Scholar] [CrossRef]
- Zemp, I.; Wild, T.; O’Donohue, M.-F.; Wandrey, F.; Widmann, B.; Gleizes, P.-E.; Kutay, U. Distinct cytoplasmic maturation steps of 40S ribosomal subunit precursors require hRio2. J. Cell Biol. 2009, 185, 1167–1180. [Google Scholar] [CrossRef] [PubMed]
- Prakash, V.; Carson, B.B.; Feenstra, J.M.; Dass, R.A.; Sekyrova, P.; Hoshino, A.; Petersen, J.; Guo, Y.; Parks, M.M.; Kurylo, C.M.; et al. Ribosome biogenesis during cell cycle arrest fuels EMT in development and disease. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Deng, M.; Li, J.; Tong, X.; Wei, Q.; Ye, X. Phosphorylation of Right Open Reading Frame 2 (Rio2) Protein Kinase by Polo-like Kinase 1 Regulates Mitotic Progression. J. Biol. Chem. 2011, 286, 36352–36360. [Google Scholar] [CrossRef] [PubMed]
- Raab, M.; Strebhardt, K.; Rudd, C.E. Immune adaptor SKAP1 acts a scaffold for Polo-like kinase 1 (PLK1) for the optimal cell cycling of T-cells. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Bostik, P.; Dodd, G.L.; Villinger, F.; Mayne, A.E.; Ansari, A.A. Dysregulation of the Polo-Like Kinase Pathway in CD4+ T Cells Is Characteristic of Pathogenic Simian Immunodeficiency Virus Infection. J. Virol. 2004, 78, 1464–1472. [Google Scholar] [CrossRef]
- Kulak, N.A.; Pichler, G.; Paron, I.; Nagaraj, N.; Mann, M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 2014, 11, 319–324. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; He, Q.Y. ReactomePA: An R/Bioconductor package for reactome pathway analysis and visualization. Mol. BioSyst. 2016, 12, 477–479. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Meguid, N.A.; Ghozlan, S.A.S.; Mohamed, M.F.; Ibrahim, M.K.; Dawood, R.M.; El Din, N.G.B.; Abdelhafez, T.H.; Hemimi, M.; El Awady, M.K. Expression of Reactive Oxygen Species—Related Transcripts in Egyptian Children With Autism. Biomark. Insights 2017, 12, 1177271917691035. [Google Scholar] [CrossRef]
- Subbannayya, Y.; Pinto, S.; Bösl, K.; Prasad, T.S.K.; Kandasamy, R.K. Dynamics of Dual Specificity Phosphatases and Their Interplay with Protein Kinases in Immune Signaling. Int. J. Mol. Sci. 2019, 20, 2086. [Google Scholar] [CrossRef]
- Doncheva, N.T.; Morris, J.H.; Gorodkin, J.; Jensen, L.J. Cytoscape StringApp: Network Analysis and Visualization of Proteomics Data. J. Proteome Res. 2019, 18, 623–632. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Subbannayya, Y.; Pinto, S.M.; Mohanty, V.; Dagamajalu, S.; Prasad, T.S.K.; Murthy, K.R. What Makes Cornea Immunologically Unique and Privileged? Mechanistic Clues from a High-Resolution Proteomic Landscape of the Human Cornea. OMICS J. Integr. Biol. 2020, 24, 129–139. [Google Scholar] [CrossRef]
- Su, G.; Morris, J.H.; Demchak, B.; Bader, G.D. Biological Network Exploration with Cytoscape 3. Curr. Protoc. Bioinform. 2014, 47, 8.13.1–8.13.24. [Google Scholar] [CrossRef] [PubMed]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [PubMed]
- Mudunuri, U.; Che, A.; Yi, M.; Stephens, R.M. bioDBnet: The biological database network. Bioinformatics 2009, 25, 555–556. [Google Scholar] [CrossRef] [PubMed]
- Coordinators, N.R. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2016, 44, D7–D19. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Vizcaino, J.A.; Deutsch, E.W.; Wang, R.; Csordas, A.; Reisinger, F.; Ríos, D.; Dianes, J.A.; Sun, Z.; Farrah, T.; Bandeira, N.; et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014, 32, 223–226. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subbannayya, Y.; Haug, M.; Pinto, S.M.; Mohanty, V.; Meås, H.Z.; Flo, T.H.; Prasad, T.S.K.; Kandasamy, R.K. The Proteomic Landscape of Resting and Activated CD4+ T Cells Reveal Insights into Cell Differentiation and Function. Int. J. Mol. Sci. 2021, 22, 275. https://doi.org/10.3390/ijms22010275
Subbannayya Y, Haug M, Pinto SM, Mohanty V, Meås HZ, Flo TH, Prasad TSK, Kandasamy RK. The Proteomic Landscape of Resting and Activated CD4+ T Cells Reveal Insights into Cell Differentiation and Function. International Journal of Molecular Sciences. 2021; 22(1):275. https://doi.org/10.3390/ijms22010275
Chicago/Turabian StyleSubbannayya, Yashwanth, Markus Haug, Sneha M. Pinto, Varshasnata Mohanty, Hany Zakaria Meås, Trude Helen Flo, T.S. Keshava Prasad, and Richard K. Kandasamy. 2021. "The Proteomic Landscape of Resting and Activated CD4+ T Cells Reveal Insights into Cell Differentiation and Function" International Journal of Molecular Sciences 22, no. 1: 275. https://doi.org/10.3390/ijms22010275
APA StyleSubbannayya, Y., Haug, M., Pinto, S. M., Mohanty, V., Meås, H. Z., Flo, T. H., Prasad, T. S. K., & Kandasamy, R. K. (2021). The Proteomic Landscape of Resting and Activated CD4+ T Cells Reveal Insights into Cell Differentiation and Function. International Journal of Molecular Sciences, 22(1), 275. https://doi.org/10.3390/ijms22010275