Transcriptomic Changes of Murine Visceral Fat Exposed to Intermittent Hypoxia at Single Cell Resolution

 ,
, 
 ,
, 
Abstract
1. Introduction
2. Results
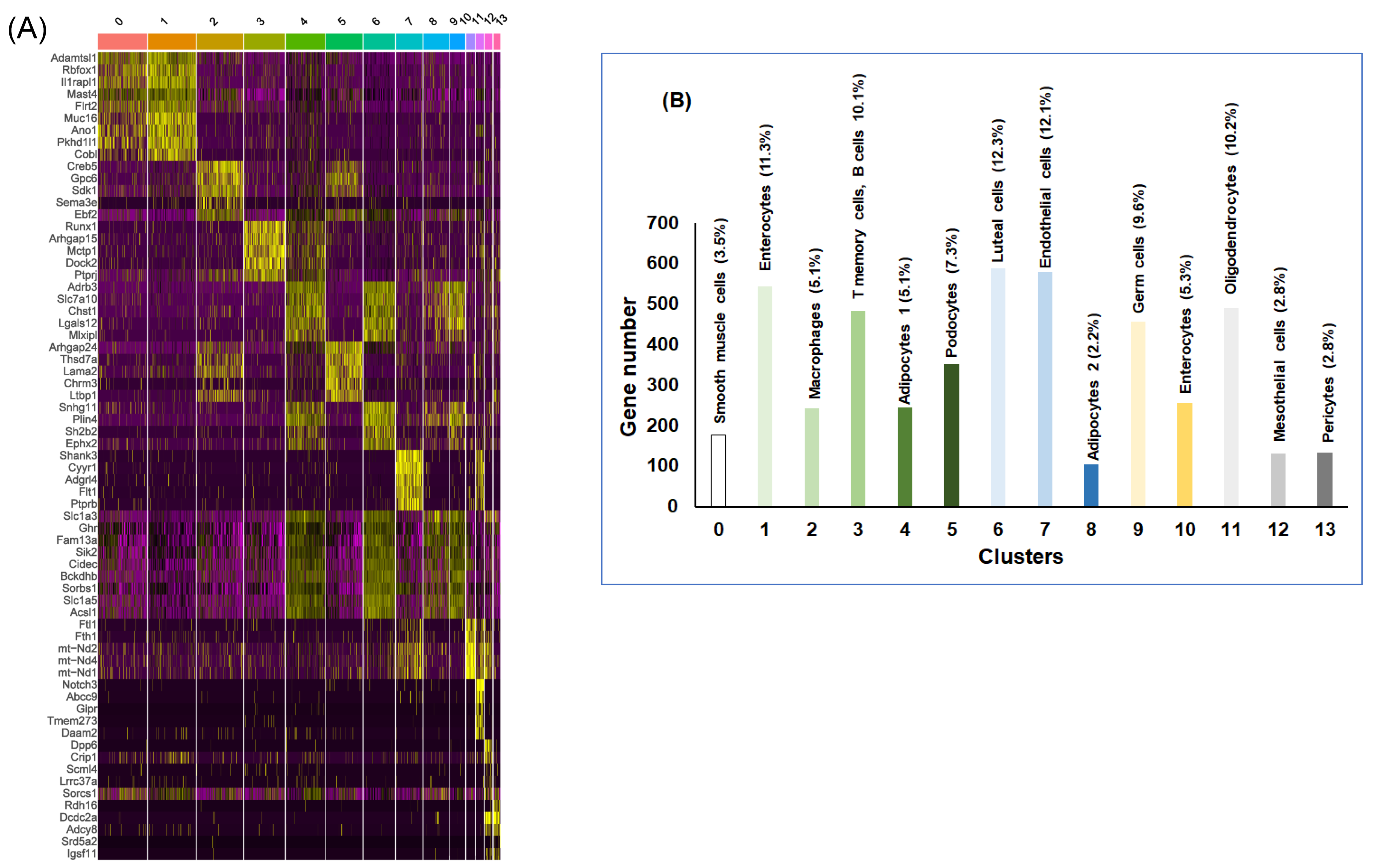
2.1. snRNA-seq Analysis
2.2. vWAT Cell Type Composition Following IH and RA Exposures
2.3. Gene Ontology and KEGG Enrichment Analyses of snRNA-seq Genes
2.4. Transcription Factors Enrichment by Cell Type
2.5. Adipocytes and Adipogenesis Genes
2.6. RNA-seq Analysis of vWAT in RA and IH Mice
2.7. Transcription Factor Analysis in Bulk RNA-Seq
2.8. Comparison of DEGs in snRNA-seq and Bulk RNA-seq
2.9. Deconvolution of Bulk RNA-seq
2.10. Verification of Bulk RNA-seq and snRNA-seq Using qRT-PCR
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Intermittent Hypoxia (IH)
4.3. Epidydimal Visceral White Adipose Tissues (vWAT)
4.4. Single Nuclei RNA-Sequencing (snRNA-seq)
4.5. snRNA-seq Bioinformatics Analysis
4.6. RNA-seq Analysis of vWAT
4.7. Functional Annotation and Gene Network Analysis
4.8. Deconvolution of RNA-seq to Cell Type Composition
4.9. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murphy, A.M.; Thomas, A.; Crinion, S.J.; Kent, B.D.; Tambuwala, M.M.; Fabre, A.; Pepin, J.L.; Roche, H.M.; Arnaud, C.; Ryan, S. Intermittent hypoxia in obstructive sleep apnoea mediates insulin resistance through adipose tissue inflammation. Eur. Respir. J. 2017, 49, 1601731. [Google Scholar] [CrossRef] [PubMed]
- Gaines, J.; Vgontzas, A.N.; Fernandez-Mendoza, J.; Bixler, E.O. Obstructive sleep apnea and the metabolic syndrome: The road to clinically-meaningful phenotyping, improved prognosis, and personalized treatment. Sleep Med. Rev. 2018, 42, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Tserenpil, G.; Gebre, M.; Zergham, A.S.; Sekhon, A.K.; Malik, B.H. Managements for Obstructive Sleep Apnea in Adults: Review. Cureus 2020, 12, e9905. [Google Scholar] [CrossRef]
- Kent, B.D.; McNicholas, W.T.; Ryan, S. Insulin resistance, glucose intolerance and diabetes mellitus in obstructive sleep apnoea. J. Thorac. Dis. 2015, 7, 1343–1357. [Google Scholar] [CrossRef]
- Pamidi, S.; Wroblewski, K.; Stepien, M.; Sharif-Sidi, K.; Kilkus, J.; Whitmore, H.; Tasali, E. Eight Hours of Nightly Continuous Positive Airway Pressure Treatment of Obstructive Sleep Apnea Improves Glucose Metabolism in Patients with Prediabetes. A Randomized Controlled Trial. Am. J. Respir. Crit. Care Med. 2015, 192, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Khalyfa, A.; Kheirandish-Gozal, L.; Gozal, D. Exosome and Macrophage Crosstalk in Sleep-Disordered Breathing-Induced Metabolic Dysfunction. Int. J. Mol. Sci. 2018, 19, 3383. [Google Scholar] [CrossRef]
- Galerneau, L.M.; Bailly, S.; Borel, J.C.; Jullian-Desayes, I.; Joyeux-Faure, M.; Benmerad, M.; Bonsignore, M.R.; Tamisier, R.; Pepin, J.L. Long-term variations of arterial stiffness in patients with obesity and obstructive sleep apnea treated with continuous positive airway pressure. PLoS ONE 2020, 15, e0236667. [Google Scholar] [CrossRef]
- Salman, L.A.; Shulman, R.; Cohen, J.B. Obstructive Sleep Apnea, Hypertension, and Cardiovascular Risk: Epidemiology, Pathophysiology, and Management. Curr. Cardiol. Rep. 2020, 22, 6. [Google Scholar] [CrossRef]
- Hassaballa, H.A.; Tulaimat, A.; Herdegen, J.J.; Mokhlesi, B. The effect of continuous positive airway pressure on glucose control in diabetic patients with severe obstructive sleep apnea. Sleep Breath. 2005, 9, 176–180. [Google Scholar] [CrossRef]
- Koren, D.; Dumin, M.; Gozal, D. Role of sleep quality in the metabolic syndrome. Diabetes Metab. Syndr. Obes. Targets Ther. 2016, 9, 281–310. [Google Scholar] [CrossRef]
- Temple, K.A.; Leproult, R.; Morselli, L.; Ehrmann, D.A.; Van Cauter, E.; Mokhlesi, B. Sex Differences in the Impact of Obstructive Sleep Apnea on Glucose Metabolism. Front. Endocrinol. 2018, 9, 376. [Google Scholar] [CrossRef]
- Hiller, N.; Schor-Bardach, R.; Gileles-Hillel, A.; Stroumsa, D.; Simanovsky, N. CT appearance of the pelvis after Cesarean delivery—What is considered normal? Clin. Imaging 2013, 37, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Gileles-Hillel, A.; Kheirandish-Gozal, L.; Gozal, D. Biological plausibility linking sleep apnoea and metabolic dysfunction. Nat. Rev. Endocrinol. 2016, 12, 290–298. [Google Scholar] [CrossRef]
- Minami, T.; Matsumoto, T.; Tabara, Y.; Gozal, D.; Smith, D.; Murase, K.; Tanizawa, K.; Takahashi, N.; Nakatsuka, Y.; Hamada, S.; et al. Impact of sleep-disordered breathing on glucose metabolism among individuals with a family history of diabetes: The Nagahama study. J. Clin. Sleep Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Murase, K.; Tabara, Y.; Gozal, D.; Smith, D.; Minami, T.; Tachikawa, R.; Tanizawa, K.; Oga, T.; Nagashima, S.; et al. Impact of sleep characteristics and obesity on diabetes and hypertension across genders and menopausal status: The Nagahama study. Sleep 2018, 41. [Google Scholar] [CrossRef]
- Louis, M.; Punjabi, N.M. Effects of acute intermittent hypoxia on glucose metabolism in awake healthy volunteers. J. Appl. Physiol. 2009, 106, 1538–1544. [Google Scholar] [CrossRef] [PubMed]
- Rasouli, N. Adipose tissue hypoxia and insulin resistance. J. Investig. Med. 2016, 64, 830–832. [Google Scholar] [CrossRef]
- Fang, Y.; Kaszuba, T.; Imoukhuede, P.I. Systems Biology Will Direct Vascular-Targeted Therapy for Obesity. Front. Physiol. 2020, 11, 831. [Google Scholar] [CrossRef]
- Ryan, S.; Arnaud, C.; Fitzpatrick, S.F.; Gaucher, J.; Tamisier, R.; Pepin, J.L. Adipose tissue as a key player in obstructive sleep apnoea. Eur. Respir. Rev. 2019, 28, 190006. [Google Scholar] [CrossRef]
- Gileles-Hillel, A.; Almendros, I.; Khalyfa, A.; Nigdelioglu, R.; Qiao, Z.; Hamanaka, R.B.; Mutlu, G.M.; Akbarpour, M.; Gozal, D. Prolonged Exposures to Intermittent Hypoxia Promote Visceral White Adipose Tissue Inflammation in a Murine Model of Severe Sleep Apnea: Effect of Normoxic Recovery. Sleep 2017, 40. [Google Scholar] [CrossRef]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Akra, S.; Aksnes, T.A.; Flaa, A.; Eggesbo, H.B.; Opstad, T.B.; Njerve, I.U.; Seljeflot, I. Markers of remodeling in subcutaneous adipose tissue are strongly associated with overweight and insulin sensitivity in healthy non-obese men. Sci. Rep. 2020, 10, 14055. [Google Scholar] [CrossRef] [PubMed]
- Almendros, I.; Farre, R.; Planas, A.M.; Torres, M.; Bonsignore, M.R.; Navajas, D.; Montserrat, J.M. Tissue oxygenation in brain, muscle, and fat in a rat model of sleep apnea: Differential effect of obstructive apneas and intermittent hypoxia. Sleep 2011, 34, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Boker, S.; Neale, M.; Maes, H.; Wilde, M.; Spiegel, M.; Brick, T.; Spies, J.; Estabrook, R.; Kenny, S.; Bates, T.; et al. OpenMx: An Open Source Extended Structural Equation Modeling Framework. Psychometrika 2011, 76, 306–317. [Google Scholar] [CrossRef]
- Glastonbury, C.A.; Couto Alves, A.; El-Sayed Moustafa, J.S.; Small, K.S. Cell-Type Heterogeneity in Adipose Tissue Is Associated with Complex Traits and Reveals Disease-Relevant Cell-Specific eQTLs. Am. J. Hum. Genet. 2019, 104, 1013–1024. [Google Scholar] [CrossRef]
- Eto, H.; Suga, H.; Matsumoto, D.; Inoue, K.; Aoi, N.; Kato, H.; Araki, J.; Yoshimura, K. Characterization of structure and cellular components of aspirated and excised adipose tissue. Plast. Reconstr. Surg. 2009, 124, 1087–1097. [Google Scholar] [CrossRef]
- Wang, Q.A.; Tao, C.; Gupta, R.K.; Scherer, P.E. Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat. Med. 2013, 19, 1338–1344. [Google Scholar] [CrossRef]
- Ryan, S. Adipose tissue inflammation by intermittent hypoxia: Mechanistic link between obstructive sleep apnoea and metabolic dysfunction. J. Physiol. 2017, 595, 2423–2430. [Google Scholar] [CrossRef]
- Ryan, S. Mechanisms of cardiovascular disease in obstructive sleep apnoea. J. Thorac. Dis. 2018, 10, S4201–S4211. [Google Scholar] [CrossRef]
- Bora, P.; Majumdar, A.S. Adipose tissue-derived stromal vascular fraction in regenerative medicine: A brief review on biology and translation. Stem Cell Res. Ther. 2017, 8, 145. [Google Scholar] [CrossRef]
- Habib, N.; Li, Y.; Heidenreich, M.; Swiech, L.; Avraham-Davidi, I.; Trombetta, J.J.; Hession, C.; Zhang, F.; Regev, A. Div-Seq: Single-nucleus RNA-Seq reveals dynamics of rare adult newborn neurons. Science 2016, 353, 925–928. [Google Scholar] [CrossRef] [PubMed]
- Lake, B.B.; Ai, R.; Kaeser, G.E.; Salathia, N.S.; Yung, Y.C.; Liu, R.; Wildberg, A.; Gao, D.; Fung, H.L.; Chen, S.; et al. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science 2016, 352, 1586–1590. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.S.; Schmitt, R.E.; Dasgupta, A.; Ducharme, A.M.; Doles, J.D. Single-cell deconstruction of post-sepsis skeletal muscle and adipose tissue microenvironments. J. Cachexia Sarcopenia Muscle 2020, 11, 1351–1363. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Song, Y. Single cell sequencing: A distinct new field. Clin. Transl. Med. 2017, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Gawad, C.; Koh, W.; Quake, S.R. Single-cell genome sequencing: Current state of the science. Nat. Rev. 2016, 17, 175–188. [Google Scholar] [CrossRef]
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour evolution inferred by single-cell sequencing. Nature 2011, 472, 90–94. [Google Scholar] [CrossRef]
- Shalek, A.K.; Satija, R.; Adiconis, X.; Gertner, R.S.; Gaublomme, J.T.; Raychowdhury, R.; Schwartz, S.; Yosef, N.; Malboeuf, C.; Lu, D.; et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature 2013, 498, 236–240. [Google Scholar] [CrossRef]
- Wills, Q.F.; Livak, K.J.; Tipping, A.J.; Enver, T.; Goldson, A.J.; Sexton, D.W.; Holmes, C. Single-cell gene expression analysis reveals genetic associations masked in whole-tissue experiments. Nat. Biotechnol. 2013, 31, 748–752. [Google Scholar] [CrossRef]
- Zeisel, A.; Munoz-Manchado, A.B.; Codeluppi, S.; Lonnerberg, P.; La Manno, G.; Jureus, A.; Marques, S.; Munguba, H.; He, L.; Betsholtz, C.; et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 2015, 347, 1138–1142. [Google Scholar] [CrossRef]
- Blakeley, P.; Fogarty, N.M.; del Valle, I.; Wamaitha, S.E.; Hu, T.X.; Elder, K.; Snell, P.; Christie, L.; Robson, P.; Niakan, K.K. Defining the three cell lineages of the human blastocyst by single-cell RNA-seq. Development 2015, 142, 3151–3165. [Google Scholar] [CrossRef]
- Venteicher, A.S.; Tirosh, I.; Hebert, C.; Yizhak, K.; Neftel, C.; Filbin, M.G.; Hovestadt, V.; Escalante, L.E.; Shaw, M.L.; Rodman, C.; et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 2017, 355, eaai8478. [Google Scholar] [CrossRef] [PubMed]
- Shalek, A.K.; Satija, R.; Shuga, J.; Trombetta, J.J.; Gennert, D.; Lu, D.; Chen, P.; Gertner, R.S.; Gaublomme, J.T.; Yosef, N.; et al. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature 2014, 510, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Torre, E.; Dueck, H.; Shaffer, S.; Gospocic, J.; Gupte, R.; Bonasio, R.; Kim, J.; Murray, J.; Raj, A. Rare Cell Detection by Single-Cell RNA Sequencing as Guided by Single-Molecule RNA FISH. Cell Syst. 2018, 6, 171–179.e175. [Google Scholar] [CrossRef] [PubMed]
- Song, A.; Dai, W.; Jang, M.J.; Medrano, L.; Li, Z.; Zhao, H.; Shao, M.; Tan, J.; Li, A.; Ning, T.; et al. Low- and high-thermogenic brown adipocyte subpopulations coexist in murine adipose tissue. J. Clin. Investig. 2020, 130, 247–257. [Google Scholar] [CrossRef]
- Rajbhandari, P.; Arneson, D.; Hart, S.K.; Ahn, I.S.; Diamante, G.; Santos, L.C.; Zaghari, N.; Feng, A.C.; Thomas, B.J.; Vergnes, L.; et al. Single cell analysis reveals immune cell-adipocyte crosstalk regulating the transcription of thermogenic adipocytes. Elife 2019, 8, e49501. [Google Scholar] [CrossRef]
- Hepler, C.; Shan, B.; Zhang, Q.; Henry, G.H.; Shao, M.; Vishvanath, L.; Ghaben, A.L.; Mobley, A.B.; Strand, D.; Hon, G.C.; et al. Identification of functionally distinct fibro-inflammatory and adipogenic stromal subpopulations in visceral adipose tissue of adult mice. Elife 2018, 7, e39636. [Google Scholar] [CrossRef]
- Vijay, J.; Gauthier, M.F.; Biswell, R.L.; Louiselle, D.A.; Johnston, J.J.; Cheung, W.A.; Belden, B.; Pramatarova, A.; Biertho, L.; Gibson, M.; et al. Single-cell analysis of human adipose tissue identifies depot and disease specific cell types. Nat. Metab. 2020, 2, 97–109. [Google Scholar] [CrossRef]
- Rondini, E.A.; Granneman, J.G. Single cell approaches to address adipose tissue stromal cell heterogeneity. Biochem. J. 2020, 477, 583–600. [Google Scholar] [CrossRef]
- Weinstock, A.; Brown, E.J.; Garabedian, M.L.; Pena, S.; Sharma, M.; Lafaille, J.; Moore, K.J.; Fisher, E.A. Single-Cell RNA Sequencing of Visceral Adipose Tissue Leukocytes Reveals that Caloric Restriction Following Obesity Promotes the Accumulation of a Distinct Macrophage Population with Features of Phagocytic Cells. Immunometabolism 2019, 1, e190008. [Google Scholar] [CrossRef]
- Burl, R.B.; Ramseyer, V.D.; Rondini, E.A.; Pique-Regi, R.; Lee, Y.H.; Granneman, J.G. Deconstructing Adipogenesis Induced by beta3-Adrenergic Receptor Activation with Single-Cell Expression Profiling. Cell Metab. 2018, 28, 300–309.e304. [Google Scholar] [CrossRef]
- Ding, J.; Adiconis, X.; Simmons, S.K.; Kowalczyk, M.S.; Hession, C.C.; Marjanovic, N.D.; Hughes, T.K.; Wadsworth, M.H.; Burks, T.; Nguyen, L.T.; et al. Systematic comparison of single-cell and single-nucleus RNA-sequencing methods. Nat. Biotechnol. 2020, 38, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Kirov, S.; Snoddy, J. WebGestalt: An integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005, 33, W741–W748. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.; Cummins, E.P.; Farre, R.; Gileles-Hillel, A.; Jun, J.C.; Oster, H.; Pepin, J.L.; Ray, D.W.; Reutrakul, S.; Sanchez-de-la-Torre, M.; et al. Understanding the pathophysiological mechanisms of cardiometabolic complications in obstructive sleep apnoea: Towards personalised treatment approaches. Eur. Respir. J. 2020, 56, 1902295. [Google Scholar] [CrossRef] [PubMed]
- Khalyfa, A.; Castro-Grattoni, A.L.; Gozal, D. Cardiovascular morbidities of obstructive sleep apnea and the role of circulating extracellular vesicles. Ther. Adv. Respir. Dis. 2019, 13, 1753466619895229. [Google Scholar] [CrossRef] [PubMed]
- Rana, D.; Torrilus, C.; Ahmad, W.; Okam, N.A.; Fatima, T.; Jahan, N. Obstructive Sleep Apnea and Cardiovascular Morbidities: A Review Article. Cureus 2020, 12, e10424. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.D.; Chen, Q.; Lin, X.J.; Chen, Q.S.; Huang, Y.Z.; Wu, R.H.; Lin, G.F.; Huang, X.Y.; Lin, Q.C. Effect of chronic intermittent hypoxia on gene expression profiles of rat liver: A better understanding of OSA-related liver disease. Sleep Breath. 2020, 24, 761–770. [Google Scholar] [CrossRef]
- Trayhurn, P.; Wang, B.; Wood, I.S. Hypoxia and the endocrine and signalling role of white adipose tissue. Arch. Physiol. Biochem. 2008, 114, 267–276. [Google Scholar] [CrossRef]
- Fitzpatrick, S.F.; King, A.D.; O’Donnell, C.; Roche, H.M.; Ryan, S. Mechanisms of intermittent hypoxia-mediated macrophage activation—Potential therapeutic targets for obstructive sleep apnoea. J. Sleep Res. 2020. [Google Scholar] [CrossRef]
- Zhang, C.; Luo, X.; Zhang, D.; Deng, B.; Tong, J.; Zhang, M.; Chen, L.; Duan, H.; Niu, W. Hypoxic adipocytes induce macrophages to release inflammatory cytokines that render skeletal muscle cells insulin resistant. Biochem. Biophys. Res. Commun. 2020, 521, 625–631. [Google Scholar] [CrossRef]
- Ge, M.Q.; Yeung, S.C.; Mak, J.C.W.; Ip, M.S.M. Differential metabolic and inflammatory responses to intermittent hypoxia in substrains of lean and obese C57BL/6 mice. Life Sci. 2019, 238, 116959. [Google Scholar] [CrossRef]
- Poulain, L.; Mathieu, H.; Thomas, A.; Borel, A.L.; Remy, C.; Levy, P.; Arnaud, C.; Dematteis, M. Intermittent hypoxia-induced insulin resistance is associated with alterations in white fat distribution. Sci. Rep. 2017, 7, 11180. [Google Scholar] [CrossRef] [PubMed]
- Gozal, D.; Gileles-Hillel, A.; Cortese, R.; Li, Y.; Almendros, I.; Qiao, Z.; Khalyfa, A.A.; Andrade, J.; Khalyfa, A. Visceral White Adipose Tissue after Chronic Intermittent and Sustained Hypoxia in Mice. Am. J. Respir. Cell Mol. Biol. 2017, 56, 477–487. [Google Scholar] [CrossRef]
- Thorn, C.E.; Knight, B.; Pastel, E.; McCulloch, L.J.; Patel, B.; Shore, A.C.; Kos, K. Adipose tissue is influenced by hypoxia of obstructive sleep apnea syndrome independent of obesity. Diabetes Metab. 2017, 43, 240–247. [Google Scholar] [CrossRef]
- Carreras, A.; Zhang, S.X.; Almendros, I.; Wang, Y.; Peris, E.; Qiao, Z.; Gozal, D. Resveratrol attenuates intermittent hypoxia-induced macrophage migration to visceral white adipose tissue and insulin resistance in male mice. Endocrinology 2015, 156, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Palmer, B.F.; Clegg, D.J. Oxygen sensing and metabolic homeostasis. Mol. Cell. Endocrinol. 2014, 397, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Packer, J.S.; Ramani, V.; Cusanovich, D.A.; Huynh, C.; Daza, R.; Qiu, X.; Lee, C.; Furlan, S.N.; Steemers, F.J.; et al. Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 2017, 357, 661–667. [Google Scholar] [CrossRef]
- Rosenberg, A.B.; Roco, C.M.; Muscat, R.A.; Kuchina, A.; Sample, P.; Yao, Z.; Graybuck, L.T.; Peeler, D.J.; Mukherjee, S.; Chen, W.; et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 2018, 360, 176–182. [Google Scholar] [CrossRef]
- Yuan, G.C.; Cai, L.; Elowitz, M.; Enver, T.; Fan, G.; Guo, G.; Irizarry, R.; Kharchenko, P.; Kim, J.; Orkin, S.; et al. Challenges and emerging directions in single-cell analysis. Genome Biol. 2017, 18, 84. [Google Scholar] [CrossRef]
- Han, X.; Chen, H.; Huang, D.; Chen, H.; Fei, L.; Cheng, C.; Huang, H.; Yuan, G.C.; Guo, G. Mapping human pluripotent stem cell differentiation pathways using high throughput single-cell RNA-sequencing. Genome Biol. 2018, 19, 47. [Google Scholar] [CrossRef]
- Koenitzer, J.R.; Wu, H.; Atkinson, J.J.; Brody, S.L.; Humphreys, B.D. Single Nucleus RNASeq Profiling of Mouse Lung: Reduced Dissociation Bias and Improved Rare Cell Type Detection Compared with Single Cell RNASeq. Am. J. Respir. Cell Mol. Biol. 2020, 63, 739–747. [Google Scholar] [CrossRef]
- Darmanis, S.; Sloan, S.A.; Zhang, Y.; Enge, M.; Caneda, C.; Shuer, L.M.; Hayden Gephart, M.G.; Barres, B.A.; Quake, S.R. A survey of human brain transcriptome diversity at the single cell level. Proc. Natl. Acad. Sci. USA 2015, 112, 7285–7290. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, K.; Linnarsson, S. Single-cell mRNA isoform diversity in the mouse brain. BMC Genom. 2017, 18, 126. [Google Scholar] [CrossRef] [PubMed]
- Villani, A.C.; Shekhar, K. Single-Cell RNA Sequencing of Human T Cells. Methods Mol. Biol. 2017, 1514, 203–239. [Google Scholar] [CrossRef] [PubMed]
- Velten, L.; Haas, S.F.; Raffel, S.; Blaszkiewicz, S.; Islam, S.; Hennig, B.P.; Hirche, C.; Lutz, C.; Buss, E.C.; Nowak, D.; et al. Human haematopoietic stem cell lineage commitment is a continuous process. Nat. Cell Biol. 2017, 19, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Cochain, C.; Vafadarnejad, E.; Arampatzi, P.; Pelisek, J.; Winkels, H.; Ley, K.; Wolf, D.; Saliba, A.E.; Zernecke, A. Single-Cell RNA-Seq Reveals the Transcriptional Landscape and Heterogeneity of Aortic Macrophages in Murine Atherosclerosis. Circ. Res. 2018, 122, 1661–1674. [Google Scholar] [CrossRef]
- Tchkonia, T.; Thomou, T.; Zhu, Y.; Karagiannides, I.; Pothoulakis, C.; Jensen, M.D.; Kirkland, J.L. Mechanisms and metabolic implications of regional differences among fat depots. Cell Metab. 2013, 17, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Schoettl, T.; Fischer, I.P.; Ussar, S. Heterogeneity of adipose tissue in development and metabolic function. J. Exp. Biol. 2018, 221, jeb162958. [Google Scholar] [CrossRef]
- Gribar, S.C.; Sodhi, C.P.; Richardson, W.M.; Anand, R.J.; Gittes, G.K.; Branca, M.F.; Jakub, A.; Shi, X.H.; Shah, S.; Ozolek, J.A.; et al. Reciprocal expression and signaling of TLR4 and TLR9 in the pathogenesis and treatment of necrotizing enterocolitis. J. Immunol. 2009, 182, 636–646. [Google Scholar] [CrossRef]
- Roberts, A.C.; Porter, K.E. Cellular and molecular mechanisms of endothelial dysfunction in diabetes. Diabetes Vasc. Dis. Res. 2013, 10, 472–482. [Google Scholar] [CrossRef]
- Baryawno, N.; Przybylski, D.; Kowalczyk, M.S.; Kfoury, Y.; Severe, N.; Gustafsson, K.; Kokkaliaris, K.D.; Mercier, F.; Tabaka, M.; Hofree, M.; et al. A Cellular Taxonomy of the Bone Marrow Stroma in Homeostasis and Leukemia. Cell 2019, 177, 1915–1932.e1916. [Google Scholar] [CrossRef] [PubMed]
- Lukowski, S.W.; Patel, J.; Andersen, S.B.; Sim, S.L.; Wong, H.Y.; Tay, J.; Winkler, I.; Powell, J.E.; Khosrotehrani, K. Single-Cell Transcriptional Profiling of Aortic Endothelium Identifies a Hierarchy from Endovascular Progenitors to Differentiated Cells. Cell Rep. 2019, 27, 2748–2758.e2743. [Google Scholar] [CrossRef] [PubMed]
- Tikhonova, A.N.; Dolgalev, I.; Hu, H.; Sivaraj, K.K.; Hoxha, E.; Cuesta-Dominguez, A.; Pinho, S.; Akhmetzyanova, I.; Gao, J.; Witkowski, M.; et al. The bone marrow microenvironment at single-cell resolution. Nature 2019, 569, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Armulik, A.; Abramsson, A.; Betsholtz, C. Endothelial/pericyte interactions. Circ. Res. 2005, 97, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Schwalie, P.C.; Dong, H.; Zachara, M.; Russeil, J.; Alpern, D.; Akchiche, N.; Caprara, C.; Sun, W.; Schlaudraff, K.U.; Soldati, G.; et al. A stromal cell population that inhibits adipogenesis in mammalian fat depots. Nature 2018, 559, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Chau, Y.Y.; Hastie, N. Wt1, the mesothelium and the origins and heterogeneity of visceral fat progenitors. Adipocyte 2015, 4, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Gupta, O.T.; Gupta, R.K. Visceral Adipose Tissue Mesothelial Cells: Living on the Edge or Just Taking Up Space? Trends Endocrinol. Metab. 2015, 26, 515–523. [Google Scholar] [CrossRef]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M., 3rd; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e1821. [Google Scholar] [CrossRef]
- Darimont, C.; Avanti, O.; Blancher, F.; Wagniere, S.; Mansourian, R.; Zbinden, I.; Leone-Vautravers, P.; Fuerholz, A.; Giusti, V.; Mace, K. Contribution of mesothelial cells in the expression of inflammatory-related factors in omental adipose tissue of obese subjects. Int. J. Obes. 2008, 32, 112–120. [Google Scholar] [CrossRef]
- Hill, D.A.; Lim, H.W.; Kim, Y.H.; Ho, W.Y.; Foong, Y.H.; Nelson, V.L.; Nguyen, H.C.B.; Chegireddy, K.; Kim, J.; Habertheuer, A.; et al. Distinct macrophage populations direct inflammatory versus physiological changes in adipose tissue. Proc. Natl. Acad. Sci. USA 2018, 115, E5096–E5105. [Google Scholar] [CrossRef]
- Lee, Y.H.; Petkova, A.P.; Mottillo, E.P.; Granneman, J.G. In vivo identification of bipotential adipocyte progenitors recruited by beta3-adrenoceptor activation and high-fat feeding. Cell Metab. 2012, 15, 480–491. [Google Scholar] [CrossRef]
- Lee, Y.H.; Petkova, A.P.; Granneman, J.G. Identification of an adipogenic niche for adipose tissue remodeling and restoration. Cell Metab. 2013, 18, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Kim, S.N.; Kwon, H.J.; Maddipati, K.R.; Granneman, J.G. Adipogenic role of alternatively activated macrophages in beta-adrenergic remodeling of white adipose tissue. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R55–R65. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Graham, T.E.; Mody, N.; Preitner, F.; Peroni, O.D.; Zabolotny, J.M.; Kotani, K.; Quadro, L.; Kahn, B.B. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature 2005, 436, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.A.; Yuen, J.J.; Jiang, H.; Kahn, B.B.; Blaner, W.S. Adipocyte-specific overexpression of retinol-binding protein 4 causes hepatic steatosis in mice. Hepatology 2016, 64, 1534–1546. [Google Scholar] [CrossRef] [PubMed]
- Zhan, S.; Li, J.; Wang, T.; Ge, W. Quantitative Proteomics Analysis of Sporadic Medullary Thyroid Cancer Reveals FN1 as a Potential Novel Candidate Prognostic Biomarker. Oncologist 2018, 23, 1415–1425. [Google Scholar] [CrossRef]
- Spencer, M.; Unal, R.; Zhu, B.; Rasouli, N.; McGehee, R.E., Jr.; Peterson, C.A.; Kern, P.A. Adipose tissue extracellular matrix and vascular abnormalities in obesity and insulin resistance. J. Clin. Endocrinol. Metab. 2011, 96, E1990–E1998. [Google Scholar] [CrossRef]
- Bouloumie, A.; Sengenes, C.; Portolan, G.; Galitzky, J.; Lafontan, M. Adipocyte produces matrix metalloproteinases 2 and 9: Involvement in adipose differentiation. Diabetes 2001, 50, 2080–2086. [Google Scholar] [CrossRef]
- Civelek, M.; Wu, Y.; Pan, C.; Raulerson, C.K.; Ko, A.; He, A.; Tilford, C.; Saleem, N.K.; Stancakova, A.; Scott, L.J.; et al. Genetic Regulation of Adipose Gene Expression and Cardio-Metabolic Traits. Am. J. Hum. Genet. 2017, 100, 428–443. [Google Scholar] [CrossRef]
- Westra, H.J.; Arends, D.; Esko, T.; Peters, M.J.; Schurmann, C.; Schramm, K.; Kettunen, J.; Yaghootkar, H.; Fairfax, B.P.; Andiappan, A.K.; et al. Cell Specific eQTL Analysis without Sorting Cells. PLoS Genet. 2015, 11, e1005223. [Google Scholar] [CrossRef]
- Baron, M.; Veres, A.; Wolock, S.L.; Faust, A.L.; Gaujoux, R.; Vetere, A.; Ryu, J.H.; Wagner, B.K.; Shen-Orr, S.S.; Klein, A.M.; et al. A Single-Cell Transcriptomic Map of the Human and Mouse Pancreas Reveals Inter- and Intra-cell Population Structure. Cell Syst. 2016, 3, 346–360.e344. [Google Scholar] [CrossRef]
- Shen-Orr, S.S.; Gaujoux, R. Computational deconvolution: Extracting cell type-specific information from heterogeneous samples. Curr. Opin. Immunol. 2013, 25, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Park, J.; Susztak, K.; Zhang, N.R.; Li, M. Bulk tissue cell type deconvolution with multi-subject single-cell expression reference. Nat. Commun. 2019, 10, 380. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Chen, Y.A.; Conejo-Garcia, J.R.; Chung, C.H.; Wang, X. Estimation of immune cell content in tumor using single-cell RNA-seq reference data. BMC Cancer 2019, 19, 715. [Google Scholar] [CrossRef]
- Avila Cobos, F.; Vandesompele, J.; Mestdagh, P.; De Preter, K. Computational deconvolution of transcriptomics data from mixed cell populations. Bioinformatics 2018, 34, 1969–1979. [Google Scholar] [CrossRef]
- Hu, P.; Zhang, W.; Xin, H.; Deng, G. Single Cell Isolation and Analysis. Front. Cell Dev. Biol. 2016, 4, 116. [Google Scholar] [CrossRef]
- Ziegenhain, C.; Vieth, B.; Parekh, S.; Reinius, B.; Guillaumet-Adkins, A.; Smets, M.; Leonhardt, H.; Heyn, H.; Hellmann, I.; Enard, W. Comparative Analysis of Single-Cell RNA Sequencing Methods. Mol. Cell 2017, 65, 631–643.e634. [Google Scholar] [CrossRef]
- La Manno, G.; Soldatov, R.; Zeisel, A.; Braun, E.; Hochgerner, H.; Petukhov, V.; Lidschreiber, K.; Kastriti, M.E.; Lonnerberg, P.; Furlan, A.; et al. RNA velocity of single cells. Nature 2018, 560, 494–498. [Google Scholar] [CrossRef]
- Jew, B.; Alvarez, M.; Rahmani, E.; Miao, Z.; Ko, A.; Garske, K.M.; Sul, J.H.; Pietilainen, K.H.; Pajukanta, P.; Halperin, E. Accurate estimation of cell composition in bulk expression through robust integration of single-cell information. Nat. Commun. 2020, 11, 1971. [Google Scholar] [CrossRef]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human Transcription Factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef] [PubMed]
- Ravasi, T.; Suzuki, H.; Cannistraci, C.V.; Katayama, S.; Bajic, V.B.; Tan, K.; Akalin, A.; Schmeier, S.; Kanamori-Katayama, M.; Bertin, N.; et al. An atlas of combinatorial transcriptional regulation in mouse and man. Cell 2010, 140, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Gertz, J.; Reddy, T.E.; Varley, K.E.; Garabedian, M.J.; Myers, R.M. Genistein and bisphenol A exposure cause estrogen receptor 1 to bind thousands of sites in a cell type-specific manner. Genome Res. 2012, 22, 2153–2162. [Google Scholar] [CrossRef] [PubMed]
- Fajas, L.; Landsberg, R.L.; Huss-Garcia, Y.; Sardet, C.; Lees, J.A.; Auwerx, J. E2Fs regulate adipocyte differentiation. Dev. Cell 2002, 3, 39–49. [Google Scholar] [CrossRef]
- Yu, X.; Shen, N.; Zhang, M.L.; Pan, F.Y.; Wang, C.; Jia, W.P.; Liu, C.; Gao, Q.; Gao, X.; Xue, B.; et al. Egr-1 decreases adipocyte insulin sensitivity by tilting PI3K/Akt and MAPK signal balance in mice. EMBO J. 2011, 30, 3754–3765. [Google Scholar] [CrossRef]
- Chakrabarti, P.; Kim, J.Y.; Singh, M.; Shin, Y.K.; Kim, J.; Kumbrink, J.; Wu, Y.; Lee, M.J.; Kirsch, K.H.; Fried, S.K.; et al. Insulin inhibits lipolysis in adipocytes via the evolutionarily conserved mTORC1-Egr1-ATGL-mediated pathway. Mol. Cell. Biol. 2013, 33, 3659–3666. [Google Scholar] [CrossRef]
- Khalyfa, A.; Gozal, D.; Masa, J.F.; Marin, J.M.; Qiao, Z.; Corral, J.; Gonzalez, M.; Marti, S.; Kheirandish-Gozal, L.; Egea, C.; et al. Sleep-disordered breathing, circulating exosomes, and insulin sensitivity in adipocytes. Int. J. Obes. 2018, 42, 1127–1139. [Google Scholar] [CrossRef]
- Khalyfa, A.; Qiao, Z.; Gileles-Hillel, A.; Khalyfa, A.A.; Akbarpour, M.; Popko, B.; Gozal, D. Activation of the Integrated Stress Response and Metabolic Dysfunction in a Murine Model of Sleep Apnea. Am. J. Respir. Cell Mol. Biol. 2017, 57, 477–486. [Google Scholar] [CrossRef]
- Cao, Y.; Zhu, J.; Jia, P.; Zhao, Z. scRNASeqDB: A Database for RNA-Seq Based Gene Expression Profiles in Human Single Cells. Genes 2017, 8, 368. [Google Scholar] [CrossRef]
- Bartlett, T.E.; Muller, S.; Diaz, A. Single-cell Co-expression Subnetwork Analysis. Sci. Rep. 2017, 7, 15066. [Google Scholar] [CrossRef]
- Satija, R.; Farrell, J.A.; Gennert, D.; Schier, A.F.; Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef]
- Ward, C.M.; To, T.H.; Pederson, S.M. ngsReports: A Bioconductor package for managing FastQC reports and other NGS related log files. Bioinformatics 2020, 36, 2587–2588. [Google Scholar] [CrossRef]
- Hafemeister, C.; Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019, 20, 296. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Franzen, O.; Gan, L.M.; Bjorkegren, J.L.M. PanglaoDB: A web server for exploration of mouse and human single-cell RNA sequencing data. Database 2019, 2019, baz046. [Google Scholar] [CrossRef] [PubMed]
- Khalyfa, A.; Mutskov, V.; Carreras, A.; Khalyfa, A.A.; Hakim, F.; Gozal, D. Sleep fragmentation during late gestation induces metabolic perturbations and epigenetic changes in adiponectin gene expression in male adult offspring mice. Diabetes 2014, 63, 3230–3241. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Chong, X.; Peng, R.; Sun, Y.; Zhang, L.; Zhang, Z. Identification of Key Genes in Gastric Cancer by Bioinformatics Analysis. BioMed Res. Int. 2020, 2020, 7658230. [Google Scholar] [CrossRef]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef]
- Gharib, S.A.; Khalyfa, A.; Abdelkarim, A.; Ramesh, V.; Buazza, M.; Kaushal, N.; Bhushan, B.; Gozal, D. Intermittent hypoxia activates temporally coordinated transcriptional programs in visceral adipose tissue. J. Mol. Med. 2012, 90, 435–445. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster | RA Cell Type | Gene Markers | IH Cell Type | Gene Markers |
|---|---|---|---|---|
| 0 | Neurons-1 | Rbfox1, lgn1, Dnajc6, xph1, Mark1 | Germ cells | Dgat2, Plin4, Pmepa1, Adrb3, Acsl1 |
| 1 | Smooth muscle cells | Gpm6a, Il1rapl1, Muc16, Adamtsl1, Rbfox1 | Podocytes | Arhgap24, Chrm3, Egfr Thsd7a, Rtl4 |
| 2 | Adipocytes | Adrb3, Slc1a3, Atp1a2, Chst1, Lipe | Retinal ganglion cells | Upk1b, Dlgap1, Nkain2, Asxl3, Lars2 |
| 3 | Fibroblasts | Nova1, Thsd7a, Lama2, Abca8a, Arhgap24 | B cells | Dock2, Runx1, Mctp1, Slc9a9, Rbpj |
| 4 | Germ cells | Adrb3, Slc27a1, Smoc1, Acsl1, Tshr | Oligodendrocyte progenitor cells | Il1rapl1, Pkhd1l1, Kcnab1, Rbfox1, Sntg1 |
| 5 | Macrophages-1 | Creb5, Fndc1, Opcml, Fbn1, Pi16 | Macrophages | Creb5, Gpc6, Fndc1, Fbn1, Opcml |
| 6 | Macrophages-2 | Mrc1, Mctp1, F13a1, Slc9a9, Rbpj | Adipocytes-1 | Adrb3, Plin4, Pmepa1, Slc1a3, Chst1 |
| 7 | Endothelial cells | Adgrl4, Cyyr1, Mecom, Cdh13, Etl4 | Endothelial cells | Cyyr1, Shank3, Adgrl4, Etl4, Cdh13 |
| 8 | Neurons-2 | Arhgap15, Dock2, Ubash3b, Ptprc, Alcam | Neurons | Muc16, Il1rapl1, Gpm6a, Nlgn1, Rbfox1 |
| 9 | Mesothelial cells | Upk3b, Gas1, Igfbp6, Igfbp5, Ap4e1 | Adipocytes-2 | Mctp1, Slc1a3, Chst1, Adrb3, Slc27a1 |
| 10 | NA | Adipocytes-3 | Pmepa1, Chst1, Adrb3, Slc1a3, D5Ertd579e | |
| 11 | NA | Adipocytes-4 | Lgals12, Vegfa, Adrb3, Slc27a1, Clstn2 | |
| 12 | NA | Pericytes | Notch3, Abcc9, Sgip1, Adgrl3, Cacna1c |
| Cluster | Cell Type | Gene Markers |
|---|---|---|
| 0 | Smooth muscle cells | Adamtsl1, Rbfox1, Il1rapl1, Mast4, Flrt2 |
| 1 | Enterocytes | Muc16, Ano1, Gpm6a, Pkhd1l1, Il1rapl1 |
| 2 | Macrophages | Creb5, Fbn1, Gpc6, Fndc1, Sdk1, Opcml |
| 3 | T memory cells, B cells | Runx1, Arhgap15, Mctp1, Dock2, Slc9a9 |
| 4 | Adipocytes-1 | Vegfa, Adrb3, Slc7a10, Usp35, Slc1a3 |
| 5 | Podocytes | Arhgap24, Thsd7a, Lama2, Abca8a, Chrm3 |
| 6 | Luteal cells | Lgals12, Slc7a10, Snhg11, Dgat2, Plin4 |
| 7 | Endothelial cells | Shank3, Cyyr1, Adgrl4, Flt1, Nova2 |
| 8 | Adipocytes-2 | Slc1a3, Adrb3, Ghr, Fam13a, Tenm4 |
| 9 | Germ cells | Adrb3, Lgals12, Tshr, Bckdhb, Sorbs1 |
| 10 | Enterocytes | Ftl1, Fth1, mt-Nd2, mt-Nd3, mt-Nd4 |
| 11 | Oligodendrocytes | Notch3, Abcc9, Gipr, Tmem273, Aspn |
| 12 | Mesothelial cells | Dpp6, Cfap77, Dnah7c, Dnah7b, Crip1 |
| 13 | Pericytes | Pkhd1, Rdh16, Dcdc2a, Adcy8, Ank3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalyfa, A.; Warren, W.; Andrade, J.; Bottoms, C.A.; Rice, E.S.; Cortese, R.; Kheirandish-Gozal, L.; Gozal, D. Transcriptomic Changes of Murine Visceral Fat Exposed to Intermittent Hypoxia at Single Cell Resolution. Int. J. Mol. Sci. 2021, 22, 261. https://doi.org/10.3390/ijms22010261
Khalyfa A, Warren W, Andrade J, Bottoms CA, Rice ES, Cortese R, Kheirandish-Gozal L, Gozal D. Transcriptomic Changes of Murine Visceral Fat Exposed to Intermittent Hypoxia at Single Cell Resolution. International Journal of Molecular Sciences. 2021; 22(1):261. https://doi.org/10.3390/ijms22010261
Chicago/Turabian StyleKhalyfa, Abdelnaby, Wesley Warren, Jorge Andrade, Christopher A. Bottoms, Edward S. Rice, Rene Cortese, Leila Kheirandish-Gozal, and David Gozal. 2021. "Transcriptomic Changes of Murine Visceral Fat Exposed to Intermittent Hypoxia at Single Cell Resolution" International Journal of Molecular Sciences 22, no. 1: 261. https://doi.org/10.3390/ijms22010261
APA StyleKhalyfa, A., Warren, W., Andrade, J., Bottoms, C. A., Rice, E. S., Cortese, R., Kheirandish-Gozal, L., & Gozal, D. (2021). Transcriptomic Changes of Murine Visceral Fat Exposed to Intermittent Hypoxia at Single Cell Resolution. International Journal of Molecular Sciences, 22(1), 261. https://doi.org/10.3390/ijms22010261