New Evidence for P-gp-Mediated Export of Amyloid-β Peptides in Molecular, Blood-Brain Barrier and Neuronal Models

 ,
, 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
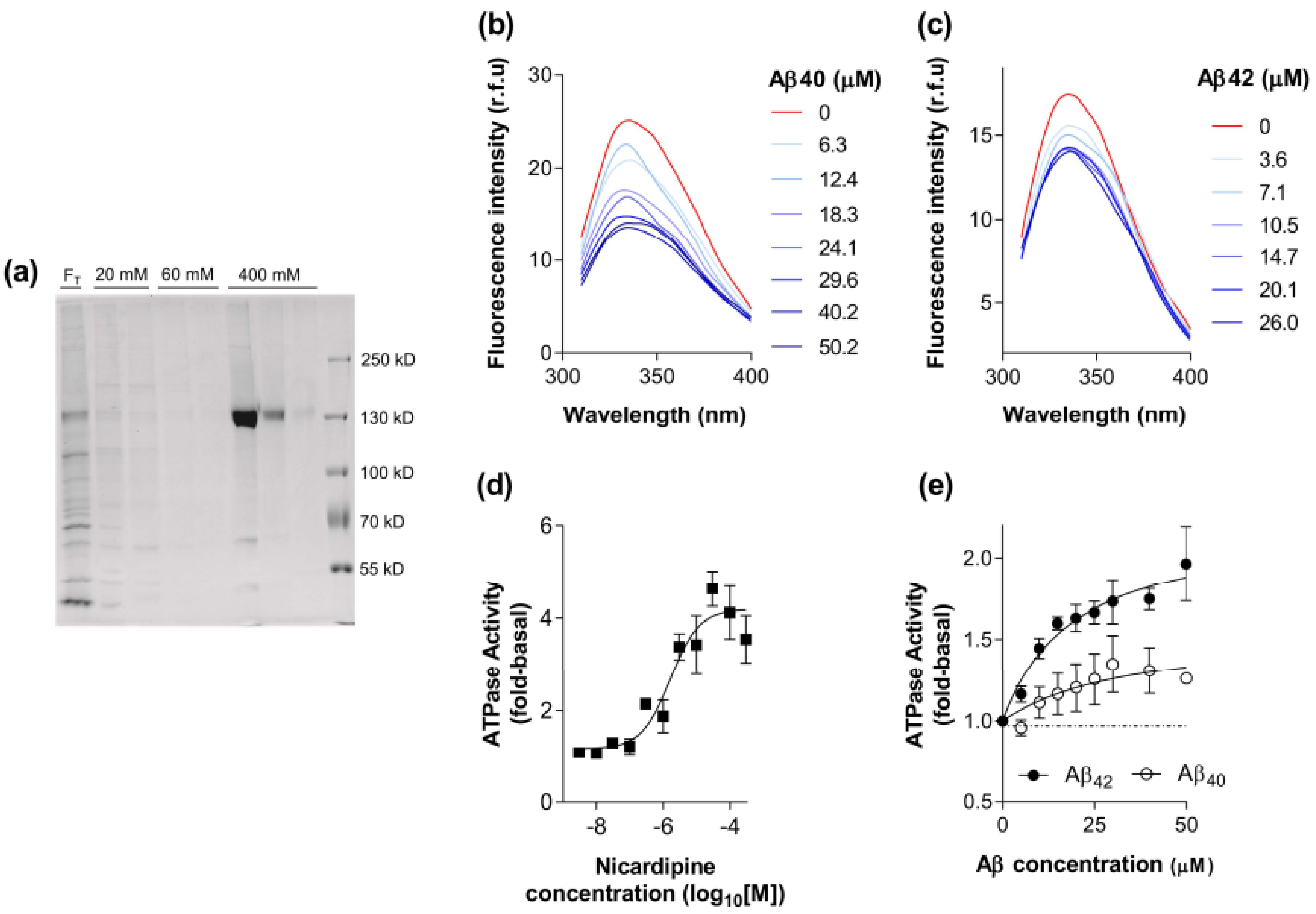
2.1. Biochemical Characterisation of the Interaction between Aβ40 and Aβ42 with P-gp
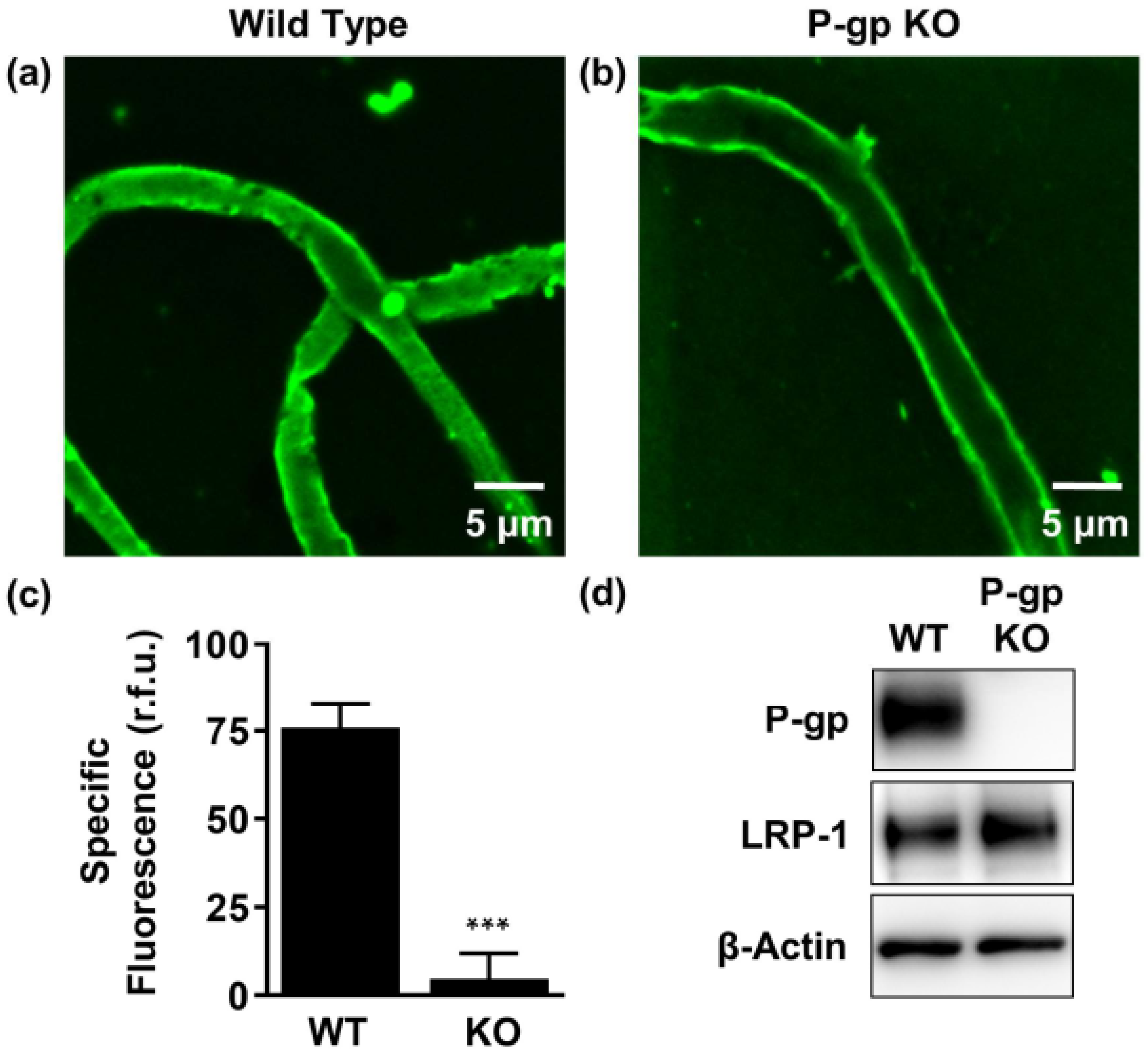
2.2. P-gp Mediates Aβ42 Transport from Brain to Capillary Lumen Ex Vivo
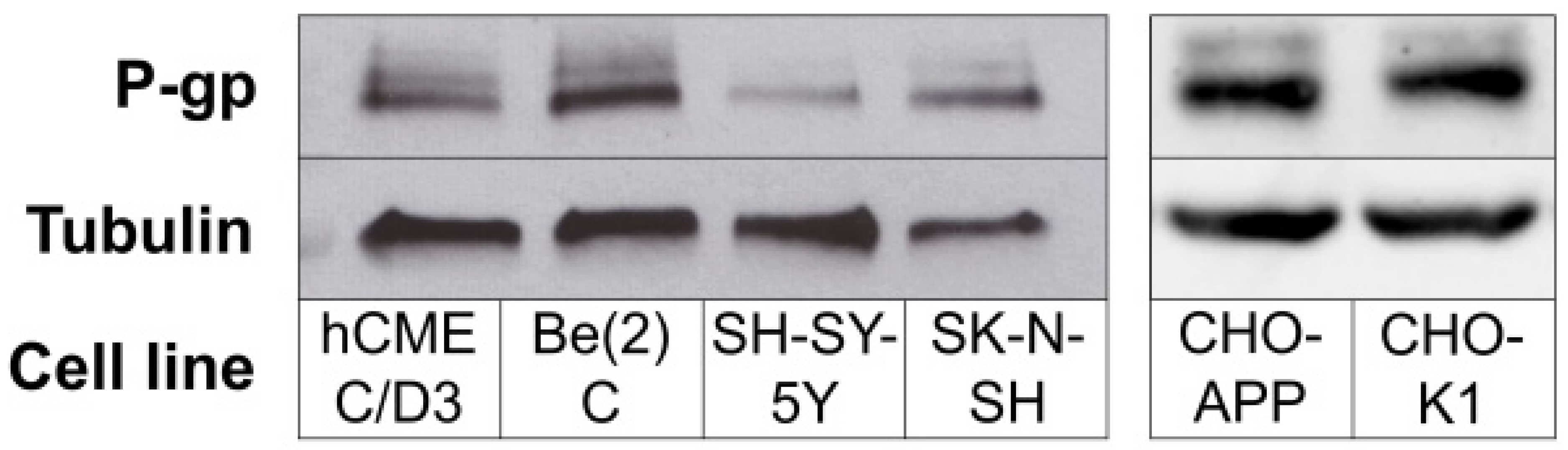
2.3. P-gp Protein Is Expressed in Human Neuroblastoma Cells
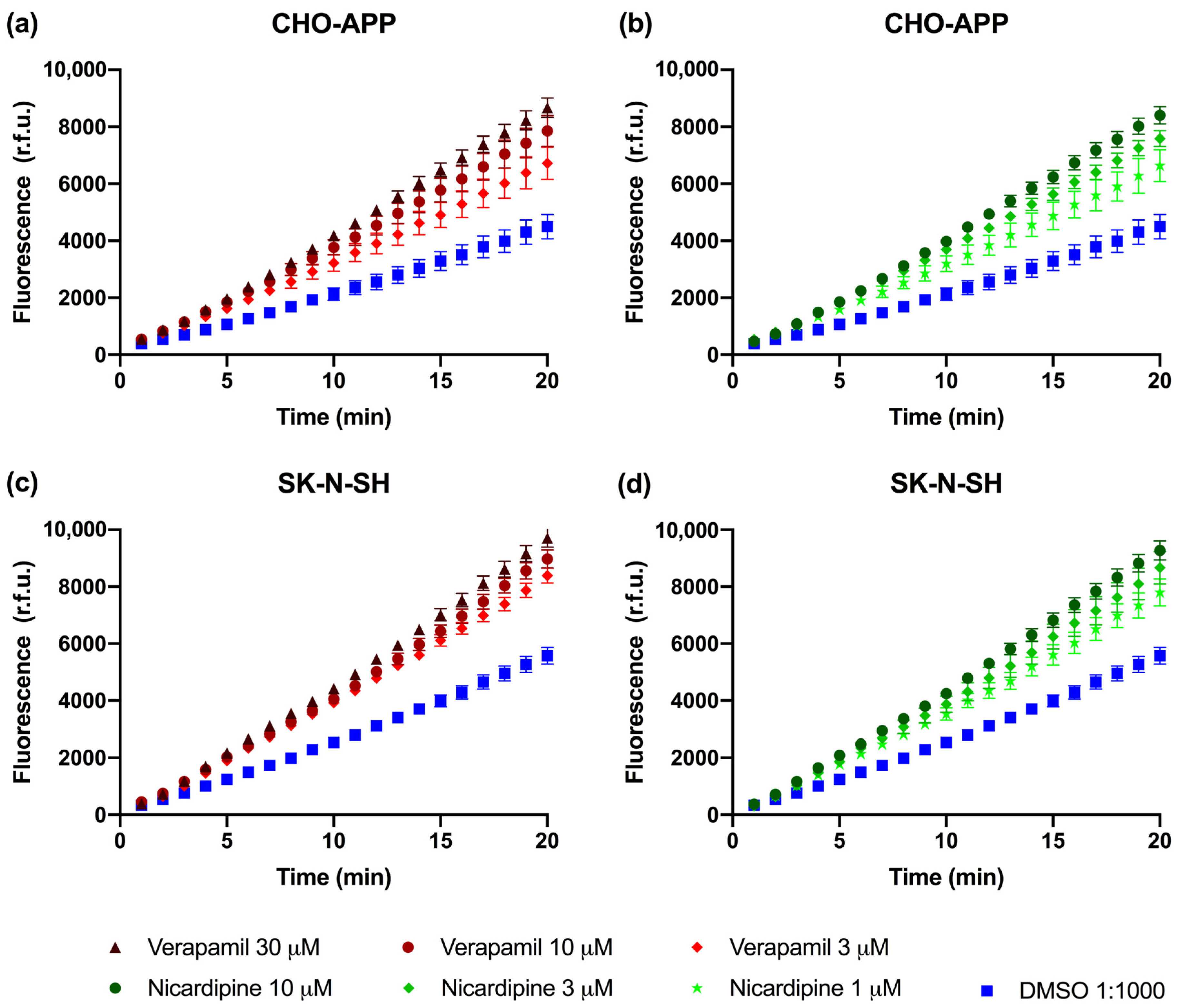
2.4. P-gp Is Active and Can Be Chemically Inhibited in CHO-APP and SK-N-SH Cells
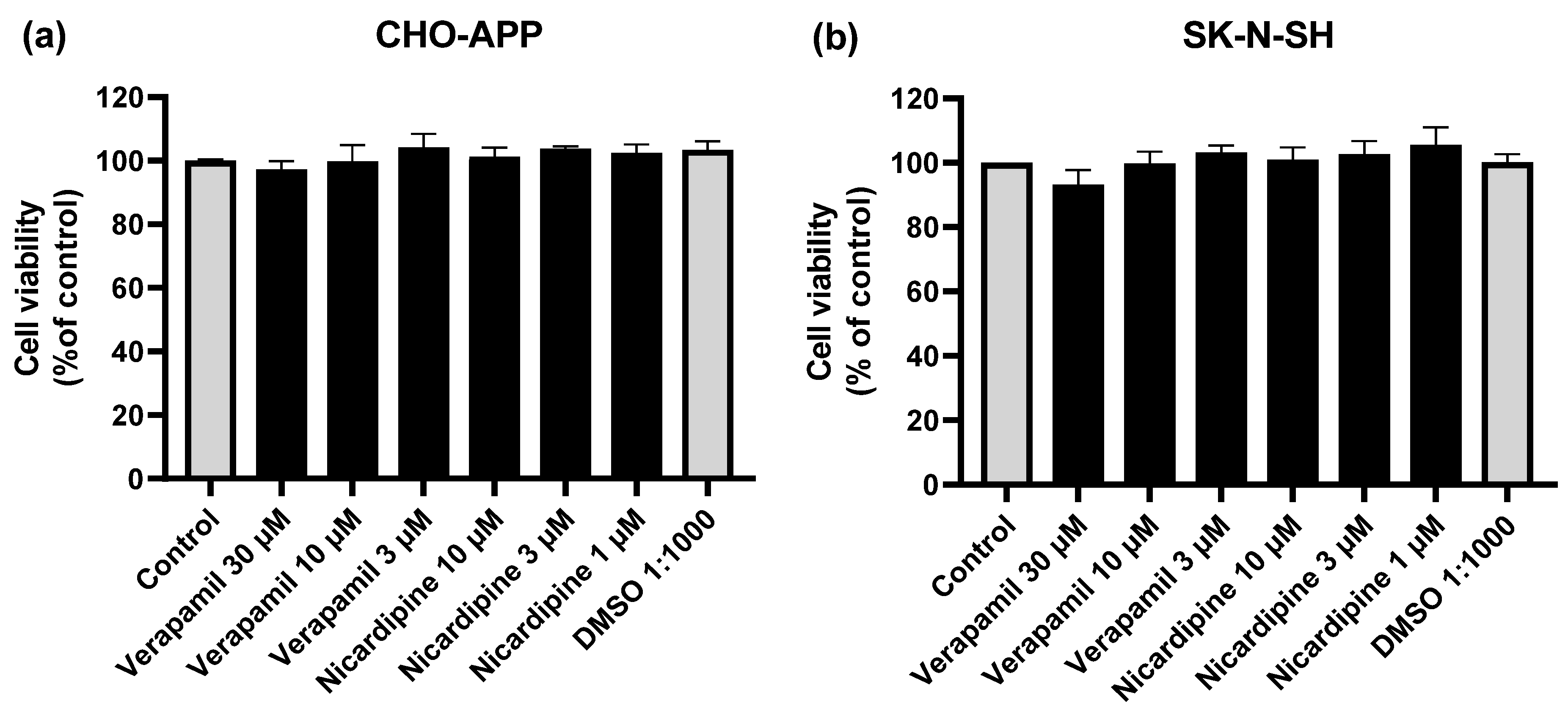
2.5. Cell viability Assays
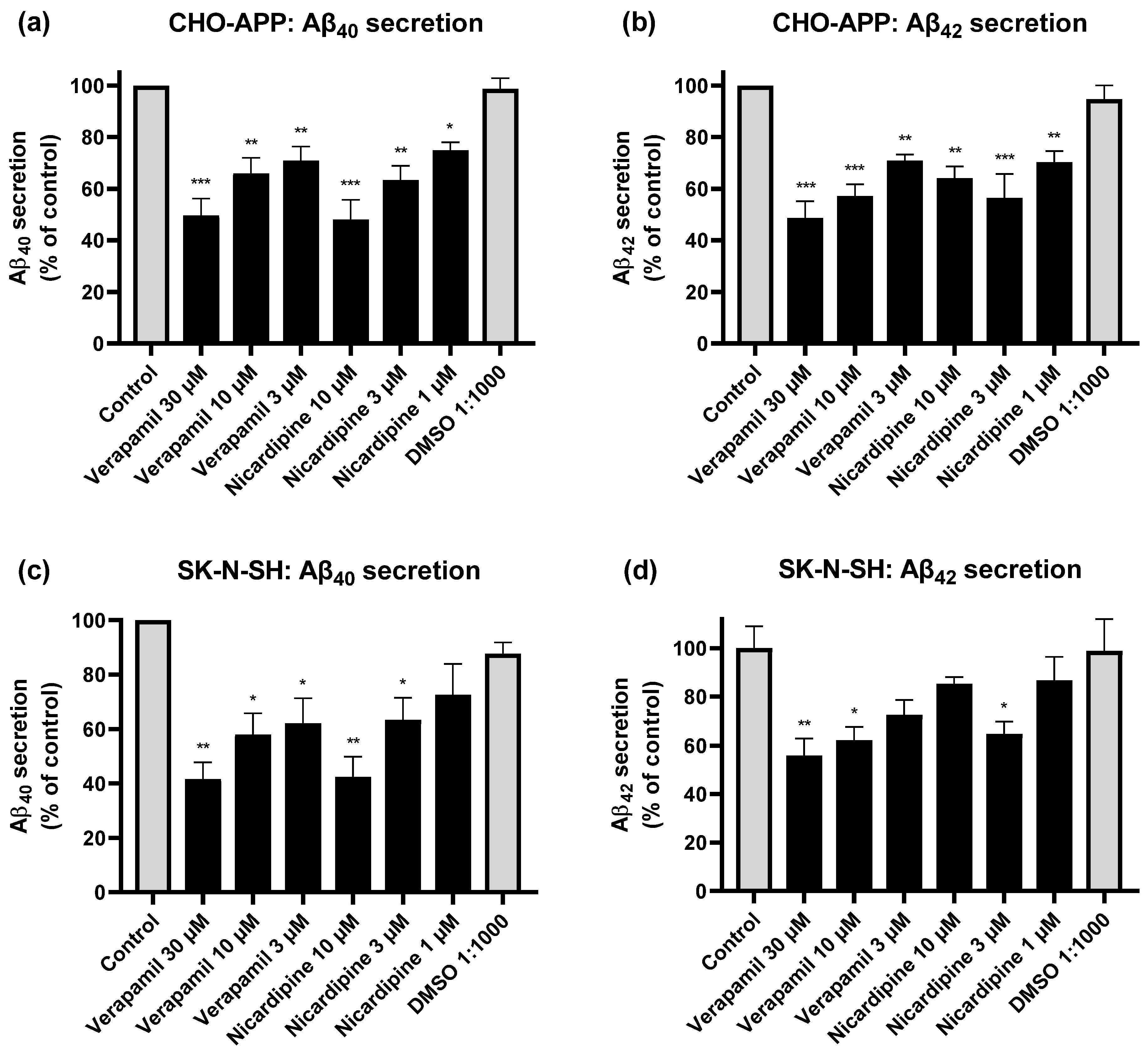
2.6. Inhibition of P-gp Reduces Aβ Secretion from CHO-APP and SK-N-SH Cells
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Materials
5.2. Antibodies
5.3. Purification and Reconstitution of P-gp
5.4. Tryptophan Fluorescence Quenching Assay for Ligand Binding to Purified, Reconstituted P-gp
5.5. ATP Hydrolysis by Purified, Reconstituted P-gp
5.6. Animals
5.7. Brain Capillary Isolation
5.8. Aβ Transport Assay in Isolated Brain Capillaries
5.9. Brain Capillary Harvest and Western Blot Analysis
5.10. Cell Culture
5.11. Cell Harvest and Western Blot Analysis
5.12. Calcein-AM Assay
5.13. MTT Assay
5.14. Preparation of Aβ Peptides
5.15. Enzyme-Linked Immunosorbent Assay (ELISA)
5.16. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Aβ | Amyloid-beta |
| ABC | ATP-binding cassette |
| AD | Alzheimer’s disease |
| APP | Amyloid precursor protein |
| ATP | Adenosine triphosphate |
| BBB | Blood-brain barrier |
| BNB | Blood-nerve-barrier |
| Calcein-AM | Calcein-acetoxymethyl ester |
| CHO | Chinese hamster ovary |
| ELISA | Enzyme-linked immunosorbent assay |
| hAβ42 | Human Aβ42 |
| KO | Knockout |
| LRP-1 | Low density lipoprotein receptor-related protein 1 |
| P-gp | P-glycoprotein |
| WT | Wild-type |
References
- Steiner, H.; Fukumori, A.; Tagami, S.; Okochi, M. Making the final cut: Pathogenic amyloid-β peptide generation by γ-secretase. Cell Stress 2018, 2, 292–310. [Google Scholar] [CrossRef]
- Murphy, M.P.; LeVine, H., 3rd. Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimers Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef]
- Zhao, J.; O’Connor, T.; Vassar, R. The contribution of activated astrocytes to Aβ production: Implications for Alzheimer’s disease pathogenesis. J. Neuroinflamm. 2011, 8, 150. [Google Scholar] [CrossRef]
- Busciglio, J.; Gabuzda, D.H.; Matsudaira, P.; Yankner, B.A. Generation of beta-amyloid in the secretory pathway in neuronal and nonneuronal cells. Proc. Natl. Acad. Sci. USA 1993, 90, 2092–2096. [Google Scholar] [CrossRef]
- Bateman, R.J.; Munsell, L.Y.; Morris, J.C.; Swarm, R.; Yarasheski, K.E.; Holtzman, D.M. Human amyloid-β synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat. Med. 2006, 12, 856–861. [Google Scholar] [CrossRef]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef]
- Viola, K.L.; Klein, W.L. Amyloid β oligomers in Alzheimer’s disease pathogenesis, treatment, and diagnosis. Acta Neuropathol. 2015, 129, 183–206. [Google Scholar] [CrossRef]
- Kayed, R.; Lasagna-Reeves, C.A. Molecular mechanisms of amyloid oligomers toxicity. J. Alzheimers Dis. 2013, 33 (Suppl. 1), S67–S78. [Google Scholar] [CrossRef]
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The Amyloid-β Oligomer Hypothesis: Beginning of the Third Decade. J. Alzheimers Dis. 2018, 64, S567–S610. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Smith, I.F.; Green, K.N.; LaFerla, F.M. A dynamic relationship between intracellular and extracellular pools of Abeta. Am. J. Pathol. 2006, 168, 184–194. [Google Scholar] [CrossRef]
- Cuello, A.C. Intracellular and Extracellular Aβ, a Tale of Two Neuropathologies. Brain Pathol. 2005, 15, 66–71. [Google Scholar] [CrossRef]
- Bayer, T.; Wirths, O. Intracellular accumulation of amyloid-beta—A predictor for synaptic dysfunction and neuron loss in Alzheimer’s disease. Front. Aging Neurosci. 2010, 2. [Google Scholar] [CrossRef]
- Mucke, L.; Selkoe, D.J. Neurotoxicity of amyloid β-protein: Synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef]
- Callaghan, R.; Gelissen, I.C.; George, A.M.; Hartz, A.M.S. Mamma Mia, P-glycoprotein binds again. FEBS Lett. 2020. [Google Scholar] [CrossRef]
- Ambudkar, S.V.; Kimchi-Sarfaty, C.; Sauna, Z.E.; Gottesman, M.M. P-glycoprotein: From genomics to mechanism. Oncogene 2003, 22, 7468–7485. [Google Scholar] [CrossRef]
- Chai, A.B.; Leung, G.K.F.; Callaghan, R.; Gelissen, I.C. P-glycoprotein: A role in the export of amyloid-β in Alzheimer’s disease? FEBS J. 2020, 287, 612–625. [Google Scholar] [CrossRef]
- Bello, I.; Salerno, M. Evidence against a role of P-glycoprotein in the clearance of the Alzheimer’s disease Aβ1-42 peptides. Cell Stress Chaperones 2015, 20, 421–430. [Google Scholar] [CrossRef]
- Hartz, A.M.; Miller, D.S.; Bauer, B. Restoring blood-brain barrier P-glycoprotein reduces brain amyloid-beta in a mouse model of Alzheimer’s disease. Mol. Pharmacol. 2010, 77, 715–723. [Google Scholar] [CrossRef]
- Mittra, R.; Pavy, M.; Subramanian, N.; George, A.M.; O’Mara, M.L.; Kerr, I.D.; Callaghan, R. Location of contact residues in pharmacologically distinct drug binding sites on P-glycoprotein. Biochem. Pharmacol. 2017, 123, 19–28. [Google Scholar] [CrossRef][Green Version]
- Crowley, E.; O’Mara, M.L.; Kerr, I.D.; Callaghan, R. Transmembrane helix 12 plays a pivotal role in coupling energy provision and drug binding in ABCB1. FEBS J. 2010, 277, 3974–3985. [Google Scholar] [CrossRef]
- Taylor, A.M.; Storm, J.; Soceneantu, L.; Linton, K.J.; Gabriel, M.; Martin, C.; Woodhouse, J.; Blott, E.; Higgins, C.F.; Callaghan, R. Detailed characterization of cysteine-less P-glycoprotein reveals subtle pharmacological differences in function from wild-type protein. Br. J. Pharmacol. 2001, 134, 1609–1618. [Google Scholar] [CrossRef] [PubMed]
- Kenakin, T. Principles: Receptor theory in pharmacology. Trends Pharmacol. Sci. 2004, 25, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Siemiarczuk, A.; Sharom, F.J. Intrinsic fluorescence of the P-glycoprotein multidrug transporter: Sensitivity of tryptophan residues to binding of drugs and nucleotides. Biochemistry 2000, 39, 14927–14938. [Google Scholar] [CrossRef] [PubMed]
- Dayan, G.; Jault, J.M.; Baubichon-Cortay, H.; Baggetto, L.G.; Renoir, J.M.; Baulieu, E.E.; Gros, P.; Di Pietro, A. Binding of steroid modulators to recombinant cytosolic domain from mouse P-glycoprotein in close proximity to the ATP site. Biochemistry 1997, 36, 15208–15215. [Google Scholar] [CrossRef]
- Qu, Q.; Chu, J.W.; Sharom, F.J. Transition state P-glycoprotein binds drugs and modulators with unchanged affinity, suggesting a concerted transport mechanism. Biochemistry 2003, 42, 1345–1353. [Google Scholar] [CrossRef]
- Lam, F.C.; Liu, R.; Lu, P.; Shapiro, A.B.; Renoir, J.-M.; Sharom, F.J.; Reiner, P.B. β-Amyloid efflux mediated by p-glycoprotein. J. Neurochem. 2001, 76, 1121–1128. [Google Scholar] [CrossRef]
- Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Substrate-induced conformational changes in the transmembrane segments of human P-glycoprotein. Direct evidence for the substrate-induced fit mechanism for drug binding. J. Biol. Chem. 2003, 278, 13603–13606. [Google Scholar] [CrossRef]
- Loo, T.W.; Clarke, D.M. Drug-stimulated ATPase activity of human P-glycoprotein requires movement between transmembrane segments 6 and 12. J. Biol. Chem. 1997, 272, 20986–20989. [Google Scholar] [CrossRef]
- Müller, M.; Bakos, E.; Welker, E.; Váradi, A.; Germann, U.A.; Gottesman, M.M.; Morse, B.S.; Roninson, I.B.; Sarkadi, B. Altered drug-stimulated ATPase activity in mutants of the human multidrug resistance protein. J. Biol. Chem. 1996, 271, 1877–1883. [Google Scholar] [CrossRef]
- Sharom, F.J.; Yu, X.; Lu, P.; Liu, R.; Chu, J.W.; Szabó, K.; Müller, M.; Hose, C.D.; Monks, A.; Váradi, A.; et al. Interaction of the P-glycoprotein multidrug transporter (MDR1) with high affinity peptide chemosensitizers in isolated membranes, reconstituted systems, and intact cells. Biochem. Pharmacol. 1999, 58, 571–586. [Google Scholar] [CrossRef]
- Orlowski, S.; Garrigos, M. Multiple recognition of various amphiphilic molecules by the multidrug resistance P-glycoprotein: Molecular mechanisms and pharmacological consequences coming from functional interactions between various drugs. Anticancer Res. 1999, 19, 3109–3123. [Google Scholar] [PubMed]
- Martin, C.; Berridge, G.; Mistry, P.; Higgins, C.; Charlton, P.; Callaghan, R. The molecular interaction of the high affinity reversal agent XR9576 with P-glycoprotein. Br. J. Pharmacol. 1999, 128, 403–411. [Google Scholar] [CrossRef]
- Schinkel, A.H. P-Glycoprotein, a gatekeeper in the blood–brain barrier. Adv. Drug Deliv. Rev. 1999, 36, 179–194. [Google Scholar] [CrossRef]
- Storck, S.E.; Hartz, A.M.S.; Bernard, J.; Wolf, A.; Kachlmeier, A.; Mahringer, A.; Weggen, S.; Pahnke, J.; Pietrzik, C.U. The concerted amyloid-beta clearance of LRP1 and ABCB1/P-gp across the blood-brain barrier is linked by PICALM. Brain Behav. Immun. 2018, 73, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Bates, S.E.; Mickley, L.A.; Chen, Y.N.; Richert, N.; Rudick, J.; Biedler, J.L.; Fojo, A.T. Expression of a drug resistance gene in human neuroblastoma cell lines: Modulation by retinoic acid-induced differentiation. Mol. Cell Biol. 1989, 9, 4337–4344. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Sun, Y.; Wang, D.; Sun, H.; Liu, X. SNHG16 promotes tumorigenesis and cisplatin resistance by regulating miR-338-3p/PLK4 pathway in neuroblastoma cells. Cancer Cell Int. 2020, 20, 236. [Google Scholar] [CrossRef] [PubMed]
- Stage, T.B.; Mortensen, C.; Khalaf, S.; Steffensen, V.; Hammer, H.S.; Xiong, C.; Nielsen, F.; Poetz, O.; Svenningsen, Å.F.; Rodriguez-Antona, C.; et al. P-Glycoprotein Inhibition Exacerbates Paclitaxel Neurotoxicity in Neurons and Patients with Cancer. Clin. Pharmacol. Ther. 2020, 108, 671–680. [Google Scholar] [CrossRef]
- Bernstein, H.-G.; Hölzl, G.; Dobrowolny, H.; Hildebrandt, J.; Trübner, K.; Krohn, M.; Bogerts, B.; Pahnke, J. Vascular and extravascular distribution of the ATP-binding cassette transporters ABCB1 and ABCC1 in aged human brain and pituitary. Mech. Ageing Dev. 2014, 141–142, 12–21. [Google Scholar] [CrossRef]
- Palladino, S.P.; Helton, E.S.; Jain, P.; Dong, C.; Crowley, M.R.; Crossman, D.K.; Ubogu, E.E. The Human Blood-Nerve Barrier Transcriptome. Sci. Rep. 2017, 7, 17477. [Google Scholar] [CrossRef]
- Balayssac, D.; Cayre, A.; Authier, N.; Bourdu, S.; Penault-Llorca, F.; Gillet, J.P.; Maublant, J.; Eschalier, A.; Coudore, F. Patterns of P-glycoprotein activity in the nervous system during vincristine-induced neuropathy in rats. J. Peripher. Nerv. Syst. 2005, 10, 301–310. [Google Scholar] [CrossRef]
- Liu, H.; Chen, Y.; Huang, L.; Sun, X.; Fu, T.; Wu, S.; Zhu, X.; Zhen, W.; Liu, J.; Lu, G.; et al. Drug Distribution into Peripheral Nerve. J. Pharmacol. Exp. Ther. 2018, 365, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Langford, D.; Grigorian, A.; Hurford, R.; Adame, A.; Ellis, R.J.; Hansen, L.; Masliah, E. Altered P-Glycoprotein Expression in AIDS Patients with HIV Encephalitis. J. Neuropathol. Exp. Neurol. 2004, 63, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Ak, H.; Ay, B.; Tanriverdi, T.; Sanus, G.Z.; Is, M.; Sar, M.; Oz, B.; Ozkara, C.; Ozyurt, E.; Uzan, M. Expression and cellular distribution of multidrug resistance-related proteins in patients with focal cortical dysplasia. Seizure 2007, 16, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Lautz, T.B.; Jie, C.; Clark, S.; Naiditch, J.A.; Jafari, N.; Qiu, Y.-Y.; Zheng, X.; Chu, F.; Madonna, M.B. The Effect of Vorinostat on the Development of Resistance to Doxorubicin in Neuroblastoma. PLoS ONE 2012, 7, e40816. [Google Scholar] [CrossRef] [PubMed]
- Chu, F.; Naiditch, J.A.; Clark, S.; Qiu, Y.-Y.; Zheng, X.; Lautz, T.B.; Holub, J.L.; Chou, P.M.; Czurylo, M.; Madonna, M.B. Midkine Mediates Intercellular Crosstalk between Drug-Resistant and Drug-Sensitive Neuroblastoma Cells In Vitro and In Vivo. ISRN Oncol. 2013, 2013, 518637. [Google Scholar] [CrossRef]
- Blanc, E.; Goldschneider, D.; Ferrandis, E.; Barrois, M.; Le Roux, G.; Leonce, S.; Douc-Rasy, S.; Bénard, J.; Raguénez, G. MYCN enhances P-gp/MDR1 gene expression in the human metastatic neuroblastoma IGR-N-91 model. Am. J. Pathol. 2003, 163, 321–331. [Google Scholar] [CrossRef]
- Sita, G.; Hrelia, P.; Tarozzi, A.; Morroni, F. P-glycoprotein (ABCB1) and Oxidative Stress: Focus on Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2017, 2017, 7905486. [Google Scholar] [CrossRef]
- Holló, Z.; Homolya, L.; Davis, C.W.; Sarkadi, B. Calcein accumulation as a fluorometric functional assay of the multidrug transporter. Biochim. Biophys. Acta Biomembr. 1994, 1191, 384–388. [Google Scholar] [CrossRef]
- Wessler, J.D.; Grip, L.T.; Mendell, J.; Giugliano, R.P. The P-Glycoprotein Transport System and Cardiovascular Drugs. J. Am. Coll. Cardiol. 2013, 61, 2495–2502. [Google Scholar] [CrossRef]
- Takara, K.; Sakaeda, T.; Tanigawara, Y.; Nishiguchi, K.; Ohmoto, N.; Horinouchi, M.; Komada, F.; Ohnishi, N.; Yokoyama, T.; Okumura, K. Effects of 12 Ca2+ antagonists on multidrug resistance, MDR1-mediated transport and MDR1 mRNA expression. Eur. J. Pharm. Sci. 2002, 16, 159–165. [Google Scholar] [CrossRef]
- Alley, G.M.; Bailey, J.A.; Chen, D.; Ray, B.; Puli, L.K.; Tanila, H.; Banerjee, P.K.; Lahiri, D.K. Memantine lowers amyloid-beta peptide levels in neuronal cultures and in APP/PS1 transgenic mice. J. Neurosci. Res. 2010, 88, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Haugabook, S.J.; Yager, D.M.; Eckman, E.A.; Golde, T.E.; Younkin, S.G.; Eckman, C.B. High throughput screens for the identification of compounds that alter the accumulation of the Alzheimer’s amyloid β peptide (Aβ). J. Neurosci. Methods 2001, 108, 171–179. [Google Scholar] [CrossRef]
- Loe, D.W.; Deeley, R.G.; Cole, S.P. Verapamil stimulates glutathione transport by the 190-kDa multidrug resistance protein 1 (MRP1). J. Pharmacol. Exp. Ther. 2000, 293, 530–538. [Google Scholar] [PubMed]
- Ivnitski-Steele, I.; Larson, R.S.; Lovato, D.M.; Khawaja, H.M.; Winter, S.S.; Oprea, T.I.; Sklar, L.A.; Edwards, B.S. High-throughput flow cytometry to detect selective inhibitors of ABCB1, ABCC1, and ABCG2 transporters. ASSAY Drug Dev. Technol. 2008, 6, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Do, T.M.; Noel-Hudson, M.S.; Ribes, S.; Besengez, C.; Smirnova, M.; Cisternino, S.; Buyse, M.; Calon, F.; Chimini, G.; Chacun, H.; et al. ABCG2- and ABCG4-mediated efflux of amyloid-beta peptide 1-40 at the mouse blood-brain barrier. J. Alzheimers Dis. 2012, 30, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Wu, Z.; Zlokovic, B.V. RAGE (yin) versus LRP (yang) balance regulates alzheimer amyloid beta-peptide clearance through transport across the blood-brain barrier. Stroke 2004, 35, 2628–2631. [Google Scholar] [CrossRef]
- Wolf, A.; Bauer, B.; Hartz, A.M. ABC Transporters and the Alzheimer’s Disease Enigma. Front. Psychiatry 2012, 3, 54. [Google Scholar] [CrossRef]
- Krohn, M.; Lange, C.; Hofrichter, J.; Scheffler, K.; Stenzel, J.; Steffen, J.; Schumacher, T.; Brüning, T.; Plath, A.-S.; Alfen, F.; et al. Cerebral amyloid-β proteostasis is regulated by the membrane transport protein ABCC1 in mice. J. Clin. Investig. 2011, 121, 3924–3931. [Google Scholar] [CrossRef]
- Zhang, Y.; Gupta, A.; Wang, H.; Zhou, L.; Vethanayagam, R.R.; Unadkat, J.D.; Mao, Q. BCRP transports dipyridamole and is inhibited by calcium channel blockers. Pharm. Res. 2005, 22, 2023–2034. [Google Scholar] [CrossRef]
- Yin, J.Y.; Huang, Q.; Yang, Y.; Zhang, J.T.; Zhong, M.Z.; Zhou, H.H.; Liu, Z.Q. Characterization and analyses of multidrug resistance-associated protein 1 (MRP1/ABCC1) polymorphisms in Chinese population. Pharm. Genom. 2009, 19, 206–216. [Google Scholar] [CrossRef]
- Blazquez, A.G.; Briz, O.; Romero, M.R.; Rosales, R.; Monte, M.J.; Vaquero, J.; Macias, R.I.; Cassio, D.; Marin, J.J. Characterization of the role of ABCG2 as a bile acid transporter in liver and placenta. Mol. Pharmacol. 2012, 81, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Chen, C.; Liu, S.; Zeng, W.; Su, J.; Wu, L.; Luo, Z.; Zhou, S.; Li, Q.; Zhang, J.; et al. CD147 promotes MTX resistance by immune cells through up-regulating ABCG2 expression and function. J. Dermatol. Sci. 2013, 70, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Kuhnke, D.; Jedlitschky, G.; Grube, M.; Krohn, M.; Jucker, M.; Mosyagin, I.; Cascorbi, I.; Walker, L.C.; Kroemer, H.K.; Warzok, R.W.; et al. MDR1-P-Glycoprotein (ABCB1) Mediates Transport of Alzheimer’s amyloid-beta peptides--implications for the mechanisms of Abeta clearance at the blood-brain barrier. Brain Pathol. 2007, 17, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Abuznait, A.H.; Cain, C.; Ingram, D.; Burk, D.; Kaddoumi, A. Up-regulation of P-glycoprotein reduces intracellular accumulation of beta amyloid: Investigation of P-glycoprotein as a novel therapeutic target for Alzheimer’s disease. J. Pharm. Pharmacol. 2011, 63, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Paris, D.; Bachmeier, C.; Patel, N.; Quadros, A.; Volmar, C.H.; Laporte, V.; Ganey, J.; Beaulieu-Abdelahad, D.; Ait-Ghezala, G.; Crawford, F.; et al. Selective antihypertensive dihydropyridines lower Aβ accumulation by targeting both the production and the clearance of Aβ across the blood-brain barrier. Mol. Med. 2011, 17, 149–162. [Google Scholar] [CrossRef]
- Murphy, M.P.; Hickman, L.J.; Eckman, C.B.; Uljon, S.N.; Wang, R.; Golde, T.E. γ-Secretase, Evidence for Multiple Proteolytic Activities and Influence of Membrane Positioning of Substrate on Generation of Amyloid β Peptides of Varying Length. J. Biol. Chem. 1999, 274, 11914–11923. [Google Scholar] [CrossRef]
- Nazer, B.; Hong, S.; Selkoe, D.J. LRP promotes endocytosis and degradation, but not transcytosis, of the amyloid-beta peptide in a blood-brain barrier in vitro model. Neurobiol. Dis. 2008, 30, 94–102. [Google Scholar] [CrossRef]
- Rajendran, L.; Honsho, M.; Zahn, T.R.; Keller, P.; Geiger, K.D.; Verkade, P.; Simons, K. Alzheimer’s disease β-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. USA 2006, 103, 11172–11177. [Google Scholar] [CrossRef]
- Yuyama, K.; Igarashi, Y. Exosomes as Carriers of Alzheimer’s Amyloid-ß. Front. Neurosci. 2017, 11. [Google Scholar] [CrossRef]
- Brothers, H.M.; Gosztyla, M.L.; Robinson, S.R. The Physiological Roles of Amyloid-β Peptide Hint at New Ways to Treat Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 118. [Google Scholar] [CrossRef]
- Nalivaeva, N.N.; Turner, A.J. The amyloid precursor protein: A biochemical enigma in brain development, function and disease. FEBS Lett. 2013, 587, 2046–2054. [Google Scholar] [CrossRef] [PubMed]
- Hartz, A.M.S.; Zhong, Y.; Shen, A.N.; Abner, E.L.; Bauer, B. Preventing P-gp Ubiquitination Lowers Aβ Brain Levels in an Alzheimer’s Disease Mouse Model. Front. Aging Neurosci. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Brenn, A.; Grube, M.; Jedlitschky, G.; Fischer, A.; Strohmeier, B.; Eiden, M.; Keller, M.; Groschup, M.H.; St. Vogelgesang, S. John’s Wort reduces beta-amyloid accumulation in a double transgenic Alzheimer’s disease mouse model-role of P-glycoprotein. Brain Pathol. 2014, 24, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Colin, M.; Dujardin, S.; Schraen-Maschke, S.; Meno-Tetang, G.; Duyckaerts, C.; Courade, J.-P.; Buée, L. From the prion-like propagation hypothesis to therapeutic strategies of anti-tau immunotherapy. Acta Neuropathol. 2020, 139, 3–25. [Google Scholar] [CrossRef]
- Stancu, I.C.; Vasconcelos, B.; Ris, L.; Wang, P.; Villers, A.; Peeraer, E.; Buist, A.; Terwel, D.; Baatsen, P.; Oyelami, T.; et al. Templated misfolding of Tau by prion-like seeding along neuronal connections impairs neuronal network function and associated behavioral outcomes in Tau transgenic mice. Acta Neuropathol. 2015, 129, 875–894. [Google Scholar] [CrossRef]
- Van Den Berge, N.; Ferreira, N.; Gram, H.; Mikkelsen, T.W.; Alstrup, A.K.O.; Casadei, N.; Tsung-Pin, P.; Riess, O.; Nyengaard, J.R.; Tamgüney, G.; et al. Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol. 2019, 138, 535–550. [Google Scholar] [CrossRef]
- Elfarrash, S.; Jensen, N.M.; Ferreira, N.; Betzer, C.; Thevathasan, J.V.; Diekmann, R.; Adel, M.; Omar, N.M.; Boraie, M.Z.; Gad, S.; et al. Organotypic slice culture model demonstrates inter-neuronal spreading of alpha-synuclein aggregates. Acta Neuropathol. Commun. 2019, 7, 213. [Google Scholar] [CrossRef]
- Walker, L.C.; Schelle, J.; Jucker, M. The Prion-Like Properties of Amyloid-β Assemblies: Implications for Alzheimer’s Disease. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef]
- Kanekiyo, T.; Xu, H.; Bu, G. ApoE and Abeta in Alzheimer’s disease: Accidental encounters or partners? Neuron 2014, 81, 740–754. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.-C.; Bu, G. Apolipoprotein E and Alzheimer disease: Pathobiology and targeting strategies. Nat. Rev. Neurol. 2019, 15, 501–518. [Google Scholar] [CrossRef]
- Richner, M.; Ferreira, N.; Dudele, A.; Jensen, T.S.; Vaegter, C.B.; Gonçalves, N.P. Functional and Structural Changes of the Blood-Nerve-Barrier in Diabetic Neuropathy. Front. Neurosci. 2018, 12, 1038. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.; Miller, M.C.; Monahan, R.; Osgood, D.P.; Stopa, E.G.; Silverberg, G.D. P-glycoprotein expression and amyloid accumulation in human aging and Alzheimer’s disease: Preliminary observations. Neurobiol. Aging 2015, 36, 2475–2482. [Google Scholar] [CrossRef] [PubMed]
- Bartels, A.L.; Kortekaas, R.; Bart, J.; Willemsen, A.T.; de Klerk, O.L.; de Vries, J.J.; van Oostrom, J.C.; Leenders, K.L. Blood-brain barrier P-glycoprotein function decreases in specific brain regions with aging: A possible role in progressive neurodegeneration. Neurobiol. Aging 2009, 30, 1818–1824. [Google Scholar] [CrossRef] [PubMed]
- Kania, K.D.; Wijesuriya, H.C.; Hladky, S.B.; Barrand, M.A. Beta amyloid effects on expression of multidrug efflux transporters in brain endothelial cells. Brain Res. 2011, 1418, 1–11. [Google Scholar] [CrossRef]
- Park, R.; Kook, S.Y.; Park, J.C.; Mook-Jung, I. Abeta1-42 reduces P-glycoprotein in the blood-brain barrier through RAGE-NF-kappaB signaling. Cell Death Dis. 2014, 5, e1299. [Google Scholar] [CrossRef]
- Hartz, A.M.S.; Zhong, Y.; Wolf, A.; LeVine, H.; Miller, D.S.; Bauer, B. Aβ40 Reduces P-Glycoprotein at the Blood–Brain Barrier through the Ubiquitin–Proteasome Pathway. J. Neurosci. 2016, 36, 1930. [Google Scholar] [CrossRef]
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K.; Hägg, S.; Athanasiu, L.; et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 2019, 51, 404–413. [Google Scholar] [CrossRef]
- van Assema, D.M.; Lubberink, M.; Rizzu, P.; van Swieten, J.C.; Schuit, R.C.; Eriksson, J.; Scheltens, P.; Koepp, M.; Lammertsma, A.A.; van Berckel, B.N. Blood-brain barrier P-glycoprotein function in healthy subjects and Alzheimer’s disease patients: Effect of polymorphisms in the ABCB1 gene. EJNMMI Res. 2012, 2, 57. [Google Scholar] [CrossRef]
- Teplow, D.B. Preparation of Amyloid β-Protein for Structural and Functional Studies. Methods Enzymol. 2006, 413, 20–33. [Google Scholar] [CrossRef]
- Chifflet, S.; Torriglia, A.; Chiesa, R.; Tolosa, S. A method for the determination of inorganic phosphate in the presence of labile organic phosphate and high concentrations of protein: Application to lens ATPases. Anal. Biochem. 1988, 168, 1–4. [Google Scholar] [CrossRef]
- Li, H.; Kim, W.S.; Guillemin, G.J.; Hill, A.F.; Evin, G.; Garner, B. Modulation of amyloid precursor protein processing by synthetic ceramide analogues. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2010, 1801, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Ray, B.; Banerjee, P.K.; Greig, N.H.; Lahiri, D.K. Memantine treatment decreases levels of secreted Alzheimer’s amyloid precursor protein (APP) and amyloid beta (A beta) peptide in the human neuroblastoma cells. Neurosci. Lett. 2010, 470, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.-S.; Liu, P.; Gao, S.-Q.; Diao, Z.-Y.; Yang, L.-L.; Xu, J.; Qu, X.; Han, E.-J. Inhibitory effect of piperlonguminine/dihydropiperlonguminine on the production of amyloid beta and APP in SK-N-SH cells. Chin. J. Physiol. 2009, 52, 160–168. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chai, A.B.; Hartz, A.M.S.; Gao, X.; Yang, A.; Callaghan, R.; Gelissen, I.C. New Evidence for P-gp-Mediated Export of Amyloid-β Peptides in Molecular, Blood-Brain Barrier and Neuronal Models. Int. J. Mol. Sci. 2021, 22, 246. https://doi.org/10.3390/ijms22010246
Chai AB, Hartz AMS, Gao X, Yang A, Callaghan R, Gelissen IC. New Evidence for P-gp-Mediated Export of Amyloid-β Peptides in Molecular, Blood-Brain Barrier and Neuronal Models. International Journal of Molecular Sciences. 2021; 22(1):246. https://doi.org/10.3390/ijms22010246
Chicago/Turabian StyleChai, Amanda B., Anika M. S. Hartz, Xuexin Gao, Alryel Yang, Richard Callaghan, and Ingrid C. Gelissen. 2021. "New Evidence for P-gp-Mediated Export of Amyloid-β Peptides in Molecular, Blood-Brain Barrier and Neuronal Models" International Journal of Molecular Sciences 22, no. 1: 246. https://doi.org/10.3390/ijms22010246
APA StyleChai, A. B., Hartz, A. M. S., Gao, X., Yang, A., Callaghan, R., & Gelissen, I. C. (2021). New Evidence for P-gp-Mediated Export of Amyloid-β Peptides in Molecular, Blood-Brain Barrier and Neuronal Models. International Journal of Molecular Sciences, 22(1), 246. https://doi.org/10.3390/ijms22010246