Hold on or Cut? Integrin- and MMP-Mediated Cell–Matrix Interactions in the Tumor Microenvironment



Abstract
1. Introduction
2. The Extracellular Matrix as a Key Component of the TME
3. Fibrillar and Non-Fibrillar ECM Components Orchestrate the Biophysical and Biochemical Properties of the TME
4. The “Give and Take” between the ECM and Cells within the TME
5. Cancer Progression is Dependent on the Proteolytic Action of MMPs on the ECM of the TME
5.1. The Metastatic Cascade
5.2. Structural and Functional Diversity of Matrix Metalloproteinases
5.3. MMP-14 Decisively Controls Tumor Progression
5.4. Regulation of MMP Expression
5.5. Regulation of MMP Activity
5.6. Invasive Cancer Cells Breach ECM Barriers with Invadopodia as Drill Heads
5.7. ECM Degradation and Remodeling Releases Bioactive Matrikines
5.8. MMPs Promote Epithelial–Mesenchymal Transition
6. Translational Assessment and Future Prospect
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADAM(TS) | a disintegrin and a metalloproteinase (with thrombospondin-1 motif) |
| BM | basement membrane |
| CAF | cancer-associated fibroblast |
| CAM | cell adhesion molecule |
| CCN | connective tissue growth factor (CTGF), cysteine-rich protein (Cyr61) and nephroblastoma overexpressed (NOV) |
| EC | endothelial cell |
| ECM | extracellular matrix |
| ED-A/B | extra domain-A/B |
| EGF(R) | epidermal growth factor (receptor) |
| EMT | epithelial–mesenchymal transition |
| GAG | glycosaminoglycan |
| GPI | glycosylphosphatidylinositol |
| HGF | hepatocyte growth factor |
| HIF | hypoxia-inducible factor |
| H/LMW-HA | high/low molecular weight hyaluronic acid |
| LOXL | lysyl oxidase-like |
| LPA1R | lysophosphatidic acid receptor 1 |
| (MT-)MMP | (membrane-type) matrix metalloproteinase |
| NOX | nicotinamide adenine dinucleotide phosphate (NADPH) oxidase |
| PAR | protease-activated receptor |
| RHAMM | receptor for hyaluronic acid-mediated migration |
| ROS | reactive oxygen species |
| SIBLING | small integrin-binding ligand N-linked glycoprotein |
| SLRP | small leucine-rich protein |
| SPARC | secreted protein acidic and rich in cysteine |
| TC | tumor cell |
| TGF-β | transforming growth factor- β |
| TIMP | tissue inhibitors of metalloproteinases |
| TLR | Toll-like receptor |
| TME | tumor microenvironment |
| VEGF | vascular endothelial cell growth factor |
References
- He, Q.; Chen, J.; Yan, J.; Cai, S.; Xiong, H.; Liu, Y.; Peng, D.; Mo, M.; Liu, Z. Tumor microenvironment responsive drug delivery systems. Asian J. Pharm. Sci. 2020, 15, 416–448. [Google Scholar] [CrossRef]
- Emon, B.; Bauer, J.; Jain, Y.; Jung, B.; Saif, T. Biophysics of Tumor Microenvironment and Cancer Metastasis—A Mini Review. Comput. Struct. Biotechnol. J. 2018, 16, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, H.; Sahai, E. Mechanisms and impact of altered tumour mechanics. Nat. Cell Biol. 2018, 20, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Iozzo, R.V.; Schaefer, L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015, 42, 11–55. [Google Scholar] [CrossRef] [PubMed]
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N.K. Extracellular matrix structure. Adv. Drug Deliv. Rev. 2016, 97, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Zhang, S.; Gong, Z.; Li, X.; Cao, K.; Deng, H.; He, Y.; et al. The role of microenvironment in tumor angiogenesis. J. Exp. Clin. Cancer Res. 2020, 39, 204. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wei, J.; Sun, J. Roles of TGF-beta signaling pathway in tumor microenvirionment and cancer therapy. Int. Immunopharmacol. 2020, 89, 107101. [Google Scholar] [CrossRef]
- Chitty, J.L.; Setargew, Y.F.I.; Cox, T.R. Targeting the lysyl oxidases in tumour desmoplasia. Biochem. Soc. Trans. 2019, 47, 1661–1678. [Google Scholar] [CrossRef]
- Gonzalez-Avila, G.; Sommer, B.; Mendoza-Posada, D.A.; Ramos, C.; Garcia-Hernandez, A.A.; Falfan-Valencia, R. Matrix metalloproteinases participation in the metastatic process and their diagnostic and therapeutic applications in cancer. Crit. Rev. Oncol. Hematol. 2019, 137, 57–83. [Google Scholar] [CrossRef]
- Amendola, P.G.; Reuten, R.; Erler, J.T. Interplay Between LOX Enzymes and Integrins in the Tumor Microenvironment. Cancers 2019, 11, 729. [Google Scholar] [CrossRef]
- Lentini, A.; Abbruzzese, A.; Provenzano, B.; Tabolacci, C.; Beninati, S. Transglutaminases: Key regulators of cancer metastasis. Amino Acids 2013, 44, 25–32. [Google Scholar] [CrossRef]
- Leight, J.L.; Drain, A.P.; Weaver, V.M. Extracellular Matrix Remodeling and Stiffening Modulate Tumor Phenotype and Treatment Response. Ann. Rev. Cancer Biol. 2017, 1, 313–334. [Google Scholar] [CrossRef]
- Martins Cavaco, A.C.; Damaso, S.; Casimiro, S.; Costa, L. Collagen biology making inroads into prognosis and treatment of cancer progression and metastasis. Cancer Metastasis Rev. 2020, 39, 603–623. [Google Scholar] [CrossRef] [PubMed]
- Bourgot, I.; Primac, I.; Louis, T.; Noel, A.; Maquoi, E. Reciprocal Interplay Between Fibrillar Collagens and Collagen-Binding Integrins: Implications in Cancer Progression and Metastasis. Front. Oncol. 2020, 10, 1488. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Xu, H.; Wang, W.; Li, S.; Li, H.; Li, T.; Zhang, W.; Yu, X.; Liu, L. The role of collagen in cancer: From bench to bedside. J. Transl. Med. 2019, 17, 309. [Google Scholar] [CrossRef] [PubMed]
- Ricard-Blum, S. The collagen family. Cold Spring Harb. Perspect. Biol. 2011, 3, a004978. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Wolf, K.; Friedl, P. Extracellular matrix determinants of proteolytic and non-proteolytic cell migration. Trends Cell Biol. 2011, 21, 736–744. [Google Scholar] [CrossRef]
- Wolf, K.; Te Lindert, M.; Krause, M.; Alexander, S.; Te Riet, J.; Willis, A.L.; Hoffman, R.M.; Figdor, C.G.; Weiss, S.J.; Friedl, P. Physical limits of cell migration: Control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J. Cell Biol. 2013, 201, 1069–1084. [Google Scholar] [CrossRef]
- Overall, C.M. Matrix metalloproteinase substrate binding domains, modules and exosites. Overview and experimental strategies. Methods Mol. Biol. 2001, 151, 79–120. [Google Scholar]
- Aumailley, M. The laminin family. Cell Adh. Migr. 2013, 7, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Halfter, W.; Oertle, P.; Monnier, C.A.; Camenzind, L.; Reyes-Lua, M.; Hu, H.; Candiello, J.; Labilloy, A.; Balasubramani, M.; Henrich, P.B.; et al. New concepts in basement membrane biology. FEBS J. 2015, 282, 4466–4479. [Google Scholar] [CrossRef] [PubMed]
- Hohenester, E.; Yurchenco, P.D. Laminins in basement membrane assembly. Cell Adh. Migr. 2013, 7, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Cavaco, A.C.M.; Rezaei, M.; Caliandro, M.F.; Lima, A.M.; Stehling, M.; Dhayat, S.A.; Haier, J.; Brakebusch, C.; Eble, J.A. The Interaction between Laminin-332 and alpha3beta1 Integrin Determines Differentiation and Maintenance of CAFs, and Supports Invasion of Pancreatic Duct Adenocarcinoma Cells. Cancers 2018, 11, 14. [Google Scholar] [CrossRef]
- Wang, Y.; Song, E.C.; Resnick, M.B. Elastin in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1272, 1–16. [Google Scholar] [CrossRef]
- Muiznieks, L.D.; Weiss, A.S.; Keeley, F.W. Structural disorder and dynamics of elastin. Biochem. Cell. Biol. 2010, 88, 239–250. [Google Scholar] [CrossRef]
- Heinz, A. Elastases and elastokines: Elastin degradation and its significance in health and disease. Crit. Rev. Biochem. Mol. Biol. 2020, 55, 252–273. [Google Scholar] [CrossRef]
- Eble, J.A.; Niland, S. The extracellular matrix in tumor progression and metastasis. Clin. Exp. Metastasis 2019, 36, 171–198. [Google Scholar] [CrossRef]
- White, E.S.; Muro, A.F. Fibronectin splice variants: Understanding their multiple roles in health and disease using engineered mouse models. IUBMB Life 2011, 63, 538–546. [Google Scholar] [CrossRef]
- Lin, T.C.; Yang, C.H.; Cheng, L.H.; Chang, W.T.; Lin, Y.R.; Cheng, H.C. Fibronectin in Cancer: Friend or Foe. Cells 2019, 9, 27. [Google Scholar] [CrossRef]
- Efthymiou, G.; Saint, A.; Ruff, M.; Rekad, Z.; Ciais, D.; Van Obberghen-Schilling, E. Shaping Up the Tumor Microenvironment With Cellular Fibronectin. Front. Oncol. 2020, 10, 641. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Lee, Y.J.; Han, H.J. Role of hypoxia-induced fibronectin-integrin beta1 expression in embryonic stem cell proliferation and migration: Involvement of PI3K/Akt and FAK. J. Cell. Physiol. 2011, 226, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Ryu, M.H.; Park, H.M.; Chung, J.; Lee, C.H.; Park, H.R. Hypoxia-inducible factor-1alpha mediates oral squamous cell carcinoma invasion via upregulation of alpha5 integrin and fibronectin. Biochem. Biophys. Res. Commun. 2010, 393, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Sollini, M.; Boni, R.; Traino, A.C.; Lazzeri, E.; Pasqualetti, F.; Modeo, L.; Mariani, G.; Petrini, M.; Erba, P.A. New approaches for imaging and therapy of solid cancer. Q. J. Nucl. Med. Mol. Imaging 2015, 59, 168–183. [Google Scholar]
- Degen, M.; Brellier, F.; Kain, R.; Ruiz, C.; Terracciano, L.; Orend, G.; Chiquet-Ehrismann, R. Tenascin-W is a novel marker for activated tumor stroma in low-grade human breast cancer and influences cell behavior. Cancer Res. 2007, 67, 9169–9179. [Google Scholar] [CrossRef]
- Midwood, K.S.; Orend, G. The role of tenascin-C in tissue injury and tumorigenesis. J. Cell Commun. Signal. 2009, 3, 287–310. [Google Scholar] [CrossRef]
- Scherberich, A.; Tucker, R.P.; Degen, M.; Brown-Luedi, M.; Andres, A.C.; Chiquet-Ehrismann, R. Tenascin-W is found in malignant mammary tumors, promotes alpha8 integrin-dependent motility and requires p38MAPK activity for BMP-2 and TNF-alpha induced expression in vitro. Oncogene 2005, 24, 1525–1532. [Google Scholar] [CrossRef]
- Brellier, F.; Tucker, R.P.; Chiquet-Ehrismann, R. Tenascins and their implications in diseases and tissue mechanics. Scand. J. Med. Sci. Sports 2009, 19, 511–519. [Google Scholar] [CrossRef]
- Oskarsson, T.; Acharyya, S.; Zhang, X.H.; Vanharanta, S.; Tavazoie, S.F.; Morris, P.G.; Downey, R.J.; Manova-Todorova, K.; Brogi, E.; Massague, J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 2011, 17, 867–874. [Google Scholar] [CrossRef]
- Degen, M.; Brellier, F.; Schenk, S.; Driscoll, R.; Zaman, K.; Stupp, R.; Tornillo, L.; Terracciano, L.; Chiquet-Ehrismann, R.; Ruegg, C.; et al. Tenascin-W, a new marker of cancer stroma, is elevated in sera of colon and breast cancer patients. Int. J. Cancer 2008, 122, 2454–2461. [Google Scholar] [CrossRef]
- Spenle, C.; Saupe, F.; Midwood, K.; Burckel, H.; Noel, G.; Orend, G. Tenascin-C: Exploitation and collateral damage in cancer management. Cell Adh. Migr. 2015, 9, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Kii, I.; Nishiyama, T.; Li, M.; Matsumoto, K.; Saito, M.; Amizuka, N.; Kudo, A. Incorporation of tenascin-C into the extracellular matrix by periostin underlies an extracellular meshwork architecture. J. Biol. Chem. 2010, 285, 2028–2039. [Google Scholar] [CrossRef] [PubMed]
- Girotti, M.R.; Salatino, M.; Dalotto-Moreno, T.; Rabinovich, G.A. Sweetening the hallmarks of cancer: Galectins as multifunctional mediators of tumor progression. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Thakur, R.; Mishra, D.P. Matrix reloaded: CCN, tenascin and SIBLING group of matricellular proteins in orchestrating cancer hallmark capabilities. Pharmacol. Ther. 2016, 168, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Leask, A. Conjunction junction, what’s the function? CCN proteins as targets in fibrosis and cancers. Am. J. Physiol. Cell Physiol. 2020, 318, C1046–C1054. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Birk, D.E. The regulatory roles of small leucine-rich proteoglycans in extracellular matrix assembly. FEBS J. 2013, 280, 2120–2137. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Li, J.P. Heparan sulfate proteoglycan—A common receptor for diverse cytokines. Cell Signal. 2019, 54, 115–121. [Google Scholar] [CrossRef]
- De Pasquale, V.; Pavone, L.M. Heparan Sulfate Proteoglycan Signaling in Tumor Microenvironment. Int. J. Mol. Sci. 2020, 21, 6588. [Google Scholar] [CrossRef]
- Mellai, M.; Casalone, C.; Corona, C.; Crociara, P.; Favole, A.; Cassoni, P.; Schiffer, D.; Boldorini, R. Chondroitin Sulphate Proteoglycans in the Tumour Microenvironment. Adv. Exp. Med. Biol. 2020, 1272, 73–92. [Google Scholar] [CrossRef]
- Iozzo, R.V.; Sanderson, R.D. Proteoglycans in cancer biology, tumour microenvironment and angiogenesis. J. Cell. Mol. Med. 2011, 15, 1013–1031. [Google Scholar] [CrossRef]
- Weniger, M.; Honselmann, K.C.; Liss, A.S. The Extracellular Matrix and Pancreatic Cancer: A Complex Relationship. Cancers 2018, 10, 316. [Google Scholar] [CrossRef] [PubMed]
- Otranto, M.; Sarrazy, V.; Bonté, F.; Hinz, B.; Gabbiani, G.; Desmoulière, A. The role of the myofibroblast in tumor stroma remodeling. Cell Adh. Migr. 2012, 6, 203–219. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, F.M.; Vitale, D.L.; Sevic, I.; Alaniz, L. Hyaluronan in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1245, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Massague, J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Ahmadi, A.; Najafi, M.; Farhood, B.; Mortezaee, K. Transforming growth factor-beta signaling: Tumorigenesis and targeting for cancer therapy. J. Cell. Physiol. 2019, 234, 12173–12187. [Google Scholar] [CrossRef]
- Itatani, Y.; Kawada, K.; Sakai, Y. Transforming Growth Factor-beta Signaling Pathway in Colorectal Cancer and Its Tumor Microenvironment. Int. J. Mol. Sci. 2019, 20, 5822. [Google Scholar] [CrossRef]
- Poltavets, V.; Kochetkova, M.; Pitson, S.M.; Samuel, M.S. The Role of the Extracellular Matrix and Its Molecular and Cellular Regulators in Cancer Cell Plasticity. Front. Oncol. 2018, 8, 431. [Google Scholar] [CrossRef]
- Brassart-Pasco, S.; Brezillon, S.; Brassart, B.; Ramont, L.; Oudart, J.B.; Monboisse, J.C. Tumor Microenvironment: Extracellular Matrix Alterations Influence Tumor Progression. Front. Oncol. 2020, 10, 397. [Google Scholar] [CrossRef]
- Najafi, M.; Farhood, B.; Mortezaee, K. Extracellular matrix (ECM) stiffness and degradation as cancer drivers. J. Cell. Biochem. 2018, 120, 2782–2790. [Google Scholar] [CrossRef]
- Bussard, K.M.; Mutkus, L.; Stumpf, K.; Gomez-Manzano, C.; Marini, F.C. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016, 18, 84. [Google Scholar] [CrossRef]
- Sobierajska, K.; Ciszewski, W.M.; Sacewicz-Hofman, I.; Niewiarowska, J. Endothelial Cells in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1234, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.; Brehm, C.U.; Gress, T.M.; Buchholz, M.; Alashkar Alhamwe, B.; von Strandmann, E.P.; Slater, E.P.; Bartsch, J.W.; Bauer, C.; Lauth, M. The Immune Microenvironment in Pancreatic Cancer. Int. J. Mol. Sci. 2020, 21, 7307. [Google Scholar] [CrossRef] [PubMed]
- Sadeghalvad, M.; Mohammadi-Motlagh, H.R.; Rezaei, N. Immune microenvironment in different molecular subtypes of ductal breast carcinoma. Breast Cancer Res. Treat. 2020, 3, 1–19. [Google Scholar] [CrossRef]
- Truffi, M.; Sorrentino, L.; Corsi, F. Fibroblasts in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1234, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, D.; Chandra, R.; Karalis, J.; Teke, M.; Aguilera, T.; Maddipati, R.; Wachsmann, M.B.; Ghersi, D.; Siravegna, G.; Zeh, H.J., III; et al. Cancer-Associated Fibroblasts: Versatile Players in the Tumor Microenvironment. Cancers 2020, 12, 2652. [Google Scholar] [CrossRef] [PubMed]
- Mhaidly, R.; Mechta-Grigoriou, F. Fibroblast heterogeneity in tumor micro-environment: Role in immunosuppression and new therapies. Semin. Immunol. 2020, 48, 101417. [Google Scholar] [CrossRef] [PubMed]
- Leask, A. A centralized communication network: Recent insights into the role of the cancer associated fibroblast in the development of drug resistance in tumors. Semin. Cell Dev. Biol. 2020, 101, 111–114. [Google Scholar] [CrossRef]
- Bu, L.; Baba, H.; Yasuda, T.; Uchihara, T.; Ishimoto, T. Functional diversity of cancer-associated fibroblasts in modulating drug resistance. Cancer Sci. 2020, 111, 3468–3477. [Google Scholar] [CrossRef]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773. [Google Scholar] [CrossRef]
- Gal, P.; Varinska, L.; Faber, L.; Novak, S.; Szabo, P.; Mitrengova, P.; Mirossay, A.; Mucaji, P.; Smetana, K. How Signaling Molecules Regulate Tumor Microenvironment: Parallels to Wound Repair. Molecules 2017, 22, 1818. [Google Scholar] [CrossRef]
- Noguchi, S.; Saito, A.; Nagase, T. YAP/TAZ Signaling as a Molecular Link between Fibrosis and Cancer. Int. J. Mol. Sci. 2018, 19, 3674. [Google Scholar] [CrossRef] [PubMed]
- Ouhtit, A.; Rizeq, B.; Saleh, H.A.; Rahman, M.M.; Zayed, H. Novel CD44-downstream signaling pathways mediating breast tumor invasion. Int. J. Biol. Sci. 2018, 14, 1782–1790. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Tolg, C.; Turley, E. Dissecting the Dual Nature of Hyaluronan in the Tumor Microenvironment. Front. Immunol. 2019, 10, 947. [Google Scholar] [CrossRef] [PubMed]
- Misra, S.; Hascall, V.C.; Markwald, R.R.; Ghatak, S. Interactions between Hyaluronan and Its Receptors (CD44, RHAMM) Regulate the Activities of Inflammation and Cancer. Front. Immunol. 2015, 6, 201. [Google Scholar] [CrossRef] [PubMed]
- Tolg, C.; McCarthy, J.B.; Yazdani, A.; Turley, E.A. Hyaluronan and RHAMM in wound repair and the “cancerization” of stromal tissues. Biomed. Res. Int. 2014, 2014, 103923. [Google Scholar] [CrossRef] [PubMed]
- Noriega-Guerra, H.; Freitas, V.M. Extracellular Matrix Influencing HGF/c-MET Signaling Pathway: Impact on Cancer Progression. Int. J. Mol. Sci. 2018, 19, 3300. [Google Scholar] [CrossRef]
- Hassan, N.; Greve, B.; Espinoza-Sánchez, N.A.; Götte, M. Cell-surface heparan sulfate proteoglycans as multifunctional integrators of signaling in cancer. Cell Signal. 2020, 77, 109822. [Google Scholar] [CrossRef]
- Kim, S.H.; Turnbull, J.; Guimond, S. Extracellular matrix and cell signalling: The dynamic cooperation of integrin, proteoglycan and growth factor receptor. J. Endocrinol. 2011, 209, 139–151. [Google Scholar] [CrossRef]
- Wang, H.; Leavitt, L.; Ramaswamy, R.; Rapraeger, A.C. Interaction of syndecan and alpha6beta4 integrin cytoplasmic domains: Regulation of ErbB2-mediated integrin activation. J. Biol. Chem. 2010, 285, 13569–13579. [Google Scholar] [CrossRef]
- Morgan, M.R.; Humphries, M.J.; Bass, M.D. Synergistic control of cell adhesion by integrins and syndecans. Nat. Rev. Mol. Cell Biol. 2007, 8, 957–969. [Google Scholar] [CrossRef]
- Vuoriluoto, K.; Jokinen, J.; Kallio, K.; Salmivirta, M.; Heino, J.; Ivaska, J. Syndecan-1 supports integrin alpha2beta1-mediated adhesion to collagen. Exp. Cell Res. 2008, 314, 3369–3381. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, L.R.; Eble, J.A. Decorin regulates endothelial cell-matrix interactions during angiogenesis. Cell Adh. Migr. 2009, 3, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Neill, T.; Schaefer, L.; Iozzo, R.V. Decorin as a multivalent therapeutic agent against cancer. Adv. Drug Deliv. Rev. 2016, 97, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, L.; Tredup, C.; Gubbiotti, M.A.; Iozzo, R.V. Proteoglycan neofunctions: Regulation of inflammation and autophagy in cancer biology. FEBS J. 2017, 284, 10–26. [Google Scholar] [CrossRef]
- Poluzzi, C.; Iozzo, R.V.; Schaefer, L. Endostatin and endorepellin: A common route of action for similar angiostatic cancer avengers. Adv. Drug Deliv. Rev. 2016, 97, 156–173. [Google Scholar] [CrossRef]
- Woodall, B.P.; Nystrom, A.; Iozzo, R.A.; Eble, J.A.; Niland, S.; Krieg, T.; Eckes, B.; Pozzi, A.; Iozzo, R.V. Integrin alpha2beta1 is the required receptor for endorepellin angiostatic activity. J. Biol. Chem. 2008, 283, 2335–2343. [Google Scholar] [CrossRef]
- Barczyk, M.; Carracedo, S.; Gullberg, D. Integrins. Cell Tissue Res. 2010, 339, 269–280. [Google Scholar] [CrossRef]
- Humphries, J.D.; Byron, A.; Humphries, M.J. Integrin ligands at a glance. J. Cell Sci. 2006, 119, 3901–3903. [Google Scholar] [CrossRef]
- Zheng, Y.; Leftheris, K. Insights into Protein-Ligand Interactions in Integrin Complexes: Advances in Structure Determinations. J. Med. Chem. 2020, 63, 5675–5696. [Google Scholar] [CrossRef]
- Jang, I.; Beningo, K.A. Integrins, CAFs and Mechanical Forces in the Progression of Cancer. Cancers 2019, 11, 721. [Google Scholar] [CrossRef]
- DiPersio, C.M.; Van De Water, L. Integrin Regulation of CAF Differentiation and Function. Cancers 2019, 11, 715. [Google Scholar] [CrossRef] [PubMed]
- Liden, A.; Karlsen, T.V.; Guss, B.; Reed, R.K.; Rubin, K. Integrin alphaV beta3 can substitute for collagen-binding beta1 -integrins in vivo to maintain a homeostatic interstitial fluid pressure. Exp. Physiol. 2018, 103, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Arnaout, M.A.; Goodman, S.L.; Xiong, J.P. Structure and mechanics of integrin-based cell adhesion. Curr. Opin. Cell Biol. 2007, 19, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Humphries, J.D.; Chastney, M.R.; Askari, J.A.; Humphries, M.J. Signal transduction via integrin adhesion complexes. Curr. Opin. Cell Biol. 2019, 56, 14–21. [Google Scholar] [CrossRef]
- Luo, B.H.; Carman, C.V.; Springer, T.A. Structural basis of integrin regulation and signaling. Annu. Rev. Immunol. 2007, 25, 619–647. [Google Scholar] [CrossRef]
- Wang, W.; Luo, B.H. Structural basis of integrin transmembrane activation. J. Cell. Biochem. 2010, 109, 447–452. [Google Scholar] [CrossRef]
- Lima, A.M.; Wegner, S.V.; Martins Cavaco, A.C.; Estevão-Costa, M.I.; Sanz-Soler, R.; Niland, S.; Nosov, G.; Klingauf, J.; Spatz, J.P.; Eble, J.A. The spatial molecular pattern of integrin recognition sites and their immobilization to colloidal nanobeads determine α2β1 integrin-dependent platelet activation. Biomaterials 2018, 167, 107–120. [Google Scholar] [CrossRef]
- Kanchanawong, P.; Shtengel, G.; Pasapera, A.M.; Ramko, E.B.; Davidson, M.W.; Hess, H.F.; Waterman, C.M. Nanoscale architecture of integrin-based cell adhesions. Nature 2010, 468, 580–584. [Google Scholar] [CrossRef]
- Zaidel-Bar, R.; Geiger, B. The switchable integrin adhesome. J. Cell Sci. 2010, 123, 1385–1388. [Google Scholar] [CrossRef]
- Li, J.; Springer, T.A. Integrin extension enables ultrasensitive regulation by cytoskeletal force. Proc. Natl. Acad. Sci. USA 2017, 114, 4685–4690. [Google Scholar] [CrossRef]
- Li, J.; Su, Y.; Xia, W.; Qin, Y.; Humphries, M.J.; Vestweber, D.; Cabanas, C.; Lu, C.; Springer, T.A. Conformational equilibria and intrinsic affinities define integrin activation. EMBO J. 2017, 36, 629–645. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Costell, M.; Fassler, R. Integrin activation by talin, kindlin and mechanical forces. Nat. Cell Biol. 2019, 21, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, I.; Eble, J.A.; Hanschmann, E.M. Thiol switches in membrane proteins—Extracellular redox regulation in cell biology. Biol. Chem. 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Chen, J. The regulation of integrin function by divalent cations. Cell Adh. Migr. 2012, 6, 20–29. [Google Scholar] [CrossRef]
- Bergerhausen, L.; Grosche, J.; Meißner, J.; Hecker, C.; Caliandro, M.F.; Westerhausen, C.; Kamenac, A.; Rezaei, M.; Mörgelin, M.; Poschmann, G.; et al. Extracellular Redox Regulation of α7β Integrin-Mediated Cell Migration Is Signaled via a Dominant Thiol-Switch. Antioxidants 2020, 9, 227. [Google Scholar] [CrossRef]
- Ng, L.; Poon, R.T.; Pang, R. Biomarkers for predicting future metastasis of human gastrointestinal tumors. Cell. Mol. Life Sci. 2013, 70, 3631–3656. [Google Scholar] [CrossRef]
- Waerzeggers, Y.; Monfared, P.; Viel, T.; Faust, A.; Kopka, K.; Schafers, M.; Tavitian, B.; Winkeler, A.; Jacobs, A. Specific biomarkers of receptors, pathways of inhibition and targeted therapies: Pre-clinical developments. Br. J. Radiol. 2011, 84, S168–S178. [Google Scholar] [CrossRef]
- Arias-Mejias, S.M.; Warda, K.Y.; Quattrocchi, E.; Alonso-Quinones, H.; Sominidi-Damodaran, S.; Meves, A. The role of integrins in melanoma: A review. Int. J. Dermatol. 2020, 59, 525–534. [Google Scholar] [CrossRef]
- Zeltz, C.; Alam, J.; Liu, H.; Erusappan, P.M.; Hoschuetzky, H.; Molven, A.; Parajuli, H.; Cukierman, E.; Costea, D.-E.; Lu, N.; et al. α11β1 Integrin is Induced in a Subset of Cancer-Associated Fibroblasts in Desmoplastic Tumor Stroma and Mediates In Vitro Cell Migration. Cancers 2019, 11, 765. [Google Scholar] [CrossRef]
- Margadant, C.; Sonnenberg, A. Integrin-TGF-beta crosstalk in fibrosis, cancer and wound healing. EMBO Rep. 2010, 11, 97–105. [Google Scholar] [CrossRef]
- Sala, M.; Ros, M.; Saltel, F. A Complex and Evolutive Character: Two Face Aspects of ECM in Tumor Progression. Front. Oncol. 2020, 10, 1620. [Google Scholar] [CrossRef] [PubMed]
- Su, C.Y.; Li, J.Q.; Zhang, L.L.; Wang, H.; Wang, F.H.; Tao, Y.W.; Wang, Y.Q.; Guo, Q.R.; Li, J.J.; Liu, Y.; et al. The Biological Functions and Clinical Applications of Integrins in Cancers. Front. Pharmacol. 2020, 11, 579068. [Google Scholar] [CrossRef]
- Webber, J.; Steadman, R.; Mason, M.D.; Tabi, Z.; Clayton, A. Cancer exosomes trigger fibroblast to myofibroblast differentiation. Cancer Res. 2010, 70, 9621–9630. [Google Scholar] [CrossRef] [PubMed]
- Weidle, U.H.; Birzele, F.; Kollmorgen, G.; Ruger, R. The Multiple Roles of Exosomes in Metastasis. Cancer Genom. Proteom. 2017, 14, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.; Wang, X.; Gong, Z.; Yu, M.; Wu, H.; Zhang, D. Exosome-mediated metabolic reprogramming: The emerging role in tumor microenvironment remodeling and its influence on cancer progression. Signal Transduct. Target Ther. 2020, 5, 242. [Google Scholar] [CrossRef]
- Yang, J.; Bahcecioglu, G.; Zorlutuna, P. The Extracellular Matrix and Vesicles Modulate the Breast Tumor Microenvironment. Bioengineering 2020, 7, 124. [Google Scholar] [CrossRef]
- Korenchan, D.E.; Flavell, R.R. Spatiotemporal pH Heterogeneity as a Promoter of Cancer Progression and Therapeutic Resistance. Cancers 2019, 11, 1026. [Google Scholar] [CrossRef]
- Pillai, S.R.; Damaghi, M.; Marunaka, Y.; Spugnini, E.P.; Fais, S.; Gillies, R.J. Causes, consequences, and therapy of tumors acidosis. Cancer Metastasis Rev. 2019, 38, 205–222. [Google Scholar] [CrossRef]
- Vaupel, P.; Multhoff, G. Accomplices of the Hypoxic Tumor Microenvironment Compromising Antitumor Immunity: Adenosine, Lactate, Acidosis, Vascular Endothelial Growth Factor, Potassium Ions, and Phosphatidylserine. Front. Immunol. 2017, 8, 1887. [Google Scholar] [CrossRef]
- Miles, F.L.; Sikes, R.A. Insidious changes in stromal matrix fuel cancer progression. Mol. Cancer Res. 2014, 12, 297–312. [Google Scholar] [CrossRef]
- Huang, L.; Xu, A.M.; Liu, W. Transglutaminase 2 in cancer. Am. J. Cancer Res. 2015, 5, 2756–2776. [Google Scholar] [PubMed]
- Kim, S.Y. New Insights into Development of Transglutaminase 2 Inhibitors as Pharmaceutical Lead Compounds. Med. Sci. 2018, 6, 87. [Google Scholar] [CrossRef] [PubMed]
- Hoye, A.M.; Erler, J.T. Structural ECM components in the premetastatic and metastatic niche. Am. J. Physiol. Cell Physiol. 2016, 310, C955–C967. [Google Scholar] [CrossRef] [PubMed]
- Ji, K.; Mayernik, L.; Moin, K.; Sloane, B.F. Acidosis and proteolysis in the tumor microenvironment. Cancer Metastasis Rev. 2019, 38, 103–112. [Google Scholar] [CrossRef]
- Wells, J.M.; Gaggar, A.; Blalock, J.E. MMP generated matrikines. Matrix Biol. 2015, 44–46, 122–129. [Google Scholar] [CrossRef]
- Mochizuki, S.; Ao, T.; Sugiura, T.; Yonemura, K.; Shiraishi, T.; Kajiwara, Y.; Okamoto, K.; Shinto, E.; Okada, Y.; Ueno, H. Expression and Function of a Disintegrin and Metalloproteinases in Cancer-Associated Fibroblasts of Colorectal Cancer. Digestion 2020, 101, 18–24. [Google Scholar] [CrossRef]
- Stern, R.; Jedrzejas, M.J. Hyaluronidases: Their genomics, structures, and mechanisms of action. Chem. Rev. 2006, 106, 818–839. [Google Scholar] [CrossRef]
- Quintero-Fabian, S.; Arreola, R.; Becerril-Villanueva, E.; Torres-Romero, J.C.; Arana-Argaez, V.; Lara-Riegos, J.; Ramirez-Camacho, M.A.; Alvarez-Sanchez, M.E. Role of Matrix Metalloproteinases in Angiogenesis and Cancer. Front. Oncol. 2019, 9, 1370. [Google Scholar] [CrossRef]
- Eatemadi, A.; Aiyelabegan, H.T.; Negahdari, B.; Mazlomi, M.A.; Daraee, H.; Daraee, N.; Eatemadi, R.; Sadroddiny, E. Role of protease and protease inhibitors in cancer pathogenesis and treatment. Biomed. Pharmacother. 2017, 86, 221–231. [Google Scholar] [CrossRef]
- Crotti, S.; Piccoli, M.; Rizzolio, F.; Giordano, A.; Nitti, D.; Agostini, M. Extracellular Matrix and Colorectal Cancer: How Surrounding Microenvironment Affects Cancer Cell Behavior? J. Cell. Physiol. 2017, 232, 967–975. [Google Scholar] [CrossRef]
- Stefanidakis, M.; Koivunen, E. Cell-surface association between matrix metalloproteinases and integrins: Role of the complexes in leukocyte migration and cancer progression. Blood 2006, 108, 1441–1450. [Google Scholar] [CrossRef] [PubMed]
- Saad, S.; Gottlieb, D.J.; Bradstock, K.F.; Overall, C.M.; Bendall, L.J. Cancer cell-associated fibronectin induces release of matrix metalloproteinase-2 from normal fibroblasts. Cancer Res. 2002, 62, 283–289. [Google Scholar] [PubMed]
- Nagase, H.; Visse, R.; Murphy, G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 2006, 69, 562–573. [Google Scholar] [CrossRef] [PubMed]
- Jacob, A.; Prekeris, R. The regulation of MMP targeting to invadopodia during cancer metastasis. Front. Cell Dev. Biol. 2015, 3, 4. [Google Scholar] [CrossRef]
- Ren, F.; Tang, R.; Zhang, X.; Madushi, W.M.; Luo, D.; Dang, Y.; Li, Z.; Wei, K.; Chen, G. Overexpression of MMP Family Members Functions as Prognostic Biomarker for Breast Cancer Patients: A Systematic Review and Meta-Analysis. PLoS ONE 2015, 10, e0135544. [Google Scholar] [CrossRef]
- Vandooren, J.; Van den Steen, P.E.; Opdenakker, G. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9): The next decade. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 222–272. [Google Scholar] [CrossRef]
- Gonzalez-Avila, G.; Sommer, B.; García-Hernández, A.A.; Ramos, C. Matrix Metalloproteinases’ Role in Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1245, 97–131. [Google Scholar] [CrossRef]
- Huang, H. Matrix Metalloproteinase-9 (MMP-9) as a Cancer Biomarker and MMP-9 Biosensors: Recent Advances. Sensors 2018, 18, 3249. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, S.; Guo, J.; Zhou, L.; You, L.; Zhang, T.; Zhao, Y. Insights into the distinct roles of MMP-11 in tumor biology and future therapeutics (Review). Int. J. Oncol. 2016, 48, 1783–1793. [Google Scholar] [CrossRef]
- Castro-Castro, A.; Marchesin, V.; Monteiro, P.; Lodillinsky, C.; Rossé, C.; Chavrier, P. Cellular and Molecular Mechanisms of MT1-MMP-Dependent Cancer Cell Invasion. Annu. Rev. Cell Dev. Biol. 2016, 32, 555–576. [Google Scholar] [CrossRef]
- Pahwa, S.; Stawikowski, M.J.; Fields, G.B. Monitoring and Inhibiting MT1-MMP during Cancer Initiation and Progression. Cancers 2014, 6, 416–435. [Google Scholar] [CrossRef] [PubMed]
- Alcantara, M.B.; Dass, C.R. Regulation of MT1-MMP and MMP-2 by the serpin PEDF: A promising new target for metastatic cancer. Cell. Physiol. Biochem. 2013, 31, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Poincloux, R.; Lizarraga, F.; Chavrier, P. Matrix invasion by tumour cells: A focus on MT1-MMP trafficking to invadopodia. J. Cell Sci. 2009, 122, 3015–3024. [Google Scholar] [CrossRef] [PubMed]
- Radisky, E.S.; Radisky, D.C. Matrix metalloproteinases as breast cancer drivers and therapeutic targets. Front. Biosci. Landmark 2015, 20, 1144–1163. [Google Scholar] [CrossRef] [PubMed]
- Parvanescu, V.; Georgescu, M.; Georgescu, I.; Surlin, V.; Patrascu, S.; Picleanu, A.M.; Georgescu, E. The Role of Matrix Metalloproteinase-9 (MMP-9) as a Prognostic Factor in Epithelial and Lymphatic Neoplasia. Chirurgia 2015, 110, 506–510. [Google Scholar]
- Fields, G.B. Biophysical studies of matrix metalloproteinase/triple-helix complexes. Adv. Protein Chem. Struct. Biol. 2014, 97, 37–48. [Google Scholar] [CrossRef]
- Tam, E.M.; Moore, T.R.; Butler, G.S.; Overall, C.M. Characterization of the distinct collagen binding, helicase and cleavage mechanisms of matrix metalloproteinase 2 and 14 (gelatinase A and MT1-MMP): The differential roles of the MMP hemopexin c domains and the MMP-2 fibronectin type II modules in collagen triple helicase activities. J. Biol. Chem. 2004, 279, 43336–43344. [Google Scholar] [CrossRef]
- Farina, A.R.; Mackay, A.R. Gelatinase B/MMP-9 in Tumour Pathogenesis and Progression. Cancers 2014, 6, 240–296. [Google Scholar] [CrossRef]
- Sato, H.; Takino, T. Coordinate action of membrane-type matrix metalloproteinase-1 (MT1-MMP) and MMP-2 enhances pericellular proteolysis and invasion. Cancer Sci. 2010, 101, 843–847. [Google Scholar] [CrossRef]
- Murphy, G.; Nagase, H. Progress in matrix metalloproteinase research. Mol. Aspects Med. 2008, 29, 290–308. [Google Scholar] [CrossRef]
- Jackson, B.C.; Nebert, D.W.; Vasiliou, V. Update of human and mouse matrix metalloproteinase families. Hum. Genom. 2010, 4, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Cui, N.; Hu, M.; Khalil, R.A. Biochemical and Biological Attributes of Matrix Metalloproteinases. Prog. Mol. Biol. Transl. Sci. 2017, 147, 1–73. [Google Scholar] [CrossRef] [PubMed]
- Mittal, R.; Patel, A.P.; Debs, L.H.; Nguyen, D.; Patel, K.; Grati, M.; Mittal, J.; Yan, D.; Chapagain, P.; Liu, X.Z. Intricate Functions of Matrix Metalloproteinases in Physiological and Pathological Conditions. J. Cell. Physiol. 2016, 231, 2599–2621. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Mandal, M.; Chakraborti, T.; Mandal, A.; Chakraborti, S. Structure and evolutionary aspects of matrix metalloproteinases: A brief overview. Mol. Cell. Biochem. 2003, 253, 31–40. [Google Scholar] [CrossRef]
- Massova, I.; Kotra, L.P.; Fridman, R.; Mobashery, S. Matrix metalloproteinases: Structures, evolution, and diversification. FASEB J. 1998, 12, 1075–1095. [Google Scholar] [CrossRef]
- Loffek, S.; Schilling, O.; Franzke, C.W. Series “matrix metalloproteinases in lung health and disease”: Biological role of matrix metalloproteinases: A critical balance. Eur. Respir. J. 2011, 38, 191–208. [Google Scholar] [CrossRef]
- Gramolelli, S.; Cheng, J.; Martinez-Corral, I.; Vähä-Koskela, M.; Elbasani, E.; Kaivanto, E.; Rantanen, V.; Tuohinto, K.; Hautaniemi, S.; Bower, M.; et al. PROX1 is a transcriptional regulator of MMP14. Sci. Rep. 2018, 8, 9531. [Google Scholar] [CrossRef]
- Hotary, K.; Allen, E.; Punturieri, A.; Yana, I.; Weiss, S.J. Regulation of cell invasion and morphogenesis in a three-dimensional type I collagen matrix by membrane-type matrix metalloproteinases 1, 2, and 3. J. Cell Biol. 2000, 149, 1309–1323. [Google Scholar] [CrossRef]
- Itoh, Y.; Seiki, M. MT1-MMP: A potent modifier of pericellular microenvironment. J. Cell. Physiol. 2006, 206, 1–8. [Google Scholar] [CrossRef]
- Sabeh, F.; Ota, I.; Holmbeck, K.; Birkedal-Hansen, H.; Soloway, P.; Balbin, M.; Lopez-Otin, C.; Shapiro, S.; Inada, M.; Krane, S.; et al. Tumor cell traffic through the extracellular matrix is controlled by the membrane-anchored collagenase MT1-MMP. J. Cell Biol. 2004, 167, 769–781. [Google Scholar] [CrossRef]
- Wolf, K.; Wu, Y.I.; Liu, Y.; Geiger, J.; Tam, E.; Overall, C.; Stack, M.S.; Friedl, P. Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat. Cell Biol. 2007, 9, 893–904. [Google Scholar] [CrossRef]
- Szabova, L.; Chrysovergis, K.; Yamada, S.S.; Holmbeck, K. MT1-MMP is required for efficient tumor dissemination in experimental metastatic disease. Oncogene 2008, 27, 3274–3281. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhou, Z.; Apte, S.S.; Soininen, R.; Cao, R.; Baaklini, G.Y.; Rauser, R.W.; Wang, J.; Cao, Y.; Tryggvason, K. Impaired endochondral ossification and angiogenesis in mice deficient in membrane-type matrix metalloproteinase I. Proc. Natl. Acad. Sci. USA 2000, 97, 4052–4057. [Google Scholar] [CrossRef] [PubMed]
- David, J.M.; Rajasekaran, A.K. Dishonorable discharge: The oncogenic roles of cleaved E-cadherin fragments. Cancer Res. 2012, 72, 2917–2923. [Google Scholar] [CrossRef] [PubMed]
- Kajita, M.; Itoh, Y.; Chiba, T.; Mori, H.; Okada, A.; Kinoh, H.; Seiki, M. Membrane-type 1 matrix metalloproteinase cleaves CD44 and promotes cell migration. J. Cell Biol. 2001, 153, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, T.; Seiki, M. Integrated functions of membrane-type 1 matrix metalloproteinase in regulating cancer malignancy: Beyond a proteinase. Cancer Sci. 2017, 108, 1095–1100. [Google Scholar] [CrossRef] [PubMed]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef]
- Gonzalez-Molina, J.; Gramolelli, S.; Liao, Z.; Carlson, J.W.; Ojala, P.M.; Lehti, K. MMP14 in Sarcoma: A Regulator of Tumor Microenvironment Communication in Connective Tissues. Cells 2019, 8, 991. [Google Scholar] [CrossRef]
- Yan, C.; Boyd, D.D. Regulation of matrix metalloproteinase gene expression. J. Cell Physiol. 2007, 211, 19–26. [Google Scholar] [CrossRef]
- Chatterjee, K.; Jana, S.; Choudhary, P.; Swarnakar, S. Triumph and tumult of matrix metalloproteinases and their crosstalk with eicosanoids in cancer. Cancer. Metastasis Rev. 2018, 37, 279–288. [Google Scholar] [CrossRef]
- Kim, Y.S.; Joh, T.H. Matrix metalloproteinases, new insights into the understanding of neurodegenerative disorders. Biomol. Ther. 2012, 20, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Chernov, A.V.; Sounni, N.E.; Remacle, A.G.; Strongin, A.Y. Epigenetic control of the invasion-promoting MT1-MMP/MMP-2/TIMP-2 axis in cancer cells. J. Biol. Chem. 2009, 284, 12727–12734. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; He, Z.; Wang, C.; Zhou, Y.; Li, F.; Pu, W.; Zhang, X.; Feng, X.; Zhang, M.; Yecheng, X.; et al. Epigenetic silencing of ZNF132 mediated by methylation-sensitive Sp1 binding promotes cancer progression in esophageal squamous cell carcinoma. Cell Death Dis. 2018, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Remacle, A.G.; Rozanov, D.V.; Fugere, M.; Day, R.; Strongin, A.Y. Furin regulates the intracellular activation and the uptake rate of cell surface-associated MT1-MMP. Oncogene 2006, 25, 5648–5655. [Google Scholar] [CrossRef]
- Williams, K.C.; Coppolino, M.G. Phosphorylation of membrane type 1-matrix metalloproteinase (MT1-MMP) and its vesicle-associated membrane protein 7 (VAMP7)-dependent trafficking facilitate cell invasion and migration. J. Biol. Chem. 2011, 286, 43405–43416. [Google Scholar] [CrossRef]
- Remacle, A.G.; Chekanov, A.V.; Golubkov, V.S.; Savinov, A.Y.; Rozanov, D.V.; Strongin, A.Y. O-glycosylation regulates autolysis of cellular membrane type-1 matrix metalloproteinase (MT1-MMP). J. Biol. Chem. 2006, 281, 16897–16905. [Google Scholar] [CrossRef]
- Kruglikov, I.L.; Joffin, N.; Scherer, P.E. The MMP14-caveolin axis and its potential relevance for lipoedema. Nat. Rev. Endocrinol. 2020, 16, 669–674. [Google Scholar] [CrossRef]
- Kim, H.N.; Chung, H.S. Caveolin-1 inhibits membrane-type 1 matrix metalloproteinase activity. BMB Rep. 2008, 41, 858–862. [Google Scholar] [CrossRef]
- Hsu, K.S.; Otsu, W.; Li, Y.; Wang, H.C.; Chen, S.; Tsang, S.H.; Chuang, J.Z.; Sung, C.H. CLIC4 regulates late endosomal trafficking and matrix degradation activity of MMP14 at focal adhesions in RPE cells. Sci. Rep. 2019, 9, 12247. [Google Scholar] [CrossRef]
- Itoh, Y. Membrane-type matrix metalloproteinases: Their functions and regulations. Matrix Biol. 2015, 44-46, 207–223. [Google Scholar] [CrossRef]
- Grafinger, O.R.; Gorshtein, G.; Stirling, T.; Brasher, M.I.; Coppolino, M.G. beta1 integrin-mediated signaling regulates MT1-MMP phosphorylation to promote tumor cell invasion. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef] [PubMed]
- Tobar, N.; Avalos, M.C.; Mendez, N.; Smith, P.C.; Bernabeu, C.; Quintanilla, M.; Martinez, J. Soluble MMP-14 produced by bone marrow-derived stromal cells sheds epithelial endoglin modulating the migratory properties of human breast cancer cells. Carcinogenesis 2014, 35, 1770–1779. [Google Scholar] [CrossRef] [PubMed]
- Osenkowski, P.; Toth, M.; Fridman, R. Processing, shedding, and endocytosis of membrane type 1-matrix metalloproteinase (MT1-MMP). J. Cell. Physiol. 2004, 200, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Lohi, J.; Lehti, K.; Westermarck, J.; Kähäri, V.M.; Keski-Oja, J. Regulation of membrane-type matrix metalloproteinase-1 expression by growth factors and phorbol 12-myristate 13-acetate. Eur. J. Biochem. 1996, 239, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Nakamura, T.; Suzuki, Y.; Imizu, T.; Matsumoto, K. 3-D collagen-dependent cell surface expression of MT1-MMP and MMP-2 activation regardless of integrin β1 function and matrix stiffness. Biochem. Biophys. Res. Commun. 2011, 412, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Haas, T.L.; Stitelman, D.; Davis, S.J.; Apte, S.S.; Madri, J.A. Egr-1 mediates extracellular matrix-driven transcription of membrane type 1 matrix metalloproteinase in endothelium. J. Biol. Chem. 1999, 274, 22679–22685. [Google Scholar] [CrossRef]
- Borrirukwanit, K.; Pavasant, P.; Blick, T.; Lafleur, M.A.; Thompson, E.W. High threshold of β1 integrin inhibition required to block collagen I-induced membrane type-1 matrix metalloproteinase (MT1-MMP) activation of matrix metalloproteinase 2 (MMP-2). Cancer Cell Int. 2014, 14, 99. [Google Scholar] [CrossRef]
- Zigrino, P.; Drescher, C.; Mauch, C. Collagen-induced proMMP-2 activation by MT1-MMP in human dermal fibroblasts and the possible role of alpha2beta1 integrins. Eur. J. Cell Biol. 2001, 80, 68–77. [Google Scholar] [CrossRef]
- Johnson, J.L.; Pillai, S.; Pernazza, D.; Sebti, S.M.; Lawrence, N.J.; Chellappan, S.P. Regulation of matrix metalloproteinase genes by E2F transcription factors: Rb-Raf-1 interaction as a novel target for metastatic disease. Cancer Res. 2012, 72, 516–526. [Google Scholar] [CrossRef]
- Elsir, T.; Smits, A.; Lindström, M.S.; Nistér, M. Transcription factor PROX1: Its role in development and cancer. Cancer Metastasis Rev. 2012, 31, 793–805. [Google Scholar] [CrossRef]
- Eiseler, T.; Döppler, H.; Yan, I.K.; Goodison, S.; Storz, P. Protein kinase D1 regulates matrix metalloproteinase expression and inhibits breast cancer cell invasion. Breast Cancer Res. 2009, 11, R13. [Google Scholar] [CrossRef] [PubMed]
- Onishi, Y.; Kawamoto, T.; Kishimoto, K.; Hara, H.; Fukase, N.; Toda, M.; Harada, R.; Kurosaka, M.; Akisue, T. PKD1 negatively regulates cell invasion, migration and proliferation ability of human osteosarcoma. Int. J. Oncol. 2012, 40, 1839–1848. [Google Scholar] [CrossRef] [PubMed]
- Jackson, H.W.; Defamie, V.; Waterhouse, P.; Khokha, R. TIMPs: Versatile extracellular regulators in cancer. Nat. Rev. Cancer 2017, 17, 38–53. [Google Scholar] [CrossRef] [PubMed]
- Murphy, G. Tissue inhibitors of metalloproteinases. Genome Biol. 2011, 12, 233. [Google Scholar] [CrossRef] [PubMed]
- Strongin, A.Y.; Collier, I.; Bannikov, G.; Marmer, B.L.; Grant, G.A.; Goldberg, G.I. Mechanism of cell surface activation of 72-kDa type IV collagenase. Isolation of the activated form of the membrane metalloprotease. J. Biol. Chem. 1995, 270, 5331–5338. [Google Scholar] [CrossRef] [PubMed]
- Ogata, Y.; Itoh, Y.; Nagase, H. Steps involved in activation of the pro-matrix metalloproteinase 9 (progelatinase B)-tissue inhibitor of metalloproteinases-1 complex by 4-aminophenylmercuric acetate and proteinases. J. Biol. Chem. 1995, 270, 18506–18511. [Google Scholar] [CrossRef]
- Mori, H.; Lo, A.T.; Inman, J.L.; Alcaraz, J.; Ghajar, C.M.; Mott, J.D.; Nelson, C.M.; Chen, C.S.; Zhang, H.; Bascom, J.L.; et al. Transmembrane/cytoplasmic, rather than catalytic, domains of Mmp14 signal to MAPK activation and mammary branching morphogenesis via binding to integrin β1. Development 2013, 140, 343–352. [Google Scholar] [CrossRef]
- Nguyen, A.T.; Chia, J.; Ros, M.; Hui, K.M.; Saltel, F.; Bard, F. Organelle Specific O-Glycosylation Drives MMP14 Activation, Tumor Growth, and Metastasis. Cancer Cell 2017, 32, 639–653. [Google Scholar] [CrossRef]
- Planchon, D.; Rios Morris, E.; Genest, M.; Comunale, F.; Vacher, S.; Bieche, I.; Denisov, E.V.; Tashireva, L.A.; Perelmuter, V.M.; Linder, S.; et al. MT1-MMP targeting to endolysosomes is mediated by upregulation of flotillins. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef]
- Infante, E.; Castagnino, A.; Ferrari, R.; Monteiro, P.; Agüera-González, S.; Paul-Gilloteaux, P.; Domingues, M.J.; Maiuri, P.; Raab, M.; Shanahan, C.M.; et al. LINC complex-Lis1 interplay controls MT1-MMP matrix digest-on-demand response for confined tumor cell migration. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Ferrari, R.; Infante, E.; Chavrier, P. Nucleus-Invadopodia Duo During Cancer Invasion. Trends Cell Biol. 2019, 29, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, N.M.; Wenzel, E.M.; Wang, L.; Antoine, S.; Chavrier, P.; Stenmark, H.; Raiborg, C. Protrudin-mediated ER-endosome contact sites promote MT1-MMP exocytosis and cell invasion. J. Cell Biol. 2020, 219. [Google Scholar] [CrossRef] [PubMed]
- Ellerbroek, S.M.; Wu, Y.I.; Overall, C.M.; Stack, M.S. Functional interplay between type I collagen and cell surface matrix metalloproteinase activity. J. Biol. Chem. 2001, 276, 24833–24842. [Google Scholar] [CrossRef] [PubMed]
- Laudański, P.; Swiatecka, J.; Kozłowski, L.; Leśniewska, M.; Wojtukiewicz, M.; Wołczyński, S. Increased serum level of membrane type 1-matrix metalloproteinase (MT1-MMP/MMP-14) in patients with breast cancer. Folia Histochem. Cytobiol. 2010, 48, 101–103. [Google Scholar] [CrossRef] [PubMed]
- Ip, Y.C.; Cheung, S.T.; Fan, S.T. Atypical localization of membrane type 1-matrix metalloproteinase in the nucleus is associated with aggressive features of hepatocellular carcinoma. Mol. Carcinog. 2007, 46, 225–230. [Google Scholar] [CrossRef]
- Mannello, F.; Medda, V. Nuclear localization of matrix metalloproteinases. Prog. Histochem. Cytochem. 2012, 47, 27–58. [Google Scholar] [CrossRef] [PubMed]
- Augoff, K.; Hryniewicz-Jankowska, A.; Tabola, R. Invadopodia: Clearing the way for cancer cell invasion. Ann. Transl. Med. 2020, 8, 902. [Google Scholar] [CrossRef]
- Branch, K.M.; Hoshino, D.; Weaver, A.M. Adhesion rings surround invadopodia and promote maturation. Biol. Open 2012, 1, 711–722. [Google Scholar] [CrossRef]
- Malik, R.; Luong, T.; Cao, X.; Han, B.; Shah, N.; Franco-Barraza, J.; Han, L.; Shenoy, V.B.; Lelkes, P.I.; Cukierman, E. Rigidity controls human desmoplastic matrix anisotropy to enable pancreatic cancer cell spread via extracellular signal-regulated kinase 2. Matrix Biol. 2019, 81, 50–69. [Google Scholar] [CrossRef]
- Peláez, R.; Pariente, A.; Pérez-Sala, Á.; Larrayoz, I.M. Integrins: Moonlighting Proteins in Invadosome Formation. Cancers 2019, 11, 615. [Google Scholar] [CrossRef]
- Pelaez, R.; Morales, X.; Salvo, E.; Garasa, S.; Ortiz de Solorzano, C.; Martinez, A.; Larrayoz, I.M.; Rouzaut, A. beta3 integrin expression is required for invadopodia-mediated ECM degradation in lung carcinoma cells. PLoS ONE 2017, 12, e0181579. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y. MT1-MMP: A key regulator of cell migration in tissue. IUBMB Life 2006, 58, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, S.; Leon, R.P.; Gribbin, K.P.; Zhang, Y.; Navarro, J.; Smith, R.; Devlin, K.; Wang, L.G.; Gibbs, S.L.; Korkola, J.; et al. Crosstalk between invadopodia and the extracellular matrix. Eur. J. Cell Biol. 2020, 99, 151122. [Google Scholar] [CrossRef] [PubMed]
- Eddy, R.J.; Weidmann, M.D.; Sharma, V.P.; Condeelis, J.S. Tumor Cell Invadopodia: Invasive Protrusions that Orchestrate Metastasis. Trends Cell Biol. 2017, 27, 595–607. [Google Scholar] [CrossRef] [PubMed]
- Revach, O.Y.; Geiger, B. The interplay between the proteolytic, invasive, and adhesive domains of invadopodia and their roles in cancer invasion. Cell Adh. Migr. 2014, 8, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Bagnato, A.; Rosano, L. Endothelin-1 receptor drives invadopodia: Exploiting how beta-arrestin-1 guides the way. Small GTPases 2018, 9, 394–398. [Google Scholar] [CrossRef]
- Harper, K.; Lavoie, R.R.; Charbonneau, M.; Brochu-Gaudreau, K.; Dubois, C.M. The hypoxic tumor microenvironment promotes invadopodia formation and metastasis through LPA1 receptor and EGFR cooperation. Mol. Cancer Res. 2018, 16, 1601–1613. [Google Scholar] [CrossRef]
- Tonisen, F.; Perrin, L.; Bayarmagnai, B.; van den Dries, K.; Cambi, A.; Gligorijevic, B. EP4 receptor promotes invadopodia and invasion in human breast cancer. Eur. J. Cell Biol. 2017. [Google Scholar] [CrossRef]
- Maziveyi, M.; Dong, S.; Baranwal, S.; Alahari, S.K. Nischarin regulates focal adhesion and Invadopodia formation in breast cancer cells. Mol. Cancer. 2018, 17, 21. [Google Scholar] [CrossRef]
- Artym, V.V.; Swatkoski, S.; Matsumoto, K.; Campbell, C.B.; Petrie, R.J.; Dimitriadis, E.K.; Li, X.; Mueller, S.C.; Bugge, T.H.; Gucek, M.; et al. Dense fibrillar collagen is a potent inducer of invadopodia via a specific signaling network. J. Cell Biol. 2015, 208, 331–350. [Google Scholar] [CrossRef]
- Alekhina, O.; Burstein, E.; Billadeau, D.D. Cellular functions of WASP family proteins at a glance. J. Cell Sci. 2017, 130, 2235–2241. [Google Scholar] [CrossRef] [PubMed]
- Frugtniet, B.; Jiang, W.G.; Martin, T.A. Role of the WASP and WAVE family proteins in breast cancer invasion and metastasis. Breast Cancer 2015, 7, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Parekh, A.; Weaver, A.M. Regulation of invadopodia by mechanical signaling. Exp. Cell Res. 2016, 343, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Jeannot, P.; Besson, A. Cortactin function in invadopodia. Small GTPases 2017, 1–15. [Google Scholar] [CrossRef]
- Pourfarhangi, K.E.; Bergman, A.; Gligorijevic, B. ECM Cross-Linking Regulates Invadopodia Dynamics. Biophys. J. 2018, 114, 1455–1466. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Ito, Y.; Miura, N.; Nagamura, Y.; Nakabo, A.; Fukami, K.; Honda, K.; Sakai, R. Actinin-1 and actinin-4 play essential but distinct roles in invadopodia formation by carcinoma cells. Eur. J. Cell Biol. 2017, 96, 685–694. [Google Scholar] [CrossRef]
- Linder, S. The matrix corroded: Podosomes and invadopodia in extracellular matrix degradation. Trends Cell Biol. 2007, 17, 107–117. [Google Scholar] [CrossRef]
- Nicholas, N.S.; Pipili, A.; Lesjak, M.S.; Wells, C.M. Differential role for PAK1 and PAK4 during the invadopodia lifecycle. Small GTPases 2017, 10, 1–7. [Google Scholar] [CrossRef]
- Suman, P.; Mishra, S.; Chander, H. High expression of FBP17 in invasive breast cancer cells promotes invadopodia formation. Med. Oncol. 2018, 35, 71. [Google Scholar] [CrossRef]
- Castagnino, A.; Castro-Castro, A.; Irondelle, M.; Guichard, A.; Lodillinsky, C.; Fuhrmann, L.; Vacher, S.; Aguera-Gonzalez, S.; Zagryazhskaya-Masson, A.; Romao, M.; et al. Coronin 1C promotes triple-negative breast cancer invasiveness through regulation of MT1-MMP traffic and invadopodia function. Oncogene 2018. [Google Scholar] [CrossRef]
- Seano, G.; Primo, L. Podosomes and invadopodia: Tools to breach vascular basement membrane. Cell Cycle 2015, 14, 1370–1374. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Deryugina, E.I.; Quigley, J.P. Tumor angiogenesis: MMP-mediated induction of intravasation- and metastasis-sustaining neovasculature. Matrix Biol. 2015, 44-46, 94–112. [Google Scholar] [CrossRef] [PubMed]
- Genis, L.; Galvez, B.G.; Gonzalo, P.; Arroyo, A.G. MT1-MMP: Universal or particular player in angiogenesis? Cancer Metastasis Rev. 2006, 25, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Binder, M.J.; McCoombe, S.; Williams, E.D.; McCulloch, D.R.; Ward, A.C. The extracellular matrix in cancer progression: Role of hyalectan proteoglycans and ADAMTS enzymes. Cancer Lett. 2017, 385, 55–64. [Google Scholar] [CrossRef]
- Kumar, S.; Das, A.; Barai, A.; Sen, S. MMP Secretion Rate and Inter-invadopodia Spacing Collectively Govern Cancer Invasiveness. Biophys. J. 2018, 114, 650–662. [Google Scholar] [CrossRef]
- Binker, M.G.; Binker-Cosen, A.A.; Gaisano, H.Y.; de Cosen, R.H.; Cosen-Binker, L.I. TGF-β1 increases invasiveness of SW1990 cells through Rac1/ROS/NF-κB/IL-6/MMP-2. Biochem. Biophys. Res. Commun. 2011, 405, 140–145. [Google Scholar] [CrossRef]
- Fan, Z.; Duan, X.; Cai, H.; Wang, L.; Li, M.; Qu, J.; Li, W.; Wang, Y.; Wang, J. Curcumin inhibits the invasion of lung cancer cells by modulating the PKCα/Nox-2/ROS/ATF-2/MMP-9 signaling pathway. Oncol. Rep. 2015, 34, 691–698. [Google Scholar] [CrossRef]
- Choi, D.H.; Kim, J.H.; Seo, J.H.; Lee, J.; Choi, W.S.; Kim, Y.S. Matrix metalloproteinase-3 causes dopaminergic neuronal death through Nox1-regenerated oxidative stress. PLoS ONE 2014, 9, e115954. [Google Scholar] [CrossRef]
- Hawk, M.A.; Schafer, Z.T. Mechanisms of redox metabolism and cancer cell survival during extracellular matrix detachment. J. Biol. Chem. 2018, 293, 7531–7537. [Google Scholar] [CrossRef]
- Monboisse, J.C.; Oudart, J.B.; Ramont, L.; Brassart-Pasco, S.; Maquart, F.X. Matrikines from basement membrane collagens: A new anti-cancer strategy. Biochim. Biophys. Acta 2014, 1840, 2589–2598. [Google Scholar] [CrossRef]
- Tran, K.T.; Lamb, P.; Deng, J.S. Matrikines and matricryptins: Implications for cutaneous cancers and skin repair. J. Dermatol. Sci. 2005, 40, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Hornebeck, W.; Maquart, F.X. Proteolyzed matrix as a template for the regulation of tumor progression. Biomed. Pharmacother. 2003, 57, 223–230. [Google Scholar] [CrossRef]
- Ricard-Blum, S.; Vallet, S.D. Fragments generated upon extracellular matrix remodeling: Biological regulators and potential drugs. Matrix Biol. 2019, 75-76, 170–189. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Fleury, C.; Jalalvand, F.; Riesbeck, K. Human pathogens utilize host extracellular matrix proteins laminin and collagen for adhesion and invasion of the host. FEMS Microbiol. Rev. 2012, 36, 1122–1180. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, D.; Camardella, L.; Ulivi, V.; Guasco, G.; Manduca, P. Trimer carboxyl propeptide of collagen I produced by mature osteoblasts is chemotactic for endothelial cells. J. Biol. Chem. 2000, 275, 32658–32663. [Google Scholar] [CrossRef]
- Palmieri, D.; Astigiano, S.; Barbieri, O.; Ferrari, N.; Marchisio, S.; Ulivi, V.; Volta, C.; Manduca, P. Procollagen I COOH-terminal fragment induces VEGF-A and CXCR4 expression in breast carcinoma cells. Exp. Cell Res. 2008, 314, 2289–2298. [Google Scholar] [CrossRef]
- Visigalli, D.; Palmieri, D.; Strangio, A.; Astigiano, S.; Barbieri, O.; Casartelli, G.; Zicca, A.; Manduca, P. The carboxyl terminal trimer of procollagen I induces pro-metastatic changes and vascularization in breast cancer cells xenografts. BMC Cancer 2009, 9, 59. [Google Scholar] [CrossRef]
- Fernandes, R.J.; Hirohata, S.; Engle, J.M.; Colige, A.; Cohn, D.H.; Eyre, D.R.; Apte, S.S. Procollagen II amino propeptide processing by ADAMTS-3. Insights on dermatosparaxis. J. Biol. Chem. 2001, 276, 31502–31509. [Google Scholar] [CrossRef]
- Wang, Z.; Bryan, J.; Franz, C.; Havlioglu, N.; Sandell, L.J. Type IIB procollagen NH(2)-propeptide induces death of tumor cells via interaction with integrins alpha(V)beta(3) and alpha(V)beta(5). J. Biol. Chem. 2010, 285, 20806–20817. [Google Scholar] [CrossRef]
- Hayashi, S.; Wang, Z.; Bryan, J.; Kobayashi, C.; Faccio, R.; Sandell, L.J. The type II collagen N-propeptide, PIIBNP, inhibits cell survival and bone resorption of osteoclasts via integrin-mediated signaling. Bone 2011, 49, 644–652. [Google Scholar] [CrossRef]
- Lipton, A.; Leitzel, K.; Ali, S.M.; Polimera, H.V.; Nagabhairu, V.; Marks, E.; Richardson, A.E.; Krecko, L.; Ali, A.; Koestler, W.; et al. High turnover of extracellular matrix reflected by specific protein fragments measured in serum is associated with poor outcomes in two metastatic breast cancer cohorts. Int. J. Cancer 2018, 143, 3027–3034. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.J.; Nedergaard, A.F.; Sun, S.; Veidal, S.S.; Larsen, L.; Zheng, Q.; Suetta, C.; Henriksen, K.; Christiansen, C.; Karsdal, M.A.; et al. The neo-epitope specific PRO-C3 ELISA measures true formation of type III collagen associated with liver and muscle parameters. Am. J. Transl. Res. 2013, 5, 303–315. [Google Scholar] [PubMed]
- Leitinger, B.; Hohenester, E. Mammalian collagen receptors. Matrix Biol. 2007, 26, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Leeming, D.; He, Y.; Veidal, S.; Nguyen, Q.; Larsen, D.; Koizumi, M.; Segovia-Silvestre, T.; Zhang, C.; Zheng, Q.; Sun, S.; et al. A novel marker for assessment of liver matrix remodeling: An enzyme-linked immunosorbent assay (ELISA) detecting a MMP generated type I collagen neo-epitope (C1M). Biomarkers 2011, 16, 616–628. [Google Scholar] [CrossRef]
- Abdul Roda, M.; Fernstrand, A.M.; Redegeld, F.A.; Blalock, J.E.; Gaggar, A.; Folkerts, G. The matrikine PGP as a potential biomarker in COPD. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L1095–L1101. [Google Scholar] [CrossRef]
- Patel, D.F.; Snelgrove, R.J. The multifaceted roles of the matrikine Pro-Gly-Pro in pulmonary health and disease. Eur. Respir. Rev. 2018, 27. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Vanderby, R., Jr. Collagen fibril morphology and organization: Implications for force transmission in ligament and tendon. Matrix Biol. 2006, 25, 71–84. [Google Scholar] [CrossRef]
- Veidal, S.S.; Vassiliadis, E.; Barascuk, N.; Zhang, C.; Segovia-Silvestre, T.; Klickstein, L.; Larsen, M.R.; Qvist, P.; Christiansen, C.; Vainer, B.; et al. Matrix metalloproteinase-9-mediated type III collagen degradation as a novel serological biochemical marker for liver fibrogenesis. Liver Int. 2010, 30, 1293–1304. [Google Scholar] [CrossRef]
- Iyengar, P.; Espina, V.; Williams, T.W.; Lin, Y.; Berry, D.; Jelicks, L.A.; Lee, H.; Temple, K.; Graves, R.; Pollard, J.; et al. Adipocyte-derived collagen VI affects early mammary tumor progression in vivo, demonstrating a critical interaction in the tumor/stroma microenvironment. J. Clin. Investig. 2005, 115, 1163–1176. [Google Scholar] [CrossRef]
- Motrescu, E.R.; Blaise, S.; Etique, N.; Messaddeq, N.; Chenard, M.P.; Stoll, I.; Tomasetto, C.; Rio, M.C. Matrix metalloproteinase-11/stromelysin-3 exhibits collagenolytic function against collagen VI under normal and malignant conditions. Oncogene 2008, 27, 6347–6355. [Google Scholar] [CrossRef]
- Park, J.; Scherer, P.E. Adipocyte-derived endotrophin promotes malignant tumor progression. J. Clin. Investig. 2012, 122, 4243–4256. [Google Scholar] [CrossRef] [PubMed]
- Willumsen, N.; Jorgensen, L.N.; Karsdal, M.A. Vastatin (the NC1 domain of human type VIII collagen a1 chain) is linked to stromal reactivity and elevated in serum from patients with colorectal cancer. Cancer Biol. Ther. 2019, 20, 692–699. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, J.; Woo, Y.M.; Shen, Z.; Yao, H.; Cai, Y.; Lin, M.C.; Poon, W.S. Enhanced expression of Vastatin inhibits angiogenesis and prolongs survival in murine orthotopic glioblastoma model. BMC Cancer 2017, 17, 126. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Yao, C.; Wang, Z.; Yue, L.; Fang, Z.; Yao, H.; Lin, F.; Zhao, H.; Sun, Y.J.; Bian, X.W.; et al. Vastatin, an Endogenous Antiangiogenesis Polypeptide That Is Lost in Hepatocellular Carcinoma, Effectively Inhibits Tumor Metastasis. Mol. Ther. 2016, 24, 1358–1368. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Yao, Z.Y.; Xin, L.; Zhang, Q.; Li, T.P.; Gan, R.B. NC1 domain of human type VIII collagen (alpha 1) inhibits bovine aortic endothelial cell proliferation and causes cell apoptosis. Biochem. Biophys. Res. Commun. 2001, 289, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Boudko, S.P.; Danylevych, N.; Hudson, B.G.; Pedchenko, V.K. Basement membrane collagen IV: Isolation of functional domains. Methods Cell Biol. 2018, 143, 171–185. [Google Scholar] [CrossRef]
- Heljasvaara, R.; Aikio, M.; Ruotsalainen, H.; Pihlajaniemi, T. Collagen XVIII in tissue homeostasis and dysregulation—Lessons learned from model organisms and human patients. Matrix Biol. 2017, 57–58, 55–75. [Google Scholar] [CrossRef]
- Mak, K.M.; Mei, R. Basement Membrane Type IV Collagen and Laminin: An Overview of Their Biology and Value as Fibrosis Biomarkers of Liver Disease. Anat. Rec. 2017, 300, 1371–1390. [Google Scholar] [CrossRef]
- Rebustini, I.T.; Myers, C.; Lassiter, K.S.; Surmak, A.; Szabova, L.; Holmbeck, K.; Pedchenko, V.; Hudson, B.G.; Hoffman, M.P. MT2-MMP-dependent release of collagen IV NC1 domains regulates submandibular gland branching morphogenesis. Dev. Cell 2009, 17, 482–493. [Google Scholar] [CrossRef]
- Sugiyama, A.; Mitsui, A.; Okada, M.; Yamawaki, H. Cathepsin S degrades arresten and canstatin in infarcted area after myocardial infarction in rats. J. Vet. Med. Sci. 2019, 81, 522–531. [Google Scholar] [CrossRef]
- Hamano, Y.; Zeisberg, M.; Sugimoto, H.; Lively, J.C.; Maeshima, Y.; Yang, C.; Hynes, R.O.; Werb, Z.; Sudhakar, A.; Kalluri, R. Physiological levels of tumstatin, a fragment of collagen IV alpha3 chain, are generated by MMP-9 proteolysis and suppress angiogenesis via alphaV beta3 integrin. Cancer Cell 2003, 3, 589–601. [Google Scholar] [CrossRef]
- Nyberg, P.; Xie, L.; Sugimoto, H.; Colorado, P.; Sund, M.; Holthaus, K.; Sudhakar, A.; Salo, T.; Kalluri, R. Characterization of the anti-angiogenic properties of arresten, an alpha1beta1 integrin-dependent collagen-derived tumor suppressor. Exp. Cell Res. 2008, 314, 3292–3305. [Google Scholar] [CrossRef] [PubMed]
- Kamphaus, G.D.; Colorado, P.C.; Panka, D.J.; Hopfer, H.; Ramchandran, R.; Torre, A.; Maeshima, Y.; Mier, J.W.; Sukhatme, V.P.; Kalluri, R. Canstatin, a novel matrix-derived inhibitor of angiogenesis and tumor growth. J. Biol. Chem. 2000, 275, 1209–1215. [Google Scholar] [CrossRef]
- Hamano, Y.; Kalluri, R. Tumstatin, the NC1 domain of alpha3 chain of type IV collagen, is an endogenous inhibitor of pathological angiogenesis and suppresses tumor growth. Biochem. Biophys. Res. Commun. 2005, 333, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, E.D.; Popel, A.S. Identification of novel short peptides derived from the alpha 4, alpha 5, and alpha 6 fibrils of type IV collagen with anti-angiogenic properties. Biochem. Biophys. Res. Commun. 2007, 354, 434–439. [Google Scholar] [CrossRef]
- Brassart-Pasco, S.; Senechal, K.; Thevenard, J.; Ramont, L.; Devy, J.; Di Stefano, L.; Dupont-Deshorgue, A.; Brezillon, S.; Feru, J.; Jazeron, J.F.; et al. Tetrastatin, the NC1 domain of the alpha4(IV) collagen chain: A novel potent anti-tumor matrikine. PLoS ONE 2012, 7, e29587. [Google Scholar] [CrossRef]
- Weckmann, M.; Moir, L.M.; Heckman, C.A.; Black, J.L.; Oliver, B.G.; Burgess, J.K. Lamstatin—A novel inhibitor of lymphangiogenesis derived from collagen IV. J. Cell. Mol. Med. 2012, 16, 3062–3073. [Google Scholar] [CrossRef]
- Magnon, C.; Galaup, A.; Mullan, B.; Rouffiac, V.; Bouquet, C.; Bidart, J.M.; Griscelli, F.; Opolon, P.; Perricaudet, M. Canstatin acts on endothelial and tumor cells via mitochondrial damage initiated through interaction with alphavbeta3 and alphavbeta5 integrins. Cancer Res. 2005, 65, 4353–4361. [Google Scholar] [CrossRef]
- Torres, P.H.; Sousa, G.L.; Pascutti, P.G. Structural analysis of the N-terminal fragment of the antiangiogenic protein endostatin: A molecular dynamics study. Proteins 2011, 79, 2684–2692. [Google Scholar] [CrossRef]
- Mundel, T.M.; Yliniemi, A.M.; Maeshima, Y.; Sugimoto, H.; Kieran, M.; Kalluri, R. Type IV collagen alpha6 chain-derived noncollagenous domain 1 (alpha6(IV)NC1) inhibits angiogenesis and tumor growth. Int. J. Cancer 2008, 122, 1738–1744. [Google Scholar] [CrossRef]
- Petitclerc, E.; Boutaud, A.; Prestayko, A.; Xu, J.; Sado, Y.; Ninomiya, Y.; Sarras, M.P., Jr.; Hudson, B.G.; Brooks, P.C. New functions for non-collagenous domains of human collagen type IV. Novel integrin ligands inhibiting angiogenesis and tumor growth in vivo. J. Biol. Chem. 2000, 275, 8051–8061. [Google Scholar] [CrossRef] [PubMed]
- Veidal, S.S.; Karsdal, M.A.; Nawrocki, A.; Larsen, M.R.; Dai, Y.; Zheng, Q.; Hägglund, P.; Vainer, B.; Skjøt-Arkil, H.; Leeming, D.J. Assessment of proteolytic degradation of the basement membrane: A fragment of type IV collagen as a biochemical marker for liver fibrosis. Fibrogenes. Tissue Repair 2011, 4, 22. [Google Scholar] [CrossRef] [PubMed]
- Heljasvaara, R.; Nyberg, P.; Luostarinen, J.; Parikka, M.; Heikkilä, P.; Rehn, M.; Sorsa, T.; Salo, T.; Pihlajaniemi, T. Generation of biologically active endostatin fragments from human collagen XVIII by distinct matrix metalloproteases. Exp. Cell Res. 2005, 307, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Veillard, F.; Saidi, A.; Burden, R.E.; Scott, C.J.; Gillet, L.; Lecaille, F.; Lalmanach, G. Cysteine cathepsins S and L modulate anti-angiogenic activities of human endostatin. J. Biol. Chem. 2011, 286, 37158–37167. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Moses, M.A.; Wiederschain, D.; Arbiser, J.L.; Folkman, J. The generation of endostatin is mediated by elastase. Cancer Res. 1999, 59, 6052–6056. [Google Scholar] [PubMed]
- Folkman, J. Antiangiogenesis in cancer therapy--endostatin and its mechanisms of action. Exp. Cell Res. 2006, 312, 594–607. [Google Scholar] [CrossRef]
- Shi, H.; Huang, Y.; Zhou, H.; Song, X.; Yuan, S.; Fu, Y.; Luo, Y. Nucleolin is a receptor that mediates antiangiogenic and antitumor activity of endostatin. Blood 2007, 110, 2899–2906. [Google Scholar] [CrossRef]
- Wickström, S.A.; Alitalo, K.; Keski-Oja, J. Endostatin associates with integrin alpha5beta1 and caveolin-1, and activates Src via a tyrosyl phosphatase-dependent pathway in human endothelial cells. Cancer Res. 2002, 62, 5580–5589. [Google Scholar]
- Kim, Y.M.; Hwang, S.; Kim, Y.M.; Pyun, B.J.; Kim, T.Y.; Lee, S.T.; Gho, Y.S.; Kwon, Y.G. Endostatin blocks vascular endothelial growth factor-mediated signaling via direct interaction with KDR/Flk-1. J. Biol. Chem. 2002, 277, 27872–27879. [Google Scholar] [CrossRef]
- Karumanchi, S.A.; Jha, V.; Ramchandran, R.; Karihaloo, A.; Tsiokas, L.; Chan, B.; Dhanabal, M.; Hanai, J.I.; Venkataraman, G.; Shriver, Z.; et al. Cell surface glypicans are low-affinity endostatin receptors. Mol. Cell. 2001, 7, 811–822. [Google Scholar] [CrossRef]
- Lin, H.C.; Chang, J.H.; Jain, S.; Gabison, E.E.; Kure, T.; Kato, T.; Fukai, N.; Azar, D.T. Matrilysin cleavage of corneal collagen type XVIII NC1 domain and generation of a 28-kDa fragment. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2517–2524. [Google Scholar]
- Chang, J.H.; Javier, J.A.; Chang, G.Y.; Oliveira, H.B.; Azar, D.T. Functional characterization of neostatins, the MMP-derived, enzymatic cleavage products of type XVIII collagen. FEBS Lett. 2005, 579, 3601–3606. [Google Scholar] [CrossRef] [PubMed]
- Arai, M.; Yada, T.; Suzuki, S.; Kimata, K. Isolation and characterization of type IX collagen-proteoglycan from the Swarm rat chondrosarcoma. Biochim. Biophys. Acta 1992, 1117, 60–70. [Google Scholar] [CrossRef]
- Oudart, J.B.; Brassart-Pasco, S.; Vautrin, A.; Sellier, C.; Machado, C.; Dupont-Deshorgue, A.; Brassart, B.; Baud, S.; Dauchez, M.; Monboisse, J.C.; et al. Plasmin releases the anti-tumor peptide from the NC1 domain of collagen XIX. Oncotarget 2015, 6, 3656–3668. [Google Scholar] [CrossRef]
- Ramont, L.; Brassart-Pasco, S.; Thevenard, J.; Deshorgue, A.; Venteo, L.; Laronze, J.Y.; Pluot, M.; Monboisse, J.C.; Maquart, F.X. The NC1 domain of type XIX collagen inhibits in vivo melanoma growth. Mol. Cancer Ther. 2007, 6, 506–514. [Google Scholar] [CrossRef]
- Oudart, J.B.; Doué, M.; Vautrin, A.; Brassart, B.; Sellier, C.; Dupont-Deshorgue, A.; Monboisse, J.C.; Maquart, F.X.; Brassart-Pasco, S.; Ramont, L. The anti-tumor NC1 domain of collagen XIX inhibits the FAK/ PI3K/Akt/mTOR signaling pathway through αvβ3 integrin interaction. Oncotarget 2016, 7, 1516–1528. [Google Scholar] [CrossRef]
- Nagase, H.; Fields, G.B. Human matrix metalloproteinase specificity studies using collagen sequence-based synthetic peptides. Biopolymers 1996, 40, 399–416. [Google Scholar] [CrossRef]
- Halper, J.; Kjaer, M. Basic components of connective tissues and extracellular matrix: Elastin, fibrillin, fibulins, fibrinogen, fibronectin, laminin, tenascins and thrombospondins. Adv. Exp. Med. Biol. 2014, 802, 31–47. [Google Scholar] [CrossRef]
- Mithieux, S.M.; Weiss, A.S. Elastin. Adv. Protein Chem. 2005, 70, 437–461. [Google Scholar] [CrossRef]
- Scandolera, A.; Odoul, L.; Salesse, S.; Guillot, A.; Blaise, S.; Kawecki, C.; Maurice, P.; El Btaouri, H.; Romier-Crouzet, B.; Martiny, L.; et al. The Elastin Receptor Complex: A Unique Matricellular Receptor with High Anti-tumoral Potential. Front. Pharmacol. 2016, 7, 32. [Google Scholar] [CrossRef]
- Duca, L.; Floquet, N.; Alix, A.J.; Haye, B.; Debelle, L. Elastin as a matrikine. Crit. Rev. Oncol. Hematol. 2004, 49, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Grahovac, J.; Wells, A. Matrikine and matricellular regulators of EGF receptor signaling on cancer cell migration and invasion. Lab. Investig. 2014, 94, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Cain, S.A.; Mularczyk, E.J.; Singh, M.; Massam-Wu, T.; Kielty, C.M. ADAMTS-10 and -6 differentially regulate cell-cell junctions and focal adhesions. Sci. Rep. 2016, 6, 35956. [Google Scholar] [CrossRef] [PubMed]
- Bax, D.V.; Mahalingam, Y.; Cain, S.; Mellody, K.; Freeman, L.; Younger, K.; Shuttleworth, C.A.; Humphries, M.J.; Couchman, J.R.; Kielty, C.M. Cell adhesion to fibrillin-1: Identification of an Arg-Gly-Asp-dependent synergy region and a heparin-binding site that regulates focal adhesion formation. J. Cell Sci. 2007, 120, 1383–1392. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, J.; Iqbal, S.; Jensen, S.; Mardon, H.; Handford, P. Fibrillin-integrin interactions in health and disease. Biochem. Soc. Trans. 2008, 36, 257–262. [Google Scholar] [CrossRef]
- Han, Z.; Lu, Z.R. Targeting Fibronectin for Cancer Imaging and Therapy. J. Mater. Chem. B 2017, 5, 639–654. [Google Scholar] [CrossRef]
- Kumra, H.; Reinhardt, D.P. Fibronectin-targeted drug delivery in cancer. Adv. Drug Deliv. Rev. 2016, 97, 101–110. [Google Scholar] [CrossRef]
- Mezzenga, R.; Mitsi, M. The Molecular Dance of Fibronectin: Conformational Flexibility Leads to Functional Versatility. Biomacromolecules 2018. [Google Scholar] [CrossRef]
- Zollinger, A.J.; Smith, M.L. Fibronectin, the extracellular glue. Matrix Biol. 2017, 60-61, 27–37. [Google Scholar] [CrossRef]
- Joshi, R.; Goihberg, E.; Ren, W.; Pilichowska, M.; Mathew, P. Proteolytic fragments of fibronectin function as matrikines driving the chemotactic affinity of prostate cancer cells to human bone marrow mesenchymal stromal cells via the alpha5beta1 integrin. Cell Adh. Migr. 2017, 11, 305–315. [Google Scholar] [CrossRef]
- Yi, M.; Ruoslahti, E. A fibronectin fragment inhibits tumor growth, angiogenesis, and metastasis. Proc. Natl. Acad. Sci. USA 2001, 98, 620–624. [Google Scholar] [CrossRef] [PubMed]
- Bossard, C.; Van den Berghe, L.; Laurell, H.; Castano, C.; Cerutti, M.; Prats, A.C.; Prats, H. Antiangiogenic properties of fibstatin, an extracellular FGF-2-binding polypeptide. Cancer Res. 2004, 64, 7507–7512. [Google Scholar] [CrossRef] [PubMed]
- Topalovski, M.; Brekken, R.A. Matrix control of pancreatic cancer: New insights into fibronectin signaling. Cancer Lett. 2016, 381, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Bachman, H.; Nicosia, J.; Dysart, M.; Barker, T.H. Utilizing Fibronectin Integrin-Binding Specificity to Control Cellular Responses. Adv. Wound Care 2015, 4, 501–511. [Google Scholar] [CrossRef]
- Wang, K.; Seo, B.R.; Fischbach, C.; Gourdon, D. Fibronectin Mechanobiology Regulates Tumorigenesis. Cell. Mol. Bioeng. 2016, 9, 1–11. [Google Scholar] [CrossRef]
- Prasad, A.; Clark, R.A. Fibronectin interaction with growth factors in the context of general ways extracellular matrix molecules regulate growth factor signaling. G. Ital. Dermatol. Venereol. 2018, 153, 361–374. [Google Scholar] [CrossRef]
- White, E.S.; Baralle, F.E.; Muro, A.F. New insights into form and function of fibronectin splice variants. J. Pathol. 2008, 216, 1–14. [Google Scholar] [CrossRef]
- Faron, G.; Balepa, L.; Parra, J.; Fils, J.F.; Gucciardo, L. The fetal fibronectin test: 25 years after its development, what is the evidence regarding its clinical utility? A systematic review and meta-analysis. J. Matern. Fetal Neonatal Med. 2018, 33, 1–31. [Google Scholar] [CrossRef]
- Sawicka, K.M.; Seeliger, M.; Musaev, T.; Macri, L.K.; Clark, R.A. Fibronectin Interaction and Enhancement of Growth Factors: Importance for Wound Healing. Adv. Wound Care 2015, 4, 469–478. [Google Scholar] [CrossRef]
- Wang, Y.; Ni, H. Fibronectin maintains the balance between hemostasis and thrombosis. Cell. Mol. Life Sci. 2016, 73, 3265–3277. [Google Scholar] [CrossRef]
- Mercuri, F.A.; Maciewicz, R.A.; Tart, J.; Last, K.; Fosang, A.J. Mutations in the interglobular domain of aggrecan alter matrix metalloproteinase and aggrecanase cleavage patterns. Evidence that matrix metalloproteinase cleavage interferes with aggrecanase activity. J. Biol. Chem. 2000, 275, 33038–33045. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, H.; Leung, T.C.; Hoffman, S.; Balsamo, J.; Lilien, J. Coordinate regulation of cadherin and integrin function by the chondroitin sulfate proteoglycan neurocan. J. Cell Biol. 2000, 149, 1275–1288. [Google Scholar] [CrossRef]
- Mohan, V.; Wyatt, E.V.; Gotthard, I.; Phend, K.D.; Diestel, S.; Duncan, B.W.; Weinberg, R.J.; Tripathy, A.; Maness, P.F. Neurocan Inhibits Semaphorin 3F Induced Dendritic Spine Remodeling Through NrCAM in Cortical Neurons. Front. Cell. Neurosci. 2018, 12, 346. [Google Scholar] [CrossRef] [PubMed]
- Viapiano, M.S.; Hockfield, S.; Matthews, R.T. BEHAB/brevican requires ADAMTS-mediated proteolytic cleavage to promote glioma invasion. J. Neurooncol. 2008, 88, 261–272. [Google Scholar] [CrossRef]
- Demircan, K.; Topcu, V.; Takigawa, T.; Akyol, S.; Yonezawa, T.; Ozturk, G.; Ugurcu, V.; Hasgul, R.; Yigitoglu, M.R.; Akyol, O.; et al. ADAMTS4 and ADAMTS5 knockout mice are protected from versican but not aggrecan or brevican proteolysis during spinal cord injury. Biomed. Res. Int. 2014, 2014, 693746. [Google Scholar] [CrossRef] [PubMed]
- Hope, C.; Emmerich, P.B.; Papadas, A.; Pagenkopf, A.; Matkowskyj, K.A.; Van De Hey, D.R.; Payne, S.N.; Clipson, L.; Callander, N.S.; Hematti, P.; et al. Versican-Derived Matrikines Regulate Batf3-Dendritic Cell Differentiation and Promote T Cell Infiltration in Colorectal Cancer. J. Immunol. 2017, 199, 1933–1941. [Google Scholar] [CrossRef] [PubMed]
- Papadas, A.; Asimakopoulos, F. Versican in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1272, 55–72. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, L.; Zheng, P.S.; Yang, B.B. beta 1-Integrin-mediated glioma cell adhesion and free radical-induced apoptosis are regulated by binding to a C-terminal domain of PG-M/versican. J. Biol. Chem. 2002, 277, 12294–12301. [Google Scholar] [CrossRef]
- Overall, C.M. Molecular determinants of metalloproteinase substrate specificity: Matrix metalloproteinase substrate binding domains, modules, and exosites. Mol. Biotechnol. 2002, 22, 51–86. [Google Scholar] [CrossRef]
- Iozzo, R.V.; Moscatello, D.K.; McQuillan, D.J.; Eichstetter, I. Decorin is a biological ligand for the epidermal growth factor receptor. J. Biol. Chem. 1999, 274, 4489–4492. [Google Scholar] [CrossRef]
- Moreth, K.; Iozzo, R.V.; Schaefer, L. Small leucine-rich proteoglycans orchestrate receptor crosstalk during inflammation. Cell Cycle 2012, 11, 2084–2091. [Google Scholar] [CrossRef] [PubMed]
- Goldoni, S.; Humphries, A.; Nystrom, A.; Sattar, S.; Owens, R.T.; McQuillan, D.J.; Ireton, K.; Iozzo, R.V. Decorin is a novel antagonistic ligand of the Met receptor. J. Cell Biol. 2009, 185, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Khan, G.A.; Girish, G.V.; Lala, N.; Di Guglielmo, G.M.; Lala, P.K. Decorin is a novel VEGFR-2-binding antagonist for the human extravillous trophoblast. Mol. Endocrinol. 2011, 25, 1431–1443. [Google Scholar] [CrossRef] [PubMed]
- Hausser, H.; Wedekind, P.; Sperber, T.; Peters, R.; Hasilik, A.; Kresse, H. Isolation and cellular localization of the decorin endocytosis receptor. Eur. J. Cell Biol. 1996, 71, 325–331. [Google Scholar]
- Nastase, M.V.; Young, M.F.; Schaefer, L. Biglycan: A multivalent proteoglycan providing structure and signals. J. Histochem. Cytochem. 2012, 60, 963–975. [Google Scholar] [CrossRef]
- Grindel, B.; Li, Q.; Arnold, R.; Petros, J.; Zayzafoon, M.; Muldoon, M.; Stave, J.; Chung, L.W.; Farach-Carson, M.C. Perlecan/HSPG2 and matrilysin/MMP-7 as indices of tissue invasion: Tissue localization and circulating perlecan fragments in a cohort of 288 radical prostatectomy patients. Oncotarget 2016, 7, 10433–10447. [Google Scholar] [CrossRef]
- Cailhier, J.F.; Sirois, I.; Laplante, P.; Lepage, S.; Raymond, M.A.; Brassard, N.; Prat, A.; Iozzo, R.V.; Pshezhetsky, A.V.; Hébert, M.J. Caspase-3 activation triggers extracellular cathepsin L release and endorepellin proteolysis. J. Biol. Chem. 2008, 283, 27220–27229. [Google Scholar] [CrossRef]
- Gubbiotti, M.A.; Neill, T.; Iozzo, R.V. A current view of perlecan in physiology and pathology: A mosaic of functions. Matrix Biol. 2017, 57-58, 285–298. [Google Scholar] [CrossRef]
- Goyal, A.; Pal, N.; Concannon, M.; Paul, M.; Doran, M.; Poluzzi, C.; Sekiguchi, K.; Whitelock, J.M.; Neill, T.; Iozzo, R.V. Endorepellin, the angiostatic module of perlecan, interacts with both the α2β1 integrin and vascular endothelial growth factor receptor 2 (VEGFR2): A dual receptor antagonism. J. Biol. Chem. 2011, 286, 25947–25962. [Google Scholar] [CrossRef]
- Gonzalez, E.M.; Reed, C.C.; Bix, G.; Fu, J.; Zhang, Y.; Gopalakrishnan, B.; Greenspan, D.S.; Iozzo, R.V. BMP-1/Tolloid-like metalloproteases process endorepellin, the angiostatic C-terminal fragment of perlecan. J. Biol. Chem. 2005, 280, 7080–7087. [Google Scholar] [CrossRef]
- Titz, B.; Dietrich, S.; Sadowski, T.; Beck, C.; Petersen, A.; Sedlacek, R. Activity of MMP-19 inhibits capillary-like formation due to processing of nidogen-1. Cell. Mol. Life Sci. 2004, 61, 1826–1833. [Google Scholar] [CrossRef] [PubMed]
- Sage, J.; Leblanc-Noblesse, E.; Nizard, C.; Sasaki, T.; Schnebert, S.; Perrier, E.; Kurfurst, R.; Brömme, D.; Lalmanach, G.; Lecaille, F. Cleavage of nidogen-1 by cathepsin S impairs its binding to basement membrane partners. PLoS ONE 2012, 7, e43494. [Google Scholar] [CrossRef] [PubMed]
- Walker, P.D.; Kaushal, G.P.; Shah, S.V. Meprin A, the major matrix degrading enzyme in renal tubules, produces a novel nidogen fragment in vitro and in vivo. Kidney Int. 1998, 53, 1673–1680. [Google Scholar] [CrossRef]
- Beck, K.; Brodsky, B. Supercoiled protein motifs: The collagen triple-helix and the alpha-helical coiled coil. J. Struct. Biol. 1998, 122, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Kikkawa, Y.; Hozumi, K.; Katagiri, F.; Nomizu, M.; Kleinman, H.K.; Koblinski, J.E. Laminin-111-derived peptides and cancer. Cell Adh. Migr. 2013, 7, 150–256. [Google Scholar] [CrossRef] [PubMed]
- Adair-Kirk, T.L.; Atkinson, J.J.; Broekelmann, T.J.; Doi, M.; Tryggvason, K.; Miner, J.H.; Mecham, R.P.; Senior, R.M. A site on laminin alpha 5, AQARSAASKVKVSMKF, induces inflammatory cell production of matrix metalloproteinase-9 and chemotaxis. J. Immunol. 2003, 171, 398–406. [Google Scholar] [CrossRef]
- Woods, A.; Longley, R.L.; Tumova, S.; Couchman, J.R. Syndecan-4 binding to the high affinity heparin-binding domain of fibronectin drives focal adhesion formation in fibroblasts. Arch. Biochem. Biophys. 2000, 374, 66–72. [Google Scholar] [CrossRef]
- Carulli, S.; Beck, K.; Dayan, G.; Boulesteix, S.; Lortat-Jacob, H.; Rousselle, P. Cell surface proteoglycans syndecan-1 and -4 bind overlapping but distinct sites in laminin alpha3 LG45 protein domain. J. Biol. Chem. 2012, 287, 12204–12216. [Google Scholar] [CrossRef]
- Bachy, S.; Letourneur, F.; Rousselle, P. Syndecan-1 interaction with the LG4/5 domain in laminin-332 is essential for keratinocyte migration. J. Cell. Physiol. 2008, 214, 238–249. [Google Scholar] [CrossRef]
- Ramovs, V.; Te Molder, L.; Sonnenberg, A. The opposing roles of laminin-binding integrins in cancer. Matrix Biol. 2017, 57-58, 213–243. [Google Scholar] [CrossRef]
- Rousselle, P.; Beck, K. Laminin 332 processing impacts cellular behavior. Cell Adh. Migr. 2013, 7, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Tsuruta, D.; Kobayashi, H.; Imanishi, H.; Sugawara, K.; Ishii, M.; Jones, J.C. Laminin-332-integrin interaction: A target for cancer therapy? Curr. Med. Chem. 2008, 15, 1968–1975. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Sekiguchi, K. Molecular Basis of Laminin-Integrin Interactions. Curr. Top. Membr. 2015, 76, 197–229. [Google Scholar] [CrossRef]
- Eble, J.A.; Wucherpfennig, K.W.; Gauthier, L.; Dersch, P.; Krukonis, E.; Isberg, R.R.; Hemler, M.E. Recombinant soluble human alpha 3 beta 1 integrin: Purification, processing, regulation, and specific binding to laminin-5 and invasin in a mutually exclusive manner. Biochemistry 1998, 37, 10945–10955. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.G.; An, H.J.; Kang, S.; Choi, Y.P.; Gao, M.Q.; Park, H.; Cho, N.H. Laminin-332-rich tumor microenvironment for tumor invasion in the interface zone of breast cancer. Am. J. Pathol. 2011, 178, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Marinkovich, M.P. Tumour microenvironment: Laminin 332 in squamous-cell carcinoma. Nat. Rev. Cancer. 2007, 7, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, W.; Wei, J.; Zhou, D.; Zhao, X.; Song, W.; Sun, Q.; Huang, P.; Zheng, S. Overexpression of beta3 Chains of Laminin-332 is Associated With Clinicopathologic Features and Decreased Survival in Patients With Pancreatic Adenocarcinoma. Appl. Immunohistochem. Mol. Morphol. 2015, 23, 516–521. [Google Scholar] [CrossRef]
- Guess, C.M.; Lafleur, B.J.; Weidow, B.L.; Quaranta, V. A decreased ratio of laminin-332 beta3 to gamma2 subunit mRNA is associated with poor prognosis in colon cancer. Cancer Epidemiol. Biomark. Prev. 2009, 18, 1584–1590. [Google Scholar] [CrossRef]
- Katayama, M.; Funakoshi, A.; Sumii, T.; Sanzen, N.; Sekiguchi, K. Laminin gamma2-chain fragment circulating level increases in patients with metastatic pancreatic ductal cell adenocarcinomas. Cancer Lett. 2005, 225, 167–176. [Google Scholar] [CrossRef]
- Hibino, S.; Shibuya, M.; Engbring, J.A.; Mochizuki, M.; Nomizu, M.; Kleinman, H.K. Identification of an active site on the laminin alpha5 chain globular domain that binds to CD44 and inhibits malignancy. Cancer Res. 2004, 64, 4810–4816. [Google Scholar] [CrossRef]
- Kusuma, N.; Anderson, R.L.; Pouliot, N. Laminin α5-derived peptides modulate the properties of metastatic breast tumour cells. Clin. Exp. Metastasis 2011, 28, 909–921. [Google Scholar] [CrossRef] [PubMed]
- Su, J.L.; Chiou, J.; Tang, C.H.; Zhao, M.; Tsai, C.H.; Chen, P.S.; Chang, Y.W.; Chien, M.H.; Peng, C.Y.; Hsiao, M.; et al. CYR61 regulates BMP-2-dependent osteoblast differentiation through the {alpha}v{beta}3 integrin/integrin-linked kinase/ERK pathway. J. Biol. Chem. 2010, 285, 31325–31336. [Google Scholar] [CrossRef] [PubMed]
- Crockett, J.C.; Schütze, N.; Tosh, D.; Jatzke, S.; Duthie, A.; Jakob, F.; Rogers, M.J. The matricellular protein CYR61 inhibits osteoclastogenesis by a mechanism independent of alphavbeta3 and alphavbeta5. Endocrinology 2007, 148, 5761–5768. [Google Scholar] [CrossRef]
- Chen, C.C.; Young, J.L.; Monzon, R.I.; Chen, N.; Todorovic, V.; Lau, L.F. Cytotoxicity of TNFalpha is regulated by integrin-mediated matrix signaling. EMBO J. 2007, 26, 1257–1267. [Google Scholar] [CrossRef] [PubMed]
- Kaasboll, O.J.; Gadicherla, A.K.; Wang, J.H.; Monsen, V.T.; Hagelin, E.M.V.; Dong, M.Q.; Attramadal, H. Connective tissue growth factor (CCN2) is a matricellular preproprotein controlled by proteolytic activation. J. Biol. Chem. 2018, 293, 17953–17970. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.C.; Chang, A.C.; Tsai, C.H.; Huang, Y.L.; Gan, L.; Chen, C.K.; Liu, S.C.; Huang, T.Y.; Fong, Y.C.; Tang, C.H. CCN2 promotes drug resistance in osteosarcoma by enhancing ABCG2 expression. J. Cell. Physiol. 2019, 234, 9297–9307. [Google Scholar] [CrossRef]
- Babic, A.M.; Chen, C.C.; Lau, L.F. Fisp12/mouse connective tissue growth factor mediates endothelial cell adhesion and migration through integrin alphavbeta3, promotes endothelial cell survival, and induces angiogenesis in vivo. Mol. Cell. Biol. 1999, 19, 2958–2966. [Google Scholar] [CrossRef]
- Hutchenreuther, J.; Vincent, K.; Norley, C.; Racanelli, M.; Gruber, S.B.; Johnson, T.M.; Fullen, D.R.; Raskin, L.; Perbal, B.; Holdsworth, D.W.; et al. Activation of cancer-associated fibroblasts is required for tumor neovascularization in a murine model of melanoma. Matrix Biol. 2018. [Google Scholar] [CrossRef]
- Qiao, L.; Liang, N.; Zhang, J.; Xie, J.; Liu, F.; Xu, D.; Yu, X.; Tian, Y. Advanced research on vasculogenic mimicry in cancer. J. Cell. Mol. Med. 2015, 19, 315–326. [Google Scholar] [CrossRef]
- Seftor, R.E.; Seftor, E.A.; Kirschmann, D.A.; Hendrix, M.J. Targeting the tumor microenvironment with chemically modified tetracyclines: Inhibition of laminin 5 gamma2 chain promigratory fragments and vasculogenic mimicry. Mol. Cancer. Ther. 2002, 1, 1173–1179. [Google Scholar]
- Velez, D.O.; Tsui, B.; Goshia, T.; Chute, C.L.; Han, A.; Carter, H.; Fraley, S.I. 3D collagen architecture induces a conserved migratory and transcriptional response linked to vasculogenic mimicry. Nat. Commun. 2017, 8, 1651. [Google Scholar] [CrossRef]
- Adams, J.C.; Chiquet-Ehrismann, R.; Tucker, R.P. The evolution of tenascins and fibronectin. Cell Adh. Migr. 2015, 9, 22–33. [Google Scholar] [CrossRef]
- Chiquet-Ehrismann, R.; Hagios, C.; Matsumoto, K. The tenascin gene family. Perspect. Dev. Neurobiol. 1994, 2, 3–7. [Google Scholar]
- Yoshida, T.; Akatsuka, T.; Imanaka-Yoshida, K. Tenascin-C and integrins in cancer. Cell Adh. Migr. 2015, 9, 96–104. [Google Scholar] [CrossRef]
- Martina, E.; Chiquet-Ehrismann, R.; Brellier, F. Tenascin-W: An extracellular matrix protein associated with osteogenesis and cancer. Int. J. Biochem. Cell. Biol. 2010, 42, 1412–1415. [Google Scholar] [CrossRef]
- Brellier, F.; Martina, E.; Degen, M.; Heuze-Vourc’h, N.; Petit, A.; Kryza, T.; Courty, Y.; Terracciano, L.; Ruiz, C.; Chiquet-Ehrismann, R. Tenascin-W is a better cancer biomarker than tenascin-C for most human solid tumors. BMC Clin. Pathol. 2012, 12, 14. [Google Scholar] [CrossRef]
- Martina, E.; Degen, M.; Ruegg, C.; Merlo, A.; Lino, M.M.; Chiquet-Ehrismann, R.; Brellier, F. Tenascin-W is a specific marker of glioma-associated blood vessels and stimulates angiogenesis in vitro. FASEB J. 2010, 24, 778–787. [Google Scholar] [CrossRef]
- Castello, L.M.; Raineri, D.; Salmi, L.; Clemente, N.; Vaschetto, R.; Quaglia, M.; Garzaro, M.; Gentilli, S.; Navalesi, P.; Cantaluppi, V.; et al. Osteopontin at the Crossroads of Inflammation and Tumor Progression. Mediators Inflamm. 2017, 2017, 4049098. [Google Scholar] [CrossRef]
- Kruger, T.E.; Miller, A.H.; Godwin, A.K.; Wang, J. Bone sialoprotein and osteopontin in bone metastasis of osteotropic cancers. Crit. Rev. Oncol. Hematol. 2014, 89, 330–341. [Google Scholar] [CrossRef]
- Gao, Y.A.; Agnihotri, R.; Vary, C.P.; Liaw, L. Expression and characterization of recombinant osteopontin peptides representing matrix metalloproteinase proteolytic fragments. Matrix Biol. 2004, 23, 457–466. [Google Scholar] [CrossRef]
- Ye, Q.H.; Qin, L.X.; Forgues, M.; He, P.; Kim, J.W.; Peng, A.C.; Simon, R.; Li, Y.; Robles, A.I.; Chen, Y.; et al. Predicting hepatitis B virus-positive metastatic hepatocellular carcinomas using gene expression profiling and supervised machine learning. Nat. Med. 2003, 9, 416–423. [Google Scholar] [CrossRef]
- Gonzalez-Gonzalez, L.; Alonso, J. Periostin: A Matricellular Protein With Multiple Functions in Cancer Development and Progression. Front. Oncol. 2018, 8, 225. [Google Scholar] [CrossRef]
- Gillan, L.; Matei, D.; Fishman, D.A.; Gerbin, C.S.; Karlan, B.Y.; Chang, D.D. Periostin secreted by epithelial ovarian carcinoma is a ligand for alpha(V)beta(3) and alpha(V)beta(5) integrins and promotes cell motility. Cancer Res. 2002, 62, 5358–5364. [Google Scholar]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Micke, P.; Ostman, A. Tumour-stroma interaction: Cancer-associated fibroblasts as novel targets in anti-cancer therapy? Lung Cancer 2004, 45 (Suppl. S2), S163–S175. [Google Scholar] [CrossRef]
- Leprini, A.; Querze, G.; Zardi, L. Tenascin isoforms: Possible targets for diagnosis and therapy of cancer and mechanisms regulating their expression. Perspect. Dev. Neurobiol. 1994, 2, 117–123. [Google Scholar]
- Nicolo, G.; Salvi, S.; Oliveri, G.; Borsi, L.; Castellani, P.; Zardi, L. Expression of tenascin and of the ED-B containing oncofetal fibronectin isoform in human cancer. Cell. Differ. Dev. 1990, 32, 401–408. [Google Scholar] [CrossRef]
- Gritsenko, P.G.; Ilina, O.; Friedl, P. Interstitial guidance of cancer invasion. J. Pathol. 2012, 226, 185–199. [Google Scholar] [CrossRef]
- Kakizaki, Y.; Makino, N.; Tozawa, T.; Honda, T.; Matsuda, A.; Ikeda, Y.; Ito, M.; Saito, Y.; Kimura, W.; Ueno, Y. Stromal Fibrosis and Expression of Matricellular Proteins Correlate With Histological Grade of Intraductal Papillary Mucinous Neoplasm of the Pancreas. Pancreas 2016, 45, 1145–1152. [Google Scholar] [CrossRef]
- Viloria, K.; Hill, N.J. Embracing the complexity of matricellular proteins: The functional and clinical significance of splice variation. Biomol. Concepts 2016, 7, 117–132. [Google Scholar] [CrossRef]
- Naschberger, E.; Liebl, A.; Schellerer, V.S.; Schütz, M.; Britzen-Laurent, N.; Kölbel, P.; Schaal, U.; Haep, L.; Regensburger, D.; Wittmann, T.; et al. Matricellular protein SPARCL1 regulates tumor microenvironment-dependent endothelial cell heterogeneity in colorectal carcinoma. J. Clin. Investig. 2016, 126, 4187–4204. [Google Scholar] [CrossRef] [PubMed]
- Kamili, N.A.; Arthur, C.M.; Gerner-Smidt, C.; Tafesse, E.; Blenda, A.; Dias-Baruffi, M.; Stowell, S.R. Key regulators of galectin-glycan interactions. Proteomics 2016, 16, 3111–3125. [Google Scholar] [CrossRef] [PubMed]
- Thijssen, V.L.; Rabinovich, G.A.; Griffioen, A.W. Vascular galectins: Regulators of tumor progression and targets for cancer therapy. Cytokine Growth Factor Rev. 2013, 24, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Huergo, S.P.; Blidner, A.G.; Rabinovich, G.A. Galectins: Emerging regulatory checkpoints linking tumor immunity and angiogenesis. Curr. Opin. Immunol. 2017, 45, 8–15. [Google Scholar] [CrossRef]
- Bellahcène, A.; Castronovo, V.; Ogbureke, K.U.; Fisher, L.W.; Fedarko, N.S. Small integrin-binding ligand N-linked glycoproteins (SIBLINGs): Multifunctional proteins in cancer. Nat. Rev. Cancer. 2008, 8, 212–226. [Google Scholar] [CrossRef]
- Barbouri, D.; Afratis, N.; Gialeli, C.; Vynios, D.H.; Theocharis, A.D.; Karamanos, N.K. Syndecans as modulators and potential pharmacological targets in cancer progression. Front. Oncol. 2014, 4, 4. [Google Scholar] [CrossRef]
- Rapraeger, A.C. Synstatin: A selective inhibitor of the syndecan-1-coupled IGF1R-αvβ3 integrin complex in tumorigenesis and angiogenesis. FEBS J. 2013, 280, 2207–2215. [Google Scholar] [CrossRef]
- Rapraeger, A.C.; Ell, B.J.; Roy, M.; Li, X.; Morrison, O.R.; Thomas, G.M.; Beauvais, D.M. Vascular endothelial-cadherin stimulates syndecan-1-coupled insulin-like growth factor-1 receptor and cross-talk between αVβ3 integrin and vascular endothelial growth factor receptor 2 at the onset of endothelial cell dissemination during angiogenesis. FEBS J. 2013, 280, 2194–2206. [Google Scholar] [CrossRef]
- Wang, H.; Jin, H.; Rapraeger, A.C. Syndecan-1 and Syndecan-4 Capture Epidermal Growth Factor Receptor Family Members and the α3β1 Integrin Via Binding Sites in Their Ectodomains: Novel synstatins prevent kinase capture and inhibit α6β4-integrin-dependent epithelial cell motility. J. Biol. Chem. 2015, 290, 26103–26113. [Google Scholar] [CrossRef]
- Beauvais, D.M.; Ell, B.J.; McWhorter, A.R.; Rapraeger, A.C. Syndecan-1 regulates alphavbeta3 and alphavbeta5 integrin activation during angiogenesis and is blocked by synstatin, a novel peptide inhibitor. J. Exp. Med. 2009, 206, 691–705. [Google Scholar] [CrossRef]
- Jung, O.; Trapp-Stamborski, V.; Purushothaman, A.; Jin, H.; Wang, H.; Sanderson, R.D.; Rapraeger, A.C. Heparanase-induced shedding of syndecan-1/CD138 in myeloma and endothelial cells activates VEGFR2 and an invasive phenotype: Prevention by novel synstatins. Oncogenesis 2016, 5, e202. [Google Scholar] [CrossRef] [PubMed]
- Stephan, A.; Mateos, J.M.; Kozlov, S.V.; Cinelli, P.; Kistler, A.D.; Hettwer, S.; Rülicke, T.; Streit, P.; Kunz, B.; Sonderegger, P. Neurotrypsin cleaves agrin locally at the synapse. FASEB J. 2008, 22, 1861–1873. [Google Scholar] [CrossRef] [PubMed]
- Monzon, M.E.; Fregien, N.; Schmid, N.; Falcon, N.S.; Campos, M.; Casalino-Matsuda, S.M.; Forteza, R.M. Reactive oxygen species and hyaluronidase 2 regulate airway epithelial hyaluronan fragmentation. J. Biol. Chem. 2010, 285, 26126–26134. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Nie, W.; Xie, Q.; Chen, G.; Li, X.; Jia, Y.; Yin, B.; Qu, X.; Li, Y.; Liang, J. Endostatin reverses immunosuppression of the tumor microenvironment in lung carcinoma. Oncol. Lett. 2018, 15, 1874–1880. [Google Scholar] [CrossRef]
- Da Silva, J.; Lameiras, P.; Beljebbar, A.; Berquand, A.; Villemin, M.; Ramont, L.; Dukic, S.; Nuzillard, J.M.; Molinari, M.; Gautier, M.; et al. Structural characterization and in vivo pro-tumor properties of a highly conserved matrikine. Oncotarget 2018, 9, 17839–17857. [Google Scholar] [CrossRef]
- Tzanakakis, G.; Kavasi, R.M.; Voudouri, K.; Berdiaki, A.; Spyridaki, I.; Tsatsakis, A.; Nikitovic, D. Role of the extracellular matrix in cancer-associated epithelial to mesenchymal transition phenomenon. Dev. Dyn. 2018, 247, 368–381. [Google Scholar] [CrossRef]
- Covic, L.; Kuliopulos, A. Protease-Activated Receptor 1 as Therapeutic Target in Breast, Lung, and Ovarian Cancer: Pepducin Approach. Int. J. Mol. Sci. 2018, 19, 2237. [Google Scholar] [CrossRef]
- Liu, X.; Yu, J.; Song, S.; Yue, X.; Li, Q. Protease-activated receptor-1 (PAR-1): A promising molecular target for cancer. Oncotarget 2017, 8, 107334–107345. [Google Scholar] [CrossRef]
- Arakaki, A.K.S.; Pan, W.A.; Trejo, J. GPCRs in Cancer: Protease-Activated Receptors, Endocytic Adaptors and Signaling. Int. J. Mol. Sci. 2018, 19, 1886. [Google Scholar] [CrossRef]
- Zara, M.; Canobbio, I.; Visconte, C.; Canino, J.; Torti, M.; Guidetti, G.F. Molecular mechanisms of platelet activation and aggregation induced by breast cancer cells. Cell Signal. 2018, 48, 45–53. [Google Scholar] [CrossRef]
- Mendonsa, A.M.; Na, T.Y.; Gumbiner, B.M. E-cadherin in contact inhibition and cancer. Oncogene 2018, 37, 4769–4780. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Ramamonjisoa, N.; Ackerstaff, E. Characterization of the Tumor Microenvironment and Tumor-Stroma Interaction by Non-invasive Preclinical Imaging. Front Oncol. 2017, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Coulson-Thomas, V.J.; Coulson-Thomas, Y.M.; Gesteira, T.F.; Andrade de Paula, C.A.; Carneiro, C.R.; Ortiz, V.; Toma, L.; Kao, W.W.; Nader, H.B. Lumican expression, localization and antitumor activity in prostate cancer. Exp. Cell Res. 2013, 319, 967–981. [Google Scholar] [CrossRef] [PubMed]
- Karamanou, K.; Franchi, M.; Vynios, D.; Brezillon, S. Epithelial-to-mesenchymal transition and invadopodia markers in breast cancer: Lumican a key regulator. Semin. Cancer. Biol. 2020, 62, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Ronca, R.; Sozzani, S.; Presta, M.; Alessi, P. Delivering cytokines at tumor site: The immunocytokine-conjugated anti-EDB-fibronectin antibody case. Immunobiology 2009, 214, 800–810. [Google Scholar] [CrossRef] [PubMed]
- Hirata, E.; Sahai, E. Tumor Microenvironment and Differential Responses to Therapy. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef]
- Arosio, D.; Casagrande, C. Advancement in integrin facilitated drug delivery. Adv. Drug Deliv. Rev. 2016, 97, 111–143. [Google Scholar] [CrossRef]
- Rodríguez-Cabello, J.C.; Arias, F.J.; Rodrigo, M.A.; Girotti, A. Elastin-like polypeptides in drug delivery. Adv. Drug Deliv. Rev. 2016, 97, 85–100. [Google Scholar] [CrossRef]
- Alexander, S.; Friedl, P. Cancer invasion and resistance: Interconnected processes of disease progression and therapy failure. Trends Mol. Med. 2012, 18, 13–26. [Google Scholar] [CrossRef]
- An, B.; Lin, Y.S.; Brodsky, B. Collagen interactions: Drug design and delivery. Adv. Drug Deliv. Rev. 2016, 97, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, A.J.; Kyriakides, T.R. Matricellular proteins in drug delivery: Therapeutic targets, active agents, and therapeutic localization. Adv. Drug Deliv. Rev. 2016, 97, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Jiang, D.; Noble, P.W. Hyaluronan as a therapeutic target in human diseases. Adv. Drug Deliv. Rev. 2016, 97, 186–203. [Google Scholar] [CrossRef] [PubMed]
- Dosio, F.; Arpicco, S.; Stella, B.; Fattal, E. Hyaluronic acid for anticancer drug and nucleic acid delivery. Adv. Drug Deliv. Rev. 2016, 97, 204–236. [Google Scholar] [CrossRef]
- Piperigkou, Z.; Manou, D.; Karamanou, K.; Theocharis, A.D. Strategies to Target Matrix Metalloproteinases as Therapeutic Approach in Cancer. Methods Mol. Biol. 2018, 1731, 325–348. [Google Scholar] [CrossRef]
- Au, J.L.; Yeung, B.Z.; Wientjes, M.G.; Lu, Z.; Wientjes, M.G. Delivery of cancer therapeutics to extracellular and intracellular targets: Determinants, barriers, challenges and opportunities. Adv. Drug Deliv. Rev. 2016, 97, 280–301. [Google Scholar] [CrossRef]
- Barker, H.E.; Cox, T.R.; Erler, J.T. The rationale for targeting the LOX family in cancer. Nat. Rev. Cancer. 2012, 12, 540–552. [Google Scholar] [CrossRef]
- Chan, N.; Willis, A.; Kornhauser, N.; Ward, M.M.; Lee, S.B.; Nackos, E.; Seo, B.R.; Chuang, E.; Cigler, T.; Moore, A.; et al. Influencing the Tumor Microenvironment: A Phase II Study of Copper Depletion Using Tetrathiomolybdate in Patients with Breast Cancer at High Risk for Recurrence and in Preclinical Models of Lung Metastases. Clin. Cancer Res. 2017, 23, 666–676. [Google Scholar] [CrossRef]
- Chan, N.; Willis, A.; Kornhauser, N.; Ward, M.M.; Lee, S.B.; Nackos, E.; Seo, B.R.; Chuang, E.; Cigler, T.; Moore, A.; et al. Correction: Influencing the Tumor Microenvironment: A Phase II Study of Copper Depletion Using Tetrathiomolybdate in Patients with Breast Cancer at High Risk for Recurrence and in Preclinical Models of Lung Metastases. Clin. Cancer Res. 2020, 26, 5051. [Google Scholar] [CrossRef]
- Hecht, J.R.; Benson, A.B., III; Vyushkov, D.; Yang, Y.; Bendell, J.; Verma, U. A Phase II, Randomized, Double-Blind, Placebo-Controlled Study of Simtuzumab in Combination with FOLFIRI for the Second-Line Treatment of Metastatic KRAS Mutant Colorectal Adenocarcinoma. Oncologist 2017, 22, 243. [Google Scholar] [CrossRef]
- Benson, A.B., III; Wainberg, Z.A.; Hecht, J.R.; Vyushkov, D.; Dong, H.; Bendell, J.; Kudrik, F. A Phase II Randomized, Double-Blind, Placebo-Controlled Study of Simtuzumab or Placebo in Combination with Gemcitabine for the First-Line Treatment of Pancreatic Adenocarcinoma. Oncologist 2017, 22, 241. [Google Scholar] [CrossRef] [PubMed]
- Barry-Hamilton, V.; Spangler, R.; Marshall, D.; McCauley, S.; Rodriguez, H.M.; Oyasu, M.; Mikels, A.; Vaysberg, M.; Ghermazien, H.; Wai, C.; et al. Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nat. Med. 2010, 16, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, H.M.; Vaysberg, M.; Mikels, A.; McCauley, S.; Velayo, A.C.; Garcia, C.; Smith, V. Modulation of lysyl oxidase-like 2 enzymatic activity by an allosteric antibody inhibitor. J. Biol. Chem. 2010, 285, 20964–20974. [Google Scholar] [CrossRef] [PubMed]
- Fields, G.B. The Rebirth of Matrix Metalloproteinase Inhibitors: Moving Beyond the Dogma. Cells 2019, 8, 984. [Google Scholar] [CrossRef] [PubMed]
- Levin, M.; Udi, Y.; Solomonov, I.; Sagi, I. Next generation matrix metalloproteinase inhibitors—Novel strategies bring new prospects. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1927–1939. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, H.S.; McCann, P.P. Matrix metalloproteinase inhibition as a novel anticancer strategy: A review with special focus on batimastat and marimastat. Pharmacol. Ther. 1997, 75, 69–75. [Google Scholar] [CrossRef]
- Cathcart, J.; Pulkoski-Gross, A.; Cao, J. Targeting Matrix Metalloproteinases in Cancer: Bringing New Life to Old Ideas. Genes Dis. 2015, 2, 26–34. [Google Scholar] [CrossRef]
- Rao, B.G. Recent developments in the design of specific Matrix Metalloproteinase inhibitors aided by structural and computational studies. Curr. Pharm. Des. 2005, 11, 295–322. [Google Scholar] [CrossRef]
- Chang, T.T.; Thakar, D.; Weaver, V.M. Force-dependent breaching of the basement membrane. Matrix Biol. 2017, 57-58, 178–189. [Google Scholar] [CrossRef]
- Vandooren, J.; Opdenakker, G.; Loadman, P.M.; Edwards, D.R. Proteases in cancer drug delivery. Adv. Drug Deliv. Rev. 2016, 97, 144–155. [Google Scholar] [CrossRef]
- Zhan, Y.; Ling, S.; Huang, H.; Zhang, Y.; Chen, G.; Huang, S.; Li, C.; Guo, W.; Wang, Q. Rapid and Nondestructive Histopathological Analysis for Intraoperative Cancer Diagnosis Using an Enzyme-Activated NIR-II Nanoprobe. Angew. Chem. Int. Ed. Engl. 2020, 59, 1–7. [Google Scholar] [CrossRef]
- Kasurinen, A.; Tervahartiala, T.; Laitinen, A.; Kokkola, A.; Sorsa, T.; Böckelman, C.; Haglund, C. High serum MMP-14 predicts worse survival in gastric cancer. PLoS ONE 2018, 13, e0208800. [Google Scholar] [CrossRef] [PubMed]
- Ingvarsen, S.; Porse, A.; Erpicum, C.; Maertens, L.; Jürgensen, H.J.; Madsen, D.H.; Melander, M.C.; Gardsvoll, H.; Hoyer-Hansen, G.; Noel, A.; et al. Targeting a single function of the multifunctional matrix metalloprotease MT1-MMP Impact on Lymphangiogenesis. J. Biol. Chem. 2013, 288, 10195–10204. [Google Scholar] [CrossRef] [PubMed]
- Paemen, L.; Martens, E.; Masure, S.; Opdenakker, G. Monoclonal antibodies specific for natural human neutrophil gelatinase B used for affinity purification, quantitation by two-site ELISA and inhibition of enzymatic activity. Eur. J. Biochem. 1995, 234, 759–765. [Google Scholar] [CrossRef]
- Martens, E.; Leyssen, A.; Van Aelst, I.; Fiten, P.; Piccard, H.; Hu, J.; Descamps, F.J.; Van den Steen, P.E.; Proost, P.; Van Damme, J.; et al. A monoclonal antibody inhibits gelatinase B/MMP-9 by selective binding to part of the catalytic domain and not to the fibronectin or zinc binding domains. Biochim. Biophys. Acta 2007, 1770, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Devy, L.; Huang, L.; Naa, L.; Yanamandra, N.; Pieters, H.; Frans, N.; Chang, E.; Tao, Q.; Vanhove, M.; Lejeune, A.; et al. Selective inhibition of matrix metalloproteinase-14 blocks tumor growth, invasion, and angiogenesis. Cancer Res. 2009, 69, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Nam, D.H.; Ge, X. Generation of Highly Selective MMP Antibody Inhibitors. Methods Mol. Biol. 2018, 1731, 307–324. [Google Scholar] [CrossRef]
- Li, X.; Ma, Z.; Wang, H.; Ren, L.; Zhang, D.; Liang, W.; Zhang, G.; Zhang, J.; Yu, D.; Fang, X. Screening, Identification, and Characterization of an Affinity Peptide Specific to MT1-MMP and Its Application in Tumor Imaging. Bioconjug. Chem. 2019, 30, 1507–1517. [Google Scholar] [CrossRef]
- Shirian, J.; Arkadash, V.; Cohen, I.; Sapir, T.; Radisky, E.S.; Papo, N.; Shifman, J.M. Converting a broad matrix metalloproteinase family inhibitor into a specific inhibitor of MMP-9 and MMP-14. FEBS Lett. 2018, 592, 1122–1134. [Google Scholar] [CrossRef]
- Wen, X.Q.; Qian, X.L.; Sun, H.K.; Zheng, L.L.; Zhu, W.Q.; Li, T.Y.; Hu, J.P. MicroRNAs: Multifaceted Regulators of Colorectal Cancer Metastasis and Clinical Applications. Onco Targets Ther. 2020, 13, 10851–10866. [Google Scholar] [CrossRef]
- D’Angelo, E.; Agostini, M. Long non-coding RNA and extracellular matrix: The hidden players in cancer-stroma cross-talk. Noncoding RNA Res. 2018, 3, 174–177. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, H. Role of microRNA-mediated MMP regulation in the treatment and diagnosis of malignant tumors. Cancer Biol. Ther. 2013, 14, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kuscu, C.; Banach, A.; Zhang, Q.; Pulkoski-Gross, A.; Kim, D.; Liu, J.; Roth, E.; Li, E.; Shroyer, K.R.; et al. miR-181a-5p Inhibits Cancer Cell Migration and Angiogenesis via Downregulation of Matrix Metalloproteinase-14. Cancer Res. 2015, 75, 2674–2685. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Wu, Y.; Wang, J.; He, J.; Han, X. Sevoflurane inhibits the migration and invasion of colorectal cancer cells through regulating ERK/MMP-9 pathway by up-regulating miR-203. Eur. J. Pharmacol. 2019, 850, 43–52. [Google Scholar] [CrossRef]
- Zhang, L.; Dong, Y.; Zhu, N.; Tsoi, H.; Zhao, Z.; Wu, C.W.; Wang, K.; Zheng, S.; Ng, S.S.; Chan, F.K.; et al. microRNA-139-5p exerts tumor suppressor function by targeting NOTCH1 in colorectal cancer. Mol. Cancer 2014, 13, 124. [Google Scholar] [CrossRef]
- Cai, H.K.; Chen, X.; Tang, Y.H.; Deng, Y.C. MicroRNA-194 modulates epithelial-mesenchymal transition in human colorectal cancer metastasis. Onco Targets Ther. 2017, 10, 1269–1278. [Google Scholar] [CrossRef]
- Yu, L.; Lu, Y.; Han, X.; Zhao, W.; Li, J.; Mao, J.; Wang, B.; Shen, J.; Fan, S.; Wang, L.; et al. microRNA -140-5p inhibits colorectal cancer invasion and metastasis by targeting ADAMTS5 and IGFBP5. Stem. Cell Res. Ther. 2016, 7, 180. [Google Scholar] [CrossRef]
- Gabriely, G.; Wurdinger, T.; Kesari, S.; Esau, C.C.; Burchard, J.; Linsley, P.S.; Krichevsky, A.M. MicroRNA 21 promotes glioma invasion by targeting matrix metalloproteinase regulators. Mol. Cell. Biol. 2008, 28, 5369–5380. [Google Scholar] [CrossRef]
- Hinderer, S.; Layland, S.L.; Schenke-Layland, K. ECM and ECM-like materials—Biomaterials for applications in regenerative medicine and cancer therapy. Adv. Drug Deliv. Rev. 2016, 97, 260–269. [Google Scholar] [CrossRef]
- Phan, T.G.; Croucher, P.I. The dormant cancer cell life cycle. Nat. Rev. Cancer. 2020, 20, 398–411. [Google Scholar] [CrossRef]
- Celià-Terrassa, T.; Kang, Y. Metastatic niche functions and therapeutic opportunities. Nat. Cell Biol. 2018, 20, 868–877. [Google Scholar] [CrossRef] [PubMed]





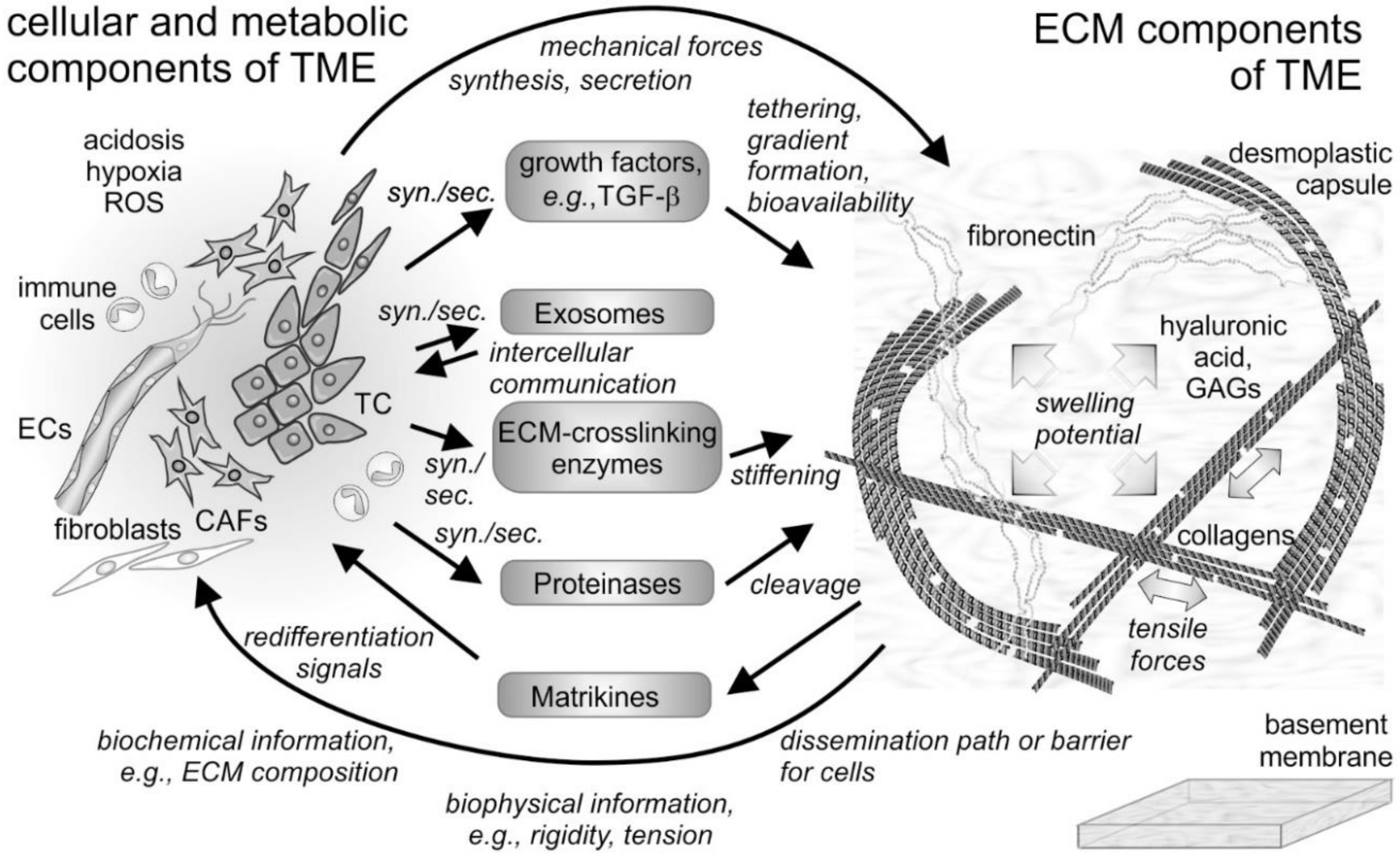{kind=link}
{kind=link}
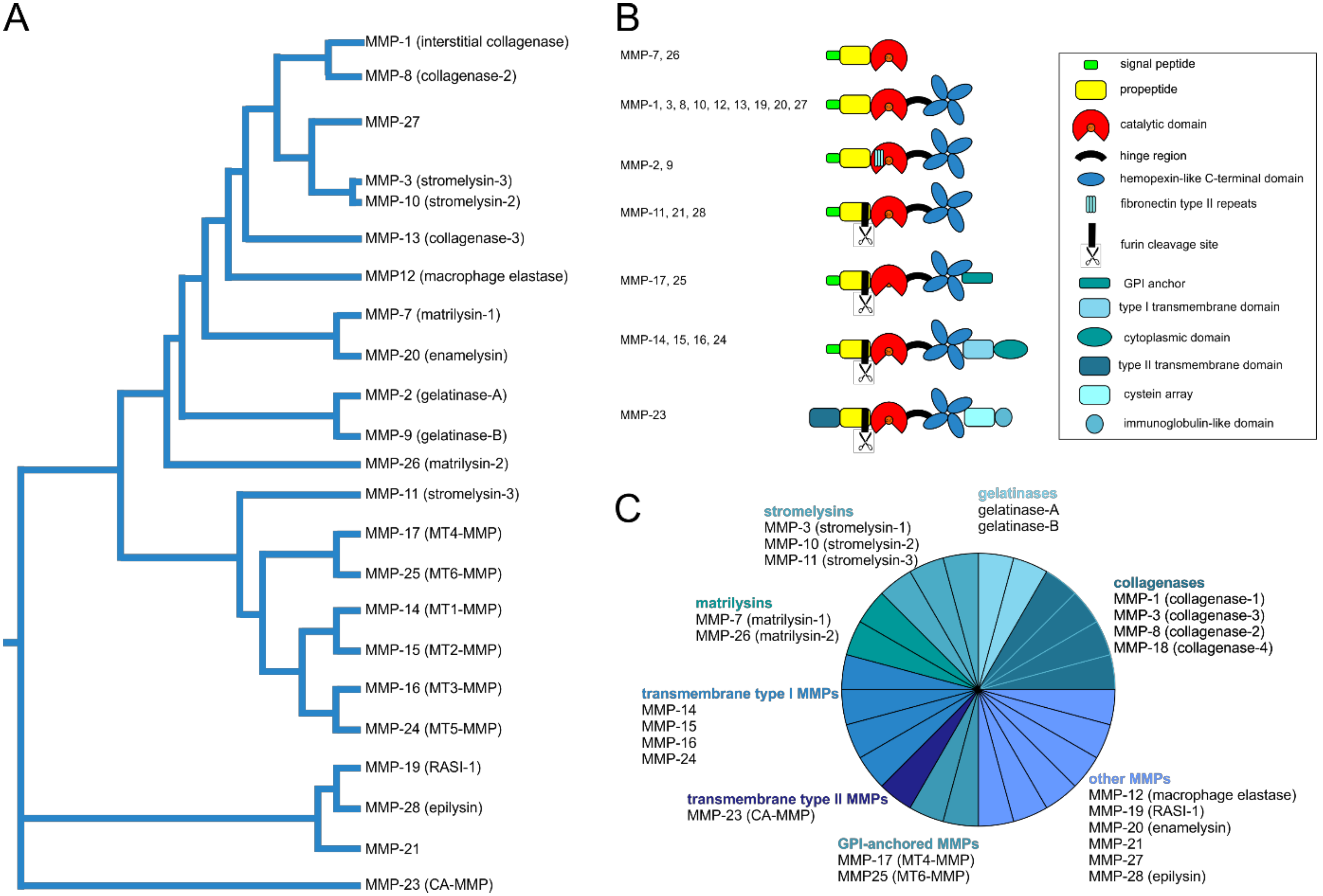{kind=link}
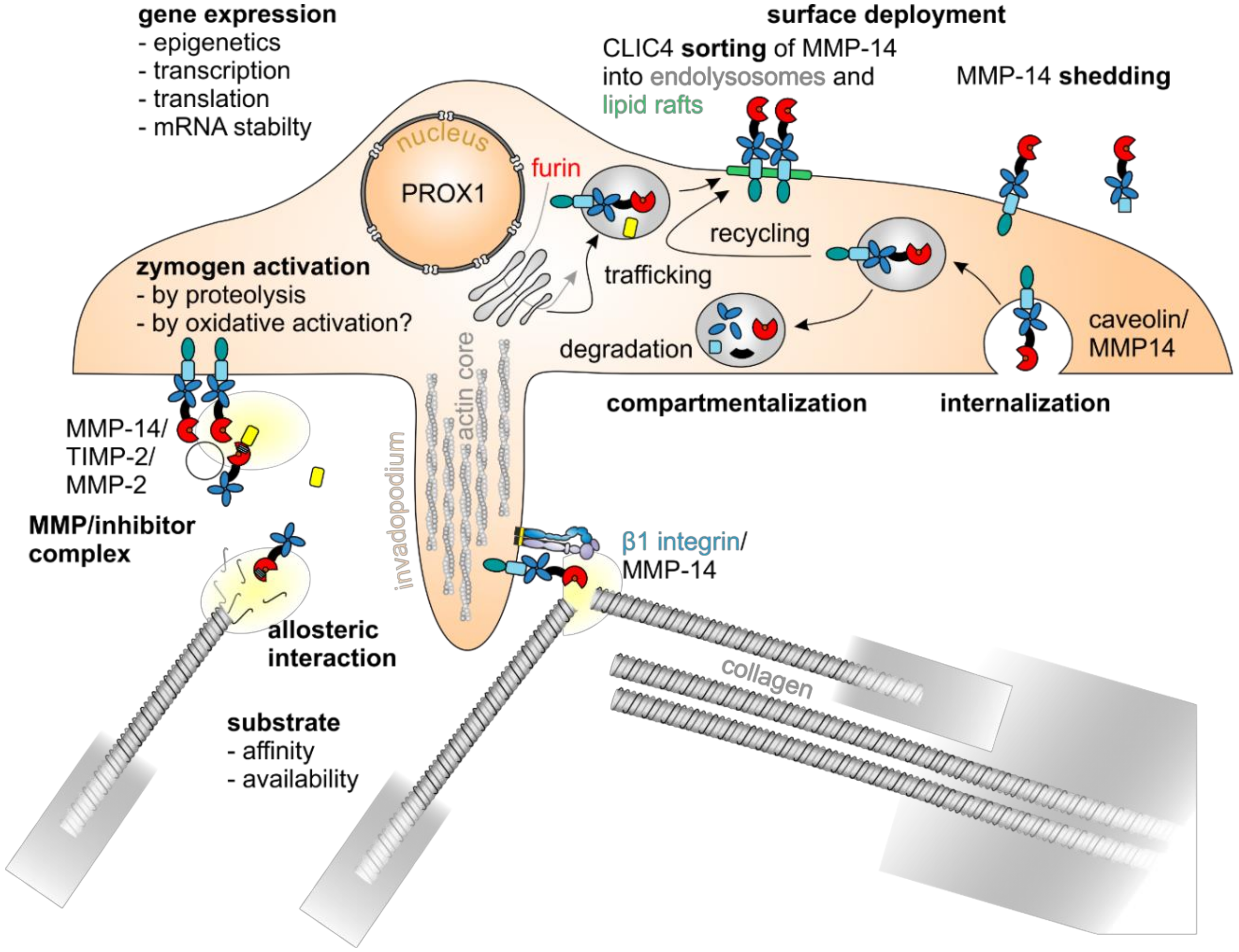{kind=link}
| Matrix Protein | BM Component | Protease | Bioactive Peptide Released | Receptor | Potential Use in Diagnostics |
|---|---|---|---|---|---|
| Collagens [5,16,244] | |||||
| Fibrillar collagens | |||||
| Procollagen I | BMP-I | P1CP [245,246,247] | |||
| Procollagen IIB | ADAMTS-3 [248] | Chondrostatin [249,250] | Integrins αvβ3, αvβ5 [249] | ||
| Procollagen III | PRO-C3 [251,252] | Associated with shorter TTP and OS in metastatic breast cancer [251] | |||
| Collagen I | Collagenases MMPs -1, -8, -14 | Integrins α1β1, α2β1, α10β1, α11β1, DDR1, DDR2, GPVI, LAIR-1, MR, PLA2R, LY75, Endo180 ([253] and references therein) | |||
| MMPs -2, -9, -13 [251] | C1M [251,254] | Associated with shorter TTP and OS in metastatic breast cancer [251] | |||
| PGP (pro–gly–pro) [255,256] | CXCR2 | ||||
| Collagen II | Gelatinases MMPs -2, -9 [135,138,145] | ||||
| Collagen III [257] | MMP-9 [251] | C3M [251,258] | Associated with shorter TTP and OS in metastatic breast cancer [251] | ||
| Collagen V [257] | |||||
| Collagen XI [257] | |||||
| Collagen VI | MMP-11 | α3(VI) endotrophin [259,260,261] | |||
| Collagen VIII | α1(VIII) vastatin [262,263,264,265] | Correlates with ECM degradation and stromal reactivity; increased in serum in colorectal cancer [262] | |||
| Network-forming collagens and multiplexins (e.g., in BMs) [266,267,268] | Yes | MT-MMPs [269], cathepsin S [270], MMPs -2, -3, 9, -13 | |||
| Collagen IV | Cathepsin S [270], MMP-14, 15 [269], MMP-9 [271] | α1(IV) arresten [272], α2(IV) canstatin [273], α3(IV) tumstatin [271,274], α4(IV) tetrastatin [275,276], α5(IV) pentastatin [275], α5(IV)NC1 lamstatin [277], α6(IV) hexastatin [275] | Integrins α1β1, α2β1, αVβ3, αvβ5 bind arresten, canstatin, tumstatin [85,240,278,279] | ||
| α6(IV)NC1 [280] | Iintegrin αVβ3 [281] | ||||
| MMPs -9, -12 [251] | C4M [251,282] | Associated with shorter TTP and OS in metastatic breast cancer [251] | |||
| Collagen XV | NC α1(XV) restin | ||||
| Collagen XVIII | MMPs -3, -9, -12, -13, -20 [283], cathepsins L, S [284], elastase [285] | α1 (XVIII) endostatin [85,240,276,283,286] | Nucleolin [287], integrins α5β1, αVβ3, αvβ5 [85], caveolin-1 [288], VEGFR2 [289], glypican-1, 2 [290] | ||
| MMP-7 [291] | Neostatin 7 [291] | ||||
| MMP-14 [292] | Neostatin 14 [292] | ||||
| FACITs (fibril-associated collagens with interrupted triple helix) | |||||
| Collagen IX [293] | |||||
| Collagen XIX | Plasmin [294] | NC-1 α1(XIX) [295] | Integrin αvβ3 [296] | ||
| Collagen of anchoring fibrils | Yes | ||||
| Collagen VII | MMP-1 [297] | NC1 | |||
| Elastic fibrils | |||||
| Elastin [298,299] | Ela-2, cathepsin G, proteinase-2, cathepsins L, S, K, V, MMPs -2, -7, -9, 12 [300] MMPs -1, -2, -8, -9, -12 [125] | Elastin-derived matrikine, VG-6 (VGVAPG), AG-9 (AGVPGLGVG) [125,241,301,302] | Elastin receptor complex (ERC) [300] | ||
| Fibrillin [298] | ADAMTS -10, -6 [303] | Integrins αVβ3, αVβ6, α5β1 [304,305] | |||
| Fibronectins [29,306,307,308,309] | Yes | MMPs -2, -3, -7, -10, -11 [297] | Fibronectin fragments (FNFr) [310], anastellin [311], fibstatin [312] | Integrins α5β1 [313], α9β1, α4β1, αv-integrins [309,314,315], growth factors and syndecans [316] | |
| Fibronectin ED-A [29] | Integrins α9β1, α4β1, α5β1, αv-integrins [29,308,317] | Marks tumor stroma [29,306,307,308,315,317,318,319,320] | |||
| Fibronectin ED-B [29] | Integrins α9β1, α4β1, α5β1, αv-integrins [29,308,317] | Marks tumor stroma [29,306,307,308,315,317,318,319,320] | |||
| Proteoglycans | |||||
| Hyalectans (Lecticans) | |||||
| Aggrecan | Aggrecanases, MMPs -1, -2, -3, 7, -8, -9, -13, -14 ([321] and references therein), ADAMTSs [234,321] | ||||
| Neurocan | NCAM-L1, indirectly N-cadherin [322] and NrCAM/Sema3F [323] | ||||
| Brevican | ADAMTSs [324] | ||||
| Versican | ADAMTS -4, -5 [325] | Versican-derived matrikine, versicine [326,327] | β1 integrins [328] | ||
| SLRPs [4,83,84] | |||||
| Decorin | MMPs -2, -3 [329] | EGFR [330], IGF-1R ([331] and references therein), MET [332], VEGFR2 [333], 51 kD receptor [334] | |||
| Biglycan | TLR-2, TLR-4, LRP6, MuSK ([335] and references therein), 51 kD receptor [334] | ||||
| Perlecan | Yes | MMP-3, -7 [329,336], cathepsin L [337] | Endorepellin [85,86] | VEGFR2/integrin α2β1 [86,338,339] | Blood levels of domain IV fragments elevated in prostate carcinoma [336] |
| BMP1/TLD-like protease [340], cathepsin L, t-PA [337] | LG3 fragment (C-terminal fragment of endorepellin) | ||||
| Glypican-3 | glypican-3 derived peptide [340] | ||||
| Nidogen-1 | Yes | MMP-19, cathepsin S, meprin A [341,342,343] | G3 domain | ||
| Laminins [21,23,344] | Yes | MMPs -2, -3, -7, -10 [297,329] | Mark BMs | ||
| α chain | α1 chain: IKVAV, RKRLQVQLSIRT (AG-73) [345] | Integrins α3β1, α6β1, syndecans 1, 2, 4 [345] | |||
| α3 chain: C-terminal fragment | |||||
| α5 chain: AQARSAASKVKVSMKF [346] | Heparan sulfate proteoglycans [50],syndecans [347,348,349] | ||||
| β chain | β1 chain: YIGSR [345] | 67 kD receptor [345] | |||
| γ chain | γ1 chain: KAFDITYVRLKF (C16) [345] | Integrins αvβ3, α5β1 [345] | |||
| Laminin-332 | Yes | MMPs -2, -9 [241,297,329] | LG3, LG4 [350,351,352,353], EGF-L repeats ([241] and references therein), γ2 chain: N-terminal fragment | Integrin α3β1 [354], EGFR ([241] and references therein) | Marks tumor stroma [352,355,356]; β3 marks tumor stroma, poor prognosis [357,358], γ 2 marks tumor stroma, poor prognosis, γ2 in blood samples [358,359] |
| Laminin-511 | RLVSYNGIIFFLK (A5G27) [360,361] | ||||
| Matricellular proteins | |||||
| CCNs [44]: | |||||
| CCN1 (CYR61) | Integrins αvβ3, αvβ5, α6β1, syndecan-4 [362,363,364] | ||||
| CCN2 (CTGF) | CCN2-fragments [365] | Integrin α6β1, αvβ3 [366,367] | Marks vasculogenic mimicry [368,369,370,371] | ||
| Tenascins [372,373] | |||||
| Tenascin C [374] | MMPs -1, -8, -13 [329] | EGF-L repeat [241] | Integrin α9β1 [37], EGFR (EGF-L) ([241] and references therein) | ||
| Tenascin W [375] | Marks tumor stroma [35,375,376,377] | ||||
| Thrombospondins [298] | CD36, αV and β1 integrins, syndecan, CD47 | ||||
| Osteopontin [378,379,380] | Marks tumor progression [381] | ||||
| Periostin [382] | Integrins αVβ3, αVβ5 [383] | Marks tumor stroma [40,358,382,384,385,386,387,388,389,390] | |||
| SPARC [391] | Abundant in healthy vessels and tumors of good prognosis [391] | ||||
| Galectins [392] | Promote tumor angiogenesis [393] and affect tumor immunology [394] | ||||
| SIBLINGs [44,395] | |||||
| Bone sialoprotein | Marks tumor progression [381] | ||||
| Dentin matrix protein I | |||||
| Sialophosphoprotein | |||||
| Matrix extracellular glycoprotein | |||||
| Syndecans [396] | |||||
| Syndecan-1 | Synstatins SSTN92-119 [397,398,399], SSTN82-130 [400], SSTN210-240 [399,401] | ||||
| Syndecan-4 | SSTN87-131 [399] | ||||
| Agrin | neurotrypsin [402] | C-terminal agrin fragment [402] | Not yet found related to the tumor microenvironment | ||
| Hyaluronan [53] | |||||
| Hyaluronic acid | HYAL2 [73,403] | HA oligosaccharides [127] | CD44, RHAMM, TLR4 [75] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niland, S.; Eble, J.A. Hold on or Cut? Integrin- and MMP-Mediated Cell–Matrix Interactions in the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 238. https://doi.org/10.3390/ijms22010238
Niland S, Eble JA. Hold on or Cut? Integrin- and MMP-Mediated Cell–Matrix Interactions in the Tumor Microenvironment. International Journal of Molecular Sciences. 2021; 22(1):238. https://doi.org/10.3390/ijms22010238
Chicago/Turabian StyleNiland, Stephan, and Johannes A. Eble. 2021. "Hold on or Cut? Integrin- and MMP-Mediated Cell–Matrix Interactions in the Tumor Microenvironment" International Journal of Molecular Sciences 22, no. 1: 238. https://doi.org/10.3390/ijms22010238
APA StyleNiland, S., & Eble, J. A. (2021). Hold on or Cut? Integrin- and MMP-Mediated Cell–Matrix Interactions in the Tumor Microenvironment. International Journal of Molecular Sciences, 22(1), 238. https://doi.org/10.3390/ijms22010238