Dynamical Correlations Reveal Allosteric Sites in G Protein-Coupled Receptors



Abstract
1. Introduction
2. Results
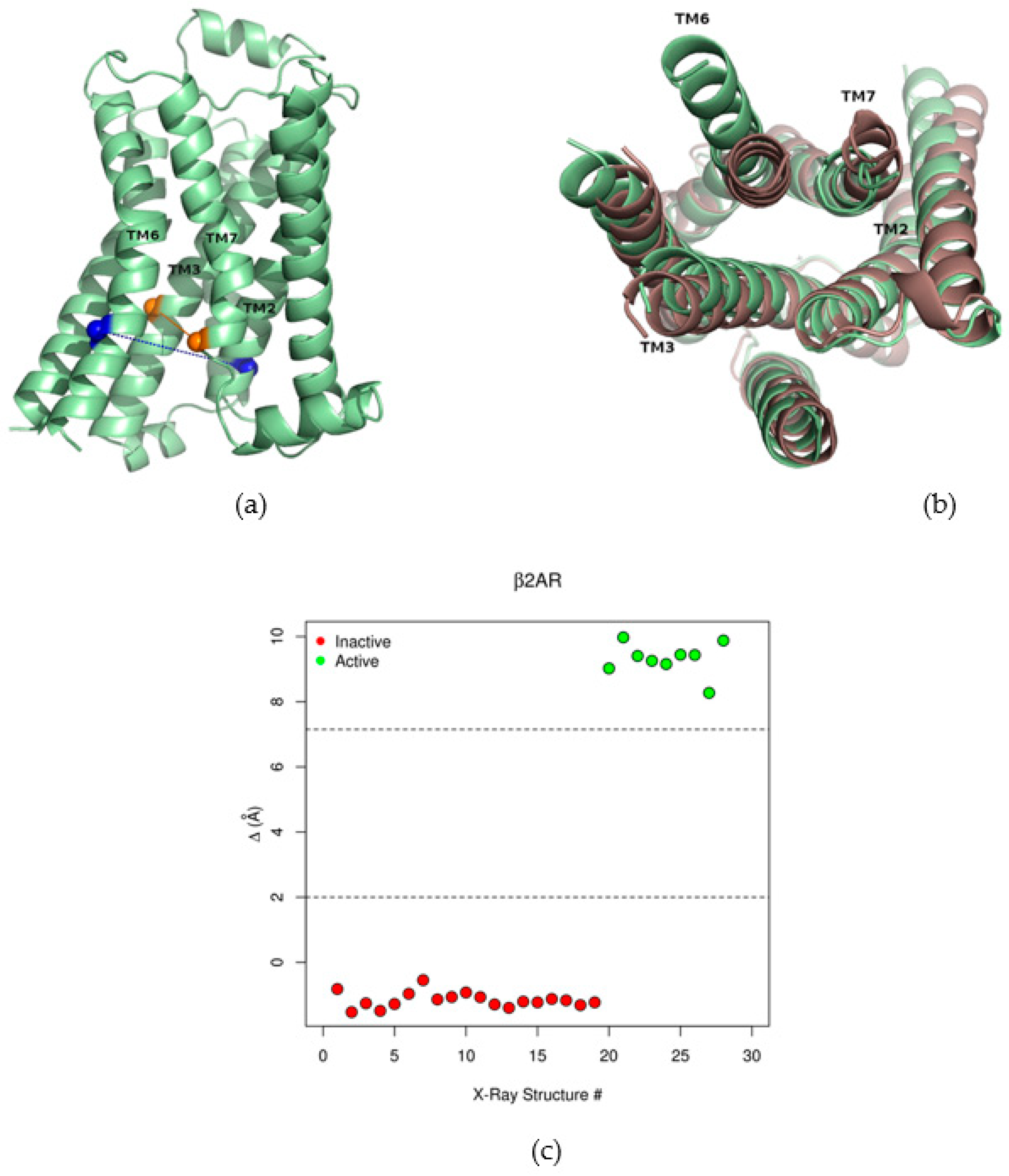
2.1. The Collective Variable Δ Discriminated between Different Conformations of the β2AR
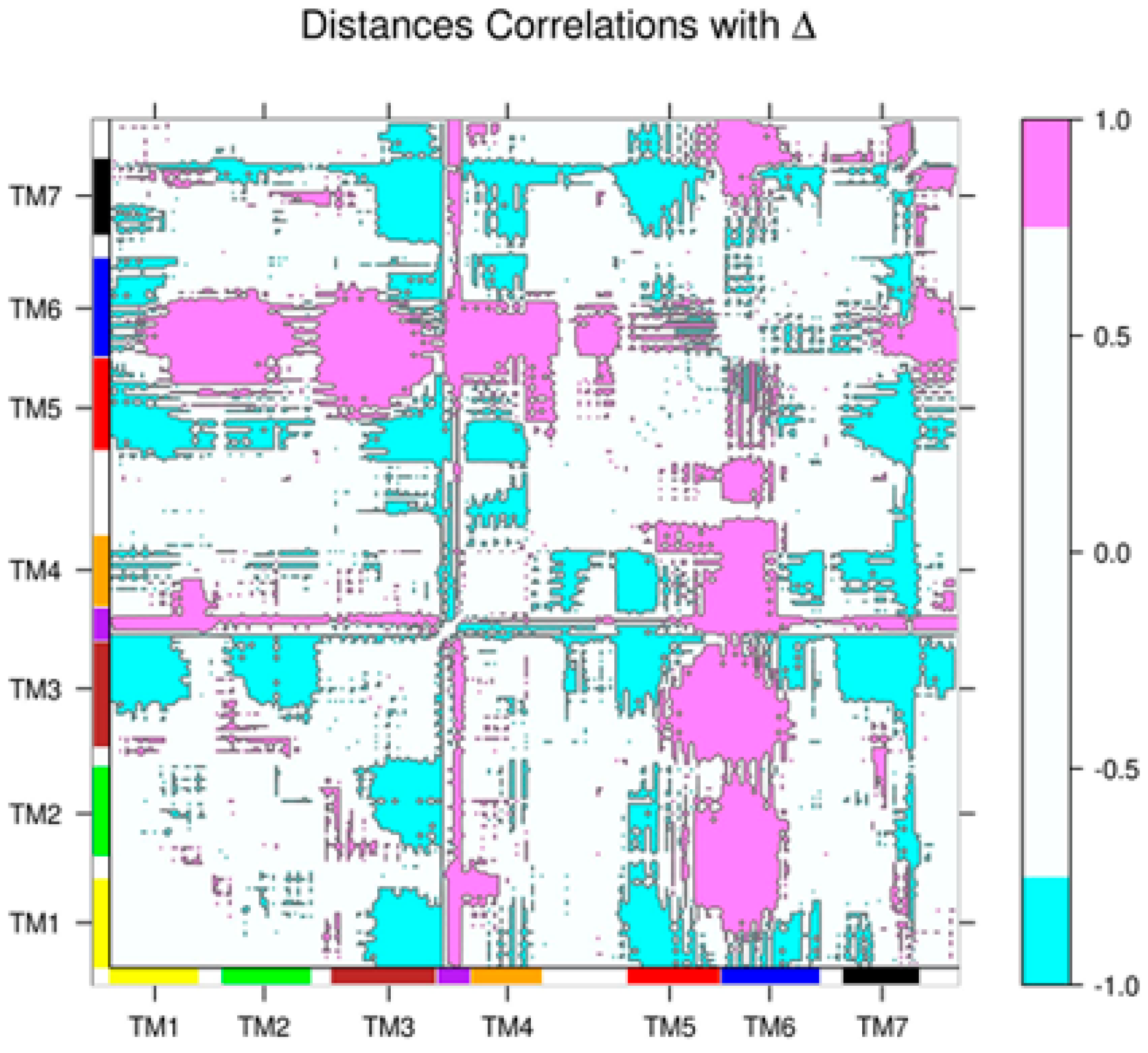
2.2. Correlations between Δ and Inter-Residue Distances in the β2AR
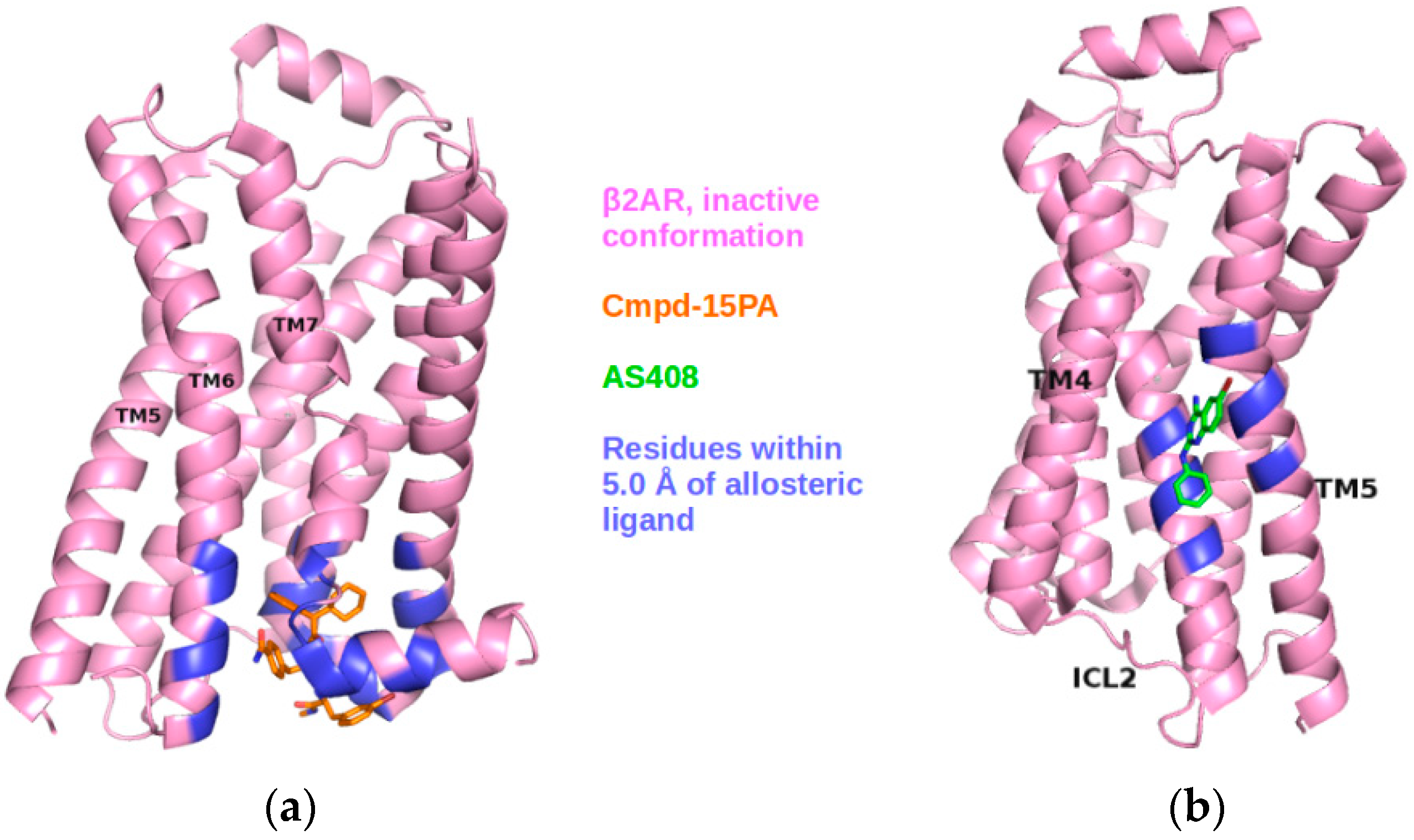
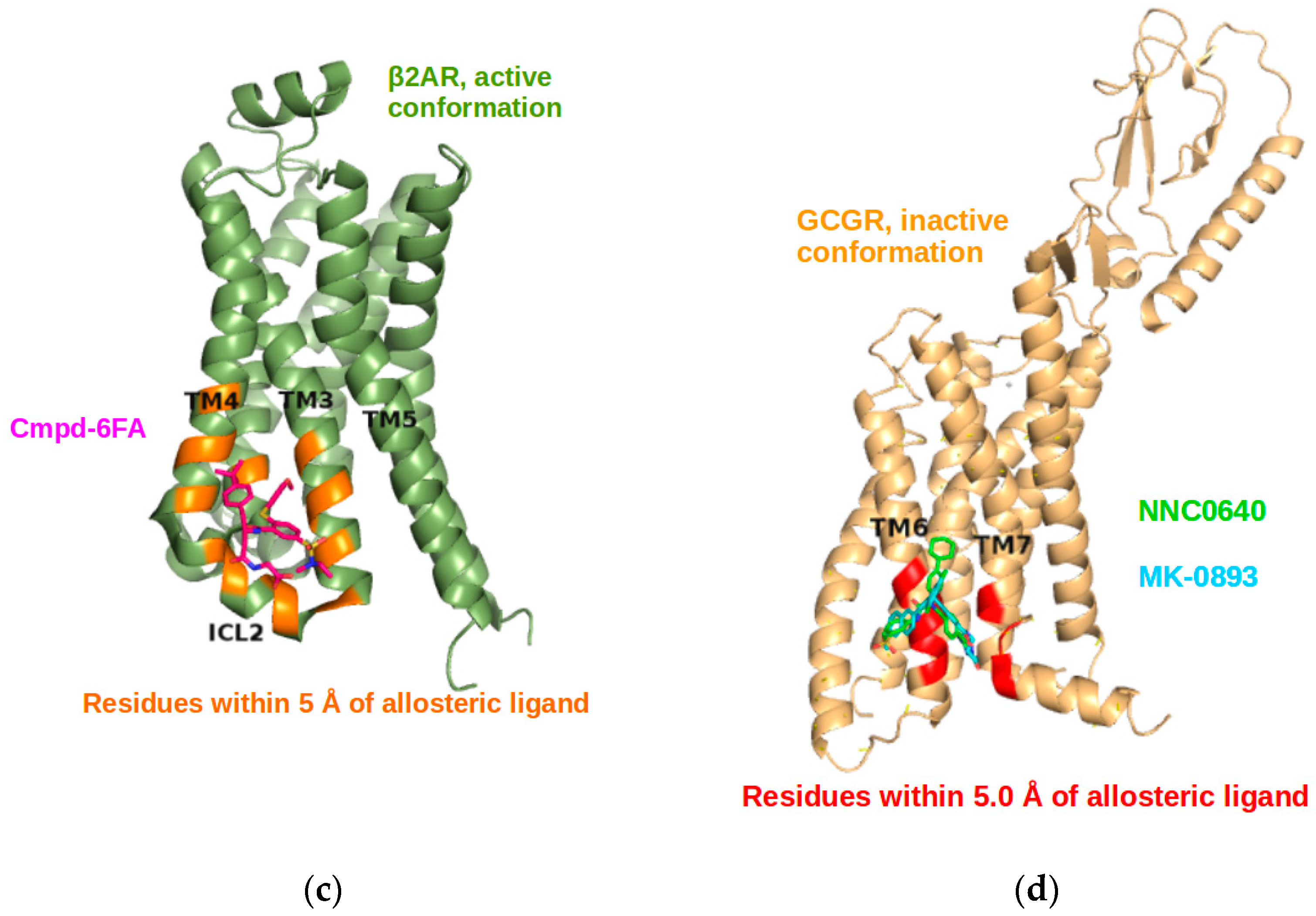
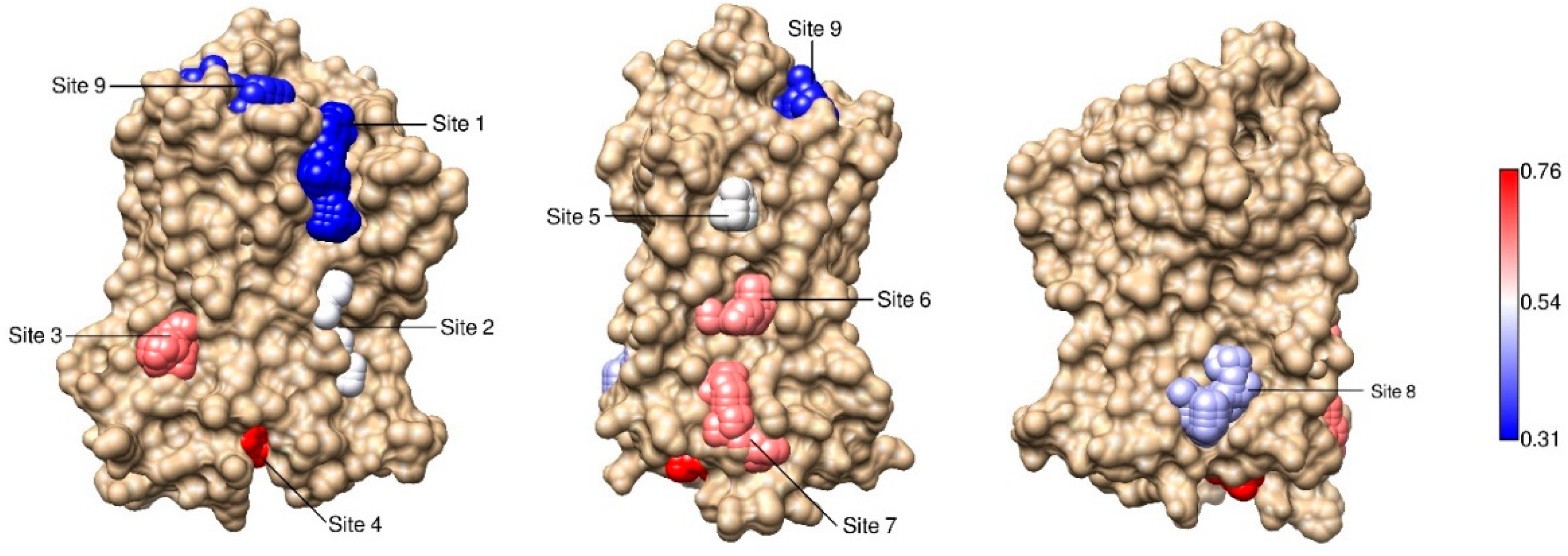
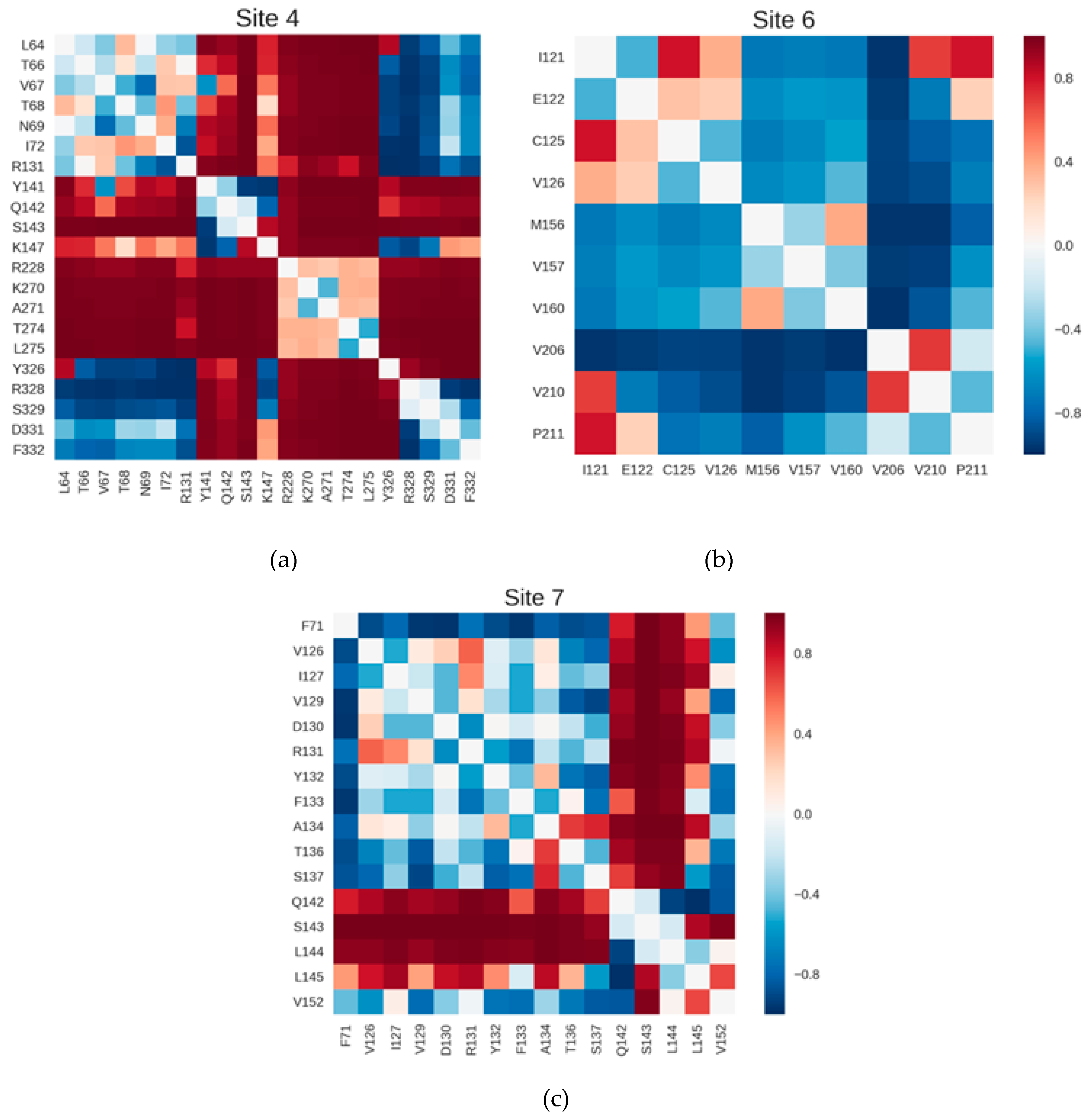
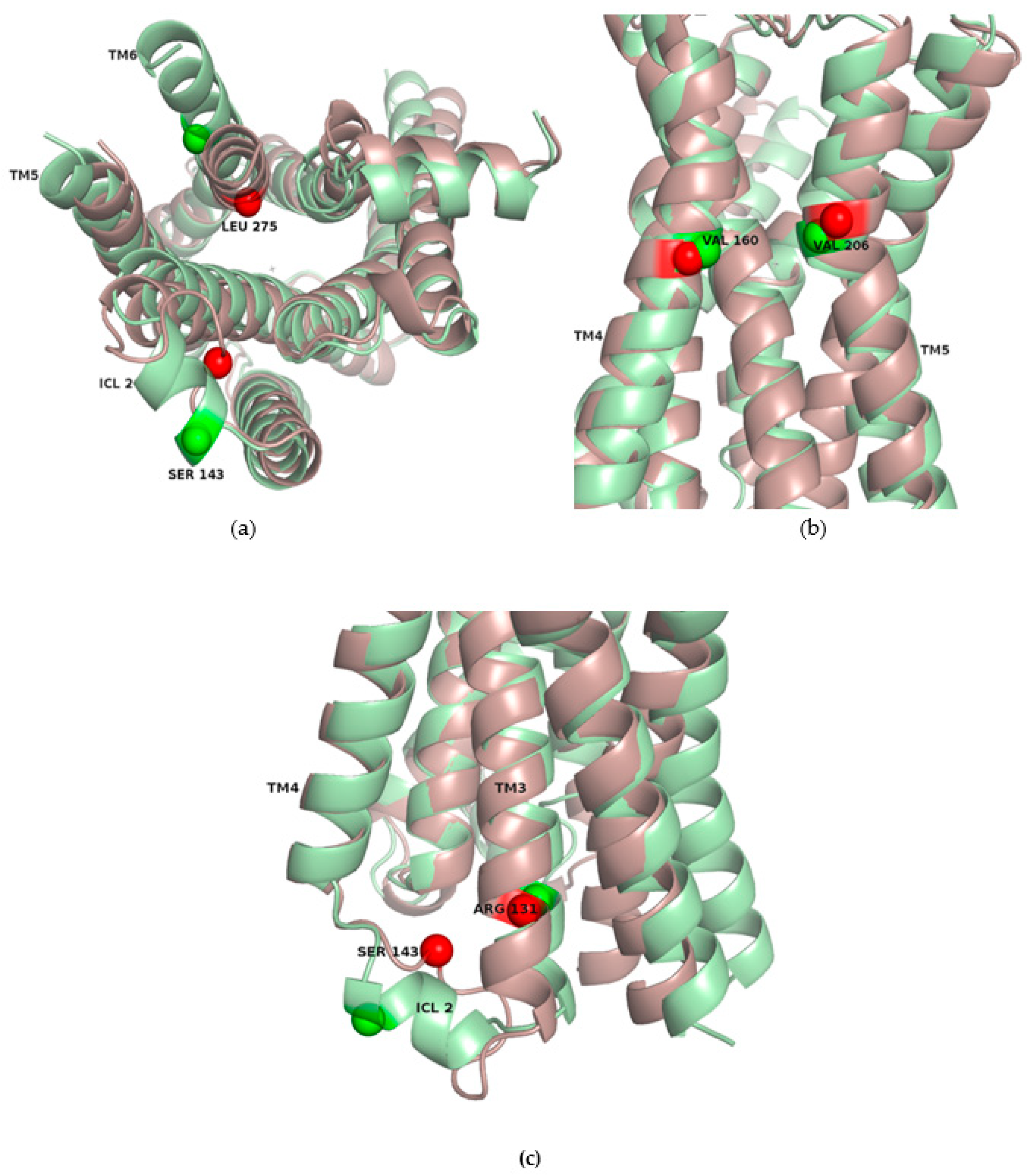
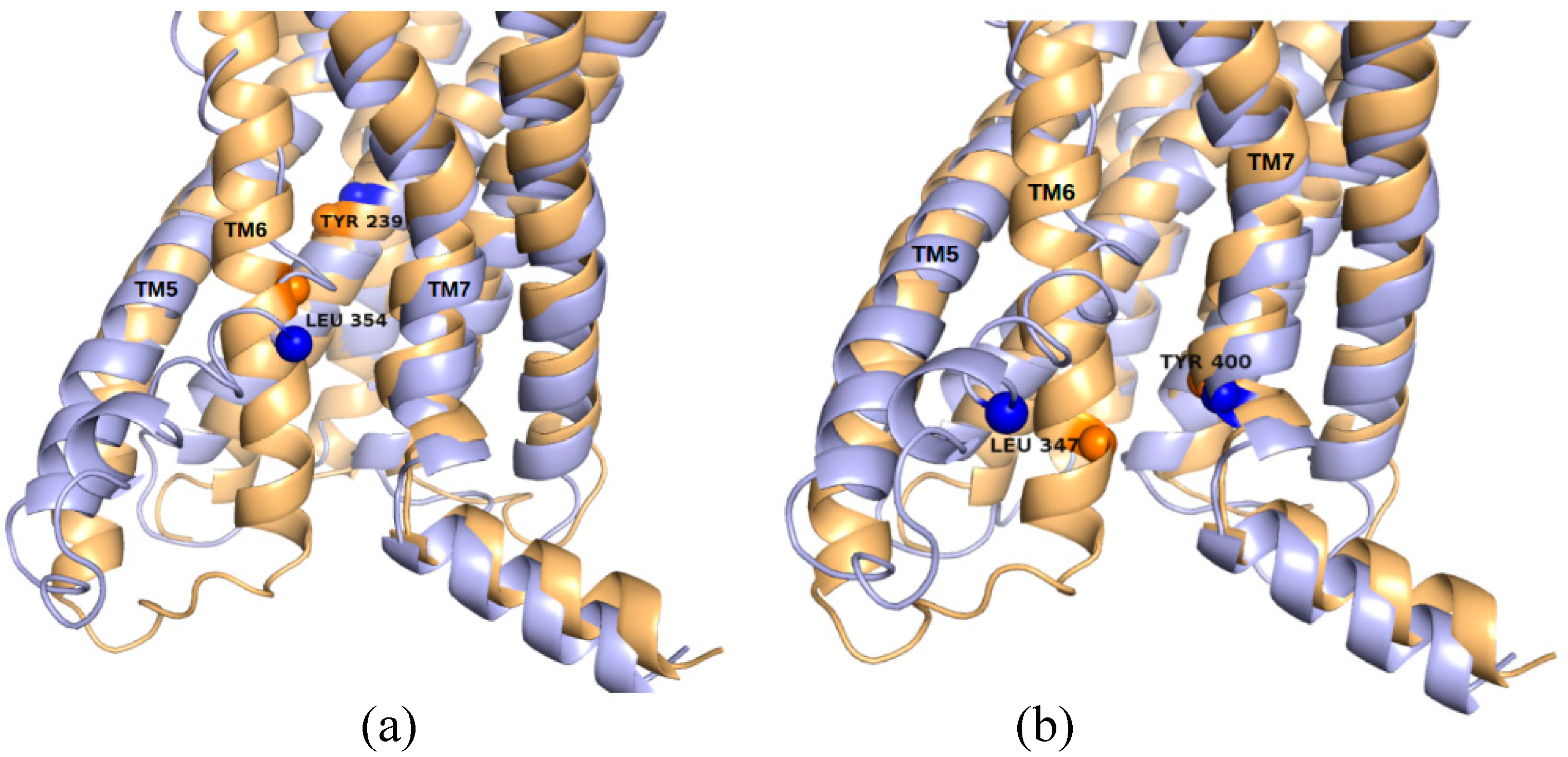
2.3. Correlations with Δ Allowed the Identification of Allosteric Sites in the β2AR
2.4. Allosteric Sites in the β2AR Could Be Detected by Normal Mode Analysis
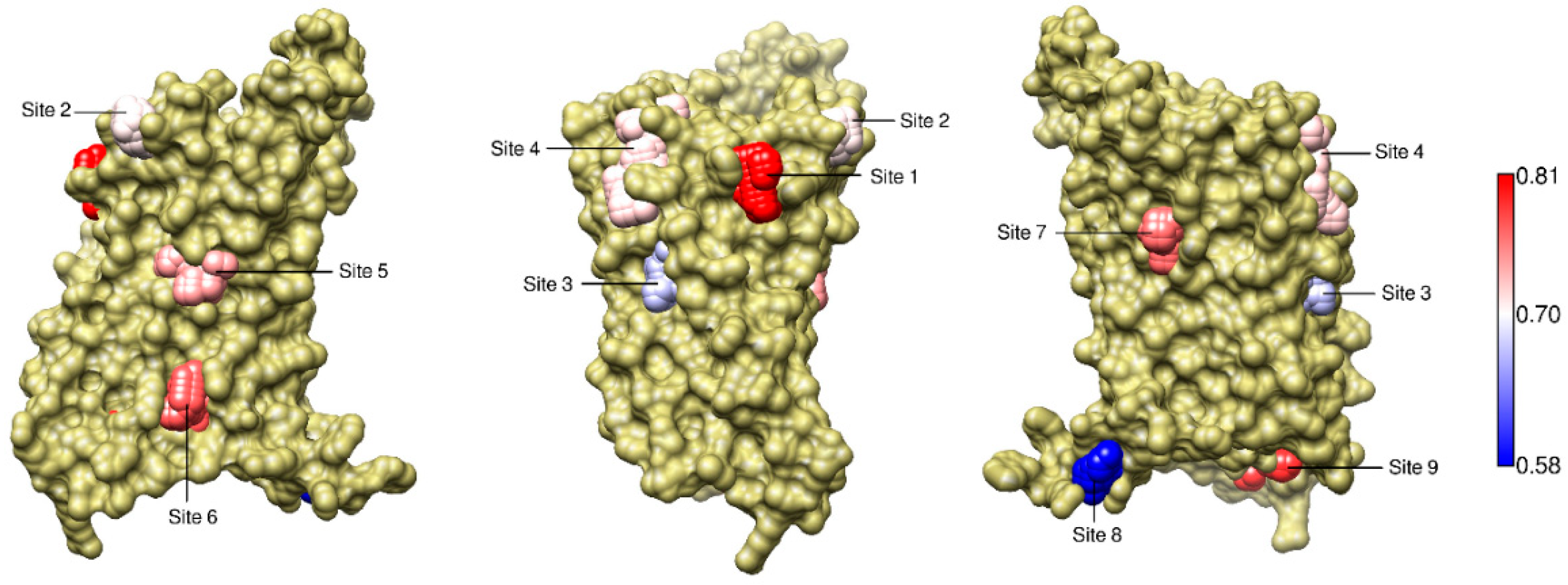
2.5. The Known Allosteric Site of GCGR Was Identified by Dynamical Correlations
2.6. Dynamical Correlations Identified a Known Allosteric Site in NMA of the M2 Muscarinic Receptor
3. Discussion
4. Materials and Methods
4.1. Structural Ensembles
4.1.1. Experimental Ensemble of the β2AR
4.1.2. Experimental Ensemble of the M2 Muscarinic Receptor
4.2. Normal Mode Analysis
4.3. Molecular Dynamics Simulations
4.4. The Collective Variable Δ
4.5. Calculation of Inter-Residue Distances
4.6. Coupling between Inter-Residue Distances and Δ
4.7. Determination of Receptors′ Sites
4.8. Coupling between Sites and Δ
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Katritch, V.; Cherezov, V.; Stevens, R.C. Structure-function of the G protein-coupled receptor superfamily. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 531–556. [Google Scholar] [CrossRef] [PubMed]
- Shimada, I.; Ueda, T.; Kofuku, Y.; Eddy, M.T.; Wüthrich, K. GPCR drug discovery: Integrating solution NMR data with crystal and cryo-EM structures. Nat. Rev. Drug Discov. 2018, 18, 59–82. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Bostock, M.J.; Solt, A.S.; Nietlispach, D. The role of NMR spectroscopy in mapping the conformational landscape of GPCRs. Curr. Opin. Struct. Biol. 2019, 57, 145–156. [Google Scholar] [CrossRef]
- Latorraca, N.R.; Venkatakrishnan, A.J.; Dror, R.O. GPCR dynamics: Structures in motion. Chem. Rev. 2017, 117, 139–155. [Google Scholar] [CrossRef]
- Wingler, L.M.; Elgeti, M.; Hilger, D.; Latorraca, N.R.; Lerch, M.T.; Staus, D.P.; Dror, R.O.; Kobilka, B.K.; Hubbell, W.L.; Lefkowitz, R.J. Angiotensin Analogs with Divergent Bias Stabilize Distinct Receptor Conformations. Cell 2019, 176, 468–478.e11. [Google Scholar] [CrossRef]
- Lee, Y.; Lazim, R.; Macalino, S.J.Y.; Choi, S. Importance of protein dynamics in the structure-based drug discovery of class A G protein-coupled receptors (GPCRs). Curr. Opin. Struct. Biol. 2019, 55, 147–153. [Google Scholar] [CrossRef]
- Chan, H.C.S.; Li, Y.; Dahoun, T.; Vogel, H.; Yuan, S. New Binding Sites, New Opportunities for GPCR Drug Discovery. Trends Biochem. Sci. 2019, 44, 312–330. [Google Scholar] [CrossRef]
- Congreve, M.; Oswald, C.; Marshall, F.H. Applying Structure-Based Drug Design Approaches to Allosteric Modulators of GPCRs. Trends Pharmacol. Sci. 2017, 38, 837–847. [Google Scholar] [CrossRef]
- Thal, D.M.; Glukhova, A.; Sexton, P.M.; Christopoulos, A. Structural insights into G-protein-coupled receptor allostery. Nature 2018, 559, 45–53. [Google Scholar] [CrossRef]
- Wacker, D.; Stevens, R.C.; Roth, B.L. How Ligands Illuminate GPCR Molecular Pharmacology. Cell 2017, 170, 414–427. [Google Scholar] [CrossRef] [PubMed]
- Lane, J.R.; May, L.T.; Parton, R.G.; Sexton, P.M.; Christopoulos, A. A kinetic view of GPCR allostery and biased agonism. Nat. Chem. Biol. 2017, 13, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Wakefield, A.E.; Mason, J.S.; Vajda, S.; Keserű, G.M. Analysis of tractable allosteric sites in G protein-coupled receptors. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Brenke, R.; Kozakov, D.; Chuang, G.Y.; Beglov, D.; Hall, D.; Landon, M.R.; Mattos, C.; Vajda, S. Fragment-based identification of druggable “hot spots” of proteins using Fourier domain correlation techniques. Bioinformatics 2009, 25, 621–627. [Google Scholar] [CrossRef]
- Ngan, C.H.; Hall, D.R.; Zerbe, B.; Grove, L.E.; Kozakov, D.; Vajda, S. FtSite: High accuracy detection of ligand binding sites on unbound protein structures. Bioinformatics 2012, 28, 286–287. [Google Scholar] [CrossRef]
- Kozakov, D.; Grove, L.E.; Hall, D.R.; Bohnuud, T.; Mottarella, S.E.; Luo, L.; Xia, B.; Beglov, D.; Vajda, S. The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat. Protoc. 2015, 10, 733–755. [Google Scholar] [CrossRef]
- Panjkovich, A.; Daura, X. PARS: A web server for the prediction of Protein Allosteric and Regulatory Sites. Bioinformatics 2014, 30, 1314–1315. [Google Scholar] [CrossRef]
- Goncearenco, A.; Mitternacht, S.; Yong, T.; Eisenhaber, B.; Eisenhaber, F.; Berezovsky, I.N. SPACER: Server for predicting allosteric communication and effects of regulation. Nucleic Acids Res. 2013, 41, 266–272. [Google Scholar] [CrossRef]
- Alfayate, A.; Caceres, C.R.; Dos Santos, H.G.; Bastolla, U. Predicted dynamical couplings of protein residues characterize catalysis, transport and allostery. Bioinformatics 2019, 35, 4971–4978. [Google Scholar] [CrossRef]
- Ma, X.; Meng, H.; Lai, L. Motions of Allosteric and Orthosteric Ligand-Binding Sites in Proteins are Highly Correlated. J. Chem. Inf. Model. 2016, 56, 1725–1733. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, X.; Peng, C.; Wang, J.; Xu, Z.; Chen, K.; Shi, J.; Zhu, W. D3Pockets: A Method and Web Server for Systematic Analysis of Protein Pocket Dynamics. J. Chem. Inf. Model. 2019, 59, 3353–3358. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ahn, S.; Kahsai, A.W.; Meng, K.C.; Latorraca, N.R.; Pani, B.; Venkatakrishnan, A.J.; Masoudi, A.; Weis, W.I.; Dror, R.O.; et al. Mechanism of intracellular allosteric β2AR antagonist revealed by X-ray crystal structure. Nature 2017, 548, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Masoudi, A.; Kahsai, A.W.; Huang, L.Y.; Pani, B.; Staus, D.P.; Shim, P.J.; Hirata, K.; Simhal, R.K.; Schwalb, A.M.; et al. Mechanism of β2AR regulation by an intracellular positive allosteric modulator. Science 2019, 364, 1283–1287. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Kaindl, J.; Korczynska, M.; Stößel, A.; Dengler, D.; Stanek, M.; Hübner, H.; Clark, M.J.; Mahoney, J.; Matt, R.A.; et al. An allosteric modulator binds to a conformational hub in the β2 adrenergic receptor. Nat. Chem. Biol. 2020, 16, 749–755. [Google Scholar] [CrossRef]
- Jazayeri, A.; Doré, A.S.; Lamb, D.; Krishnamurthy, H.; Southall, S.M.; Baig, A.H.; Bortolato, A.; Koglin, M.; Robertson, N.J.; Errey, J.C.; et al. Extra-helical binding site of a glucagon receptor antagonist. Nature 2016, 533, 274–277. [Google Scholar] [CrossRef]
- Zhang, H.; Qiao, A.; Yang, D.; Yang, L.; Dai, A.; De Graaf, C.; Reedtz-Runge, S.; Dharmarajan, V.; Zhang, H.; Han, G.W.; et al. Structure of the full-length glucagon class B G-protein-coupled receptor. Nature 2017, 546, 259–264. [Google Scholar] [CrossRef]
- Pándy-Szekeres, G.; Munk, C.; Tsonkov, T.M.; Mordalski, S.; Harpsøe, K.; Hauser, A.S.; Bojarski, A.J.; Gloriam, D.E. GPCRdb in 2018: Adding GPCR structure models and ligands. Nucleic Acids Res. 2018, 46, D440–D446. [Google Scholar] [CrossRef]
- Schmidtke, P.; Bidon-chanal, A.; Luque, F.J.; Barril, X. MDpocket: Open-source cavity detection and characterization on molecular dynamics trajectories. Bioinformatics 2011, 27, 3276–3285. [Google Scholar] [CrossRef]
- Skjaerven, L.; Hollup, S.M.; Reuter, N. Normal mode analysis for proteins. J. Mol. Struct. THEOCHEM 2009, 898, 42–48. [Google Scholar] [CrossRef]
- Chen, K.-Y.M.; Keri, D.; Barth, P. Computational design of G Protein-Coupled Receptor allosteric signal transductions. Nat. Chem. Biol. 2020, 16, 77–86. [Google Scholar] [CrossRef]
- Cherezov, V.; Rosenbaum, D.M.; Hanson, M.A.; Rasmussen, S.G.F.; Thian, F.S.; Kobilka, T.S.; Choi, H.-J.; Kuhn, P.; Weis, W.I.; Kobilka, B.K.; et al. High-Resolution Crystal Structure of an Engineered Human 2-Adrenergic G Protein-Coupled Receptor. Science 2007, 318, 1258–1265. [Google Scholar] [CrossRef] [PubMed]
- Ring, A.M.; Manglik, A.; Kruse, A.C.; Enos, M.D.; Weis, W.I.; Garcia, K.C.; Kobilka, B.K. Adrenaline-activated structure of β2-adrenoceptor stabilized by an engineered nanobody. Nature 2013, 502, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Kruse, A.C.; Ring, A.M.; Manglik, A.; Hu, J.; Hu, K.; Eitel, K.; Hübner, H.; Pardon, E.; Valant, C.; Sexton, P.M.; et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 2013, 504, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Chen, Y.; Wang, Z.L.; Liu, Z. Fluctuation correlations as major determinants of structure- and dynamics-driven allosteric effects. Phys. Chem. Chem. Phys. 2019, 21, 5200–5214. [Google Scholar] [CrossRef]
- Tang, Q.Y.; Kaneko, K. Long-range correlation in protein dynamics: Confirmation by structural data and normal mode analysis. PLoS Comput. Biol. 2020, 16, 1–17. [Google Scholar] [CrossRef]
- La Sala, G.; Decherchi, S.; De Vivo, M.; Rocchia, W. Allosteric Communication Networks in Proteins Revealed through Pocket Crosstalk Analysis. ACS Cent. Sci. 2017, 3, 949–960. [Google Scholar] [CrossRef]
- Clarke, D.; Sethi, A.; Li, S.; Kumar, S.; Chang, R.W.F.; Chen, J.; Gerstein, M. Identifying Allosteric Hotspots with Dynamics: Application to Inter- and Intra-species Conservation. Structure 2016, 24, 826–837. [Google Scholar] [CrossRef]
- Greener, J.G.; Filippis, I.; Sternberg, M.J.E. Predicting Protein Dynamics and Allostery Using Multi-Protein Atomic Distance Constraints. Structure 2017, 25, 546–558. [Google Scholar] [CrossRef]
- Song, K.; Liu, X.; Huang, W.; Lu, S.; Shen, Q.; Zhang, L.; Zhang, J. Improved Method for the Identification and Validation of Allosteric Sites. J. Chem. Inf. Model. 2017, 57, 2358–2363. [Google Scholar] [CrossRef]
- Weis, W.I.; Kobilka, B.K. The Molecular Basis of G Protein–Coupled Receptor Activation. Annu. Rev. Biochem. 2018, 87, 897–919. [Google Scholar] [CrossRef]
- Lamim Ribeiro, J.M.; Filizola, M. Allostery in G protein-coupled receptors investigated by molecular dynamics simulations. Curr. Opin. Struct. Biol. 2019, 55, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Whitten, S.T.; García-Moreno, E.B.; Hilser, V.J. Local conformational fluctuations can modulate the coupling between proton binding and global structural transitions in proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 4282–4287. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, R.J.; McLeish, T.C.B. Coupling of global and local vibrational modes in dynamic allostery of proteins. Biophys. J. 2006, 91, 2055–2062. [Google Scholar] [CrossRef] [PubMed]
- Wenthur, C.J.; Gentry, P.R.; Mathews, T.P.; Lindsley, C.W. Drugs for allosteric sites on receptors. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 165–184. [Google Scholar] [CrossRef]
- Jeffrey Conn, P.; Christopoulos, A.; Lindsley, C.W. Allosteric modulators of GPCRs: A novel approach for the treatment of CNS disorders. Nat. Rev. Drug Discov. 2009, 8, 41–54. [Google Scholar] [CrossRef]
- Van Der Westhuizen, E.T.; Valant, C.; Sexton, P.M.; Christopoulos, A. Endogenous allosteric modulators of G protein-coupled receptors. J. Pharmacol. Exp. Ther. 2015, 353, 246–260. [Google Scholar] [CrossRef]
- Miller-Gallacher, J.L.; Nehmé, R.; Warne, T.; Edwards, P.C.; Schertler, G.F.X.; Leslie, A.G.W.; Tate, C.G. The 2.1 Å resolution structure of cyanopindolol-bound β1-adrenoceptor identifies an intramembrane Na+ ion that stabilises the ligand-free receptor. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Fenalti, G.; Giguere, P.M.; Katritch, V.; Huang, X.P.; Thompson, A.A.; Cherezov, V.; Roth, B.L.; Stevens, R.C. Molecular control of δ-opioid receptor signalling. Nature 2014, 506, 191–196. [Google Scholar] [CrossRef]
- Swaminath, G.; Steenhuis, J.; Kobilka, B.; Lee, T.W. Allosteric modulation of β2-adrenergic receptor by Zn2+. Mol. Pharmacol. 2002, 61, 65–72. [Google Scholar] [CrossRef]
- Swaminath, G.; Lee, T.W.; Kobilka, B. Identification of an allosteric binding site for Zn2+ on the β2 adrenergic receptor. J. Biol. Chem. 2003, 278, 352–356. [Google Scholar] [CrossRef]
- Díaz, Ó.; Dalton, J.A.R.; Giraldo, J. Revealing the Mechanism of Agonist-Mediated Cannabinoid Receptor 1 (CB1) Activation and Phospholipid-Mediated Allosteric Modulation. J. Med. Chem. 2019, 62, 5638–5654. [Google Scholar] [CrossRef] [PubMed]
- Bruzzese, A.; Dalton, J.A.R.; Giraldo, J. Insights into adenosine A2A receptor activation through cooperative modulation of agonist and allosteric lipid interactions. PLoS Comput. Biol. 2020, 16, e1007818. [Google Scholar] [CrossRef]
- Bruzzese, A.; Gil, C.; Dalton, J.A.R.; Giraldo, J. Structural insights into positive and negative allosteric regulation of a G protein-coupled receptor through protein-lipid interactions. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sejdiu, B.I.; Tieleman, D.P. Lipid-Protein Interactions Are a Unique Property and Defining Feature of G Protein-Coupled Receptors. Biophys. J. 2020, 118, 1887–1900. [Google Scholar] [CrossRef]
- Bhattarai, A.; Wang, J.; Miao, Y. G-Protein-Coupled Receptor–Membrane Interactions Depend on the Receptor Activation State. J. Comput. Chem. 2020, 41, 460–471. [Google Scholar] [CrossRef]
- Hanson, M.A.; Cherezov, V.; Griffith, M.T.; Roth, C.B.; Jaakola, V.-P.; Chien, E.Y.T.; Velasquez, J.; Kuhn, P.; Stevens, R.C. A Specific Cholesterol Binding Site Is Established by the 2.8 Å Structure of the Human β2-Adrenergic Receptor. Structure 2008, 16, 897–905. [Google Scholar] [CrossRef]
- Bartuzi, D.; Kaczor, A.A.; Matosiuk, D. Interplay between Two Allosteric Sites and Their Influence on Agonist Binding in Human μ Opioid Receptor. J. Chem. Inf. Model. 2016, 56, 563–570. [Google Scholar] [CrossRef]
- Alhadeff, R.; Vorobyov, I.; Yoon, H.W.; Warshel, A. Exploring the free-energy landscape of GPCR activation. Proc. Natl. Acad. Sci. USA 2018, 115, 10327–10332. [Google Scholar] [CrossRef]
- Šali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Qiao, A.; Han, S.; Li, X.; Li, Z.; Zhao, P.; Dai, A.; Chang, R.; Tai, L.; Tan, Q.; Chu, X.; et al. Structural basis of Gs and Gi recognition by the human glucagon receptor. Science 2020, 367, 1346–1352. [Google Scholar] [CrossRef] [PubMed]
- Haga, K.; Kruse, A.C.; Asada, H.; Yurugi-Kobayashi, T.; Shiroishi, M.; Zhang, C.; Weis, W.I.; Okada, T.; Kobilka, B.K.; Haga, T.; et al. Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature 2012, 482, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Louet, M.; Karakas, E.; Perret, A.; Perahia, D.; Martinez, J.; Floquet, N. Conformational restriction of G-proteins Coupled Receptors (GPCRs) upon complexation to G-proteins: A putative activation mode of GPCRs? FEBS Lett. 2013, 587, 2656–2661. [Google Scholar] [CrossRef] [PubMed]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ1 and χ2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef]
- Klauda, J.; Venable, R.; Freites, J.; O’Connor, J.; Tobias, D.; Mondragon-Ramirez, C.; Vorobyov, I.; Mackerell, A.; Pastor, R. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef]
- Lomize, M.A.; Lomize, A.L.; Pogozheva, I.D.; Mosberg, H.I. OPM: Orientations of proteins in membranes database. Bioinformatics 2006, 22, 623–625. [Google Scholar] [CrossRef]
- Doerr, S.; Harvey, M.J.; Noé, F.; De Fabritiis, G. HTMD: High-Throughput Molecular Dynamics for Molecular Discovery. J. Chem. Theory Comput. 2016, 12, 1845–1852. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Søndergaard, C.R.; Olsson, M.H.M.; Rostkowski, M.; Jensen, J.H. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef]
- Olsson, M.H.M.; SØndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Harvey, M.J.; Giupponi, G.; De Fabritiis, G. ACEMD: Accelerating biomolecular dynamics in the microsecond time scale. J. Chem. Theory Comput. 2009, 5, 1632–1639. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089. [Google Scholar] [CrossRef]
- Isberg, V.; De Graaf, C.; Bortolato, A.; Cherezov, V.; Katritch, V.; Marshall, F.H.; Mordalski, S.; Pin, J.P.; Stevens, R.C.; Vriend, G.; et al. Generic GPCR residue numbers-Aligning topology maps while minding the gaps. Trends Pharmacol. Sci. 2015, 36, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Grant, B.J.; Rodrigues, A.P.C.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S.D. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef]
- Wacker, D.; Fenalti, G.; Brown, M.A.; Katritch, V.; Abagyan, R.; Cherezov, V.; Stevens, R.C. Conserved Binding Mode of Human β2 Adrenergic Receptor Inverse Agonists and Antagonist Revealed by X-ray Crystallography. J. Am. Chem. Soc. 2010, 132, 11443–11445. [Google Scholar] [CrossRef]
- Rosenbaum, D.M.; Zhang, C.; Lyons, J.A.; Holl, R.; Aragao, D.; Arlow, D.H.; Rasmussen, S.G.F.; Choi, H.-J.; DeVree, B.T.; Sunahara, R.K.; et al. Structure and function of an irreversible agonist-β2 adrenoceptor complex. Nature 2011, 469, 236–240. [Google Scholar] [CrossRef]
- Zou, Y.; Weis, W.I.; Kobilka, B.K. N-Terminal T4 Lysozyme Fusion Facilitates Crystallization of a G Protein Coupled Receptor. PLoS ONE 2012, 7, e46039. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Olieric, V.; Ma, P.; Howe, N.; Vogeley, L.; Liu, X.; Warshamanage, R.; Weinert, T.; Panepucci, E.; Kobilka, B.; et al. In meso in situ serial X-ray crystallography of soluble and membrane proteins at cryogenic temperatures. Acta Crystallogr. Sect. D Struct. Biol. 2016, 72, 93–112. [Google Scholar] [CrossRef]
- Ma, P.; Weichert, D.; Aleksandrov, L.A.; Jensen, T.J.; Riordan, J.R.; Liu, X.; Kobilka, B.K.; Caffrey, M. The cubicon method for concentrating membrane proteins in the cubic mesophase. Nat. Protoc. 2017, 12, 1745–1762. [Google Scholar] [CrossRef]
- Staus, D.P.; Strachan, R.T.; Manglik, A.; Pani, B.; Kahsai, A.W.; Kim, T.H.; Wingler, L.M.; Ahn, S.; Chatterjee, A.; Masoudi, A.; et al. Allosteric nanobodies reveal the dynamic range and diverse mechanisms of G-protein-coupled receptor activation. Nature 2016, 535, 448–452. [Google Scholar] [CrossRef]
- Ishchenko, A.; Stauch, B.; Han, G.W.; Batyuk, A.; Shiriaeva, A.; Li, C.; Zatsepin, N.; Weierstall, U.; Liu, W.; Nango, E.; et al. Toward G protein-coupled receptor structure-based drug design using X-ray lasers. IUCrJ 2019, 6, 1106–1119. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.G.F.; Choi, H.-J.; Fung, J.J.; Pardon, E.; Casarosa, P.; Chae, P.S.; DeVree, B.T.; Rosenbaum, D.M.; Thian, F.S.; Kobilka, T.S.; et al. Structure of a nanobody-stabilized active state of the β2 adrenoceptor. Nature 2011, 469, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.G.F.; Devree, B.T.; Zou, Y.; Kruse, A.C.; Chung, K.Y.; Kobilka, T.S.; Thian, F.S.; Chae, P.S.; Pardon, E.; Calinski, D.; et al. Crystal structure of the β2adrenergic receptor-Gs protein complex. Nature 2011, 477, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Weichert, D.; Kruse, A.C.; Manglik, A.; Hiller, C.; Zhang, C.; Hubner, H.; Kobilka, B.K.; Gmeiner, P. Covalent agonists for studying G protein-coupled receptor activation. Proc. Natl. Acad. Sci. USA 2014, 111, 10744–10748. [Google Scholar] [CrossRef]
- Liu, X.; Xu, X.; Hilger, D.; Aschauer, P.; Tiemann, J.K.S.; Du, Y.; Liu, H.; Hirata, K.; Sun, X.; Guixà-González, R.; et al. Structural Insights into the Process of GPCR-G Protein Complex Formation. Cell 2019, 177, 1243–1251.e12. [Google Scholar] [CrossRef]
- Masureel, M.; Zou, Y.; Picard, L.-P.; van der Westhuizen, E.; Mahoney, J.P.; Rodrigues, J.P.G.L.M.; Mildorf, T.J.; Dror, R.O.; Shaw, D.E.; Bouvier, M.; et al. Structural insights into binding specificity, efficacy and bias of a β2AR partial agonist. Nat. Chem. Biol. 2018, 14, 1059–1066. [Google Scholar] [CrossRef]
- Nguyen, A.H.; Thomsen, A.R.B.; Cahill, T.J.; Huang, R.; Huang, L.-Y.; Marcink, T.; Clarke, O.B.; Heissel, S.; Masoudi, A.; Ben-Hail, D.; et al. Structure of an endosomal signaling GPCR–G protein–β-arrestin megacomplex. Nat. Struct. Mol. Biol. 2019, 26, 1123–1131. [Google Scholar] [CrossRef]
- Suno, R.; Lee, S.; Maeda, S.; Yasuda, S.; Yamashita, K.; Hirata, K.; Horita, S.; Tawaramoto, M.S.; Tsujimoto, H.; Murata, T.; et al. Structural insights into the subtype-selective antagonist binding to the M2 muscarinic receptor. Nat. Chem. Biol. 2018, 14, 1150–1158. [Google Scholar] [CrossRef]
- Maeda, S.; Qu, Q.; Robertson, M.J.; Skiniotis, G.; Kobilka, B.K. Structures of the M1 and M2 muscarinic acetylcholine receptor/G-protein complexes. Science 2019, 364, 552–557. [Google Scholar] [CrossRef]
- Staus, D.P.; Hu, H.; Robertson, M.J.; Kleinhenz, A.L.W.; Wingler, L.M.; Capel, W.D.; Latorraca, N.R.; Lefkowitz, R.J.; Skiniotis, G. Structure of the M2 muscarinic receptor–β-arrestin complex in a lipid nanodisc. Nature 2020, 579, 297–302. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site Label | Coupling with Δ (Csite) |
|---|---|
| 4 * | 0.76 |
| 3 | 0.61 |
| 7 * | 0.60 |
| 6 * | 0.59 |
| 5 | 0.50 |
| 2 | 0.49 |
| 8 | 0.44 |
| 9 | 0.33 |
| 1 | 0.31 |
| Site Label | Coupling with Δ (Csite) |
|---|---|
| 1 | 0.81 |
| 9 | 0.79 |
| 6 * | 0.77 |
| 7 | 0.76 |
| 5 * | 0.74 |
| 4 | 0.72 |
| 2 | 0.71 |
| 3 | 0.67 |
| 1 | 0.58 |
| Receptor | TM2 | TM6 | TM3 | TM7 |
|---|---|---|---|---|
| β2AR | Y70 | G276 | C125 | I325 |
| GCGR | A175 | L347 | L243 | V398 |
| M2 | Y60 | L390 | L115 | C439 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Renault, P.; Giraldo, J. Dynamical Correlations Reveal Allosteric Sites in G Protein-Coupled Receptors. Int. J. Mol. Sci. 2021, 22, 187. https://doi.org/10.3390/ijms22010187
Renault P, Giraldo J. Dynamical Correlations Reveal Allosteric Sites in G Protein-Coupled Receptors. International Journal of Molecular Sciences. 2021; 22(1):187. https://doi.org/10.3390/ijms22010187
Chicago/Turabian StyleRenault, Pedro, and Jesús Giraldo. 2021. "Dynamical Correlations Reveal Allosteric Sites in G Protein-Coupled Receptors" International Journal of Molecular Sciences 22, no. 1: 187. https://doi.org/10.3390/ijms22010187
APA StyleRenault, P., & Giraldo, J. (2021). Dynamical Correlations Reveal Allosteric Sites in G Protein-Coupled Receptors. International Journal of Molecular Sciences, 22(1), 187. https://doi.org/10.3390/ijms22010187