Molecular Recognition of the HPLC Whelk-O1 Selector towards the Conformational Enantiomers of Nevirapine and Oxcarbazepine

 , ,
, ,  ,
,  and
and 
Abstract
1. Introduction
2. Results and Discussion
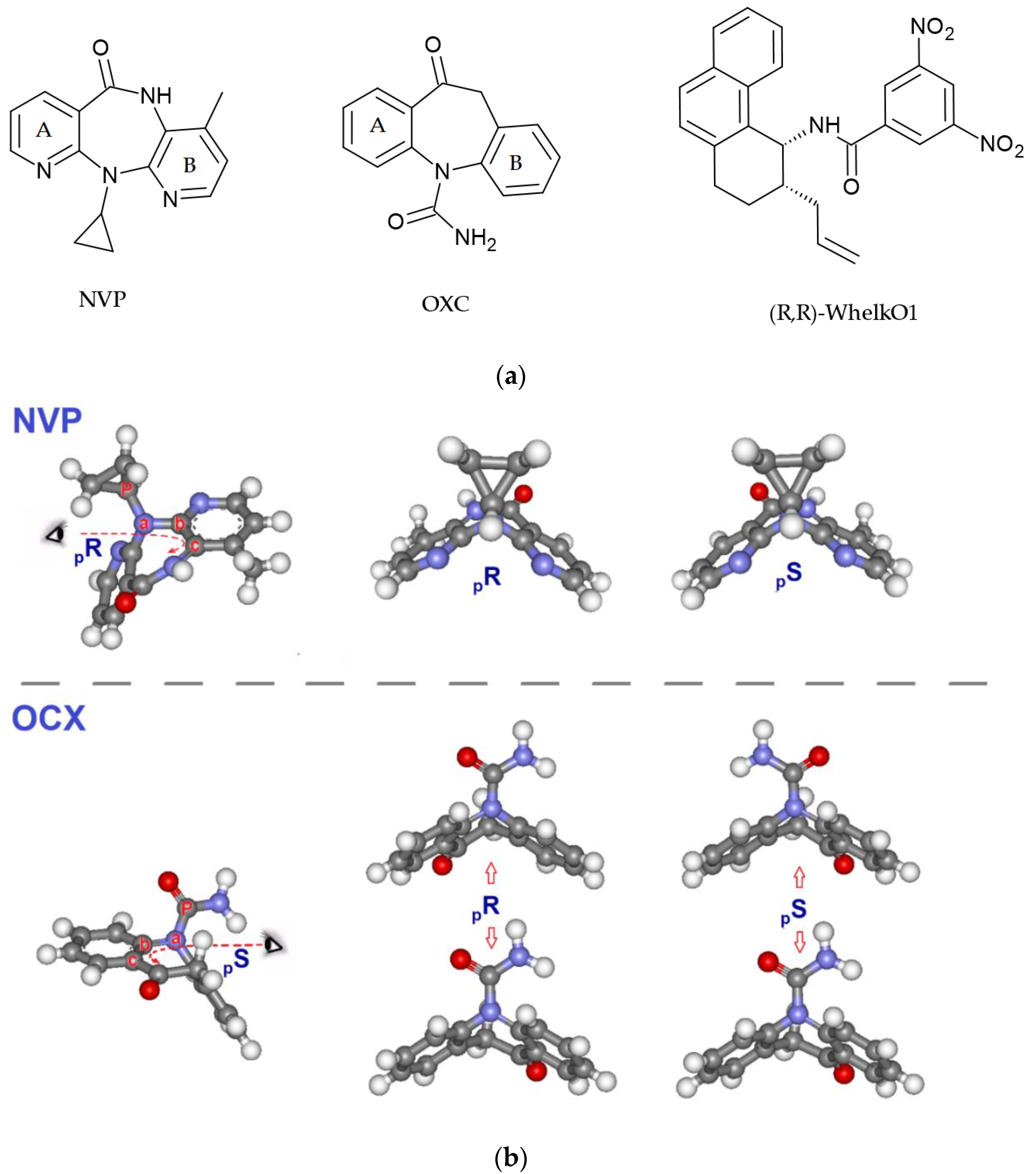
2.1. Chromatographic Retention and Enantioselectivity
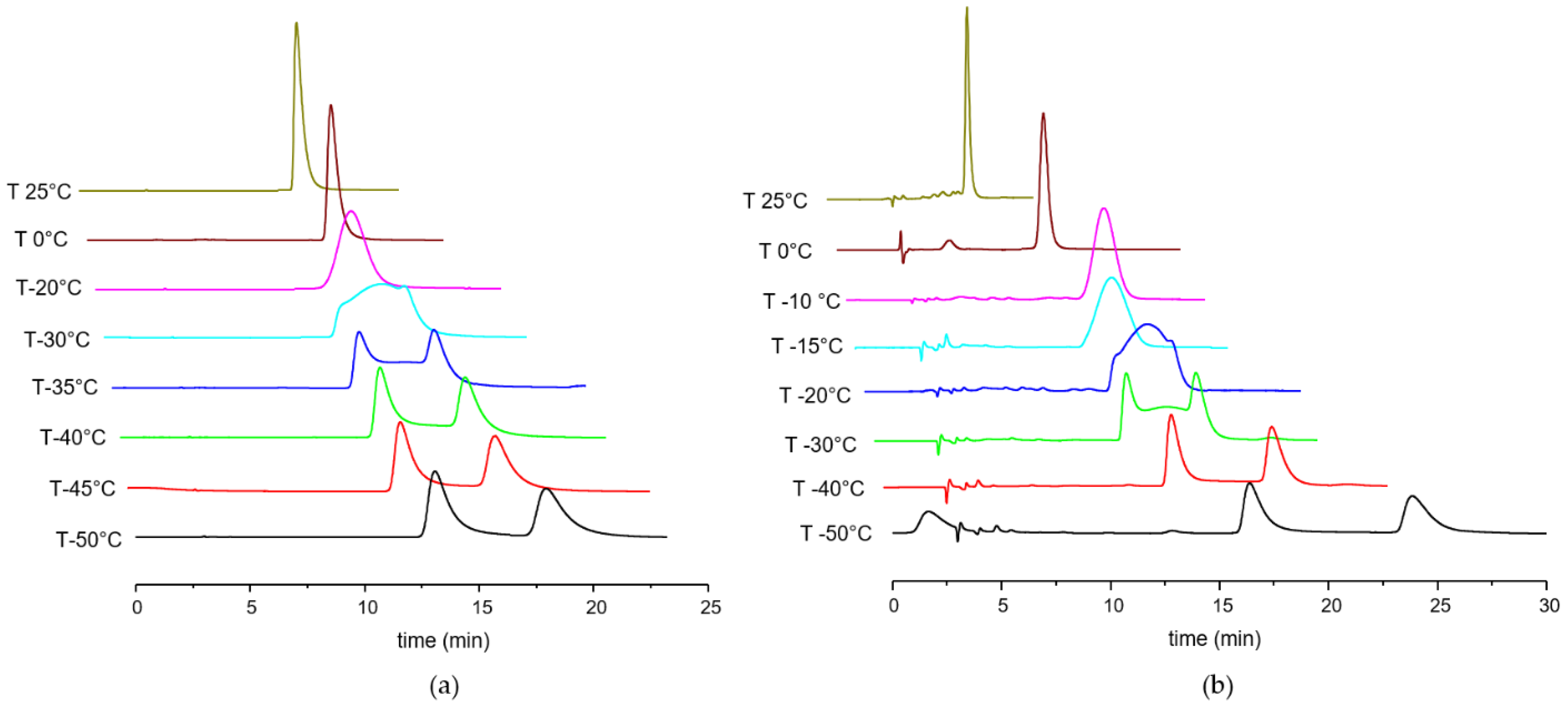
2.2. Variable Temperature HPLC and Enantiomerization Barriers
- Semiempirical AM1 level of calculation of the energetic profile that leads to the change of configuration of such compounds, from the initial (pR) to the final (pS) condition;
- Optimization performed at the ab initio B3LYP/6−31G* density functional level of theory (DFT), of the transition state (TS) geometries of the enantiomerization processes suggested by the energy profile achieved in the previous step;
- The ground states (GS) of NVP and OXC obtained by the conformational search described in Section 2.4.1: “Conformational Analysis of NVP and OXC”;
- Single point energy calculations performed at the wB97X-D/6-311++G** level of theory on the GS and TS geometries obtained for NVP and OXC in the second step.
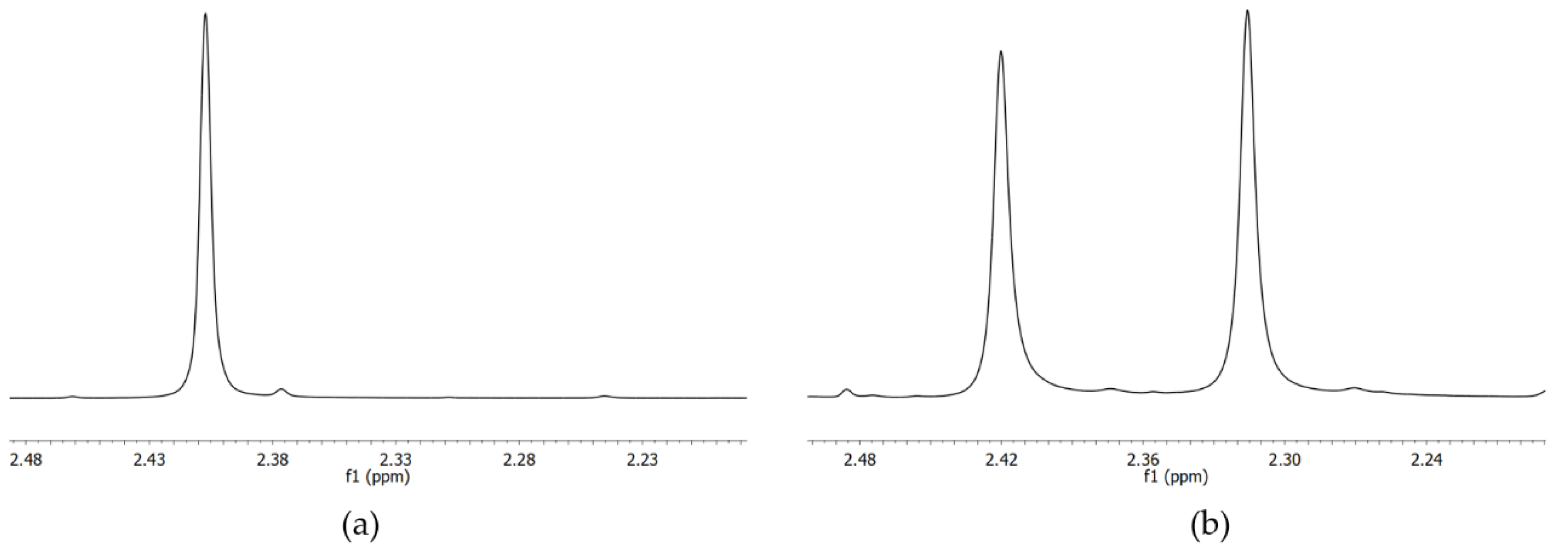
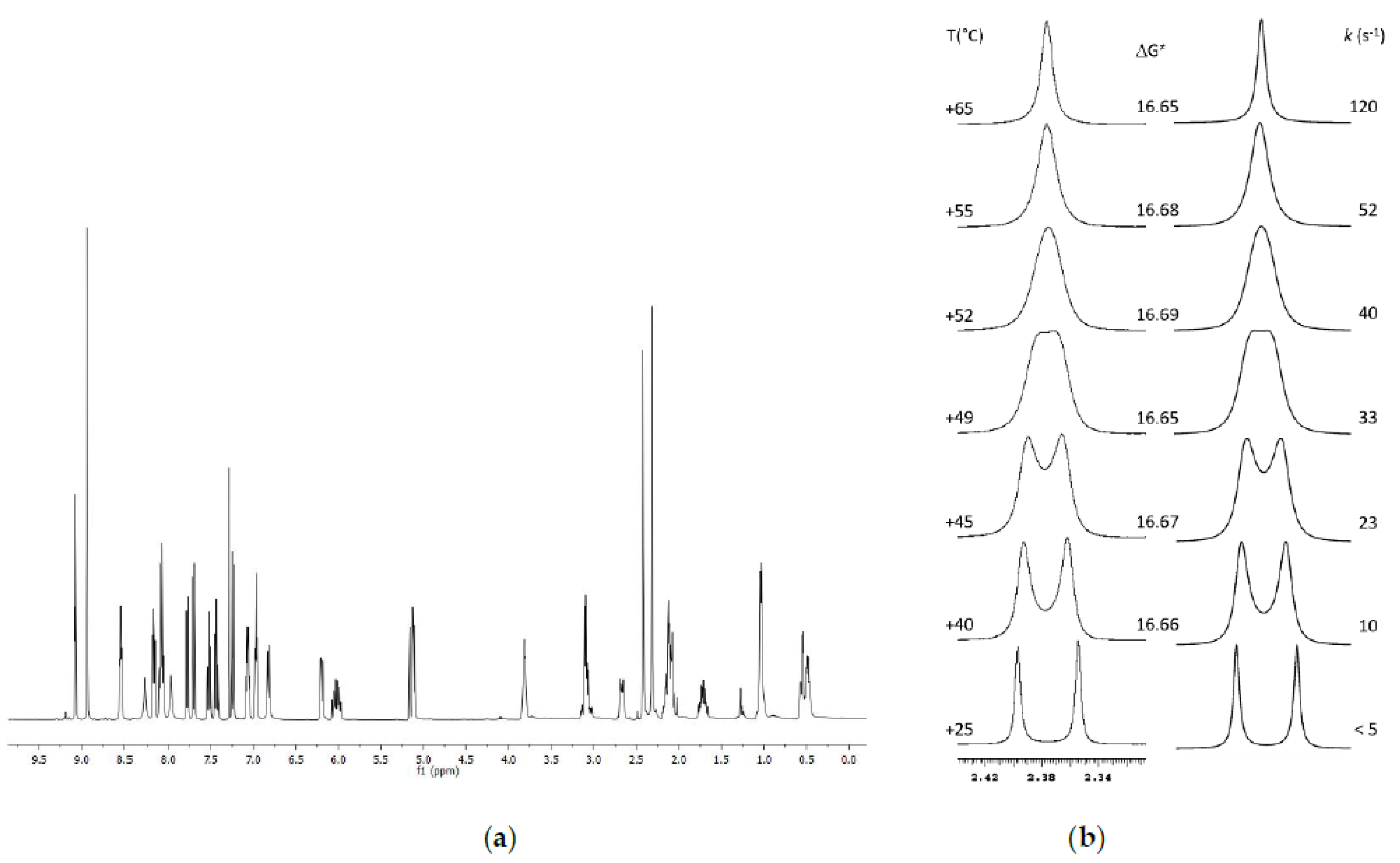
2.3. Variable Temperature NMR
2.3.1. VT-NMR Nevirapine in the Presence of (R,R)-Whelk-O1 as Chiral Solvating Agent
2.3.2. VT-NMR of Oxcarbazepine
2.4. Computational Analysis of the Host–Guest Realtionship between Whelk-O1 and NVP/OXC
2.4.1. Conformational Analysis of NVP and OXC
2.4.2. “Quasi Flexible” Docking Analysis of the Host–Guest System
3. Materials and Methods
3.1. Chemicals and Materials
3.2. Computational Analysis
3.3. HPLC Measurements
3.3.1. Chromatographic Apparatus and Column
3.3.2. Simulation of Dynamic Chromatograms
3.4. NMR Measurements
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Calcaterra, A.; D’Acquarica, I. The market of chiral drugs: Chiral switches versus de novo enantiomerically pure compounds. J. Pharm. Biomed. Anal. 2018, 147, 323–340. [Google Scholar] [CrossRef]
- Nguyen, L.A.; He, H.; Pham-Huy, C. Chiral Drugs: An overview. Int. J. Biomed. Sci. 2006, 2, 85–100. [Google Scholar]
- FDA. FDA’s Policy Statement for the Development of New Stereoisomeric Drugs. Chirality 1992, 4, 338–340. [Google Scholar] [CrossRef]
- Glunz, P.W. Recent encounters with atropisomerism in drug discovery. Bioorg. Med. Chem. Lett. 2018, 28, 53–60. [Google Scholar] [CrossRef]
- LaPlante, S.R.; Fader, D.; Fandrick, K.R.; Fandrick, D.R.; Hucke, O.; Kemper, R.; Miller, S.P.F.; Edwards, P.J. Assessing Atropisomer Axial Chirality in Drug Discovery and Development. J. Med. Chem. 2011, 54, 7005–7022. [Google Scholar] [CrossRef]
- Smyth, J.E.; Butler, N.M.; Keller, P.A. A twist of nature–the significance of atropisomersin biological systems. Nat. Prod. Rep. 2015, 32, 1562–1583. [Google Scholar] [CrossRef]
- Clayden, J.; Moran, W.J.; Edwards, P.J.; LaPlante, S.R. The Challenge of Atropisomerism in Drug Discovery. Angew. Chem. Int. Ed. 2009, 48, 6398–6401. [Google Scholar] [CrossRef]
- Simonyi, M. The Concept of Chiral Conformers and its Significance in Molecular Pharmacology. Adv. Drug Res. 1997, 30, 73–110. [Google Scholar]
- Baker, G.B.; Prior, T.I. Stereochemistry and drug efficacy and development: Relevance of chirality to antidepressant and antipsychotic drugs. Ann. Med. 2002, 34, 537–543. [Google Scholar] [CrossRef]
- Abram, M.; Jakubiec, M.; Kaminski, K. Chirality as an Important Factor for the Development of New Antiepileptic Drugs. ChemMedChem 2019, 14, 1744–1761. [Google Scholar] [CrossRef]
- Eveleigh, P.; Hulme, E.C.; Schudt, C.; Birdsall, N.J. The existence of stable enantiomers of telenzepine and their stereoselective interaction with muscarinic receptor subtypes. Mol. Pharmacol. 1989, 35, 477–483. [Google Scholar]
- LaPlante, S.R.; Forgione, P.; Boucher, C.; Coulombe, R.; Gillard, J.; Hucke, O.; Jakalian, A.; Joly, M.; Kukolj, G.; Lemke, C.; et al. Enantiomeric Atropisomers Inhibit HCV Polymerase and/or HIV Matrix: Characterizing Hindered Bond Rotations and Target Selectivity. J. Med. Chem. 2014, 57, 1944–1951. [Google Scholar] [CrossRef]
- Yan, T.Q.; Riley, F.; Philippe, L.; Davoren, J.; Cox, L.; Orozco, C.; Rai, B.; Hardink, M. Chromatographic resolution of atropisomers for toxicity and biotransformation studies in pharmaceutical research. J. Chromatogr. A 2015, 1398, 108–120. [Google Scholar] [CrossRef]
- Zhang, L.; Hu, Y.; Galella, E.; Tomasella, F.P.; Fish, W.P. Separation of atropisomers by chiral liquid chromatography and thermodynamic analysis of separation mechanism. J. Pharm. Anal. 2017, 7, 156–162. [Google Scholar] [CrossRef]
- Dai, J.; Wang, C.; Traeger, S.C.; Discenza, L.; Obermeier, M.T.; Tymiak, A.A.; Zhang, Y. The role of chromatographic and chiroptical spectroscopic techniques and methodologies in support of drug discovery for atropisomeric drug inhibitors of Bruton’s tyrosine kinase. J. Chromatogr. A 2017, 1487, 116–128. [Google Scholar] [CrossRef]
- Salvadori, P.; Bertucci, C.; Ascoli, G.; Uccello-Barretta, G.; Rossi, E. Direct resolution, characterization, and stereospecific binding properties of an atropisomeric 1,4-benzodiazepine. Chirality 1997, 9, 495–505. [Google Scholar] [CrossRef]
- Costil, R.; Sterling, A.J.; Duarte, F.; Clayden, J. Atropisomerism in Diarylamines: Structural Requirements and Mechanisms of Conformational Interconversion. Angew. Chem. 2020, 132, 2–11. [Google Scholar] [CrossRef]
- Ciogli, A.; Kumar, S.V.; Mancinelli, M.; Mazzanti, A.; Perumal, S.; Severi, C.; Villani, C. Atropisomerism in 3-arylthiazolidine-2-thiones. A combined dynamic NMR and dynamic HPLC study. Org. Biomol. Chem. 2016, 14, 11137–11147. [Google Scholar] [CrossRef]
- Casarini, D.; Lunazzi, L.; Alcaro, S.; Gasparrini, F.; Villani, C. Atropisomerism in Hindered Naphthyl Sulfones Investigated by Dynamic NMR and Dynamic HPLC Techniques. J. Org. Chem. 1995, 60, 5515–5519. [Google Scholar] [CrossRef]
- Gasparrini, F.; Lunazzi, L.; Misiti, D.; Villani, C. Organic Stereochemistry and Conformational Analysis from Enantioselective Chromatography and Dynamic Nuclear Magnetic Resonance Measurements. Acc. Chem. Res. 1995, 28, 163–170. [Google Scholar] [CrossRef]
- Mortera, S.L.; Sabia, R.; Pierini, M.; Gasparrini, F.; Villani, C. The dynamic chromatographic behavior of tri-o-thymotide on HPLC chiral stationary phases. Chem. Commun. 2012, 48, 3167–3169. [Google Scholar] [CrossRef]
- Ward, T.J.; Ward, K.D. Chiral Separations: A Review of Current Topics and Trends. Anal. Chem. 2012, 84, 626–635. [Google Scholar] [CrossRef]
- Fernandes, C.; Phyo, Y.Z.; Silva, A.S.; Tiritan, M.E.; Kijjoa, A.; Pinto, M.M.M. Chiral Stationary Phases Based on Small Molecules: An Update of the Last 17 Years. Sep. Purif. Rev. 2018, 47, 89–123. [Google Scholar] [CrossRef]
- Lämmerhofer, M. Chiral recognition by enantioselective liquid chromatography: Mechanisms and modern chiral stationary phases. J. Chromatogr. A 2010, 1217, 814–856. [Google Scholar] [CrossRef]
- Lipkowitz, K.B. Theoretical studies of brush-type chiral stationary phases. J. Chromatogr. A 1994, 666, 493–503. [Google Scholar] [CrossRef]
- Lipkowitz, K.B. Atomistic modeling of enantioselection in chromatography. J. Chromatogr. A 2001, 906, 417–442. [Google Scholar] [CrossRef]
- Del Rio, A. Exploring enantioselective molecular recognition mechanisms with chemoinformatic techniques. J. Sep. Sci. 2009, 32, 1566–1584. [Google Scholar] [CrossRef]
- Berthod, A. Chiral recognition mechanism. Anal. Chem. 2006, 78, 2093–2099. [Google Scholar] [CrossRef]
- Gasparrini, F.; Misiti, D.; Still, C.W.; Villani, C.; Wennemers, H. Enantioselective and Diastereoselective Binding Study of Silica Bound Macrobicyclic Receptors by HPLC. J. Org. Chem. 1997, 62, 8221–8224. [Google Scholar] [CrossRef]
- Alcaro, S.; Gasparrini, F.; Incani, O.; Mecucci, S.; Misiti, D.; Pierini, M.; Villani, C. A “quasi-flexible” automatic docking processing for studying stereoselective recognition mechanisms. Part I. Protocol validation. J. Comput. Chem. 2000, 21, 515–530. [Google Scholar] [CrossRef]
- Yang, L.; Wenzel, T.; Williamson, R.T.; Christensen, M.; Schafer, W.; Welch, C.J. Expedited Selection of NMR Chiral Solvating Agents for Determination of Enantiopurity. ACS Cent. Sci. 2016, 2, 332–340. [Google Scholar] [CrossRef]
- Casarini, D.; Davalli, S.; Lunazzi, L.; Macciantelli, D. Conformation studies by dynamic NMR. 37. Monitoring the stereomutations of symmetric amines by a chiral auxiliary agent. J. Org. Chem. 1989, 54, 4616–4619. [Google Scholar] [CrossRef]
- Pirkle, W.H.; Welch, C.J. Chromatographic and ‘H NMR support for a proposed chiral recognition model. J. Chromatogr. A 1994, 483, 347–353. [Google Scholar] [CrossRef]
- Paizs, B.; Simonyi, M. Ring inversion barrier of diazepam and derivatives: An ab Initio study. Chirality 1999, 11, 651–658. [Google Scholar] [CrossRef]
- Tabata, H.; Wada, N.; Takada, Y.; Oshitari, T.; Takahashi, H.; Natsugari, H. Isolation and Characterization of Atropisomers of Seven-Membered-Ring Benzolactams. J. Org. Chem. 2011, 76, 5123–5131. [Google Scholar] [CrossRef]
- Kanase, Y.; Kuniyoshi, M.; Tabata, H.; Takahashi, Y.; Kayama, S.; SWakamtsu, S.; Oshitari, T.; Natsugari, H.; Takahashi, H. Freezing the Butterfly Motion of Carbamazepine Derivatives. Synthesis 2015, 47, 3907–3913. [Google Scholar]
- Pirkle, W.H.; Welch, C.J.; Lamm, B. Design, Synthesis, and Evaluation of an Improved Enantioselective Naproxen Selector. J. Org. Chem. 1992, 57, 3854–3860. [Google Scholar] [CrossRef]
- Fernandes, C.; Palmeira, A.; Santos, A.; Tiritan, M.E.; Afonso, C.; Pinto, M.M. Enantioresolution of Chiral Derivatives of Xanthones on (S,S)-Whelk-O1 and L-Phenylglycine Stationary Phases and Chiral Recognition Mechanism by Docking Approach for (S,S)-Whelk-O1. Chirality 2013, 25, 89–100. [Google Scholar] [CrossRef]
- Zhao, C.F.; Cann, N.M. Molecular Dynamics Study of Chiral Recognition for the Whelk-O1 Chiral Stationary Phase. Anal. Chem. 2008, 80, 2426–2438. [Google Scholar] [CrossRef]
- Trapp, O.; Schoetz, G.; Schurig, V. Determination of enantiomerization barriers by dynamic and stopped-flow chromatographic methods. Chirality 2001, 13, 403–414. [Google Scholar] [CrossRef]
- Wolf, C. Stereolabile chiral compounds: Analysis by dynamic chromatography and stopped-flow methods. Chem. Soc. Rev. 2005, 34, 595–608. [Google Scholar] [CrossRef] [PubMed]
- D’Acquarica, I.; Gasparrini, F.; Pierini, M.; Villani, C.; Zappia, G. Dynamic HPLC on chiral stationary phases: A powerful tool for the investigation of stereomutation processes. J. Sep. Sci. 2006, 29, 1508–1516. [Google Scholar] [CrossRef] [PubMed]
- Sabia, R.; Ciogli, A.; Pierini, M.; Gasparrini, F.; Villani, C. Dynamic high performance liquid chromatography on chiral stationary phases. Low temperature separation of the interconverting enantiomers of diazepam, flunitrazepam, prazepam and tetrazepam. J. Chromatogr. A 2014, 1363, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Alcaro, S.; Gasparrini, F.; Incani, O.; Caglioti, L.; Pierini, M.; Villani, C. “Quasi flexible” automatic docking processing for studying stereoselective recognition mechanisms, part 2: Prediction of ΔΔG of complexation and 1H-NMR NOE correlation. J. Comput. Chem. 2007, 28, 1119–1128. [Google Scholar] [CrossRef] [PubMed]
- Keller, R.A.; Giddings, J.C. Multiple zones and spots in chromatography. J. Chromatogr. A 1960, 3, 205–220. [Google Scholar] [CrossRef]
- Kramer, R. Simultan-reaktionsgaschromatographie mit reversible reaktion erster ordnung I. J. Chromatogr. A 1975, 107, 241–252. [Google Scholar] [CrossRef]
- Jung, M. Simul: A general program for the simulation of chromatograms using the theoretical plate model. QCPE Bull. 1992, 12, 1–52. [Google Scholar]
- Cabrera, K.; Jung, M.; Fluck, M.; Schurig, V. Determination of enantiomerization barriers by computer simulation of experimental elution profiles obtained by high-performance liquid chromatography on a chiral stationary phase. J. Chromatogr. A 1996, 731, 315–321. [Google Scholar] [CrossRef]
- Schurig, V.; Bürkle, W. Extending the scope of enantiomer resolution by complexation gas chromatography. J. Am. Chem. Soc. 1982, 104, 7573–7580. [Google Scholar] [CrossRef]
- Bürkle, W.; Karfunkel, H.; Schurig, V. Dynamic phenomena during enantiomer resolution by complexation gas chromatography: A kinetic study of enantiomerization. J. Chromatogr. A 1984, 288, 1–14. [Google Scholar] [CrossRef]
- Veciana, J.; Crespo, M.I. Dynamic HPLC: A method for determining rate constants, energy barriers, and equilibrium constants of molecular dynamic processes. Angew. Chem. Int. Ed. 1991, 30, 74–76. [Google Scholar] [CrossRef]
- Gasparrini, F.; Misiti, D.; Pierini, M.; Villani, C. Enantiomerization barriers by dynamic HPLC. Stationary phase effects. Tetrahedron Asymmetry 1997, 8, 2069–2073. [Google Scholar] [CrossRef]
- Oxelbark, J.; Allenmark, S. Barriers to stereoinversion of N-aryl-1,3,2-benzodithiazole 1-oxides studied by dynamic enantioselective liquid chromatography. J. Org. Chem. 1999, 64, 1483–1486. [Google Scholar] [CrossRef] [PubMed]
- Gasparrini, F.; D’Acquarica, I.; Pierini, M.; Villani, C. Chromatographic resolution and enantiomerization barriers of axially chiral 1-naphthamides. J. Sep. Sci. 2001, 24, 941–946. [Google Scholar] [CrossRef]
- Wolf, C. Dynamic Stereochemistry of Chiral Compounds: Principles and Applications; RSC Publishing: London, UK, 2008. [Google Scholar]
- Trapp, O. Unified equation for access to rate constants of first-order reactions in dynamic and on-column reaction chromatography. Anal. Chem. 2006, 78, 189–198. [Google Scholar] [CrossRef]
- Gasparrini, F.; Lunazzi, L.; Mazzanti, A.; Pierini, M.; Pietrusiewicz, K.M.; Villani, C. Comparison of dynamic HPLC and dynamic NMR in the study of conformational stereodynamics: Case of the enantiomers of a hindered secondary phosphine oxide. J. Am. Chem. Soc. 2000, 122, 4735–4738. [Google Scholar] [CrossRef]
- Dalla Cort, A.; Gasparrini, F.; Lunazzi, L.; Mandolini, L.; Mazzanti, A.; Pasquini, C.; Pierini, M.; Rompietti, R.; Schiaffino, L. Stereomutations of atropisomers of sterically hindered salophen ligands. J. Org. Chem. 2005, 70, 8877–8883. [Google Scholar] [CrossRef]
- Dell’Erba, C.; Gasparrini, F.; Grilli, S.; Lunazzi, L.; Mazzanti, A.; Novi, M.; Pierini, M.; Tavani, C.; Villani, C. Conformational studies by dynamic NMR. 86. Structure, stereodynamics and cryogenic enantioseparation of the stereolabile isomers of o-dinaphthylphenyl derivatives. J. Org. Chem. 2002, 67, 1663–1668. [Google Scholar]
- Cirilli, R.; Ferretti, R.; La Torre, F.; Secci, D.; Bolasco, A.; Carradori, S.; Pierini, M. High-performance liquid chromatographic separation of enantiomers and diastereomers of 2-methylcyclohexanone thiosemicarbazone, and determination of absolute configuration and configurational stability. J. Chromatogr. A 2007, 1172, 160–169. [Google Scholar] [CrossRef]
- Carradori, S.; Cirilli, R.; Dei Cicchi, S.; Ferretti, R.; Menta, S.; Pierini, M.; Secci, D. 3-Methylcyclohexanone thiosemicarbazone: Determination of E/Z isomerization barrier by dynamic high-performance liquid chromatography, configuration assignment and theoretical study of the mechanisms involved by the spontaneous, acid and base catalyzed processes. J. Chromatogr. A 2012, 1269, 168–177. [Google Scholar]
- Cirilli, R.; Costi, R.; Di Santo, R.; La Torre, F.; Pierini, M.; Siani, G. Perturbing effects of chiral stationary phase on enantiomerization second-order rate constants determined by enantioselective dynamic high-performance liquid chromatography: A practical tool to quantify the accessible acid and basic catalytic sites bonded on chromatographic supports. Anal. Chem. 2009, 81, 3560–3570. [Google Scholar] [PubMed]
- Cabri, W.; D’Acquarica, I.; Simone, P.; Di Iorio, M.; Di Mattia, M.; Gasparrini, F.; Giorgi, F.; Mazzanti, A.; Pierini, M.; Quaglia, M.; et al. Stereolability of dihydroartemisinin, an antimalarial drug: A comprehensive kinetic investigation. Part 2. J. Org. Chem. 2011, 76, 4831–4840. [Google Scholar] [CrossRef]
- Lunazzi, L.; Mancinelli, M.; Mazzanti, A.; Pierini, M. Stereomutation of axially chiral coumarins. J. Org. Chem. 2010, 75, 5927–5933. [Google Scholar] [CrossRef] [PubMed]
- Gasparrini, F.; Pierini, M.; Villani, C.; De Maria, P.; Fontana, A.; Ballini, R. Enantiomerization study of some α-nitroketones by dynamic high resolution gas chromatography. J. Org. Chem. 2003, 68, 3173–3177. [Google Scholar] [CrossRef]
- Uray, G.; Jahangir, S.; Fabian, W.M.F. On- and off column enantiomerizations of 4,4-bisquinolin-2-ones: A comparison of Auto-DHPLC-y2k and DCXplorer calculated thermodynamic data generated by dynamic high, performance liquid chromatography with theoretically calculated data. J. Chromatogr. A 2010, 1217, 1017–1023. [Google Scholar] [CrossRef]
- Villani, C.; Gasparrini, F.; Pierini, M.; Mortera, S.L.; D’Acquarica, I.; Ciogli, A.; Zappia, G. Dynamic HPLC of stereolabile iron(II) complexes on chiral stationary phases. Chirality 2009, 21, 97–103. [Google Scholar] [CrossRef]
- Ciogli, A.; Bicker, W.; Lindner, W. Determination of enantiomerizations barriers of hypericin and pseudo hypericin by dynamic high-performance liquid chromatography on immobilized polysaccharide-type chiral stationary phases and off-column racemization experiments. Chirality 2010, 22, 463–471. [Google Scholar]
- Rizzo, S.; Benincori, T.; Bonometti, V.; Cirilli, R.; Mussini, P.R.; Pierini, M.; Pilati, T.; Sannicolò, F. Steric and electronic effects on the configurational stability of residual chiral phosphorus-centered three-bladed propellers: Tris-aryl phosphanes. Chem. Eur. J. 2013, 19, 182–194. [Google Scholar] [CrossRef]
- Casarini, D.; Lunazzi, L.; Mazzanti, A. Recent Advances in Stereodynamics and Conformational Analysis by Dynamic NMR and Theoretical Calculations. Eur. J. Org. Chem. 2010, 2010, 2035–2056. [Google Scholar] [CrossRef]
- Burke, E.W.D.; Morris, G.A.; Vincent, M.A.; Hillier, I.H.; Clayden, J. Is nevirapine atropisomeric? Experimental and computational evidence for rapid conformational inversion. Org. Biomol. Chem. 2012, 10, 716–719. [Google Scholar] [CrossRef]
- Brown, J.H.; Bushweller, C.H. QCPE Bulletin, A Windows-based PC version of program QCPE 633 is also available from the authors (A.M.); Quantum Chemistry Program Exchange (QCPE): Bloomington, IN, USA, 1983; Volume 3, pp. 102–103. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | T Column °C | k’1,2 | k’2,1 | α |
|---|---|---|---|---|
| NVP 1 | −50 | 3.36 | 4.99 | 1.48 |
| OXC 2 | −50 | 4.47 | 6.95 | 1.55 |
| Compound | T Column °C | ΔGⱡ12 kcal/mol | ΔGⱡ21 kcal/mol |
|---|---|---|---|
| NVP | −30 | 16.82 | 16.96 |
| −35 | 17.01 | 17.14 | |
| −40 | 17.08 | 17.22 | |
| OXC | −20 | 17.55 | 17.68 |
| −30 | 17.30 | 17.42 | |
| −40 | 17.39 | 17.53 |
| Compound | ΔG‡ NMR (kcal/mol) | T (°C) | ΔG‡ HPLC (kcal/mol) | T (°C) | ΔG‡ DFT (kcal/mol) | T (°C) |
|---|---|---|---|---|---|---|
| Nevirapine (NVP) | 16.7 | 49–52 | ΔG‡1→2 16.82 | −30 | 15.9 | 25 |
| ΔG‡2→1 16.96 | −30 | |||||
| Oxcarbazepine (OXC) | 17.7 | 33–133 | ΔG‡1→2 17.55 | −30 | 16.9 | 25 |
| DG‡2→1 17.68 | −30 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franzini, R.; Pierini, M.; Mazzanti, A.; Iazzetti, A.; Ciogli, A.; Villani, C. Molecular Recognition of the HPLC Whelk-O1 Selector towards the Conformational Enantiomers of Nevirapine and Oxcarbazepine. Int. J. Mol. Sci. 2021, 22, 144. https://doi.org/10.3390/ijms22010144
Franzini R, Pierini M, Mazzanti A, Iazzetti A, Ciogli A, Villani C. Molecular Recognition of the HPLC Whelk-O1 Selector towards the Conformational Enantiomers of Nevirapine and Oxcarbazepine. International Journal of Molecular Sciences. 2021; 22(1):144. https://doi.org/10.3390/ijms22010144
Chicago/Turabian StyleFranzini, Roberta, Marco Pierini, Andrea Mazzanti, Antonia Iazzetti, Alessia Ciogli, and Claudio Villani. 2021. "Molecular Recognition of the HPLC Whelk-O1 Selector towards the Conformational Enantiomers of Nevirapine and Oxcarbazepine" International Journal of Molecular Sciences 22, no. 1: 144. https://doi.org/10.3390/ijms22010144
APA StyleFranzini, R., Pierini, M., Mazzanti, A., Iazzetti, A., Ciogli, A., & Villani, C. (2021). Molecular Recognition of the HPLC Whelk-O1 Selector towards the Conformational Enantiomers of Nevirapine and Oxcarbazepine. International Journal of Molecular Sciences, 22(1), 144. https://doi.org/10.3390/ijms22010144