Chronic Effects of a High Sucrose Diet on Murine Gastrointestinal Nutrient Sensor Gene and Protein Expression Levels and Lipid Metabolism

 , ,
, ,
Abstract
1. Introduction
2. Results
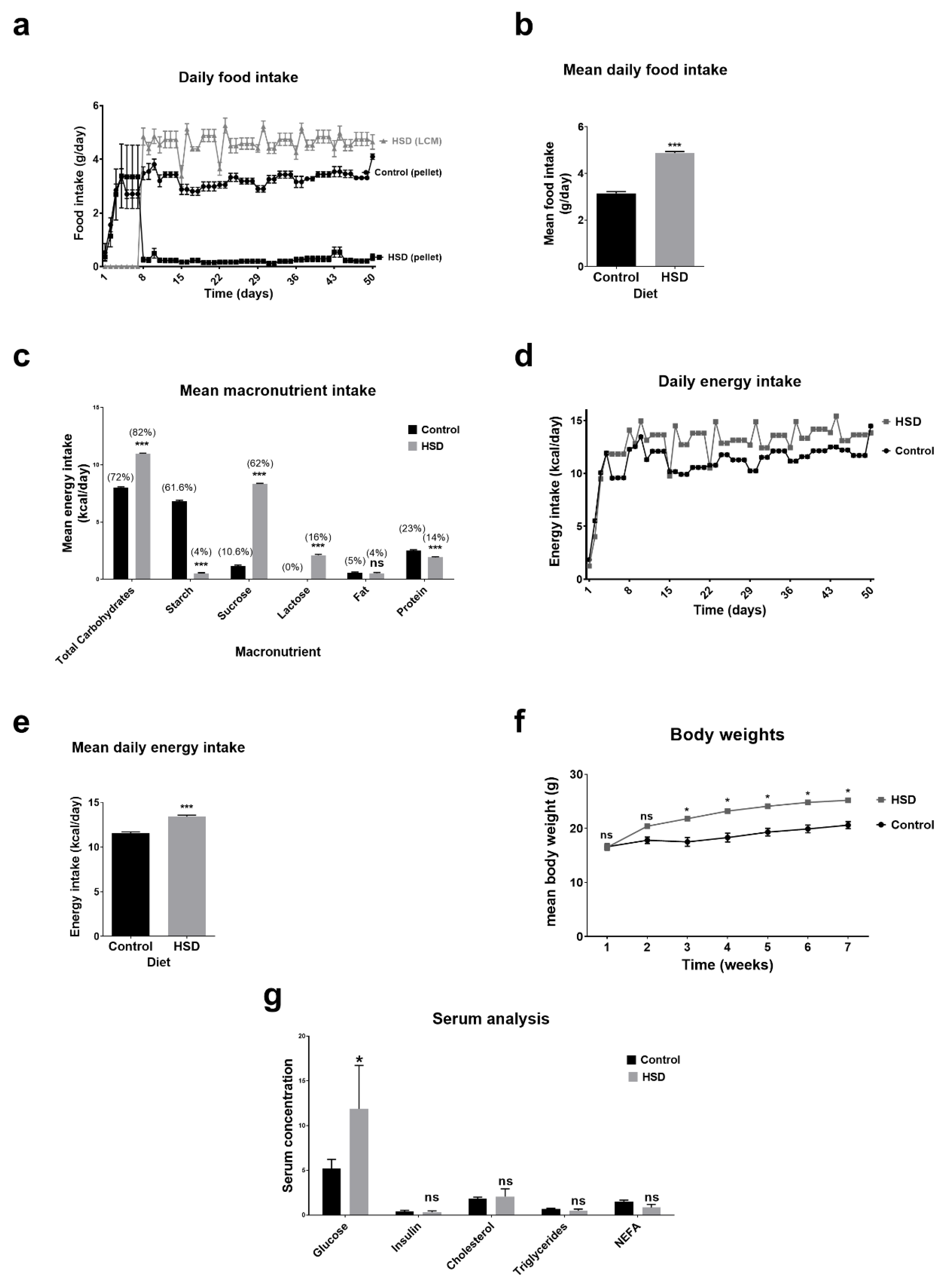
2.1. Dietary Intake, Anthropometry and Blood Biochemistry Analysis of Control and Sucrose Fed Mice
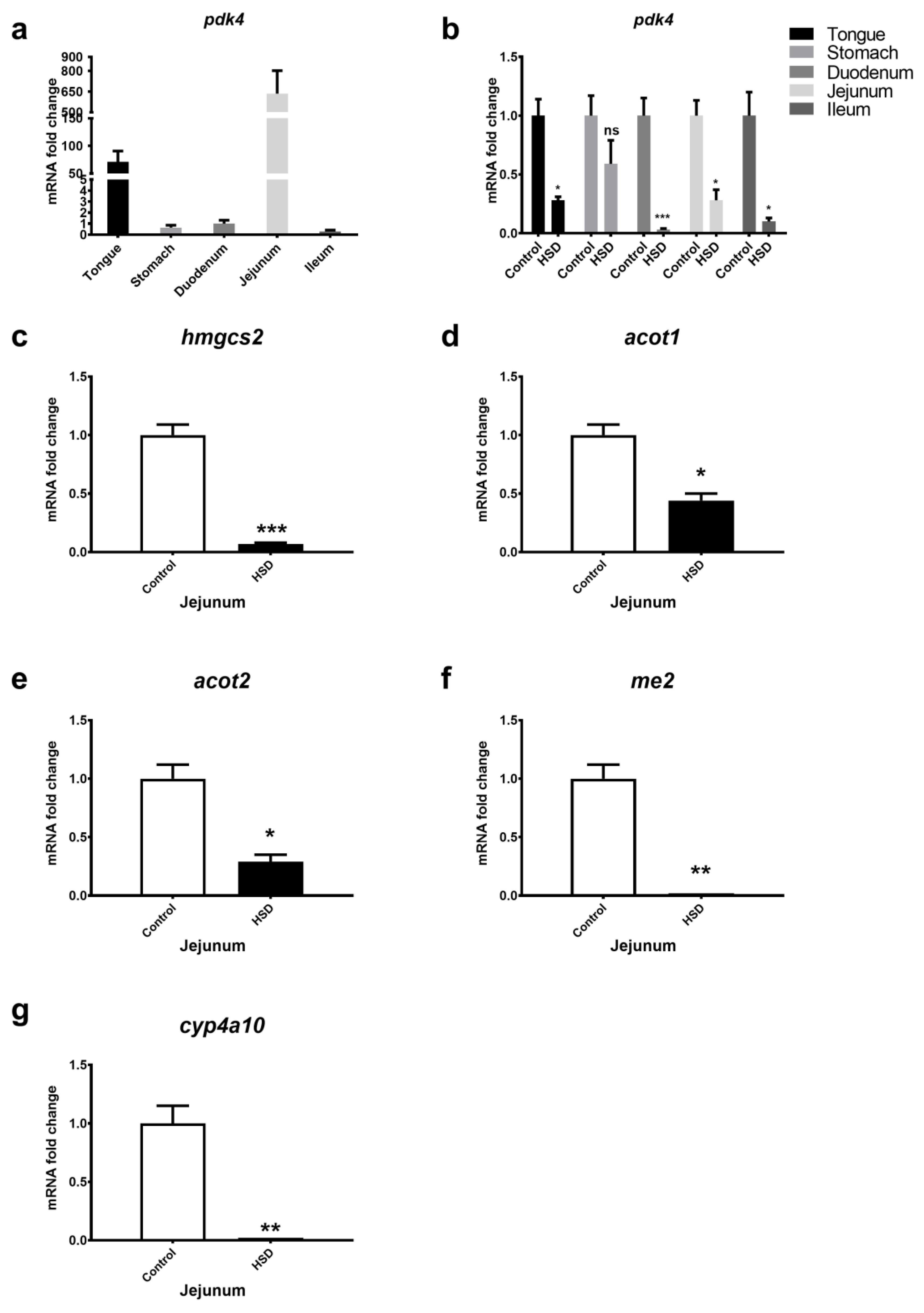
2.2. qPCR Analysis of Small Intestinal Fat Oxidation Genes
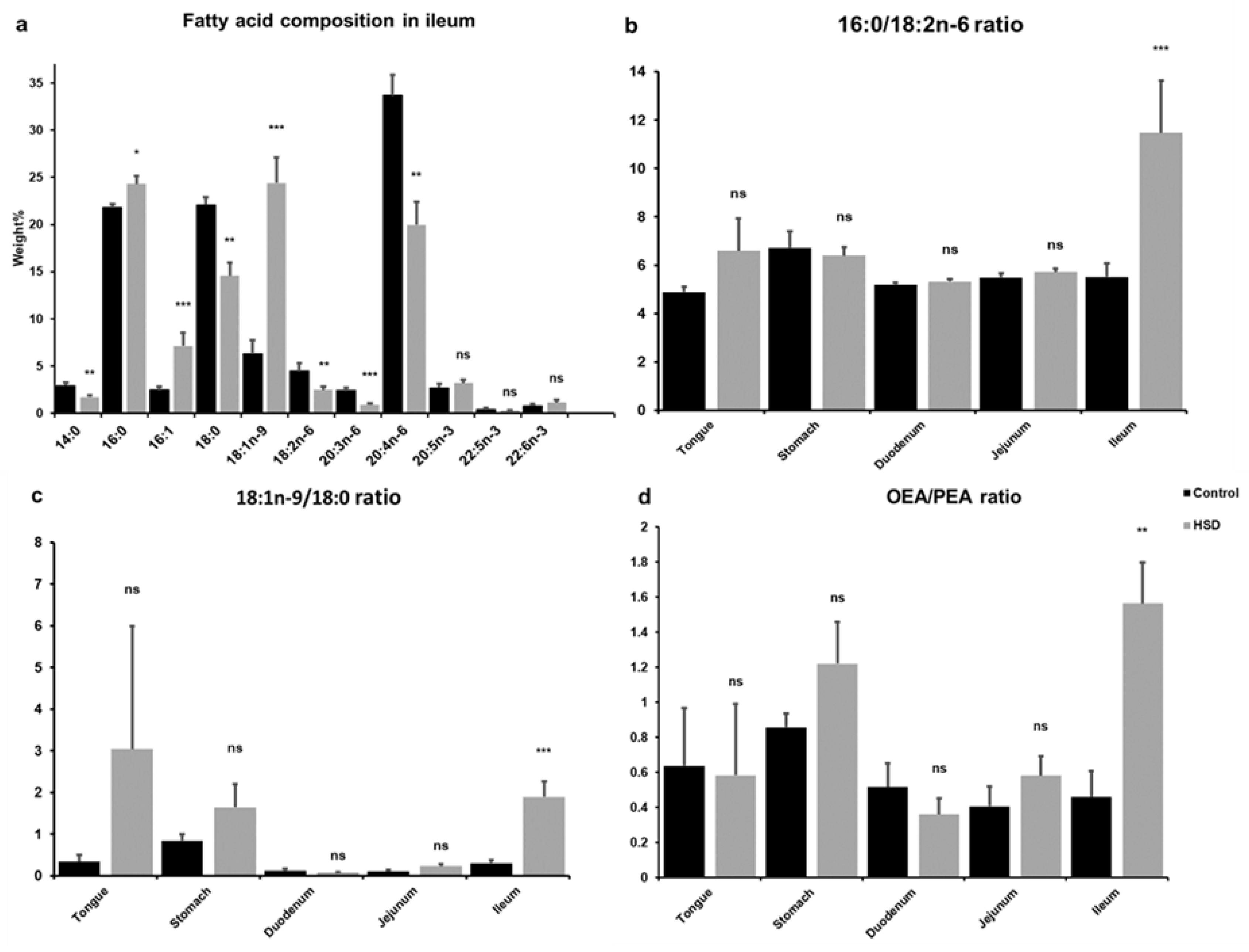
2.3. Analysis of Intestinal Lipid Metabolites
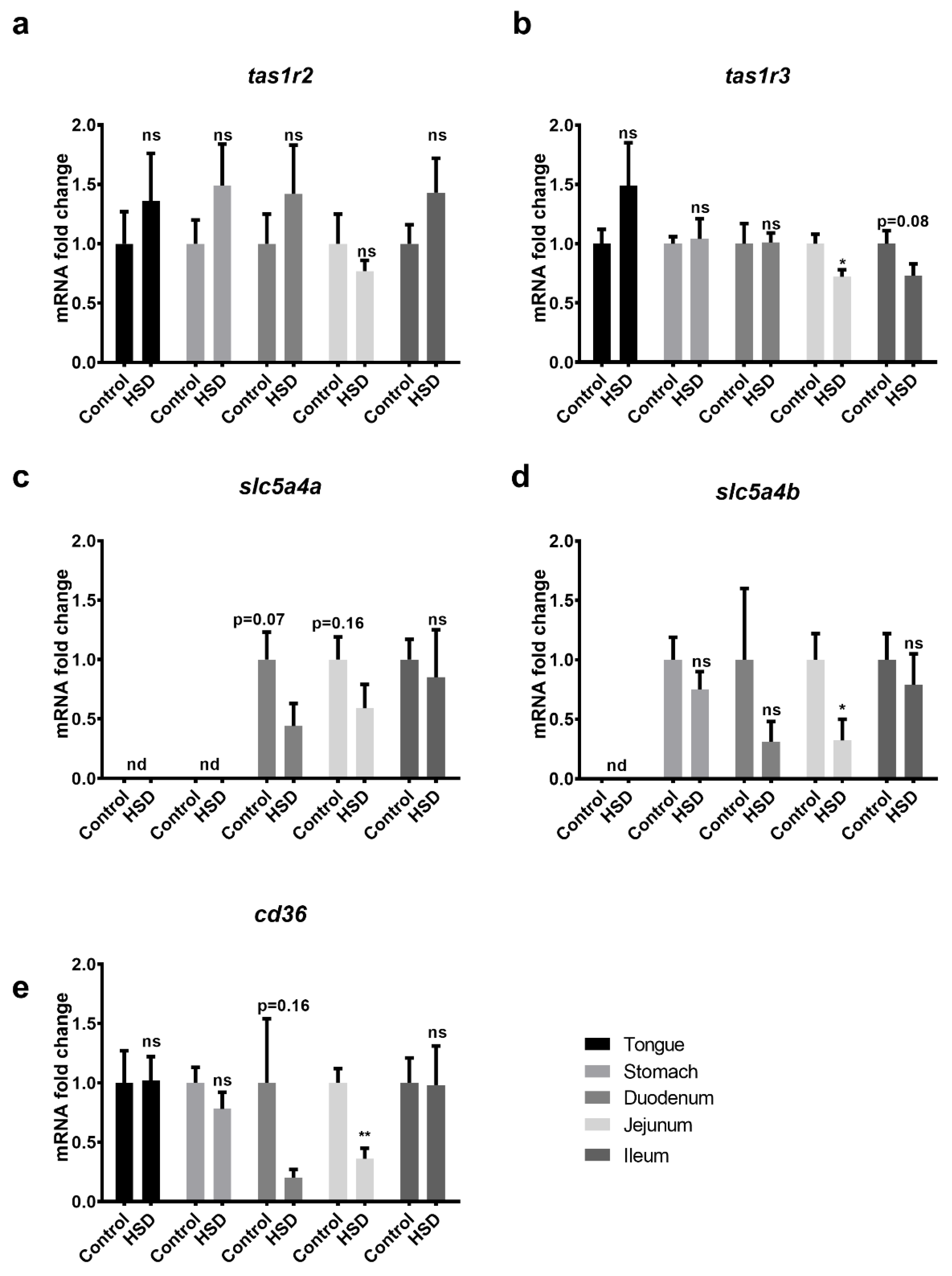
2.4. Validation of Microarray Data Obtained from the Small Intestines of Control and Sucrose Fed Mice
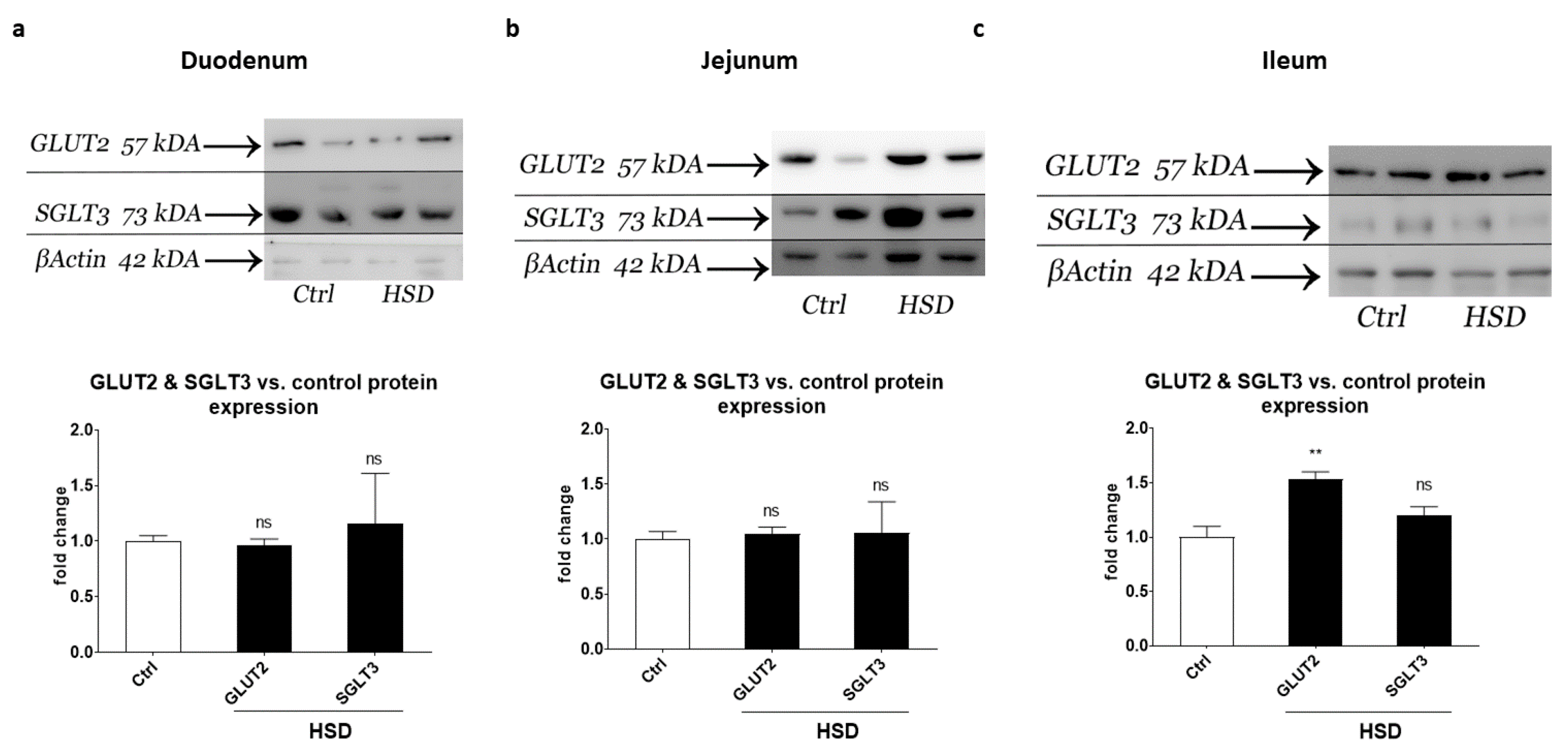
2.5. Western Blot Analysis of Small Intestinal Sugar Sensors and Transporters
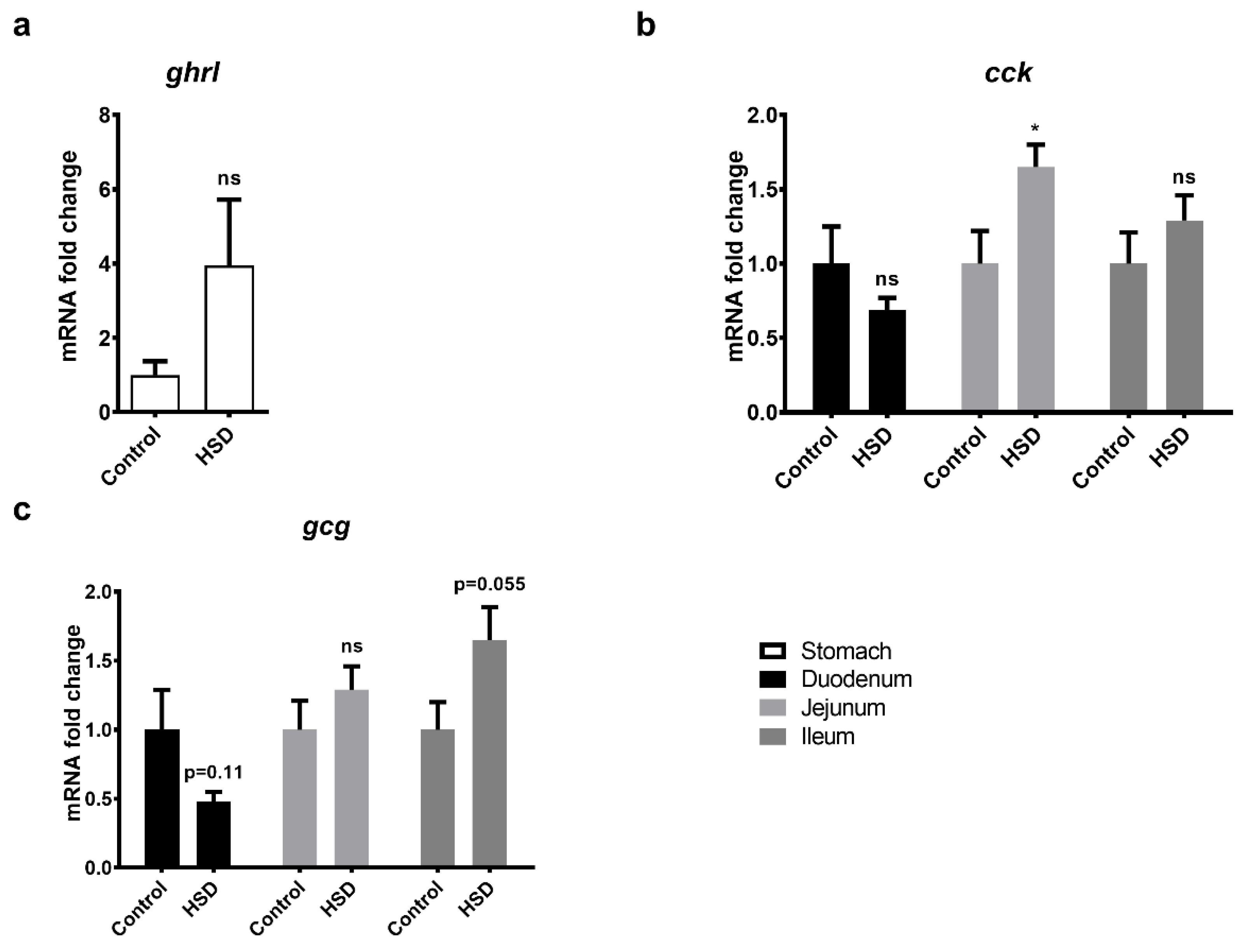
2.6. qPCR Analysis of Gut Peptide Gene Expression Levels
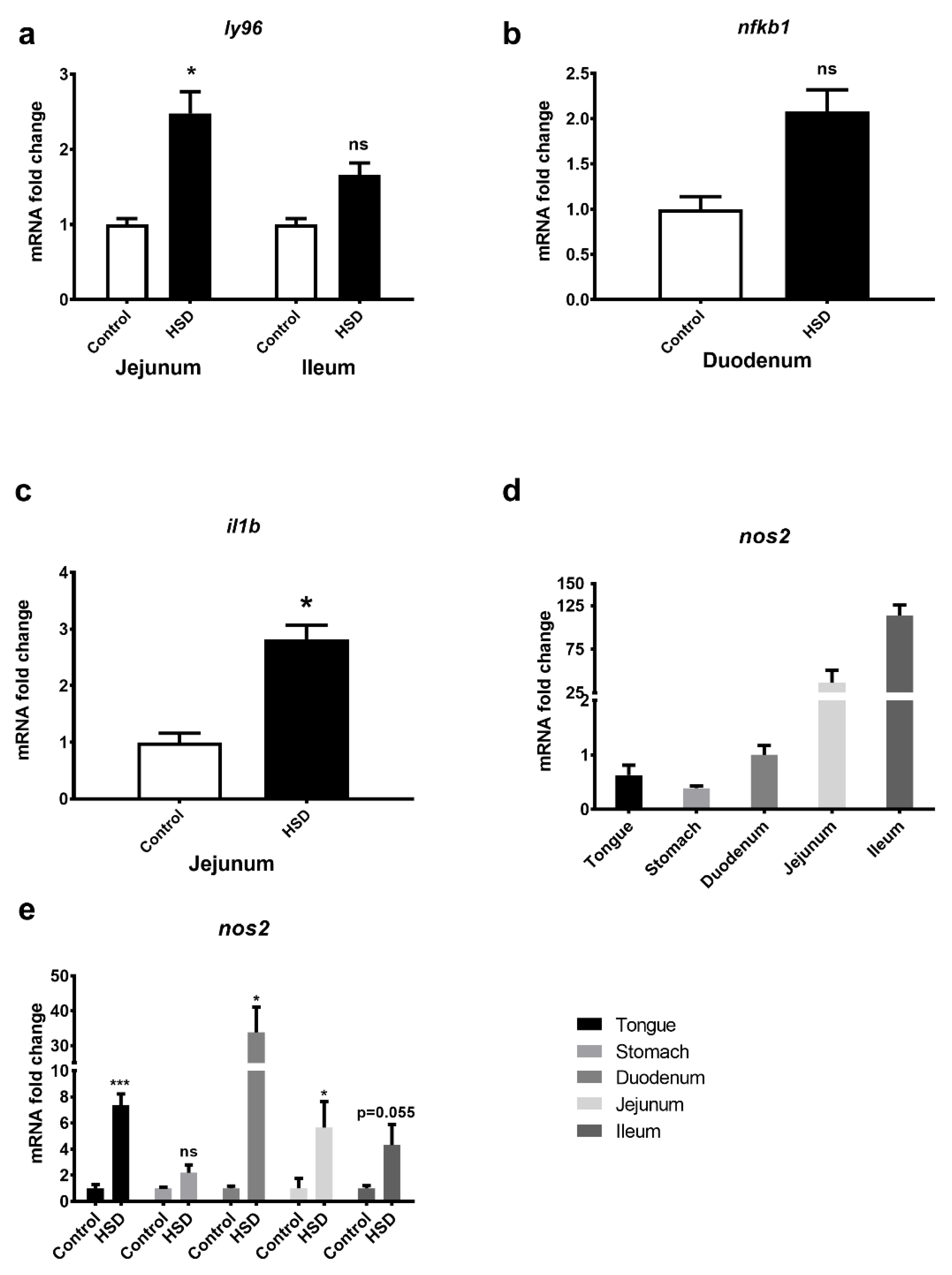
2.7. Identification of an LPS Mediated Inflammatory Pathway by GeneGo Metacore Pathway Analysis of Microarray Data and Validation by qPCR
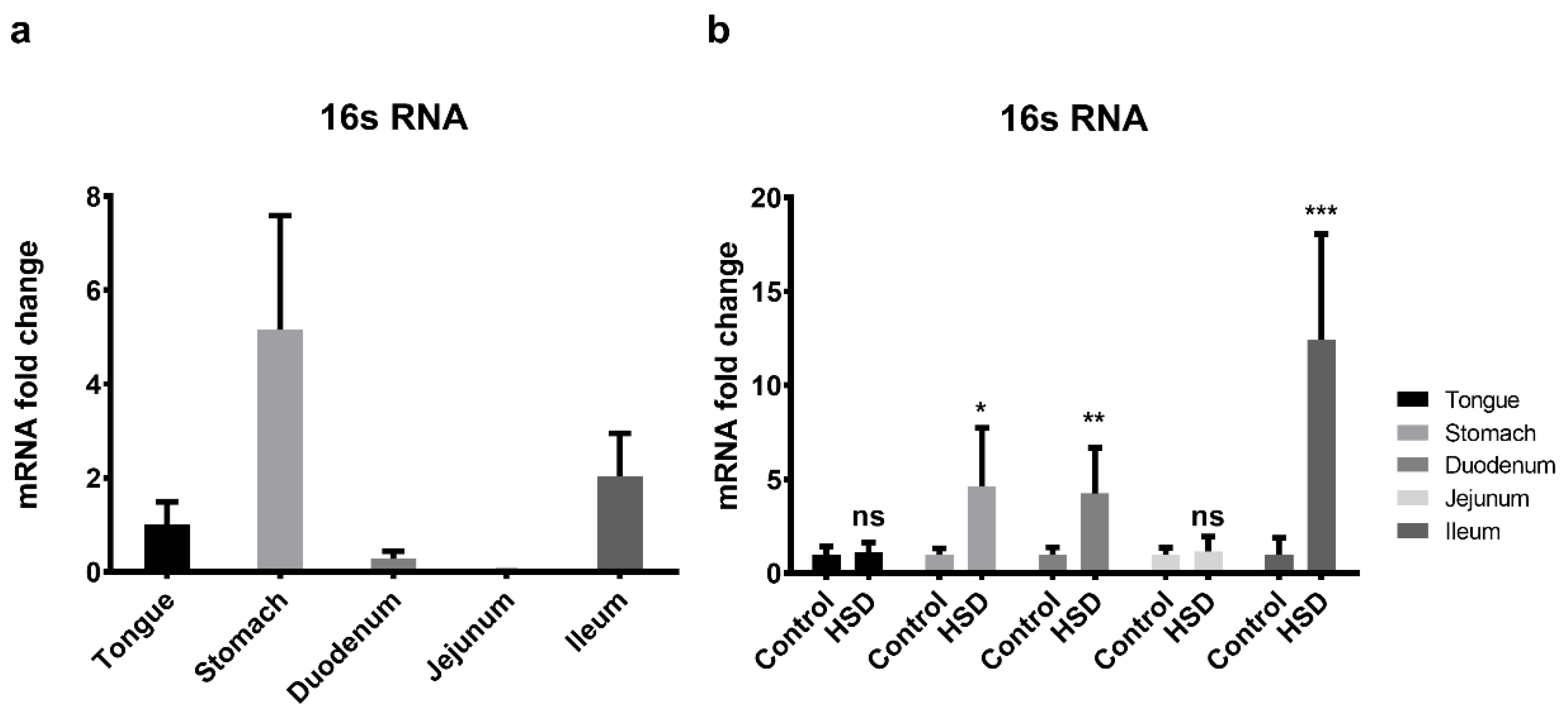
2.8. qPCR of the Small Intestinal Microflora
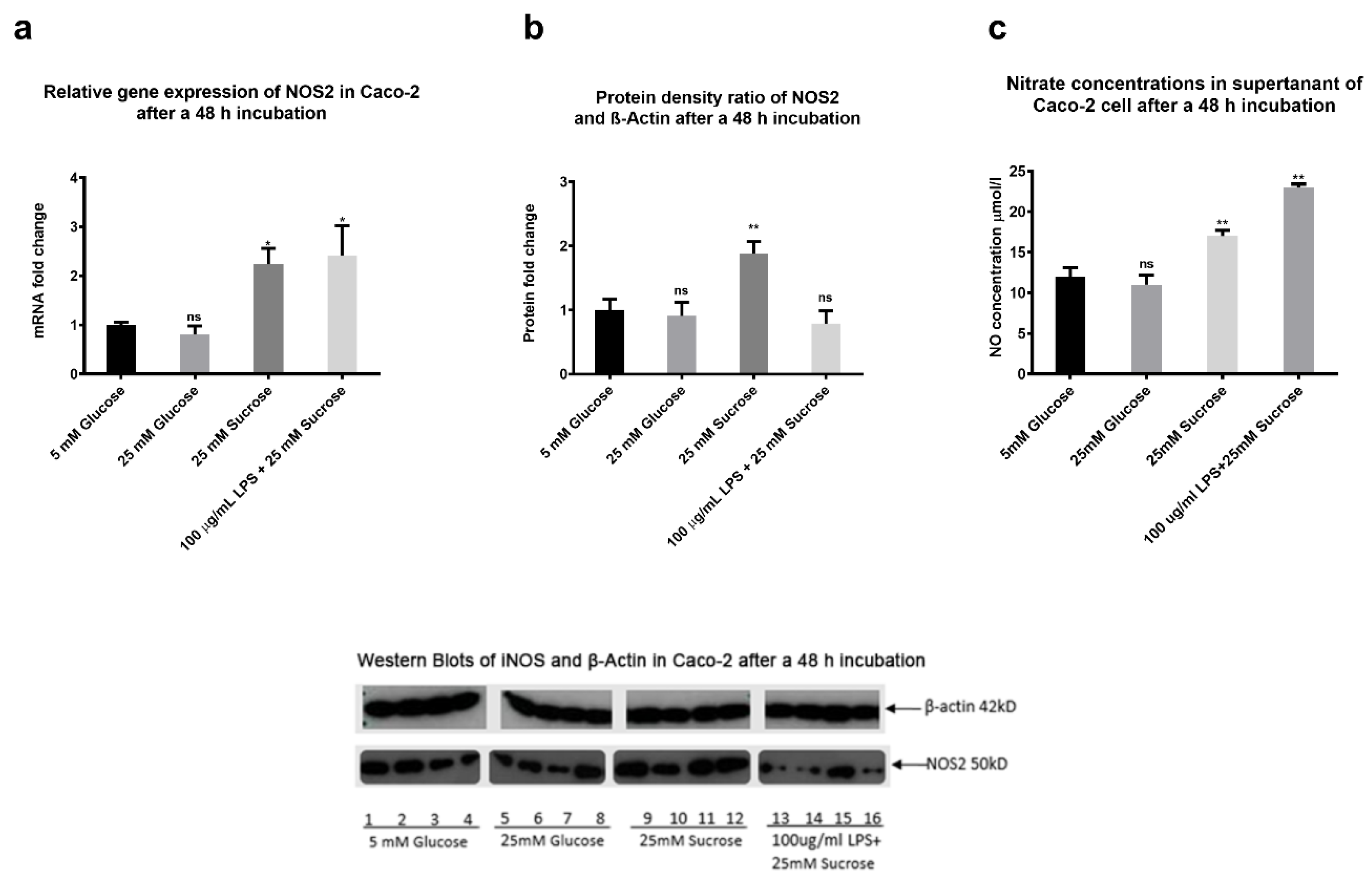
2.9. The Effects of Sugars and LPS on Nitric Oxide Production In Vitro
3. Discussion
4. Materials and Methods
4.1. Laboratory Animals and Study Protocol
4.2. Serum Analysis
4.3. Cell Culture
4.4. RNA Extraction
4.5. Microarray Analysis
4.6. Quantitative Polymerase Chain Reaction (qPCR)
4.7. Protein Extractions
4.8. Western Blotting
4.9. Nitric-Oxide Detection
4.10. Lipids Analysis
4.11. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Jarolimova, J.; Tagoni, J.; Stern, T.A. Obesity: Its epidemiology, comorbidities, and management. Prim. Care Companion CNS Disord. 2013, 15. [Google Scholar] [CrossRef] [PubMed]
- Bluher, M. Obesity: Global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar] [CrossRef]
- Rosinger, A.; Herrick, K.; Gahche, J.; Park, S. Sugar-sweetened Beverage Consumption among U.S. Adults, 2011–2014. NCHS Data Brief. 2017, 270, 1–8. [Google Scholar]
- Park, S.; Pan, L.; Sherry, B.; Blanck, H.M. Consumption of sugar-sweetened beverages among US adults in 6 states: Behavioral Risk Factor Surveillance System, 2011. Prev. Chronic Dis. 2014, 11, E65. [Google Scholar] [CrossRef] [PubMed]
- Momin, S.R.; Wood, A.C. Sugar-Sweetened Beverages and Child Health: Implications for Policy. Curr. Nutr. Rep. 2018, 7, 286–293. [Google Scholar] [CrossRef]
- Malik, V.S.; Pan, A.; Willett, W.C.; Hu, F.B. Sugar-sweetened beverages and weight gain in children and adults: A systematic review and meta-analysis. Am. J. Clin. Nutr. 2013, 98, 1084–1102. [Google Scholar] [CrossRef]
- Malik, V.S.; Popkin, B.M.; Bray, G.A.; Despres, J.P.; Willett, W.C.; Hu, F.B. Sugar-sweetened beverages and risk of metabolic syndrome and type 2 diabetes: A meta-analysis. Diabetes Care 2010, 33, 2477–2483. [Google Scholar] [CrossRef]
- Lewis, A.S.; McCourt, H.J.; Ennis, C.N.; Bell, P.M.; Courtney, C.H.; McKinley, M.C.; Young, I.S.; Hunter, S.J. Comparison of 5% versus 15% sucrose intakes as part of a eucaloric diet in overweight and obese subjects: Effects on insulin sensitivity, glucose metabolism, vascular compliance, body composition and lipid profile. A randomised controlled trial. Metabolism 2013, 62, 694–702. [Google Scholar] [CrossRef]
- Khan, T.A.; Sievenpiper, J.L. Controversies about sugars: Results from systematic reviews and meta-analyses on obesity, cardiometabolic disease and diabetes. Eur. J. Nutr. 2016, 55, 25–43. [Google Scholar] [CrossRef] [PubMed]
- Limb, M. Halve sugar intake to 5% of daily calories, says advisory group. BMJ 2014, 348, g4310. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Guideline: Sugars Intake for Adults and Children; World Health Organization: Geneva, Switzerland, 2015. [Google Scholar]
- Woods, S.C.; May-Zhang, A.A.; Begg, D.P. How and why do gastrointestinal peptides influence food intake? Physiol. Behav. 2018, 193, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Cummings, D.E.; Overduin, J. Gastrointestinal regulation of food intake. J. Clin. Investig. 2007, 117, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Calanna, S.; Christensen, M.; Holst, J.J.; Laferrere, B.; Gluud, L.L.; Vilsboll, T.; Knop, F.K. Secretion of glucose-dependent insulinotropic polypeptide in patients with type 2 diabetes: Systematic review and meta-analysis of clinical studies. Diabetes Care 2013, 36, 3346–3352. [Google Scholar] [CrossRef] [PubMed]
- Calanna, S.; Christensen, M.; Holst, J.J.; Laferrere, B.; Gluud, L.L.; Vilsboll, T.; Knop, F.K. Secretion of glucagon-like peptide-1 in patients with type 2 diabetes mellitus: Systematic review and meta-analyses of clinical studies. Diabetologia 2013, 56, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Breen, D.M.; Rasmussen, B.A.; Cote, C.D.; Jackson, V.M.; Lam, T.K. Nutrient-sensing mechanisms in the gut as therapeutic targets for diabetes. Diabetes 2013, 62, 3005–3013. [Google Scholar] [CrossRef] [PubMed]
- Choo, E.; Wong, L.; Chau, P.; Bushnell, J.; Dando, R. Offspring of obese mice display enhanced intake and sensitivity for palatable stimuli, with altered expression of taste signaling elements. Sci. Rep. 2020, 10, 12776. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.Y.; Caspi, L.; Lam, C.K.; Chari, M.; Li, X.; Light, P.E.; Gutierrez-Juarez, R.; Ang, M.; Schwartz, G.J.; Lam, T.K. Upper intestinal lipids trigger a gut-brain-liver axis to regulate glucose production. Nature 2008, 452, 1012–1016. [Google Scholar] [CrossRef]
- Ferolla, S.M.; Armiliato, G.N.; Couto, C.A.; Ferrari, T.C. The role of intestinal bacteria overgrowth in obesity-related nonalcoholic fatty liver disease. Nutrients 2014, 6, 5583–5599. [Google Scholar] [CrossRef]
- Madrid, A.M.; Poniachik, J.; Quera, R.; Defilippi, C. Small intestinal clustered contractions and bacterial overgrowth: A frequent finding in obese patients. Dig. Dis. Sci. 2011, 56, 155–160. [Google Scholar] [CrossRef]
- Ierardi, E.; Losurdo, G.; Sorrentino, C.; Giorgio, F.; Rossi, G.; Marinaro, A.; Romagno, K.R.; Di Leo, A.; Principi, M. Macronutrient intakes in obese subjects with or without small intestinal bacterial overgrowth: An alimentary survey. Scand. J. Gastroenterol. 2016, 51, 277–280. [Google Scholar] [CrossRef]
- Hoffman, R.A.; Zhang, G.; Nussler, N.C.; Gleixner, S.L.; Ford, H.R.; Simmons, R.L.; Watkins, S.C. Constitutive expression of inducible nitric oxide synthase in the mouse ileal mucosa. Am. J. Physiol. 1997, 272, G383–G392. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.S.; Kwon, M.Y.; Kim, K.H.; Lee, Y.; Lyoo, I.K.; Kim, J.E.; Oh, E.S.; Han, I.O. Lipopolysaccharide (LPS)-stimulated iNOS Induction Is Increased by Glucosamine under Normal Glucose Conditions but Is Inhibited by Glucosamine under High Glucose Conditions in Macrophage Cells. J. Biol. Chem. 2017, 292, 1724–1736. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, S.; Sharafi, M.; Spiecker, M.; Buerke, M.; Fisch, A.; Grosser, T.; Veit, K.; Gierer, C.; Ibe, W.; Meyer, J.; et al. NO reduces PMN adhesion to human vascular endothelial cells due to downregulation of ICAM-1 mRNA and surface expression. Thromb. Res. 2000, 97, 113–123. [Google Scholar] [CrossRef]
- Wilms, H.; Sievers, J.; Rickert, U.; Rostami-Yazdi, M.; Mrowietz, U.; Lucius, R. Dimethylfumarate inhibits microglial and astrocytic inflammation by suppressing the synthesis of nitric oxide, IL-1beta, TNF-alpha and IL-6 in an in-vitro model of brain inflammation. J. Neuroinflamm. 2010, 7, 30. [Google Scholar] [CrossRef] [PubMed]
- Tiso, M.; Schechter, A.N. Nitrate reduction to nitrite, nitric oxide and ammonia by gut bacteria under physiological conditions. PLoS ONE 2015, 10, e0119712. [Google Scholar] [CrossRef]
- Nakamura, T.; Murata, Y.; Mashiko, M.; Okano, K.; Satoh, H.; Ozaki, M.; Amakawa, T. The nitric oxide-cyclic GMP cascade in sugar receptor cells of the blowfly, Phormia regina. Chem. Senses 2005, 30 (Suppl. 1), i281–i282. [Google Scholar] [CrossRef]
- Morley, J.E.; Flood, J.F. Competitive antagonism of nitric oxide synthetase causes weight loss in mice. Life Sci. 1992, 51, 1285–1289. [Google Scholar] [CrossRef]
- Mulders, R.J.; de Git, K.C.G.; Schele, E.; Dickson, S.L.; Sanz, Y.; Adan, R.A.H. Microbiota in obesity: Interactions with enteroendocrine, immune and central nervous systems. Obes. Rev. 2018, 19, 435–451. [Google Scholar] [CrossRef]
- Vecchini, F.; Pringault, E.; Billiar, T.R.; Geller, D.A.; Hausel, P.; Felley-Bosco, E. Decreased activity of inducible nitric oxide synthase type 2 and modulation of the expression of glutathione S-transferase alpha, bcl-2, and metallothioneins during the differentiation of CaCo-2 cells. Cell Growth Differ. 1997, 8, 261–268. [Google Scholar]
- Bernal, S.Y.; Dostova, I.; Kest, A.; Abayev, Y.; Kandova, E.; Touzani, K.; Sclafani, A.; Bodnar, R.J. Role of dopamine D1 and D2 receptors in the nucleus accumbens shell on the acquisition and expression of fructose-conditioned flavor-flavor preferences in rats. Behav. Brain Res. 2008, 190, 59–66. [Google Scholar] [CrossRef]
- Lane, M.D.; Cha, S.H. Effect of glucose and fructose on food intake via malonyl-CoA signaling in the brain. Biochem. Biophys. Res. Commun. 2009, 382, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Roncal-Jimenez, C.A.; Lanaspa, M.A.; Rivard, C.J.; Nakagawa, T.; Sanchez-Lozada, L.G.; Jalal, D.; Andres-Hernando, A.; Tanabe, K.; Madero, M.; Li, N.; et al. Sucrose induces fatty liver and pancreatic inflammation in male breeder rats independent of excess energy intake. Metabolism 2011, 60, 1259–1270. [Google Scholar] [CrossRef] [PubMed]
- Bocarsly, M.E.; Powell, E.S.; Avena, N.M.; Hoebel, B.G. High-fructose corn syrup causes characteristics of obesity in rats: Increased body weight, body fat and triglyceride levels. Pharmacol. Biochem. Behav. 2010, 97, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Cummings, B.P.; Stanhope, K.L.; Graham, J.L.; Evans, J.L.; Baskin, D.G.; Griffen, S.C.; Havel, P.J. Dietary fructose accelerates the development of diabetes in UCD-T2DM rats: Amelioration by the antioxidant, alpha-lipoic acid. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1343–R1350. [Google Scholar] [CrossRef]
- Marques, A.M.; Linhares, B.S.; Dias Novaes, R.; Freitas, M.B.; Sarandy, M.M.; Goncalves, R.V. Effects of the amount and type of carbohydrates used in type 2 diabetes diets in animal models: A systematic review. PLoS ONE 2020, 15, e0233364. [Google Scholar] [CrossRef]
- Levin, R.J. Digestion and absorption of carbohydrates—From molecules and membranes to humans. Am. J. Clin. Nutr. 1994, 59, 690S–698S. [Google Scholar] [CrossRef]
- Crossley, J.N.; Macdonald, I. The influence in male baboons, of a high sucrose diet on the portal and arterial levels of glucose and fructose following a sucrose meal. Nutr. Metab. 1970, 12, 171–178. [Google Scholar] [CrossRef]
- Johnson, R.J.; Nakagawa, T.; Sanchez-Lozada, L.G.; Shafiu, M.; Sundaram, S.; Le, M.; Ishimoto, T.; Sautin, Y.Y.; Lanaspa, M.A. Sugar, uric acid, and the etiology of diabetes and obesity. Diabetes 2013, 62, 3307–3315. [Google Scholar] [CrossRef]
- Taskinen, M.R.; Packard, C.J.; Boren, J. Dietary Fructose and the Metabolic Syndrome. Nutrients 2019, 11, 1987. [Google Scholar] [CrossRef]
- Jang, C.; Hui, S.; Lu, W.; Cowan, A.J.; Morscher, R.J.; Lee, G.; Liu, W.; Tesz, G.J.; Birnbaum, M.J.; Rabinowitz, J.D. The Small Intestine Converts Dietary Fructose into Glucose and Organic Acids. Cell Metab. 2018, 27, 351–361.e353. [Google Scholar] [CrossRef]
- Haidari, M.; Leung, N.; Mahbub, F.; Uffelman, K.D.; Kohen-Avramoglu, R.; Lewis, G.F.; Adeli, K. Fasting and postprandial overproduction of intestinally derived lipoproteins in an animal model of insulin resistance. Evidence that chronic fructose feeding in the hamster is accompanied by enhanced intestinal de novo lipogenesis and ApoB48-containing lipoprotein overproduction. J. Biol. Chem. 2002, 277, 31646–31655. [Google Scholar] [CrossRef]
- Kelly, T.J.; Holt, P.R.; Wu, A.L. Effect of sucrose on intestinal very low-density lipoprotein production. Am. J. Clin. Nutr. 1980, 33, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.H.; et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, M.W.; Berk, P.D. Lipid metabolism in hepatic steatosis. Clin. Liver Dis. 2004, 8, 639–671. [Google Scholar] [CrossRef] [PubMed]
- Piomelli, D. A fatty gut feeling. Trends Endocrinol. Metab. 2013, 24, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Gaetani, S.; Oveisi, F.; Lo Verme, J.; Serrano, A.; Rodriguez De Fonseca, F.; Rosengarth, A.; Luecke, H.; Di Giacomo, B.; Tarzia, G.; et al. Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-alpha. Nature 2003, 425, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Naughton, S.S.; Mathai, M.L.; Hryciw, D.H.; McAinch, A.J. Fatty Acid modulation of the endocannabinoid system and the effect on food intake and metabolism. Int. J. Endocrinol. 2013, 2013, 361895. [Google Scholar] [CrossRef]
- Kaji, I.; Kaunitz, J.D. Luminal chemosensing in the gastroduodenal mucosa. Curr. Opin. Gastroenterol. 2017, 33, 439–445. [Google Scholar] [CrossRef]
- Li, T.; Zhao, M.; Raza, A.; Guo, J.; He, T.; Zou, T.; Song, H. The effect of taste and taste perception on satiation/satiety: A review. Food Funct. 2020, 11, 2838–2847. [Google Scholar] [CrossRef]
- Daniel, H.; Zietek, T. Taste and move: Glucose and peptide transporters in the gastrointestinal tract. Exp. Physiol. 2015, 100, 1441–1450. [Google Scholar] [CrossRef]
- Sotak, M.; Marks, J.; Unwin, R.J. Putative tissue location and function of the SLC5 family member SGLT3. Exp. Physiol. 2017, 102, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.M. Role of amino acid transporters in amino acid sensing. Am. J. Clin. Nutr. 2014, 99, 223S–230S. [Google Scholar] [CrossRef] [PubMed]
- Noel, C.A.; Sugrue, M.; Dando, R. Participants with pharmacologically impaired taste function seek out more intense, higher calorie stimuli. Appetite 2017, 117, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Wise, P.M.; Nattress, L.; Flammer, L.J.; Beauchamp, G.K. Reduced dietary intake of simple sugars alters perceived sweet taste intensity but not perceived pleasantness. Am. J. Clin. Nutr. 2016, 103, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Sukumaran, S.K.; Yee, K.K.; Iwata, S.; Kotha, R.; Quezada-Calvillo, R.; Nichols, B.L.; Mohan, S.; Pinto, B.M.; Shigemura, N.; Ninomiya, Y.; et al. Taste cell-expressed alpha-glucosidase enzymes contribute to gustatory responses to disaccharides. Proc. Natl. Acad. Sci. USA 2016, 113, 6035–6040. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, P.; Hewett, R.; Corpe, C. Sugar sensor genes in the murine gastrointestinal tract display a cephalocaudal axis of expression and a diurnal rhythm. Physiol. Genom. 2018, 50, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.E.; Keast, R.S. Recent fat intake modulates fat taste sensitivity in lean and overweight subjects. Int. J. Obes. (Lond.) 2012, 36, 834–842. [Google Scholar] [CrossRef]
- Martin, C.; Passilly-Degrace, P.; Gaillard, D.; Merlin, J.F.; Chevrot, M.; Besnard, P. The lipid-sensor candidates CD36 and GPR120 are differentially regulated by dietary lipids in mouse taste buds: Impact on spontaneous fat preference. PLoS ONE 2011, 6, e24014. [Google Scholar] [CrossRef]
- Zhang, X.J.; Zhou, L.H.; Ban, X.; Liu, D.X.; Jiang, W.; Liu, X.M. Decreased expression of CD36 in circumvallate taste buds of high-fat diet induced obese rats. Acta Histochem. 2011, 113, 663–667. [Google Scholar] [CrossRef]
- Campbell, S.E.; Tandon, N.N.; Woldegiorgis, G.; Luiken, J.J.; Glatz, J.F.; Bonen, A. A novel function for fatty acid translocase (FAT)/CD36: Involvement in long chain fatty acid transfer into the mitochondria. J. Biol. Chem. 2004, 279, 36235–36241. [Google Scholar] [CrossRef]
- Widmayer, P.; Goldschmid, H.; Henkel, H.; Kuper, M.; Konigsrainer, A.; Breer, H. High fat feeding affects the number of GPR120 cells and enteroendocrine cells in the mouse stomach. Front. Physiol. 2015, 6, 53. [Google Scholar] [CrossRef] [PubMed]
- Poykko, S.M.; Kellokoski, E.; Horkko, S.; Kauma, H.; Kesaniemi, Y.A.; Ukkola, O. Low plasma ghrelin is associated with insulin resistance, hypertension, and the prevalence of type 2 diabetes. Diabetes 2003, 52, 2546–2553. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Lopez, M.; Rahmouni, K. The cellular and molecular bases of leptin and ghrelin resistance in obesity. Nat. Rev. Endocrinol. 2017, 13, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Ritze, Y.; Hengelhaupt, C.; Bardos, G.; Ernst, B.; Thurnheer, M.; D’Haese, J.G.; Bischoff, S.C.; Schultes, B. Altered intestinal neuroendocrine gene expression in humans with obesity. Obesity (Silver Spring) 2015, 23, 2278–2285. [Google Scholar] [CrossRef]
- Dockray, G.J. Cholecystokinin. Curr. Opin. Endocrinol. Diabetes Obes. 2012, 19, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Ritze, Y.; Bardos, G.; D’Haese, J.G.; Ernst, B.; Thurnheer, M.; Schultes, B.; Bischoff, S.C. Effect of high sugar intake on glucose transporter and weight regulating hormones in mice and humans. PLoS ONE 2014, 9, e101702. [Google Scholar] [CrossRef] [PubMed]
- Bellissimo, N.; Anderson, G.H. Cholecystokinin—A receptors are involved in food intake suppression in rats after intake of all fats and carbohydrates tested. J. Nutr. 2003, 133, 2319–2325. [Google Scholar] [CrossRef]
- Richards, P.; Pais, R.; Habib, A.M.; Brighton, C.A.; Yeo, G.S.; Reimann, F.; Gribble, F.M. High fat diet impairs the function of glucagon-like peptide-1 producing L-cells. Peptides 2016, 77, 21–27. [Google Scholar] [CrossRef]
- Nauck, M.; Stockmann, F.; Ebert, R.; Creutzfeldt, W. Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia 1986, 29, 46–52. [Google Scholar] [CrossRef]
- Rubino, F. Is type 2 diabetes an operable intestinal disease? A provocative yet reasonable hypothesis. Diabetes Care 2008, 31 (Suppl. 2), S290–S296. [Google Scholar] [CrossRef]
- Graham, C.; Mullen, A.; Whelan, K. Obesity and the gastrointestinal microbiota: A review of associations and mechanisms. Nutr. Rev. 2015, 73, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Madrid, A.M.; Defilippi, C.C.; Defilippi, G.C.; Slimming, A.J.; Quera, P.R. Small intestinal bacterial overgrowth in patients with functional gastrointestinal diseases. Rev. Med. Child 2007, 135, 1245–1252. [Google Scholar] [CrossRef]
- Virally-Monod, M.; Tielmans, D.; Kevorkian, J.P.; Bouhnik, Y.; Flourie, B.; Porokhov, B.; Ajzenberg, C.; Warnet, A.; Guillausseau, P.J. Chronic diarrhoea and diabetes mellitus: Prevalence of small intestinal bacterial overgrowth. Diabetes Metab. 1998, 24, 530–536. [Google Scholar] [PubMed]
- Lach, G.; Schellekens, H.; Dinan, T.G.; Cryan, J.F. Anxiety, Depression, and the Microbiome: A Role for Gut Peptides. Neurotherapeutics 2018, 15, 36–59. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef]
- Ngkelo, A.; Meja, K.; Yeadon, M.; Adcock, I.; Kirkham, P.A. LPS induced inflammatory responses in human peripheral blood mononuclear cells is mediated through NOX4 and Gialpha dependent PI-3kinase signalling. J. Inflamm. (Lond.) 2012, 9, 1. [Google Scholar] [CrossRef]
- Garcia-Herrera, J.; Marca, M.C.; Brot-Laroche, E.; Guillen, N.; Acin, S.; Navarro, M.A.; Osada, J.; Rodriguez-Yoldi, M.J. Protein kinases, TNF-α, and proteasome contribute in the inhibition of fructose intestinal transport by sepsis in vivo. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G155–G164. [Google Scholar] [CrossRef]
- Garcia-Barrios, A.; Guillen, N.; Gascon, S.; Osada, J.; Vazquez, C.M.; Miguel-Carrasco, J.L.; Rodriguez-Yoldi, M.J. Nitric oxide involved in the IL-1beta-induced inhibition of fructose intestinal transport. J. Cell Biochem. 2010, 111, 1321–1329. [Google Scholar] [CrossRef]
- Morley, J.E.; Flood, J.F. Effect of competitive antagonism of NO synthetase on weight and food intake in obese and diabetic mice. Am. J. Physiol. 1994, 266, R164–R168. [Google Scholar] [CrossRef]
- Morley, J.E.; Farr, S.A.; Suarez, M.D.; Flood, J.F. Nitric oxide synthase inhibition and food intake: Effects on motivation to eat and in female mice. Pharmacol. Biochem. Behav. 1995, 50, 369–373. [Google Scholar] [CrossRef]
- O’Brien, P.; Corpe, C.P. Acute Effects of Sugars and Artificial Sweeteners on Small Intestinal Sugar Transport: A Study Using CaCo-2 Cells As an In Vitro Model of the Human Enterocyte. PLoS ONE 2016, 11, e0167785. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.Q.; Zhang, Y.; Hoon, M.A.; Chandrashekar, J.; Erlenbach, I.; Ryba, N.J.; Zuker, C.S. The receptors for mammalian sweet and umami taste. Cell 2003, 115, 255–266. [Google Scholar] [CrossRef]
- Chantret, I.; Rodolosse, A.; Barbat, A.; Dussaulx, E.; Brot-Laroche, E.; Zweibaum, A.; Rousset, M. Differential expression of sucrase-isomaltase in clones isolated from early and late passages of the cell line Caco-2: Evidence for glucose-dependent negative regulation. J. Cell Sci. 1994, 107 Pt 1, 213–225. [Google Scholar]
- Bagul, P.K.; Middela, H.; Matapally, S.; Padiya, R.; Bastia, T.; Madhusudana, K.; Reddy, B.R.; Chakravarty, S.; Banerjee, S.K. Attenuation of insulin resistance, metabolic syndrome and hepatic oxidative stress by resveratrol in fructose-fed rats. Pharmacol. Res. 2012, 66, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Babacanoglu, C.; Yildirim, N.; Sadi, G.; Pektas, M.B.; Akar, F. Resveratrol prevents high-fructose corn syrup-induced vascular insulin resistance and dysfunction in rats. Food Chem. Toxicol. 2013, 60, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.V.; Kiat, H. The mechanisms underlying fructose-induced hypertension: A review. J. Hypertens. 2015, 33, 912–920. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, K.; Bruick, R.K.; Liang, G.; Horton, J.D.; Uyeda, K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc. Natl. Acad. Sci. USA 2004, 101, 7281–7286. [Google Scholar] [CrossRef] [PubMed]
- Dotimas, J.R.; Lee, A.W.; Schmider, A.B.; Carroll, S.H.; Shah, A.; Bilen, J.; Elliott, K.R.; Myers, R.B.; Soberman, R.J.; Yoshioka, J.; et al. Diabetes regulates fructose absorption through thioredoxin-interacting protein. Elife 2016, 5. [Google Scholar] [CrossRef]
- Zwarts, I.; van Zutphen, T.; Kruit, J.K.; Liu, W.; Oosterveer, M.H.; Verkade, H.J.; Uhlenhaut, N.H.; Jonker, J.W. Identification of the fructose transporter GLUT5 (SLC2A5) as a novel target of nuclear receptor LXR. Sci. Rep. 2019, 9, 9299. [Google Scholar] [CrossRef]
- Lanaspa, M.A.; Sanchez-Lozada, L.G.; Choi, Y.J.; Cicerchi, C.; Kanbay, M.; Roncal-Jimenez, C.A.; Ishimoto, T.; Li, N.; Marek, G.; Duranay, M.; et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: Potential role in fructose-dependent and -independent fatty liver. J. Biol. Chem. 2012, 287, 40732–40744. [Google Scholar] [CrossRef]
- Lanaspa, M.A.; Sanchez-Lozada, L.G.; Cicerchi, C.; Li, N.; Roncal-Jimenez, C.A.; Ishimoto, T.; Le, M.; Garcia, G.E.; Thomas, J.B.; Rivard, C.J.; et al. Uric acid stimulates fructokinase and accelerates fructose metabolism in the development of fatty liver. PLoS ONE 2012, 7, e47948. [Google Scholar] [CrossRef] [PubMed]
- Lanaspa, M.A.; Cicerchi, C.; Garcia, G.; Li, N.; Roncal-Jimenez, C.A.; Rivard, C.J.; Hunter, B.; Andres-Hernando, A.; Ishimoto, T.; Sanchez-Lozada, L.G.; et al. Counteracting roles of AMP deaminase and AMP kinase in the development of fatty liver. PLoS ONE 2012, 7, e48801. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Gene Name | Fold Change Ctrl vs. HSD | |||
|---|---|---|---|---|---|
| Duo | Jej | Ile | |||
| Sweet | T1R2 | tas1r2 | 1.15 | −1.03 | 1.12 |
| T1R3 | tas1r3 | 1.13 | 1.07 | −1.05 | |
| SGLT3a | slc5a4a | −2.61 | −1.73 | 1.19 | |
| SGLT3b | slc5a4b | −4.23 | −3.0 | −1.13 | |
| Fat | CD36/FAT | cd36 | −4.62 | −3.12 | −1.41 |
| FFAR2 | ffar2 | 1.02 | 1.04 | 1.09 | |
| GPR120 | gpr120 | −1.10 | −1.21 | −1.13 | |
| Umami | mGluR4 | grm4 | −1.03 | −1.13 | −1.01 |
| T1R1 | tas1r1 | 1.11 | −1.07 | −1.15 | |
| Sour | TRPP3 | pkd2l1 | 1.15 | 1.00 | −1.13 |
| ENaCα | scnn1a | −1.09 | −1.15 | −1.06 | |
| ENaCβ | scnn1b | 1.00 | −1.31 | −1.07 | |
| ENaCγ | scnn1c | NA | NA | NA | |
| Bitter | T2R102–T2R143 | tas2r102–tas2r143 | −1.43–1.33 | −1.59–1.22 | −1.27–1.06 |
| Signalling | αGustducin | gnat3 | 1.28 | 1.31 | 1.29 |
| αTransducing | gnat1 | 1.19 | −1.44 | −1.74 | |
| TRPM5 | trpm5 | 1.66 | 1.20 | 1.14 | |
| Phospholipaseβ2 | plcb2 | 1.52 | 1.18 | −1.04 | |
| Key Gene | Protein Encoded | Fold Changes | Effect |
| MD-2 | Lymphocyte antigen 96 | Duodenum: 1.17 Jejunum: 1.64 Ileum: 1.76 | MD-2 associates with toll-like receptor 4 on the cell surface and responds to LPS, thus providing a link between the receptor and LPS signalling. |
| I-kB | Nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, epsilon (NFKBIE) | Duodenum: 1.63 Jejunum: 1.29 Ileum: 1.4 | This protein complex controls DNA transcription, cytokine production and cell survival, playing a key role in mediating the immune response to infection. |
| IL-1β | Interleukin 1 beta | Duodenum: 1.22 Jejunum: 1.77 Ileum: 1.4 | IL-1β is a member of the interleukin 1 cytokine family, an important regulator of the inflammatory response, and is involved in a variety of cellular activities, including cell proliferation, differentiation and apoptosis. |
| iNOS | Nitric oxide synthase 2 | Duodenum: 4.74 Jejunum: 1.95 Ileum: 1.2 | This protein is induced by a combination of lipopolysaccharide and certain cytokines, and NO gas, a mediator of several biological processes, is released. |
| 2% Corn Oil Chow | LCM + 1% Corn Oil | |||
|---|---|---|---|---|
| g/100 g | kcal/100 g | g/100 g | kcal/100 g | |
| Total carbohydrate | 68 | 255 | 59.5 | 223.1 |
| Starch | 58 | 217.5 | 0 | 0 |
| Sucrose | 10 | 37.5 | 47.3 | 177.4 |
| Lactose | 0 | 0 | 12.2 | 45.7 |
| Fat | 2 | 18 | 1.2 | 10.8 |
| Protein | 20 | 80 | 9.3 | 37.2 |
| Total | 100 | 353 | 100 | 271.1 |
| Gene | Forward Primer: 5′-3′ | Reverse Primer: 5′-3′ |
|---|---|---|
| 18s | gcaattattccccatgaacg | gggacttaatcaacgcaagc |
| hprt1 | tcctcctcagaccgctttt | cctggttcatcatcgctaatc |
| hmbs | tccctgaaggatgtgcctac | aagggttttcccgtttgc |
| slc5a1 | ctggcaggccgaagtatg | ttccaatgttactggcaaagag |
| 16s | cgtgccagccgcggtaatacg | gggttgcgctcgttgcgggacttaacccaacat |
| slc5a4a | aaacccattcccgatgttc | tcgattctttcctccttactgttc |
| slc5a4b | ccgattcctgatgttcacct | atccgctcctctgtgttgtt |
| gcg | cacgcccttcaagacacag | gtcctcatgcgcttctgtc |
| cck | tgatttccccatccaaagc | gcttctgcagggactaccg |
| ghrl | ccagaggacagaggacaagc | catcgaagggagcattgaac |
| tas1r2 | aagcatcgcctcctactcc | ggctggcaactcttagaacac |
| tas1r3 | gaagcatccagatgacttca | gggaacagaaggacactgag |
| cd36 | ttgtacctatactgtggctaaatgaga | cttgtgttttgaacatttctgctt |
| ly96 | gccttctcagtcttggtggt | tctttccacggagattctgg |
| nfkb1 | ctgacctgagcctctggac | gcaggctattgctcatcaca |
| il1b | gcccatcctctgtgactcat | aggccacaggtattttgtcg |
| nos2 | ctttgccacggacgagac | tcattgtactctgagggctgac |
| pdk4 | cgcttagtgaacactccttcg | cttctgggctcttctcatgg |
| hmgcs2 | ctgtggcaatgctgatcg | tccatgtgagttcccctca |
| acot1 | ggaggttggggaaaggtacaa | actccattcccagcccttga |
| acot2 | tttctctgcggaaccgagg | tgctctcaggacagcgaaag |
| me2 | gcagctcttcgaataaccaag | aagtgagcaatccccaagg |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Brien, P.; Han, G.; Ganpathy, P.; Pitre, S.; Zhang, Y.; Ryan, J.; Sim, P.Y.; Harding, S.V.; Gray, R.; Preedy, V.R.; et al. Chronic Effects of a High Sucrose Diet on Murine Gastrointestinal Nutrient Sensor Gene and Protein Expression Levels and Lipid Metabolism. Int. J. Mol. Sci. 2021, 22, 137. https://doi.org/10.3390/ijms22010137
O’Brien P, Han G, Ganpathy P, Pitre S, Zhang Y, Ryan J, Sim PY, Harding SV, Gray R, Preedy VR, et al. Chronic Effects of a High Sucrose Diet on Murine Gastrointestinal Nutrient Sensor Gene and Protein Expression Levels and Lipid Metabolism. International Journal of Molecular Sciences. 2021; 22(1):137. https://doi.org/10.3390/ijms22010137
Chicago/Turabian StyleO’Brien, Patrick, Ge Han, Priya Ganpathy, Shweta Pitre, Yi Zhang, John Ryan, Pei Ying Sim, Scott V. Harding, Robert Gray, Victor R. Preedy, and et al. 2021. "Chronic Effects of a High Sucrose Diet on Murine Gastrointestinal Nutrient Sensor Gene and Protein Expression Levels and Lipid Metabolism" International Journal of Molecular Sciences 22, no. 1: 137. https://doi.org/10.3390/ijms22010137
APA StyleO’Brien, P., Han, G., Ganpathy, P., Pitre, S., Zhang, Y., Ryan, J., Sim, P. Y., Harding, S. V., Gray, R., Preedy, V. R., Sanders, T. A. B., & Corpe, C. P. (2021). Chronic Effects of a High Sucrose Diet on Murine Gastrointestinal Nutrient Sensor Gene and Protein Expression Levels and Lipid Metabolism. International Journal of Molecular Sciences, 22(1), 137. https://doi.org/10.3390/ijms22010137