Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease: Current Insights into the Disease Pathophysiology, Diagnosis and Management


Abstract
1. Introduction
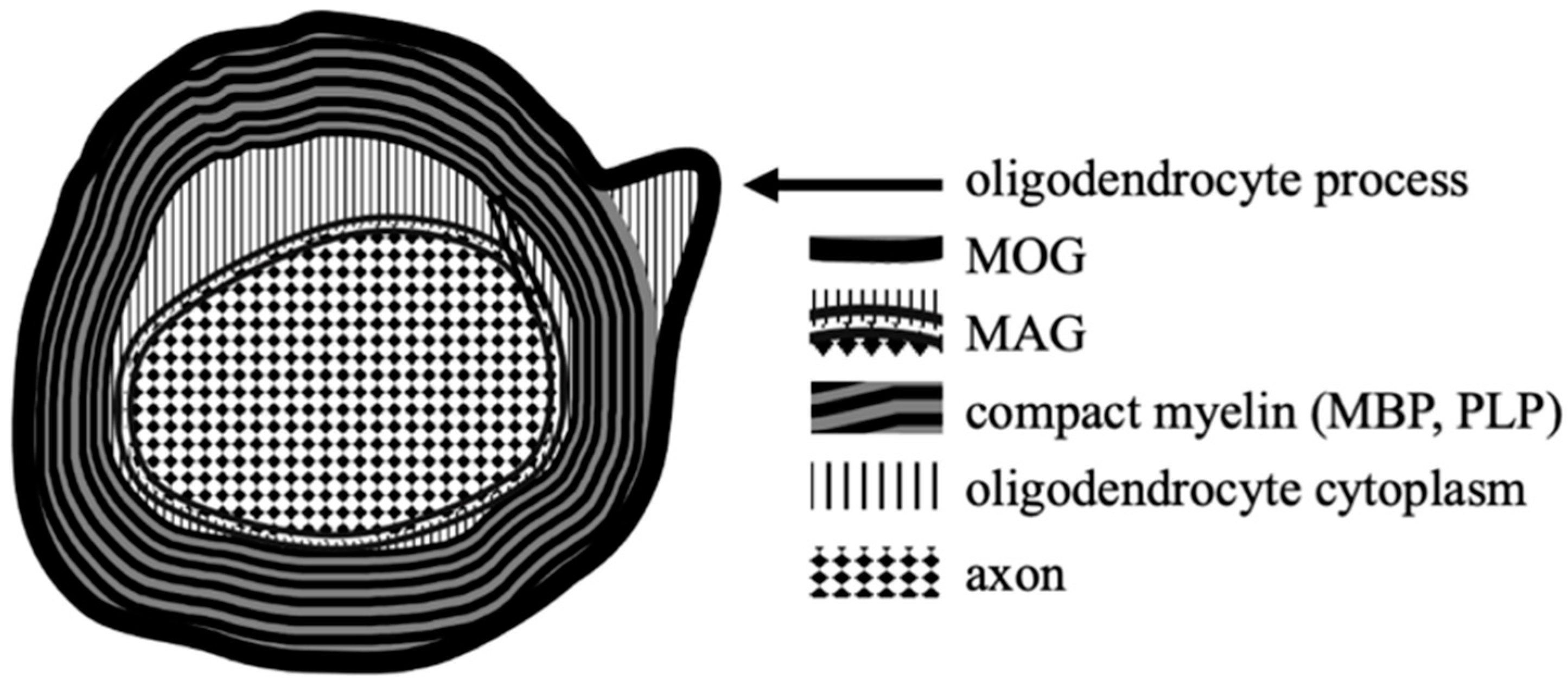
2. Myelin Oligodendrocyte Glycoprotein–Structure and Function
3. Clinical Picture
3.1. ADEM Presentation
3.2. Optic Neuritis Presentation
3.3. Transverse Myelitis Presentation
3.4. Other Manifestations (Brainstem and Cortical Encephalitis)
- -
- clinical picture,
- -
- neuroimaging (MRI) or neurophysiological exam (in ON optical coherence tomography or visual evoked potentials) findings indicating demyelinating injury within CNS,
- -
- biochemical (the positive result of MOG IgG test performed with modern cell-based assay).
4. Disease Course and Epidemiology
5. Neuropathology
6. Neuroimaging
7. MOGAD Diagnosis
8. MOGAD Treatment
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schumacher, G.A.; Beebe, G.; Kibler, R.F.; Kurland, L.T.; Kurtzke, J.F.; McDowell, F.; Nagler, B.; Sibley, W.A.; Tourtellotte, W.W.; Willmon, T.L. Problems of experimental trials of therapy in multiple sclerosis: Report by the panel on the evaluation of experimental trials of therapy in multiple sclerosis. Ann. N. Y. Acad. Sci. 1965, 122, 552–568. [Google Scholar] [CrossRef] [PubMed]
- Poser, C.M.; Paty, D.W.; Scheinberg, L.; McDonald, W.I.; Davis, F.A.; Ebers, G.C.; Johnson, K.P.; Sibley, W.A.; Silberberg, D.H.; Tourtellotte, W.W. New diagnostic criteria for multiple sclerosis: Guidelines for research protocols. Ann. Neurol. 1983, 1393, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.J.; Banwell, B.L.; Barkhof, F.; Carroll, W.M.; Coetzee, T.; Comi, G.; Correale, J.; Fazekas, F.; Filippi, M.; Freedman, M.S.; et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018, 17, 162–173. [Google Scholar] [CrossRef]
- Wingerchuk, D.M.; Banwell, B.; Bennett, J.L.; Cabre, P.; Carroll, W.; Chitnis, T.; De Seze, J.; Fujihara, K.; Greenberg, B.; Jacob, A.; et al. International consensus diagnostic criteria for neuromyelitis opstica spectrum disorders. Neurology 2015, 85, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Jasiak-Zatonska, M.; Kalinowska-Lyszczarz, A.; Michalak, S.; Kozubski, W. The immunology of neuromyelitis optica—Current knowledge, clinical implications, controversies and future perspectives. Int. J. Mol. Sci. 2016, 17, 273. [Google Scholar] [CrossRef]
- Sato, D.K.; Callegaro, D.; Lana-Peixoto, M.A.; Waters, P.J.; De Haidar Jorge, F.M.; Takahashi, T.; Nakashima, I.; Apostolos- Pereira, S.L.; Simm, N.T.R.F.; Lino, A.M.; et al. Distinction between MOG antibody positive and AQP4 antibody-positive NMO spectrum disorders. Neurology 2014, 82, 474–481. [Google Scholar] [CrossRef]
- Jarius, S.; Paul, F.; Aktas, O.; Asgari, N.; Dale, R.C.; de Seze, J.; Franciotta, D.; Fujihara, K.; Jacob, A.; Kim, H.J.; et al. MOG Encephalomyelitis: International Recommendations on Diagnosis and Antibody Testing. J. Neuroinflamm. 2018, 15, 134. [Google Scholar] [CrossRef]
- Quarles, R.H. Myelin Sheaths: Glycoproteins Involved in Their Formation, Maintenance and Degeneration. Cell. Mol. Life Sci. 2002, 59, 1851–1871. [Google Scholar] [CrossRef]
- Lebar, R.; Boutry, J.M.; Vincent, C.; Robineaux, R.; Voisin, G.A. Studies on Autoimmune Encephalomyelitis in the Guinea Pig. II. An in Vitro Investigation on the Nature, Properties, and Specificity of the Serum Demyelinating Factor. J. Immunol. 1976, 116, 1439–1446. [Google Scholar]
- Linnington, C.; Webb, M.; Woodhams, P.L. A Novel Myelin-Associated Glycoprotein Defined by a Mouse Monoclonal Antibody. J. Neuroimmunol. 1984, 59, 1851–1857. [Google Scholar] [CrossRef]
- Lebar, R.; Baudrimont, M.; Vincent, C. Chronic Experimental Autoimmune Encephalomyelitis in the Guinea Pig. Presence of Anti-M2 Antibodies in Central Nervous System Tissue and the Possible Role of M2 Autoantigen in the Induction of the Disease. J. Autoimmun. 1989, 2, 115–132. [Google Scholar] [CrossRef]
- Hilton, A.A.; Slavin, A.J.; Hilton, D.J.; Bernard, C.C.A. Characterization of CDNA and Genomic Clones Encoding Human Myelin Oligodendrocyte Glycoprotein. J. Neurochem. 1995, 65, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Kroepfl, J.F.; Viise, L.R.; Charron, A.J.; Linington, C.; Gardinier, M.V. Investigation of Myelin/Oligodendrocyte Glycoprotein Membrane Topology. J. Neurochem. 2002, 67, 2219–2222. [Google Scholar] [CrossRef] [PubMed]
- Barkovich, A.J. Concepts of Myelin and Myelination in Neuroradiology. Am. J. Neuroradiol. 2000, 67, 2219–2222. [Google Scholar]
- Linington, C.; Kaushansky, N.; Chapple, K.; Ben-Nun, A. Myelin Oligodendrocyte Glycoprotein (MOG): An Archetypal Target for Demyelinating Autoantibodies in the Central Nervous System. Autoantibodies 2014, 73, 617–627. [Google Scholar]
- Lopez, P.H.H. Role of Myelin-Associated Glycoprotein (Siglec-4a) in the Nervous System. Adv. Neurobiol. 2014, 9, 245–262. [Google Scholar]
- Pagany, M.; Jagodic, M.; Schubart, A.; Pham-Dinh, D.; Bachelin, C.; Van Evercooren, A.B.; Lachapelle, F.; Olsson, T.; Linington, C. Myelin Oligodendrocyte Glycoprotein Is Expressed in the Peripheral Nervous System of Rodents and Primates. Neurosci. Lett. 2003, 67, 2219–2222. [Google Scholar] [CrossRef]
- Do Campo, R.V.; Stephens, A.; Marin Collazo, I.V.; Rubin, D.I. MOG Antibodies in Combined Central and Peripheral Demyelination Syndromes. Neurol. Neuroimmunol. NeuroInflamm. 2018, 5, e503. [Google Scholar] [CrossRef]
- Harauz, G.; Ladizhansky, V.; Boggs, J.M. Structural Polymorphism and Multifunctionality of Myelin Basic Protein. Biochemistry 2009, 48, 8094–8104. [Google Scholar] [CrossRef]
- Menon, K.K.; Piddlesden, S.J.; Bernard, C.C.A. Demyelinating Antibodies to Myelin Oligodendrocyte Glycoprotein and Galactocerebroside Induce Degradation of Myelin Basic Protein in Isolated Human Myelin. J. Neurochem. 2002, 69, 214–222. [Google Scholar] [CrossRef]
- Dale, R.C.; Tantsis, E.M.; Merheb, V.; Kumaran, R.Y.A.; Sinmaz, N.; Pathmanandavel, K.; Ramanathan, S.; Booth, D.R.; Wienholt, L.A.; Prelog, K.; et al. Antibodies to MOG Have a Demyelination Phenotype and Affect Oligodendrocyte Cytoskeleton. Neurol. Neuroimmunol. NeuroInflamm. 2014, 1, e12. [Google Scholar] [CrossRef] [PubMed]
- Morise, J.; Takematsu, H.; Oka, S. The Role of Human Natural Killer-1 (HNK-1) Carbohydrate in Neuronal Plasticity and Disease. Biochim. et Biophys. Acta Gen. Subj. 2017, 1861, 2455–2461. [Google Scholar] [CrossRef] [PubMed]
- Burger, D.; Steck, A.J.; Bernard, C.C.A.; de Rosbo, N.K. Human Myelin/Oligodendrocyte Glycoprotein: A New Member of the L2/HNK-1 Family. J. Neurochem. 1993, 61, 1822–1827. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.H.; Freeman, G.J. The B7-CD28 Superfamily. Nature Reviews Immunology 2002, 1861, 2455–2461. [Google Scholar] [CrossRef] [PubMed]
- Jégou, J.-F.; Chan, P.; Schouft, M.-T.; Griffiths, M.R.; Neal, J.W.; Gasque, P.; Vaudry, H.; Fontaine, M. C3d Binding to the Myelin Oligodendrocyte Glycoprotein Results in an Exacerbated Experimental Autoimmune Encephalomyelitis. J. Immunol. 2007, 178, 3323–3331. [Google Scholar] [CrossRef] [PubMed]
- Weissert, R.; Wallström, E.; Storch, M.; Stefferl, A.; Lorentzen, J.; Lassmann, H.; Linington, C.; Olsson, T. MHC haplotype-dependent regulation of MOG-induced EAE in rats. J. Clin. Invest. 1998, 102, 1265–1273. [Google Scholar] [CrossRef]
- Gold, R.; Linington, C.; Lassmann, H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain 2006, 129, 1953–1971. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Pagany, M.; Weiner, H.L.; Linington, C.; Sobel, R.A.; Kuchroo, V.K. Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J. Exp. Med. 2003, 197, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Metz, I.; König, F.B.; Ruprecht, K.; Reindl, M.; Paul, F.; Brück, W.; Wildemann, B. Screening for MOG-IgG and 27 Other Anti-Glial and Anti-Neuronal Autoantibodies in “pattern II Multiple Sclerosis” and Brain Biopsy Findings in a MOG-IgG-Positive Case. Mult. Scler. 2016, 22, 1541–1549. [Google Scholar] [CrossRef]
- Reindl, M.; Linington, C.; Brehm, U.; Egg, R.; Dilitz, E.; Deisenhammer, F.; Poewe, W.; Berger, T. Antibodies against the Myelin Oligodendrocyte Glycoprotein and the Myelin Basic Protein in Multiple Sclerosis and Other Neurological Diseases: A Comparative Study. Brain 1999, 122, 2047–2056. [Google Scholar] [CrossRef]
- Egg, R.; Reindl, M.; Deisenhammer, F.; Linington, C.; Berger, T. Anti-MOG and Anti MBP Antibody Subclasses in Multiple Sclerosis. Mult. Scler. J. 2001, 7, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Berger, T.; Rubner, P.; Schautzer, F.; Egg, R.; Ulmer, H.; Mayringer, I.; Dilitz, E.; Deisenhammer, F.; Reindl, M. Antimyelin Antibodies as a Predictor of Clinically Definite Multiple Sclerosis after a First Demyelinating Event. N. Engl. J. Med. 2003, 349, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Lampasona, V.; Franciotta, D.; Furlan, R.; Zanaboni, S.; Fazio, R.; Bonifacio, E.; Comi, G.; Martino, G. Similar Low Frequency of Anti-MOG IgG and IgM in MS Patients and Healthy Subjects. Neurology 2004, 62, 2092–2094. [Google Scholar] [CrossRef] [PubMed]
- Kuhle, J.; Pohl, C.; Mehling, M.; Edan, G.; Freedman, M.S.; Hartung, H.P.; Polman, C.H.; Miller, D.H.; Montalban, X.; Barkhof, F.; et al. Lack of Association between Antimyelin Antibodies and Progression to Multiple Sclerosis. N. Engl. J. Med. 2007, 356, 371–378. [Google Scholar] [CrossRef]
- Cobo-Calvo, Á.; d’Indy, H.; Ruiz, A.; Collongues, N.; Kremer, L.; Durand-Dubief, F.; Rollot, F.; Casey, R.; Vukusic, S.; De Seze, J.; et al. Frequency of Myelin Oligodendrocyte Glycoprotein Antibody in Multiple Sclerosis: A Multicenter Cross-Sectional Study. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, 1–5. [Google Scholar] [CrossRef]
- Takai, Y.; Misu, T.; Kaneko, K.; Chihara, N.; Narikawa, K.; Tsuchida, S.; Nishida, H.; Komori, T.; Seki, M.; Komatsu, T.; et al. Myelin Oligodendrocyte Glycoprotein Antibody Associated Disease: An Immunopathological Study. Brain 2020, 143, 1431–1446. [Google Scholar] [CrossRef]
- de Graaf, K.L.; Albert, M.; Weissert, R. Autoantigen conformation influences both B- and T-cell responses and encephalitogenicity. J. Biol. Chem. 2012, 287, 17206–17213. [Google Scholar] [CrossRef]
- Waters, P.J.; Komorowski, L.; Woodhall, M.; Lederer, S.; Majed, M.; Fryer, J.; Mills, J.; Flanagan, E.P.; Irani, S.R.; Kunchok, A.C.; et al. A Multicenter Comparison of MOG-IgG Cell-Based Assays. Neurology 2019, 92, e1250–e1255. [Google Scholar] [CrossRef]
- O’Connor, K.C.; McLaughlin, K.A.; De Jager, P.L.; Chitnis, T.; Bettelli, E.; Xu, C.; Robinson, W.H.; Cherry, S.V.; Bar-Or, A.; Banwell, B.; et al. Self-Antigen Tetramers Discriminate between Myelin Autoantibodies to Native or Denatured Protein. Nat. Med. 2007, 13, 211–217. [Google Scholar] [CrossRef]
- Reindl, M.; Schanda, K.; Woodhall, M.; Tea, F.; Ramanathan, S.; Sagen, J.; Fryer, J.P.; Mills, J.; Teegen, B.; Mindorf, S.; et al. International Multicenter Examination of MOG Antibody Assays. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e674. [Google Scholar] [CrossRef]
- López-Chiriboga, A.S.; Majed, M.; Fryer, J.; Dubey, D.; McKeon, A.; Flanagan, E.P.; Jitprapaikulsan, J.; Kothapalli, N.; Tillema, J.M.; Chen, J.; et al. Association of MOG-IgG Serostatus with Relapse after Acute Disseminated Encephalomyelitis and Proposed Diagnostic Criteria for MOG-IgG-Associated Disorders. JAMA Neurol. 2018, 75, 1355–1363. [Google Scholar]
- Brilot, F.; Dale, R.C.; Selter, R.C.; Grummel, V.; Kalluri, S.R.; Aslam, M.; Phil, M.; Busch, V.; Zhou, D.; Cepok, S.; et al. Antibodies to Native Myelin Oligodendrocyte Glycoprotein in Children with Inflammatory Demyelinating Central Nervous System Disease. Ann. Neurol. 2009, 66, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Ketelslegers, I.A.; Van Pelt, D.E.; Bryde, S.; Neuteboom, R.F.; Catsman-Berrevoets, C.E.; Hamann, D.; Hintzen, R.Q. Anti-MOG Antibodies Plead against MS Diagnosis in an Acquired Demyelinating Syndromes Cohort. Mult. Scler. 2015, 21, 1513–1520. [Google Scholar] [CrossRef] [PubMed]
- Cobo-Calvo, Á.; Ruiz, A.; D’Indy, H.; Poulat, A.L.; Carneiro, M.; Philippe, N.; Durand Dubief, F.; Deiva, K.; Vukusic, S.; Desportes, V.; et al. MOG Antibody-Related Disorders: Common Features and Uncommon Presentations. J. Neurol. 2017, 264, 1945–1955. [Google Scholar] [CrossRef]
- Jurynczyk, M.; Messina, S.; Woodhall, M.R.; Raza, N.; Everett, R.; Roca-Fernandez, A.; Tackley, G.; Hamid, S.; Sheard, A.; Reynolds, G.; et al. Clinical Presentation and Prognosis in MOG-Antibody Disease: A UK Study. Brain 2017, 140, 3128–3138. [Google Scholar] [CrossRef]
- Ciotti, J.; Eby, N.; Wu, G.; Naismith, R.; Chahin, S.; Cross, A. Clinical and laboratory features distinguishing MOG antibody disease from multiple sclerosis and AQP4 antibody-positive neuromyelitis optica. Mult. Scler. Relat. Disord. 2020, 45, 102399. [Google Scholar] [CrossRef]
- Armangue, T.; Olivé-Cirera, G.; Martínez-Hernandez, E.; Sepulveda, M.; Ruiz-Garcia, R.; Muñoz-Batista, M.; Ariño, H.; González-Álvarez, V.; Felipe-Rucián, A.; Jesús Martínez-González, M.; et al. Associations of paediatric demyelinating and encephalitic syndromes with myelin oligodendrocyte glycoprotein antibodies: A multicentre observational study. Lancet Neurol. 2020, 19, 234–246. [Google Scholar] [CrossRef]
- Cobo-Calvo, A.; Ruiz, A.; Maillart, E.; Audoin, B.; Zephir, H.; Bourre, B.; Ciron, J.; Collongues, N.; Brassat, D.; Cotton, F.; et al. Clinical Spectrum and Prognostic Value of CNS MOG Autoimmunity in Adults: The MOGADOR Study. Neurology 2018, 90, e1858–e1869. [Google Scholar] [CrossRef]
- Jarius, S.; Ruprecht, K.; Kleiter, I.; Borisow, N.; Asgari, N.; Pitarokoili, K.; Pache, F.; Stich, O.; Beume, L.A.; Hümmert, M.; et al. MOG-IgG in NMO and Related Disorders: A Multicenter Study of 50 Patients. Part 2: Epidemiology, Clinical Presentation, Radiological and Laboratory Features, Treatment Responses, and Long-Term Outcome. J. Neuroinflamm. 2016, 13, 280. [Google Scholar] [CrossRef]
- Petzold, A.; Plant, G.T. Chronic Relapsing Inflammatory Optic Neuropathy: A Systematic Review of 122 Cases Reported. J. Neurol. 2014, 261, 17–26. [Google Scholar] [CrossRef]
- Netravathi, M.; Venkappayya Holla, V.; Nalini, A.; Yadav, R.; Vengalil, S.; Oommen, A.; Shaik Reshma, S.; Kamble, N.; Thomas, P.; Maya, B.; et al. Myelin oligodendrocyte glycoprotein-antibody-associated disorder: A new inflammatory CNS demyelinating disorder. J. Neurol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Kleiter, I.; Ruprecht, K.; Asgari, N.; Pitarokoili, K.; Borisow, N.; Hümmert, M.W.; Trebst, C.; Pache, F.; Winkelmann, A.; et al. MOG-IgG in NMO and Related Disorders: A Multicenter Study of 50 Patients. Part 3: Brainstem Involvement—Frequency, Presentation and Outcome. J. Neuroinflamm. 2016, 13, 281. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.H.; Zheng, Y.; Cai, M.T.; Yang, F.; Fang, W.; Zhang, Y.X.; Ding, M.P. Seizure Occurrence in Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease: A Systematic Review and Meta-Analysis. Mult. Scler. Relat. Disord. 2020, 42, 102057. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.; Uyttenboogaart, M.; Polman, S.; De Keyser, J. Seizures in Multiple Sclerosis. Epilepsia 2008, 49, 948–953. [Google Scholar] [CrossRef]
- Hochmeister, S.; Gattringer, T.; Asslaber, M.; Stangl, V.; Haindl, M.T.; Enzinger, C.; Höftberger, R. A Fulminant Case of Demyelinating Encephalitis with Extensive Cortical Involvement Associated with Anti-MOG Antibodies. Front. Neurol. 2020, 11, 31. [Google Scholar] [CrossRef]
- Wingerchuk, D.M.; Hogancamp, W.F.; O’Brien, P.C.; Weinshenker, B.G. The clinical course of neuromyelitis optica (Devic’s syndrome). Neurology 1999, 53, 1107–1114. [Google Scholar] [CrossRef]
- Wingerchuk, D.M.; Lennon, V.A.; Lucchinetti, C.F.; Pittock, S.; Weinshenker, B. The spectrum of neuromyelitis optica. Lancet Neurol. 2007, 6, 805–815. [Google Scholar] [CrossRef]
- Wingerchuk, D.M. Neuromyelitis spectrum disorders. Contin. Lifelong Learn. Neurol. 2010, 16, 105–121. [Google Scholar] [CrossRef]
- Jarius, S.; Pellkofer, H.; Siebert, N.; Korporal-Kuhnke, M.; Hümmert, M.W.; Ringelstein, M.; Rommer, P.S.; Ayzenberg, I.; Ruprecht, K. In cooperation with the Neuromyelitis Optica Study Group (NEMOS). Cerebrospinal fluid findings in patients with myelin oligodendrocyte glycoprotein (MOG) antibodies. Part 1: Results from 163 lumbar punctures in 100 adult patients. J. Neuroinflamm. 2020, 17, 261. [Google Scholar] [CrossRef]
- Höftberger, R.; Guo, Y.; Flanagan, E.P.; Lopez-Chiriboga, A.S.; Endmayr, V.; Hochmeister, S.; Joldic, D.; Pittock, S.J.; Tillema, J.M.; Gorman, M.; et al. The pathology of central nervous system inflammatory demyelinating disease accompanying myelin oligodendrocyte glycoprotein autoantibody. Acta Neuropathol. 2020, 139, 875–892. [Google Scholar] [CrossRef]
- Biotti, D.; Bonneville, F.; Tournaire, E.; Ayrignac, X.; Dallière, C.C.; Mahieu, L.; Vignal, C.; Dulau, C.; Brochet, B.; Ruet, A.; et al. Optic Neuritis in Patients with Anti-MOG Antibodies Spectrum Disorder: MRI and Clinical Features from a Large Multicentric Cohort in France. J. Neurol. 2017, 264, 2173–2175. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, S.; Prelog, K.; Barnes, E.H.; Tantsis, E.M.; Reddel, S.W.; Henderson, A.P.D.; Vucic, S.; Gorman, M.P.; Benson, L.A.; Alper, G.; et al. Radiological Differentiation of Optic Neuritis with Myelin Oligodendrocyte Glycoprotein Antibodies, Aquaporin-4 Antibodies, and Multiple Sclerosis. Mult. Scler. 2016, 22, 470–482. [Google Scholar] [CrossRef] [PubMed]
- Dubey, D.; Pittock, S.J.; Krecke, K.N.; Morris, P.; Sechi, E.; Zalewski, N.; Weinshenker, B.D.; Shosha, E.; Lucchinetti, C. Clinical, Radiologic, and Prognostic Features of Myelitis Associated with Myelin Oligodendrocyte Glycoprotein Autoantibody. JAMA Neurol. 2019, 76, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Baumann, M.; Grams, A.; Djurdjevic, T.; Wendel, E.M.; Lechner, C.; Behring, B.; Blaschek, A.; Diepold, K.; Eisenkölbl, A.; Fluss, J.; et al. MRI of the First Event in Pediatric Acquired Demyelinating Syndromes with Antibodies to Myelin Oligodendrocyte Glycoprotein. J. Neurol. 2018, 265, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Budhram, A.; Mirian, A.; Le, C.; Hosseini-Moghaddam, S.M.; Sharma, M.; Nicolle, M.W. Unilateral Cortical FLAIR-Hyperintense Lesions in Anti-MOG-Associated Encephalitis with Seizures (FLAMES): Characterization of a Distinct Clinico-Radiographic Syndrome. J. Neurol. 2019, 266, 2481–2487. [Google Scholar] [CrossRef]
- Budhram, A.; Kunchok, A.; Flanagan, E. Adding FUEL to the FLAMES: FLAIR-Variable Unilateral Enhancement of the Leptomeninges (FUEL) in MOG-IgG-Associated Disease. Neurology 2020, 94, 862. [Google Scholar]
- Juryńczyk, M.; Tackley, G.; Kong, Y.; Geraldes, R.; Matthews, L.; Woodhall, M.; Waters, P.; Kuker, W.; Craner, M.; Weir, A.; et al. Brain Lesion Distribution Criteria Distinguish MS from AQP4-Antibody NMOSD and MOG-Antibody Disease. J. Neurol. Neurosurg. Psychiatry 2017, 88, 132–136. [Google Scholar] [CrossRef]
- Yang, L.; Li, H.; Xia, W.; Quan, C.; Zhou, L.; Geng, D.; Li, Y. Quantitative Brain Lesion Distribution May Distinguish MOG-Ab and AQP4-Ab Neuromyelitis Optica Spectrum Disorders. Eur. Radiol. 2020, 30, 1470–1479. [Google Scholar] [CrossRef]
- Wynford-Thomas, R.; Jacob, A.; Tomassini, V. Neurological update: MOG antibody disease. J. Neurol. 2019, 266, 1280–1286. [Google Scholar] [CrossRef]
- Ogawa, R.; Nakashima, I.; Takahashi, T.; Kaneko, K.; Akaishi, T.; Takai, Y.; Sato, D.K.; Nishiyama, S.; Misu, T.; Kuroda, H.; et al. MOG Antibody-Positive, Benign, Unilateral, Cerebral Cortical Encephalitis with Epilepsy. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e322. [Google Scholar] [CrossRef]
- Wang, L.; ZhangBao, J.; Zhou, L.; Zhang, Y.; Li, H.; Li, Y.; Huang, Y.; Wang, M.; Lu, C.; Lu, J.; et al. Encephalitis Is an Important Clinical Component of Myelin Oligodendrocyte Glycoprotein Antibody Associated Demyelination: A Single-Center Cohort Study in Shanghai, China. Eur. J. Neurol. 2019, 26, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Cobo-Calvo, A.; Sepúlveda, M.; Rollot, F.; Armangué, T.; Ruiz, A.; Maillart, E.; Papeix, C.; Audoin, B.; Zephir, H.; Biotti, D.; et al. Evaluation of Treatment Response in Adults with Relapsing MOG-Ab-Associated Disease. J. Neuroinflamm. 2019, 16, 134. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, S.; Mohammad, S.; Tantsis, E.; Nguyen, T.K.; Merheb, V.; Fung, V.S.C.; White, O.B.; Broadley, S.; Lechner-Scott, J.; Vucic, S.; et al. Clinical Course, Therapeutic Responses and Outcomes in Relapsing MOG Antibody-Associated Demyelination. J. Neurol. Neurosurg. Psychiatry 2018, 89, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Hacohen, Y.; Wong, Y.Y.; Lechner, C.; Jurynczyk, M.; Wright, S.; Konuskan, B.; Kalser, J.; Poulat, A.L.; Maurey, H.; Ganelin-Cohen, E.; et al. Disease Course and Treatment Responses in Children with Relapsing Myelin Oligodendrocyte Glycoprotein Antibody–Associated Disease. JAMA Neurol. 2018, 75, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ren, H.; Xu, Y.; Xu, T.; Zhang, Y.; Yin, H.; Zhang, W.; Li, J.; Ren, X.; Fang, F.; et al. Long-Term Efficacy of Mycophenolate Mofetil in Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disorders: A Prospective Study. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e705. [Google Scholar] [CrossRef] [PubMed]
- Durozard, P.; Rico, A.; Boutiere, C.; Maarouf, A.; Lacroix, R.; Cointe, S.; Fritz, S.; Brunet, C.; Pelletier, J.; Marignier, R.; et al. Comparison of the Response to Rituximab between Myelin Oligodendrocyte Glycoprotein and Aquaporin-4 Antibody Diseases. Ann. Neurol. 2020, 87, 256–266. [Google Scholar] [CrossRef]
- Domingo-Santos, Á.; Sepúlveda, M.; Matarazzo, M.; Calleja-Castaño, P.; Ramos González, A.; Saiz, A.; Benito-León, J. Intravenous Immunoglobulin Therapy in a Patient with Anti-Myelin Oligodendrocyte Glycoprotein-Seropositive Neuromyelitis Optica. Clin. Neuropharmacol. 2016, 39, 332–334. [Google Scholar] [CrossRef]
- Chen, J.J.; Flanagan, E.P.; Bhatti, M.T.; Jitprapaikulsan, J.; Dubey, D.; Chiriboga, A.S.; Fryer, J.P.; Weinshenker, B.G.; McKeon, A. Steroid Sparing Maintenance Immunotherapy for MOG-IgG Associated Disorder. Neurology 2020, 95, e111–e120. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NMO | MS | MOGAD | |
|---|---|---|---|
| Oligoclonal bands (OCB) | Typically absent (present in approx. 15–30% of NMO patients, they can occur transiently) | Positive in approx. 85–90% of MS patients (they do not disappear or change in the course of the disease or following treatment) | CSF-restricted OCB unusual, positive in a minority of samples (13.2%) |
| IgG index | Usually elevated | Elevated > 0.7 (typically > 1.7) in approx. 70% of MS patients (decreases following steroid treatment) | elevated in a minority of samples (8%) |
| Total protein | Elevated (100–500 mg/dL) in approx. 25–30% of NMO patients | Within normal limits or > 40 mg/dL in approx. 15% of MS patients | elevated in approx. 44% of samples (range 45.3–176 mg/dL) |
| Cytosis | >50/mm3 (at the time of the attack in approx. 30–80% of NMO patients) | >5/mm3 (rarely above 50/mm3) in approx. 30% of patients | Pleocytosis present at least once in> 57% of samples, > 50 cells/mm3 in about 19% of cases |
| Cell type | Neutrophil-predominant pleocytosis, with the presence of eosinophils | Mononuclear cells; lymphocyte-predominant | Lymphocytes and monocytes, neutrophils present in at least 43% of cases; eosinophils and basophils–very rare |
| Clinical | |
|---|---|
| Optic neuritis (ON) |
|
| Myelitis |
|
| ADEM brainstem/cortical encephalitis |
|
| Fundoscopy |
|
| Others |
|
| Laboratory | |
| MRI image |
|
| CSF |
|
| Histopathology |
|
| Treatment response |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ambrosius, W.; Michalak, S.; Kozubski, W.; Kalinowska, A. Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease: Current Insights into the Disease Pathophysiology, Diagnosis and Management. Int. J. Mol. Sci. 2021, 22, 100. https://doi.org/10.3390/ijms22010100
Ambrosius W, Michalak S, Kozubski W, Kalinowska A. Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease: Current Insights into the Disease Pathophysiology, Diagnosis and Management. International Journal of Molecular Sciences. 2021; 22(1):100. https://doi.org/10.3390/ijms22010100
Chicago/Turabian StyleAmbrosius, Wojciech, Sławomir Michalak, Wojciech Kozubski, and Alicja Kalinowska. 2021. "Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease: Current Insights into the Disease Pathophysiology, Diagnosis and Management" International Journal of Molecular Sciences 22, no. 1: 100. https://doi.org/10.3390/ijms22010100
APA StyleAmbrosius, W., Michalak, S., Kozubski, W., & Kalinowska, A. (2021). Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease: Current Insights into the Disease Pathophysiology, Diagnosis and Management. International Journal of Molecular Sciences, 22(1), 100. https://doi.org/10.3390/ijms22010100