Identification and Functional Study of Chitin Metabolism and Detoxification-Related Genes in Glyphodes pyloalis Walker (Lepidoptera: Pyralidae) Based on Transcriptome Analysis


 ,
,
Abstract
1. Introduction
2. Results
2.1. Overview of G. pyloalis Transcriptome
2.2. Unigenes Annotation and Classification
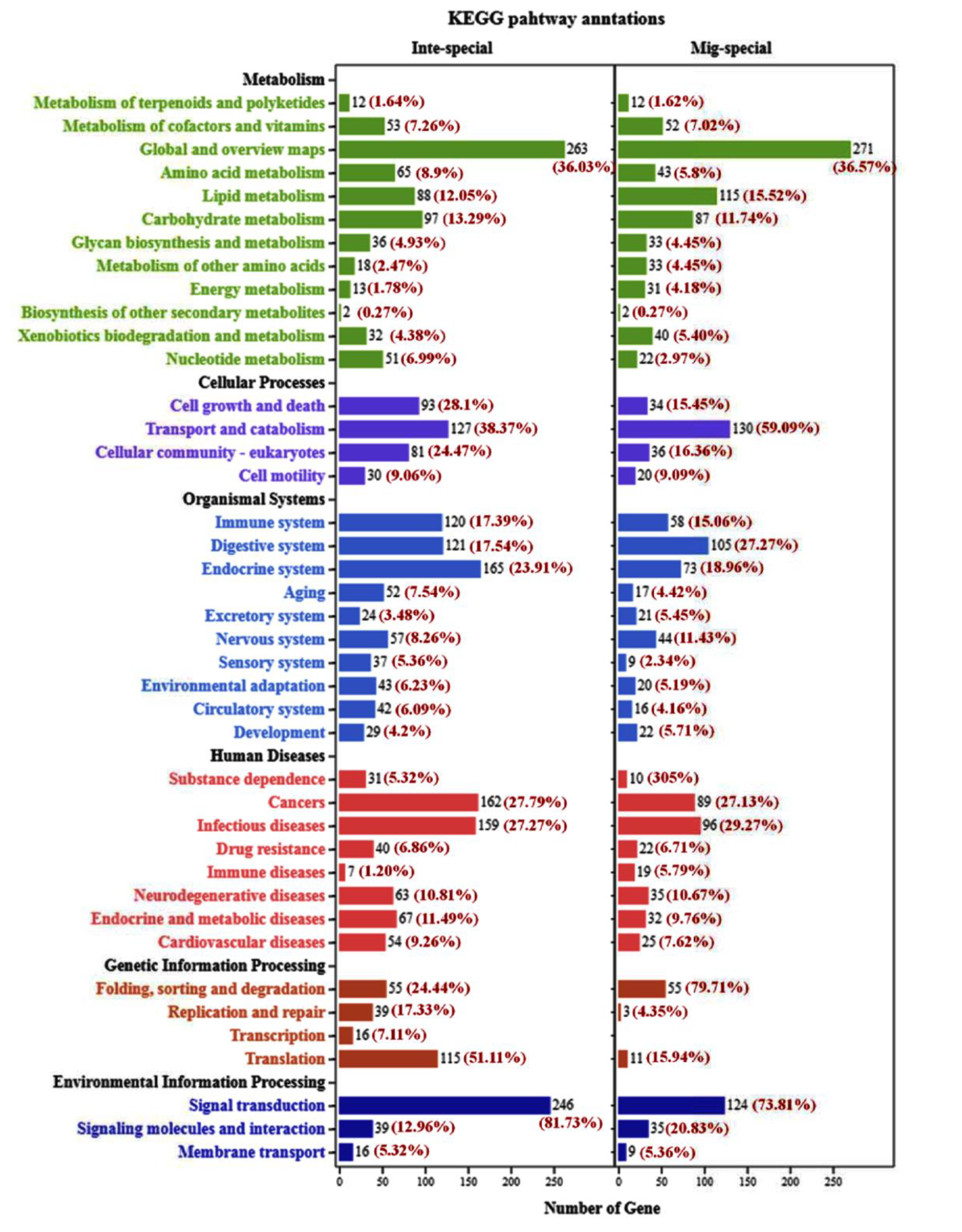
2.3. The Analysis of KEGG Pathway and GO Annotation between Integument and Midgut
2.4. The Validation of Transcriptome Data by RT-qPCR
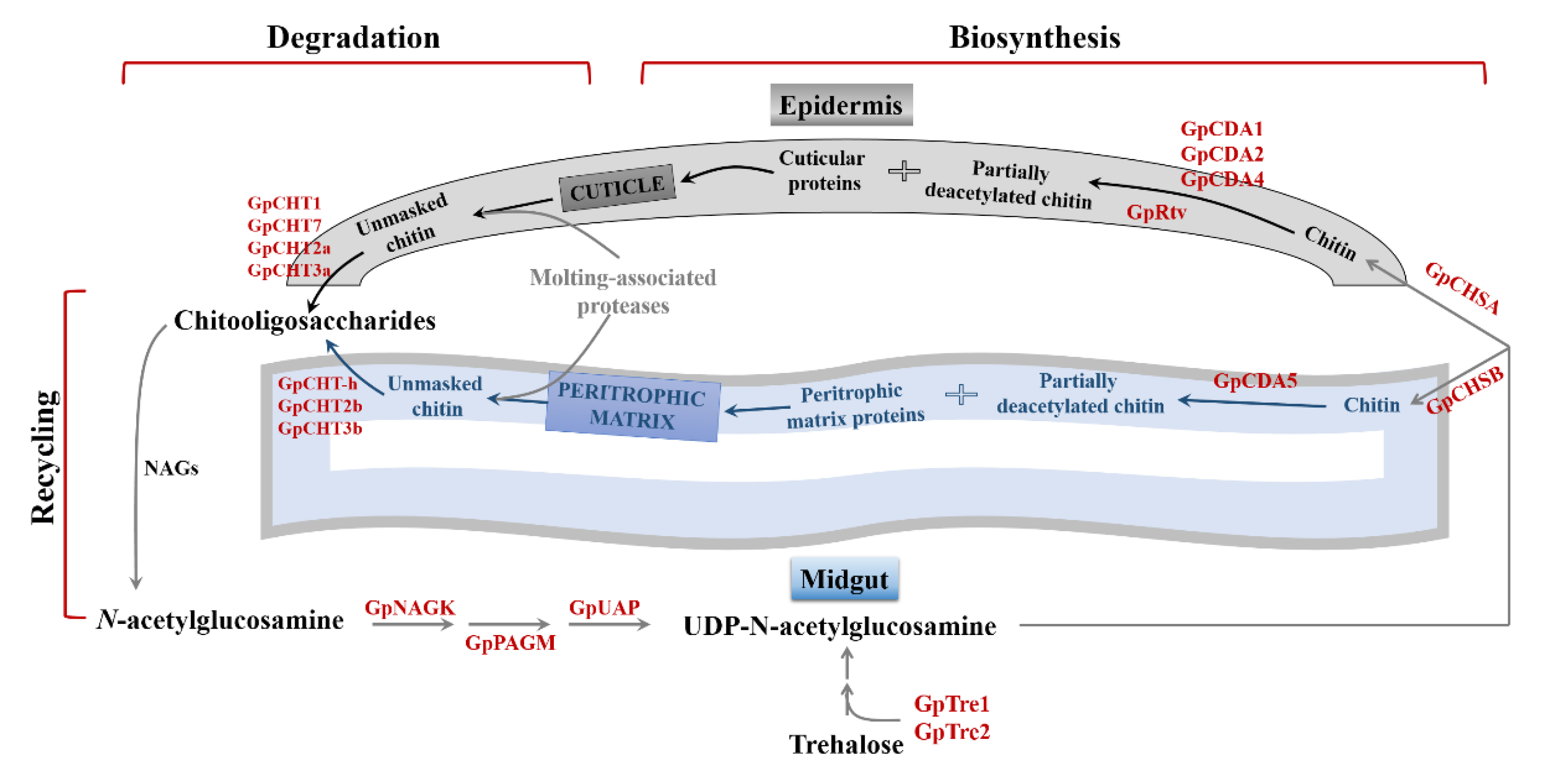
2.5. Transcripts Encoding Chitin Metabolism Enzymes
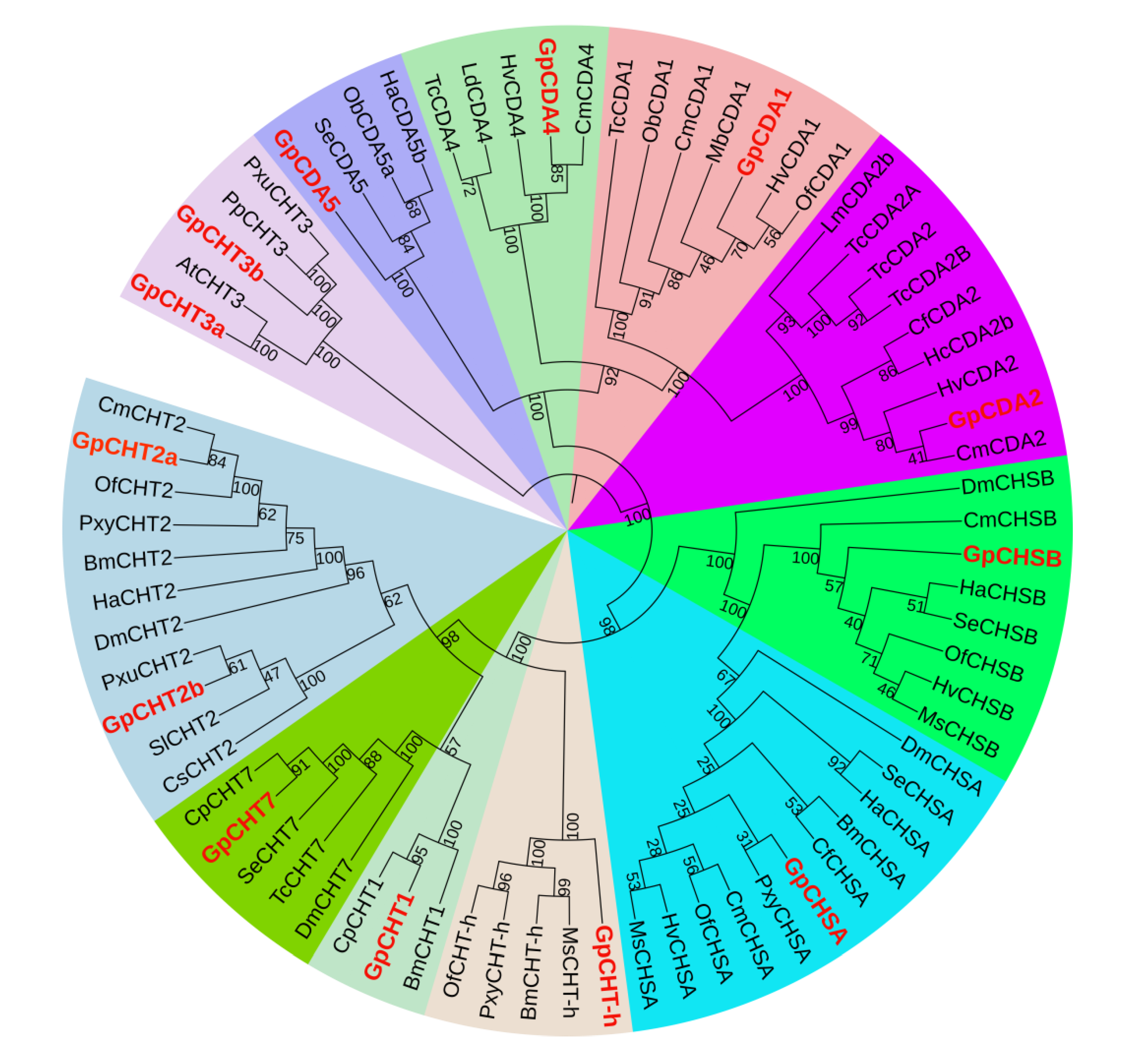
2.6. Chitin-Metabolizing Enzyme Networks of G. pyloalis
2.7. Identification of Detoxification-Related Genes
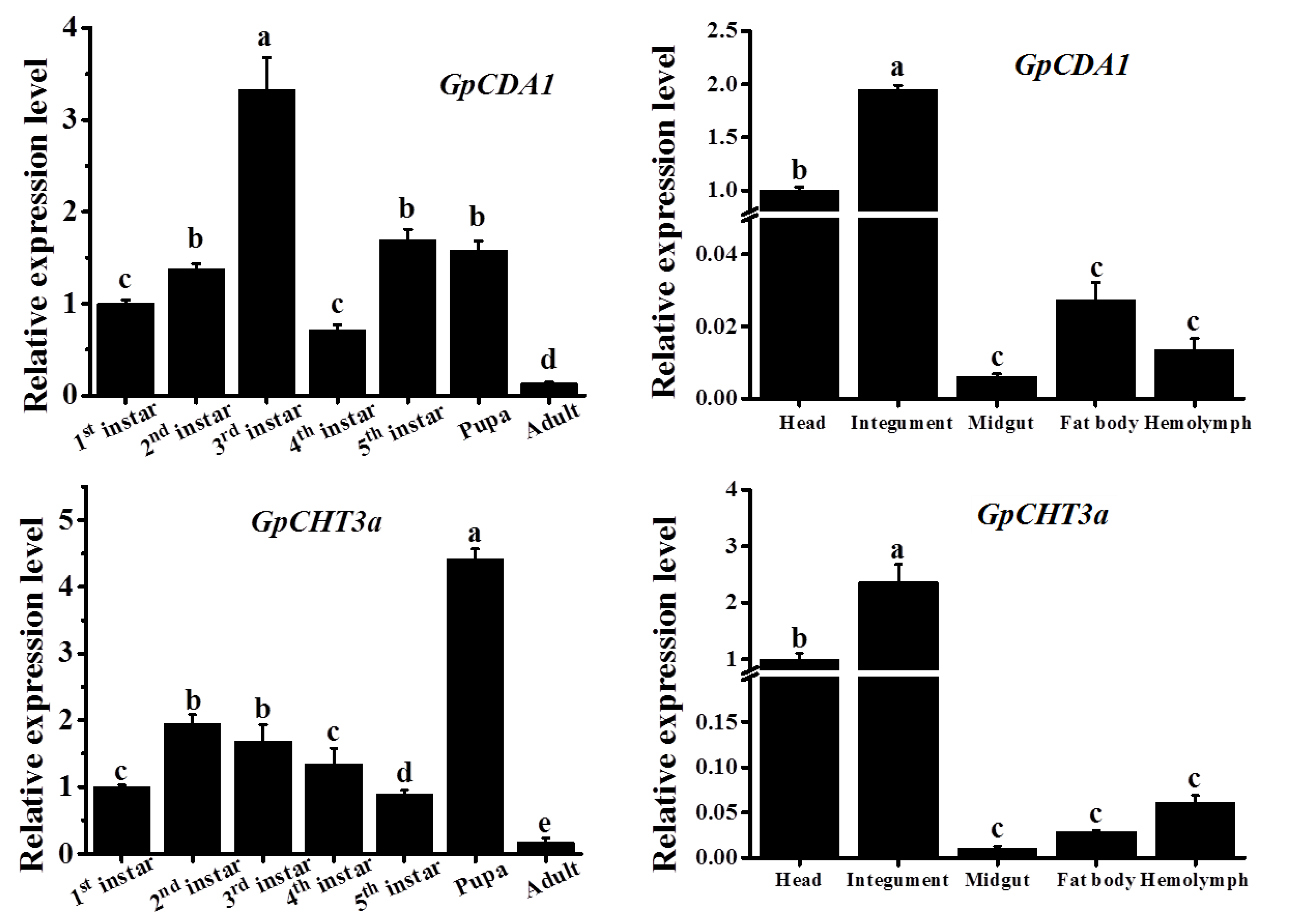
2.8. The Spatiotemporal Expression Pattern of GpCDA1 and GpCHT3a
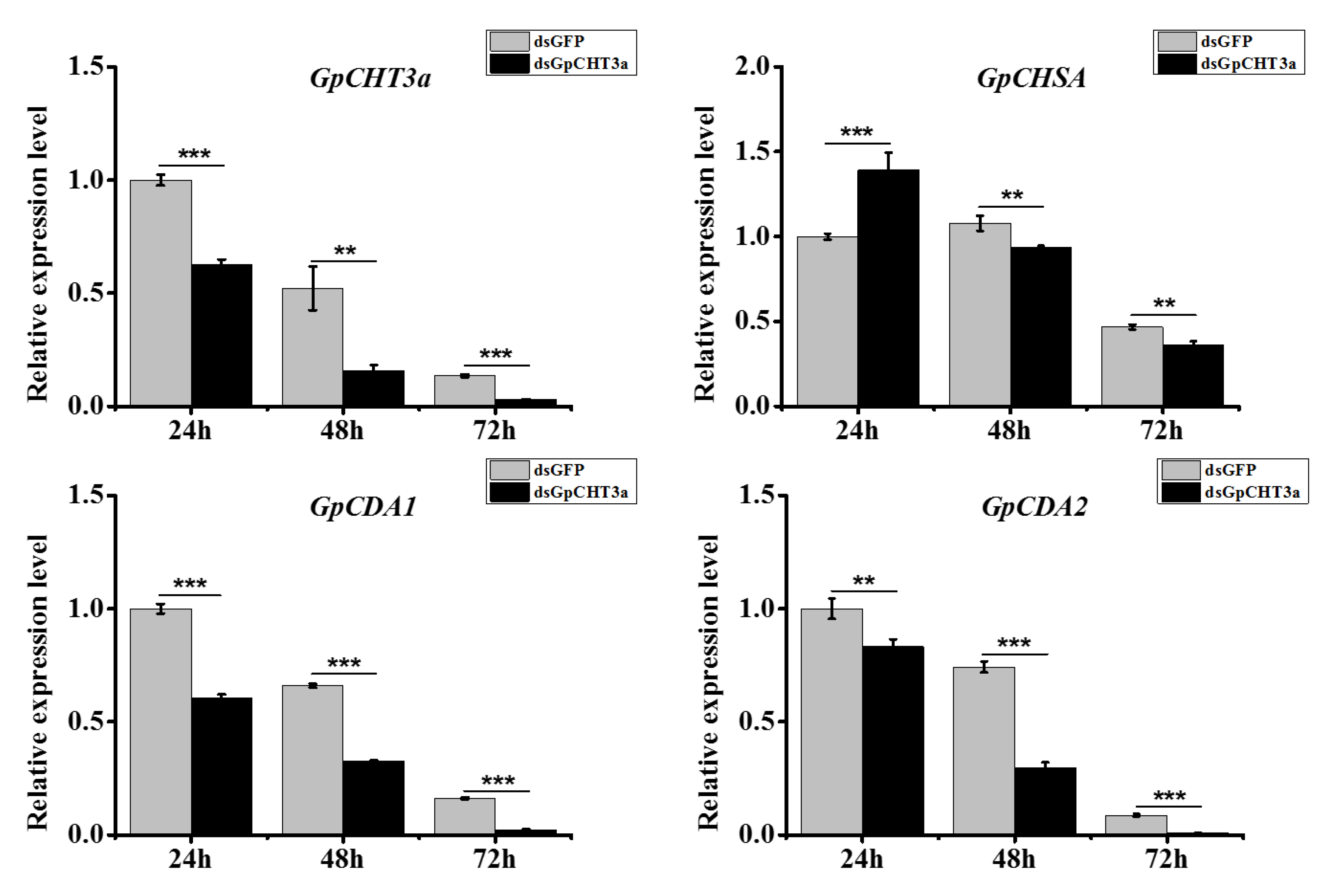
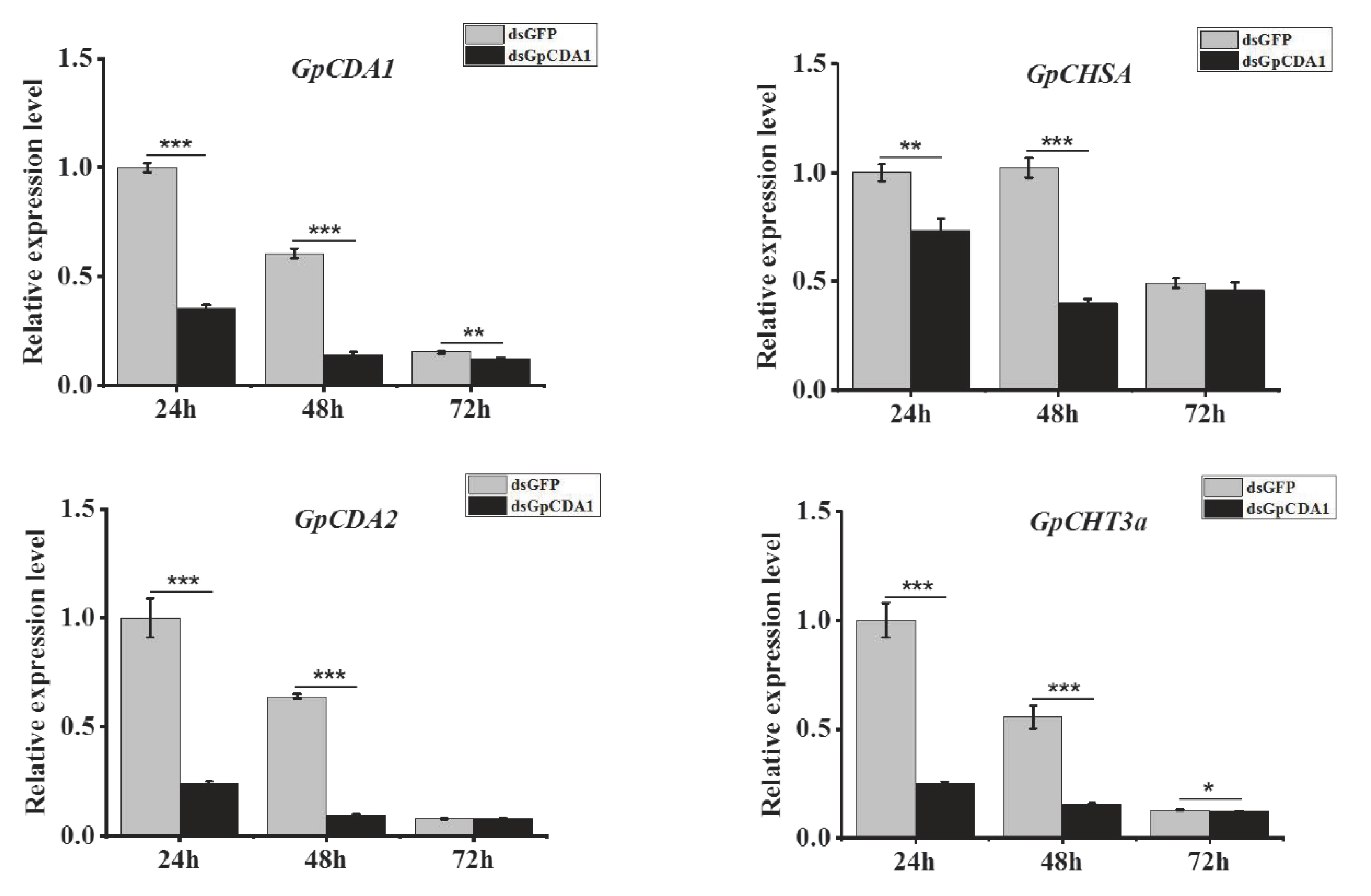
2.9. The Analysis of the Function of GpCHT3a in Chitin Metabolism Using dsRNA
2.10. The Analysis of the Function of GpCDA1 in Chitin Metabolism Using dsRNA
3. Discussion
4. Materials and Methods
4.1. G. pyloalis Rearing and Sample Preparation
4.2. RNA Extraction and Quality Analysis
4.3. Library Construction, Sequencing, and Assembly
4.4. Functional Annotation
4.5. Real-Time Quantitative PCR (RT-qPCR)
4.6. Bioinformatics Analysis
4.7. The Synthesis of dsRNA and Quality Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Khosravi, R.; Sendi, J.J. Biology and demography of Glyphodes pyloalis Walker (Lepidoptera: Pyralidae) on mulberry. J. Asia-Pac. Entomol. 2010, 13, 273–276. [Google Scholar] [CrossRef]
- Watanabe, H.; Kurihara, Y.; Wang, Y.X.; Shimizu, T. Mulberry pyralid, Glyphodes pyloalis: Habitual host of nonoccluded viruses pathogenic to the silkworm, Bombyx mori. J. Invertebr. Pathol. 1988, 52, 401–408. [Google Scholar] [CrossRef]
- Su, H.; Gao, Y.; Liu, Y.; Li, X.; Liang, Y.; Dai, X.; Xu, Y.; Zhou, Y.; Wang, H. Comparative transcriptome profiling reveals candidate genes related to insecticide resistance of Glyphodes pyloalis. Bull. Entomol. Res. 2019, 110, 57–67. [Google Scholar] [CrossRef]
- Marguerite, R. Chitin and chitosan: Properties and applications. Prog. Polym. Sci. 2006, 31, 603–632. [Google Scholar]
- Rahman, M.A.; Halfar, J. First evidence of chitin in calcified coralline algae: New insights into the calcification process of Clathromorphum compactum. Sci. Rep. 2014, 4, 6162. [Google Scholar] [CrossRef]
- Rahman, M.A.; Halfar, J.; Adey, W.H.; Nash, M.; Paulo, C.; Dittrich, M. The role of chitin-rich skeletal organic matrix on the crystallization of calcium carbonate in the crustose coralline alga Leptophytum foecundum. Sci. Rep. 2019, 9, 11869. [Google Scholar] [CrossRef]
- Kramer, K.J.; Muthukrishnan, S. Insect Chitinases: Molecular Biology and Potential Use as Biopesticides. Insect. Biochem. Mol. Biol. 1997, 27, 887–900. [Google Scholar] [CrossRef]
- Merzendorfer, H. The cellular basis of chitin synthesis in fungi and insects: Common principles and differences. Eur. J. Cell. Biol. 2011, 90, 759–769. [Google Scholar] [CrossRef]
- Zhu, K.Y.; Merzendorfer, H.; Zhang, W.; Zhang, J.; Muthukrishnan, S. Biosynthesis, Turnover, and Functions of Chitin in Insects. Annu. Rev. Entomol. 2016, 61, 177–196. [Google Scholar] [CrossRef]
- Arakane, Y.; Dixit, R.; Begum, K.; Park, Y.; Specht, C.A.; Merzendorfer, H.; Kramer, K.J.; Muthukrishnan, S.; Beeman, R.W. Analysis of functions of the chitin deacetylase gene family in Tribolium castaneum. Insect. Biochem. Mol. Biol. 2009, 39, 355–365. [Google Scholar] [CrossRef]
- Toprak, U.; Hegedus, D.D.; Baldwin, D.; Coutu, C.; Erlandson, M. Spatial and temporal synthesis of Mamestra configurata peritrophic matrix through a larval stadium. Insect. Biochem. Mol. Biol. 2014, 54, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Arakane, Y.; Muthukrishnan, S. Insect chitinase and chitinase-like proteins. Cell. Mol. Life. Sci. 2010, 67, 201–216. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Tsuji, N.; Miyoshi, T.; Motobu, M.; Islam, M.K.; Alim, M.A.; Fujisaki, K. Characterization of glutamine: Fructose-6-phosphate aminotransferase from the ixodid tick, Haemaphysalis longicornis, and its critical role in host blood feeding. Int. J. Parasitol. 2007, 37, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, H.; Li, S.; Zhu, K.Y.; Ma, E.; Zhang, J. Characterization of a midgut-specific chitin synthase gene (LmCHS2) responsible for biosynthesis of chitin of peritrophic matrix in Locusta migratoria. Insect. Biochem. Mol. Biol. 2012, 42, 902–910. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.Z.; Wen, D.F.; Wang, W.L.; Geng, L.; Zhang, Y.; Xu, J.P. Identification of Genes Putatively Involved in Chitin Metabolism and Insecticide Detoxification in the Rice Leaf Folder (Cnaphalocrocis medinalis) Larvae through Transcriptomic Analysis. Int. J. Mol. Sci. 2015, 16, 21873–21896. [Google Scholar] [CrossRef]
- Liu, N.N. Insecticide Resistance in Mosquitoes: Impact, Mechanisms, and Research Directions. Annu. Rev. Entomol. 2015, 60, 537–559. [Google Scholar] [CrossRef]
- Ranson, H.; Claudianos, C.; Ortelli, F.; Abgrall, C.; Hemingway, J.; Sharakhova, M.V.; Unger, M.F.; Collins, F.H.; Feyereisen, R. Evolution of Supergene Families Associated with Insecticide Resistance. Science 2002, 298, 179–181. [Google Scholar] [CrossRef]
- Lee, H.; Yoon, K.L.; Kwon, H.J.; Clark, A.M. A point mutation in a glutamate-gated chloride channel confers abamectin resistance in the two-spotted spider mite, Tetranychus urticae Koch. Insect. Mol. Biol. 2010, 19, 583–591. [Google Scholar]
- Khajehali, J.; Leeuwen, T.V.; Grispou, M.; Morou, E.; Alout, H.; Weill, M.; Tirry, L.; Vontas, J.; Tsagkarakou, A. Acetylcholinesterase point mutations in European strains of Tetranychus urticae (Acari: Tetranychidae) resistant to organophosphates. Pest. Manag. Sci. 2010, 66, 220–228. [Google Scholar]
- Xia, W.K.; Ding, T.B.; Niu, J.Z.; Liao, C.Y.; Zhong, R.; Yang, W.J.; Liu, B.; Dou, W.; Wang, J.J. Exposure to diflubenzuron results in an up-regulation of a chitin synthase 1 gene in citrus red mite, Panonychus citri (Acari: Tetranychidae). Int. J. Mol. Sci. 2014, 15, 3711–3728. [Google Scholar] [CrossRef]
- Wang, Z.; Zhou, C.; Long, G.Y.; Yang, H.; Jin, D.C. Sublethal effects of buprofezin on development, reproduction, and chitin synthase 1 gene (SfCHS1) expression in the white-backed planthopper, Sogatella furcifera (Hemiptera: Delphacidae). J. Asia-Pac. Entomol. 2018, 21, 585–591. [Google Scholar] [CrossRef]
- Harethardottir, H.M.; Male, R.; Nilsen, F.; Dalvin, S. Effects of chitin synthesis inhibitor treatment on Lepeophtheirus salmonis (Copepoda, Caligidae) larvae. PLoS ONE 2019, 14, e0222520. [Google Scholar]
- Tetreau, G.; Cao, X.; Chen, Y.R.; Muthukrishnan, S.; Jiang, H.; Blissard, G.W.; Kanost, M.R.; Wang, P. Overview of chitin metabolism enzymes in Manduca sexta: Identification, domain organization, phylogenetic analysis and gene expression. Insect. Biochem. Molec. 2015, 62, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.R.d.; Santos, R.d.C. Pest Control: Can Chitinases Help To Reduce Pesticide Use? J. Agric. Food. Chem. 2019, 67, 8071–8073. [Google Scholar] [CrossRef]
- Bansal, R.; Mian, M.A.; Mittapalli, O.; Michel, A.P. Characterization of a chitin synthase encoding gene and effect of diflubenzuron in soybean aphid, Aphis glycines. Int. J. Biol. Sci. 2012, 8, 1323–1334. [Google Scholar] [CrossRef]
- Ye, C.; Jiang, Y.D.; An, X.; Yang, L.; Shang, F.; Niu, J.; Wang, J.J. Effects of RNAi-based silencing of chitin synthase gene on moulting and fecundity in pea aphids (Acyrthosiphon pisum). Sci. Rep. 2019, 9, 3694. [Google Scholar] [CrossRef]
- Wu, J.J.; Mu, L.L.; Chen, Z.C.; Fu, K.Y.; Guo, W.C.; Li, C.; Li, G.Q. Disruption of ecdysis in Leptinotarsa decemlineata by knockdown of chitin deacetylase 1. J. Asia-Pac. Entomol. 2019, 22, 443–452. [Google Scholar] [CrossRef]
- Yu, R.R.; Liu, W.M.; Zhao, X.M.; Zhang, M.; Li, D.Q.; Zuber, R.; Ma, E.B.; Zhu, K.Y.; Moussian, B.; Zhang, J.Z. LmCDA1 organizes the cuticle by chitin deacetylation in Locusta migratoria. Insect. Mol. Biol. 2019, 28, 301–312. [Google Scholar] [CrossRef]
- Wu, J.J.; Chen, Z.C.; Wang, Y.W.; Fu, K.Y.; Guo, W.C.; Li, G.Q. Silencing chitin deacetylase 2 impairs larval-pupal and pupal-adult molts in Leptinotarsa decemlineata. Insect. Mol. Biol. 2019, 28, 52–64. [Google Scholar] [CrossRef]
- Zhu, B.; Shan, J.Q.; Li, R.; Liang, P.; Gao, X.W. Identification and RNAi-based function analysis of chitinase family genes in diamondback moth, Plutella xylostella. Pest. Manag. Sci. 2019, 75, 1951–1961. [Google Scholar] [CrossRef]
- Shen, Q.D.; Yang, M.M.; Xie, G.Q.; Wang, H.J.; Zhang, L.; Qiu, L.Y.; Wang, S.G.; Tang, B. Excess trehalose and glucose affects chitin metabolism in brown planthopper (Nilaparvata lugens). J. Asia-Pac. Entomol. 2017, 20, 449–455. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, X.; Katsuma, S.; Xu, Y.; Shi, L.; Shimada, T.; Wang, H. Duplication and diversification of trehalase confers evolutionary advantages on lepidopteran insects. Mol. Ecol. 2019, 28, 5282–5298. [Google Scholar] [CrossRef]
- Chaudhari, S.S.; Noh, M.Y.; Moussian, B.; Specht, C.A.; Kramer, K.J.; Beeman, R.W.; Arakane, Y.; Muthukrishnan, S. Knickkopf and retroactive proteins are required for formation of laminar serosal procuticle during embryonic development of Tribolium castaneum. Insect. Biochem. Mol. Biol. 2015, 60, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, S.S.; Arakane, Y.; Specht, C.A.; Moussian, B.; Kramer, K.J.; Muthukrishnan, S.; Beeman, R.W. Retroactive maintains cuticle integrity by promoting the trafficking of Knickkopf into the procuticle of Tribolium castaneum. PLoS. Genet. 2013, 9, e1003268. [Google Scholar] [CrossRef] [PubMed]
- Shaoya, L.; Dan, Z.; Jing, L.; Xiaotong, S.; Wei, G.; Xiujun, L. Response of CDA5 in Hyphantria cunea to Bt toxin ingestion and Knockdown in transfected Sf9 cells. J. Appl. Entomol. 2017, 141, 308–314. [Google Scholar] [CrossRef]
- Neitzel, L.R.; Spencer, Z.T.; Nayak, A.; Cselenyi, C.S.; Benchabane, H.; Youngblood, C.Q.; Zouaoui, A.; Ng, V.; Stephens, L.; Hann, T.; et al. Developmental regulation of Wnt signaling by Nagk and the UDP-GlcNAc salvage pathway. Mech. Dev. 2019, 156, 20–31. [Google Scholar] [CrossRef]
- Muthukrishnan, S.; Merzendorfer, H.; Arakane, Y.; Kramer, K.J. Chitin Metabolism in Insects. Insect. Biochem. Mol. 2012, 193–235. [Google Scholar]
- Merzendorfer, H. Chitin metabolism in insects: Structure, function and regulation of chitin synthases and chitinases. J. Exp. Biol. 2003, 206, 4393–4412. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Li, R.Q.; Yu, C.; Li, Y.R.; Lam, T.W.; Yiu, S.M.; Kristiansen, K.; Wang, J. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar] [CrossRef]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talon, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic. Acids. Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Iseli, C.; Jongeneel, C.V.; Bucher, P. ESTScan: A program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1999, 99, 138–148. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-△△Ct method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Data (bp) | Clean Data (bp) | Q20 (%) | Q30 (%) | GC Content (%) |
|---|---|---|---|---|---|
| Inte-1 | 7,986,373,500 | 7,947,476,345 | 97.65% | 93.45% | 52.98% |
| Inte-2 | 7,641,577,800 | 7,605,711,075 | 97.88% | 94.01% | 52.77% |
| Inte-3 | 7,992,333,000 | 7,956,556,516 | 97.83% | 93.86% | 53.66% |
| Midg-1 | 9,036,411,000 | 8,924,294,465 | 97.06% | 92.52% | 53.10% |
| Midg-2 | 7,794,233,100 | 7,716,963,009 | 97.36% | 93.11% | 52.75% |
| Midg-3 | 7,476,890,700 | 7,412,795,894 | 97.38% | 93.10% | 52.58% |
| Larv-1 | 5,629,523,700 | 5,593,818,739 | 97.84% | 93.94% | 52.24% |
| Larv-2 | 6,382,601,400 | 6,344,754,317 | 97.61% | 93.40% | 51.73% |
| Larv-3 | 7,023,010,500 | 6,978,558,841 | 97.56% | 93.33% | 51.66% |
| Gene Name | Accession No. | BLASTX Best Hit | Integument (RPKM) | Midgut (RPKM) | Larva (RPKM) | |||
|---|---|---|---|---|---|---|---|---|
| Species | Gene Description | Accession No. | E-value | |||||
| GpCHS A | MN915086 | Ostrinia furnacalis | Chitin synthase A | ACB13821.1 | 0.0 | 24.39 | 0.75 | 5.66 |
| GpCHS B | MN915087 | Cnaphalocrocis medinalis | Chitin synthase B | AJG44539.1 | 0.0 | 1.99 | 265.97 | 88.34 |
| GpCDA1 | MN915088 | Ostrinia furnacalis | Chitin deacetylase 1 | QDZ05988.1 | 0.0 | 899.05 | 3.79 | 226.69 |
| GpCDA2 | MN915096 | Choristoneura fumiferana | CDA2 isoform B | AGT28749.1 | 0.0 | 558.26 | 2.54 | 144.88 |
| GpCDA4 | MN915090 | Cnaphalocrocis medinalis | Chitin deacetylase 4 | AJG44548.1 | 0.0 | 31.24 | 0.82 | 7.73 |
| GpCDA5 | MN915091 | Operophtera brumata | Chitin deacetylase 5a, partial | KOB56571.1 | 6e-152 | 70.44 | 4961.48 | 1818.85 |
| GpCHT1 | MN915094 | Conogethes punctiferalis | Chitinase 1 | ASM94206.1 | 0.0 | 10.11 | 0.18 | 2.52 |
| GpCHT2a | MN915093 | Cnaphalocrocis medinalis | Chitinase 2 | AJG44542.1 | 0.0 | 20.32 | 7.39 | 9.71 |
| GpCHT2b | MN915098 | Amyelois transitella | PREDICTED: probable chitinase 2 | XP_013190968.1 | 0.0 | 0.04 | 30.74 | 15.47 |
| GpCHT3a | MN915089 | Amyelois transitella | PREDICTED: probable chitinase 3 | XP_013183423.1 | 4e-169 | 249.83 | 1.75 | 61.94 |
| GpCHT3b | MN915097 | Papilio polytes | PREDICTED: probable chitinase 3 | KPI96666.1 | 2e-47 | 0.10 | 236.62 | 79.34 |
| GpCHT-h | MN915092 | Samia cynthia | Chitinase | BAE16586.1 | 0.0 | 2.63 | 55.59 | 19.26 |
| GpCHT7 | MN915095 | Conogethes punctiferalis | Chitinase 7 | ASM94207.1 | 0.0 | 49.42 | 0.18 | 11.75 |
| GpPAGM | MN915104 | Cnaphalocrocis medinalis | N-phosphoacetylglucosamine mutase | AJG44540.1 | 0.0 | 7.42 | 22.07 | 10.55 |
| GpNAGK | MN915100 | Omphisa fuscidentalis | N-acetyl-d-glucosamine kinase | KPJ05170.1 | 2e-159 | 2.94 | 9.39 | 4.51 |
| GpUAP | MN915103 | Cnaphalocrocis medinalis | UDP-N-acetylglucosamine pyrophosphorylase | AKO90063.1 | 0.0 | 15.16 | 142.19 | 45.74 |
| GpTre1 | MN915101 | Omphisa fuscidentalis | Soluble trehalase | ANY30160.1 | 0.0 | 71.42 | 39.16 | 30.21 |
| GpTre2 | MN915102 | Omphisa fuscidentalis | Trehalase-2 | ABO20845.1 | 0.0 | 27.55 | 21.91 | 17.49 |
| GpRtv | MN915099 | Papilio xuthus | retroactive | BAM18479.1 | 3e-61 | 9.45 | 1.35 | 4.69 |
| Genes | Number (Percentage) |
|---|---|
| Cytochrome P450 | 92 (32.6%) |
| Glutathione S-transferase | 37 (13.1%) |
| Carboxylesterase | 59 (21%) |
| Nicotinic acetylcholine receptor | 7 (2.5%) |
| Aquaporin | 19 (6.7%) |
| Chloride channel | 3 (1.1%) |
| Methoprene-tolerant protein | 2 (0.7%) |
| Serine protease inhibitor | 22 (7.8%) |
| Sodium channel | 11 (3.9%) |
| Calcium channel | 30 (10.6%) |
| Gene Names | Forward Primer (5′-3′) | Reverse Primer (5′-3′) |
|---|---|---|
| GpCHSA | TACGCTTTCCACATCACCGC | ACGGGCCTTCTCTTCCTTGT |
| GpCHSB | ACTTGGCTTTGGGCAGCTTT | GGTCCCTCGTCAACGCATTT |
| GpCDA1 | TTCAAGCCATTCGCTGTCCC | CCAGGAAGCCATCTTGGCAG |
| GpCDA5 | GTGCTTCCCTCCTAACACGC | CCCTTTCGATGGCAGGGTTC |
| GpCDA2 | TTGGTGTGCGTGCTCCTTAC | TATGGGCGTTACCGTTGCAC |
| GpCHT-h | GCGACCCTTACAGAGGCAAC | TTTTCGCTTCACCGCATCGT |
| GpCHT7 | GGAGGAGTCTGTGGTGGGAA | ACGCTACTGAGGCCCAATCT |
| GpCDA4 | GCCCTATACCAACAACGCCC | AGAAAGTCCCGCGTATGGGA |
| GpCHT3a | TTCAACGACTACAGCCCCGA | GAGAAGTAGCCGTTCAGGCG |
| GpCYP304 | GGCCGTGAATGGACCCAAAT | GCCACGTGCCAACTCATACA |
| GpGST | GGGCACTCAACCTGAACCTC | TCGCGTATAGGGAGCCGATG |
| GpCarE | ATTGTGCGCATAGAGCAGGG | ACGGCATCCAAAACCGACAA |
| GpRpl32 | CGATCACCTTCCGCTTCT | TGCTACCCAATGGCTTCC |
| Primer Names | Sequence (5′-3′) |
|---|---|
| GpCDA1-1-Olig-1 | GATCACTAATACGACTCACTATAGGGGCAGACTTGTGACTGGAAATT |
| GpCDA1-1-Olig-2 | AATTTCCAGTCACAAGTCTGCCCCTATAGTGAGTCGTATTAGTGATC |
| GpCDA1-1-Olig-3 | AAGCAGACTTGTGACTGGAAACCCTATAGTGAGTCGTATTAGTGATC |
| GpCDA1-1-Olig-4 | GATCACTAATACGACTCACTATAGGGTTTCCAGTCACAAGTCTGCTT |
| GpCDA1-2-Olig-1 | GATCACTAATACGACTCACTATAGGGGCGACATTAAAGCCACCTTTT |
| GpCDA1-2-Olig-2 | AAAAGGTGGCTTTAATGTCGCCCCTATAGTGAGTCGTATTAGTGATC |
| GpCDA1-2-Olig-3 | AAGCGACATTAAAGCCACCTTCCCTATAGTGAGTCGTATTAGTGATC |
| GpCDA1-2-Olig-4 | GATCACTAATACGACTCACTATAGGGAAGGTGGCTTTAATGTCGCTT |
| GpCHT3a-1-Olig-1 | GATCACTAATACGACTCACTATAGGGGCAGTGCGACAAGTACTATTT |
| GpCHT3a-1-Olig-2 | AAATAGTACTTGTCGCACTGCCCCTATAGTGAGTCGTATTAGTGATC |
| GpCHT3a-1-Olig-3 | AAGCAGTGCGACAAGTACTATCCCTATAGTGAGTCGTATTAGTGATC |
| GpCHT3a-1-Olig-4 | GATCACTAATACGACTCACTATAGGGATAGTACTTGTCGCACTGCTT |
| GpCHT3a-2-Olig-1 | GATCACTAATACGACTCACTATAGGGCCCTTCAACATCGACTGTTTT |
| GpCHT3a-2-Olig-2 | AAAACAGTCGATGTTGAAGGGCCCTATAGTGAGTCGTATTAGTGATC |
| GpCHT3a-2-Olig-3 | AACCCTTCAACATCGACTGTTCCCTATAGTGAGTCGTATTAGTGATC |
| GpCHT3a-2-Olig-4 | GATCACTAATACGACTCACTATAGGGAACAGTCGATGTTGAAGGGTT |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shao, Z.-m.; Li, Y.-j.; Zhang, X.-r.; Chu, J.; Ma, J.-h.; Liu, Z.-x.; Wang, J.; Sheng, S.; Wu, F.-a. Identification and Functional Study of Chitin Metabolism and Detoxification-Related Genes in Glyphodes pyloalis Walker (Lepidoptera: Pyralidae) Based on Transcriptome Analysis. Int. J. Mol. Sci. 2020, 21, 1904. https://doi.org/10.3390/ijms21051904
Shao Z-m, Li Y-j, Zhang X-r, Chu J, Ma J-h, Liu Z-x, Wang J, Sheng S, Wu F-a. Identification and Functional Study of Chitin Metabolism and Detoxification-Related Genes in Glyphodes pyloalis Walker (Lepidoptera: Pyralidae) Based on Transcriptome Analysis. International Journal of Molecular Sciences. 2020; 21(5):1904. https://doi.org/10.3390/ijms21051904
Chicago/Turabian StyleShao, Zuo-min, Yi-jiangcheng Li, Xiao-rui Zhang, Jie Chu, Jia-hui Ma, Zhi-xiang Liu, Jun Wang, Sheng Sheng, and Fu-an Wu. 2020. "Identification and Functional Study of Chitin Metabolism and Detoxification-Related Genes in Glyphodes pyloalis Walker (Lepidoptera: Pyralidae) Based on Transcriptome Analysis" International Journal of Molecular Sciences 21, no. 5: 1904. https://doi.org/10.3390/ijms21051904
APA StyleShao, Z.-m., Li, Y.-j., Zhang, X.-r., Chu, J., Ma, J.-h., Liu, Z.-x., Wang, J., Sheng, S., & Wu, F.-a. (2020). Identification and Functional Study of Chitin Metabolism and Detoxification-Related Genes in Glyphodes pyloalis Walker (Lepidoptera: Pyralidae) Based on Transcriptome Analysis. International Journal of Molecular Sciences, 21(5), 1904. https://doi.org/10.3390/ijms21051904