The GPR17 Receptor—A Promising Goal for Therapy and a Potential Marker of the Neurodegenerative Process in Multiple Sclerosis




Abstract
1. Introduction
2. General Characteristics of G Protein-Coupled Receptors
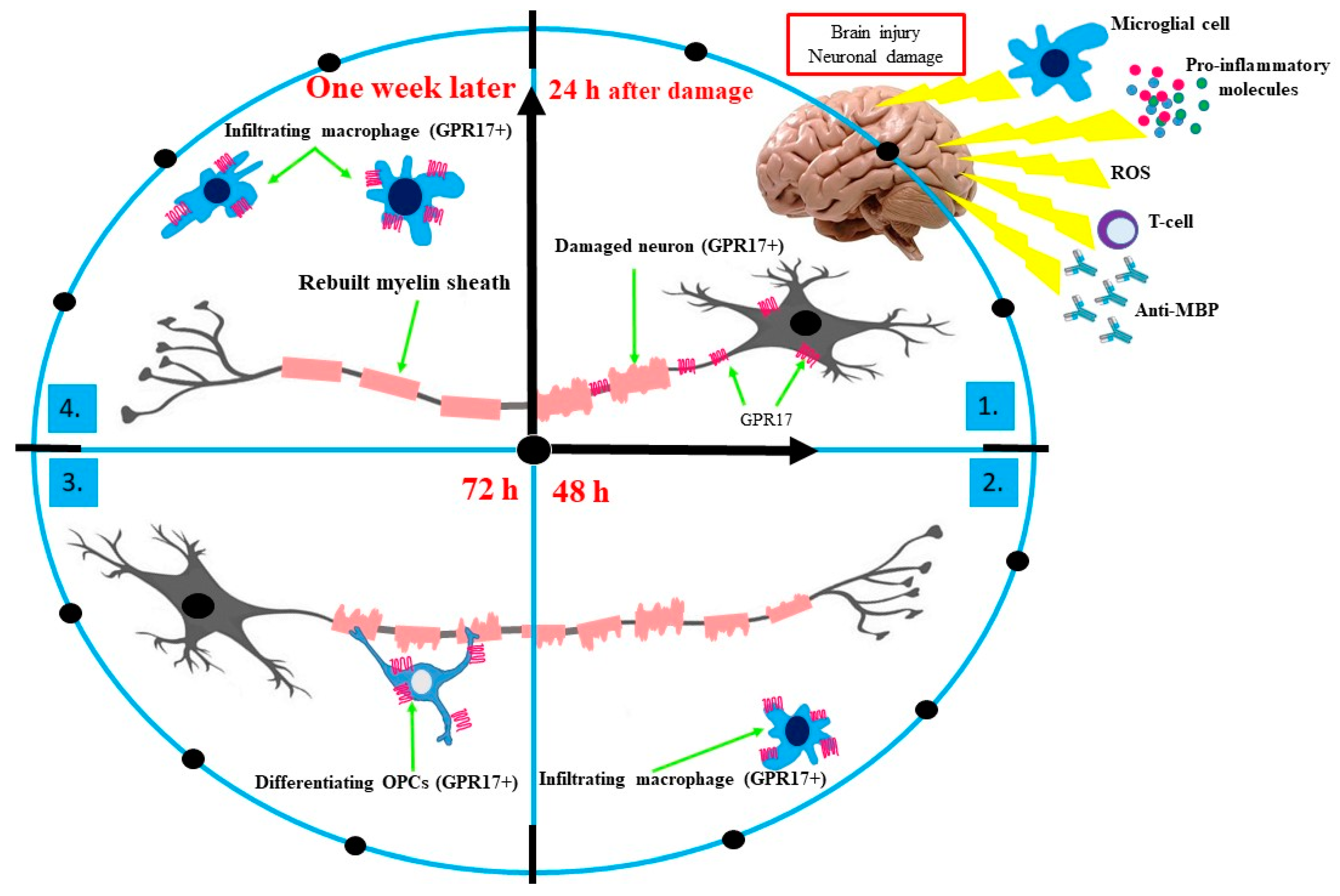
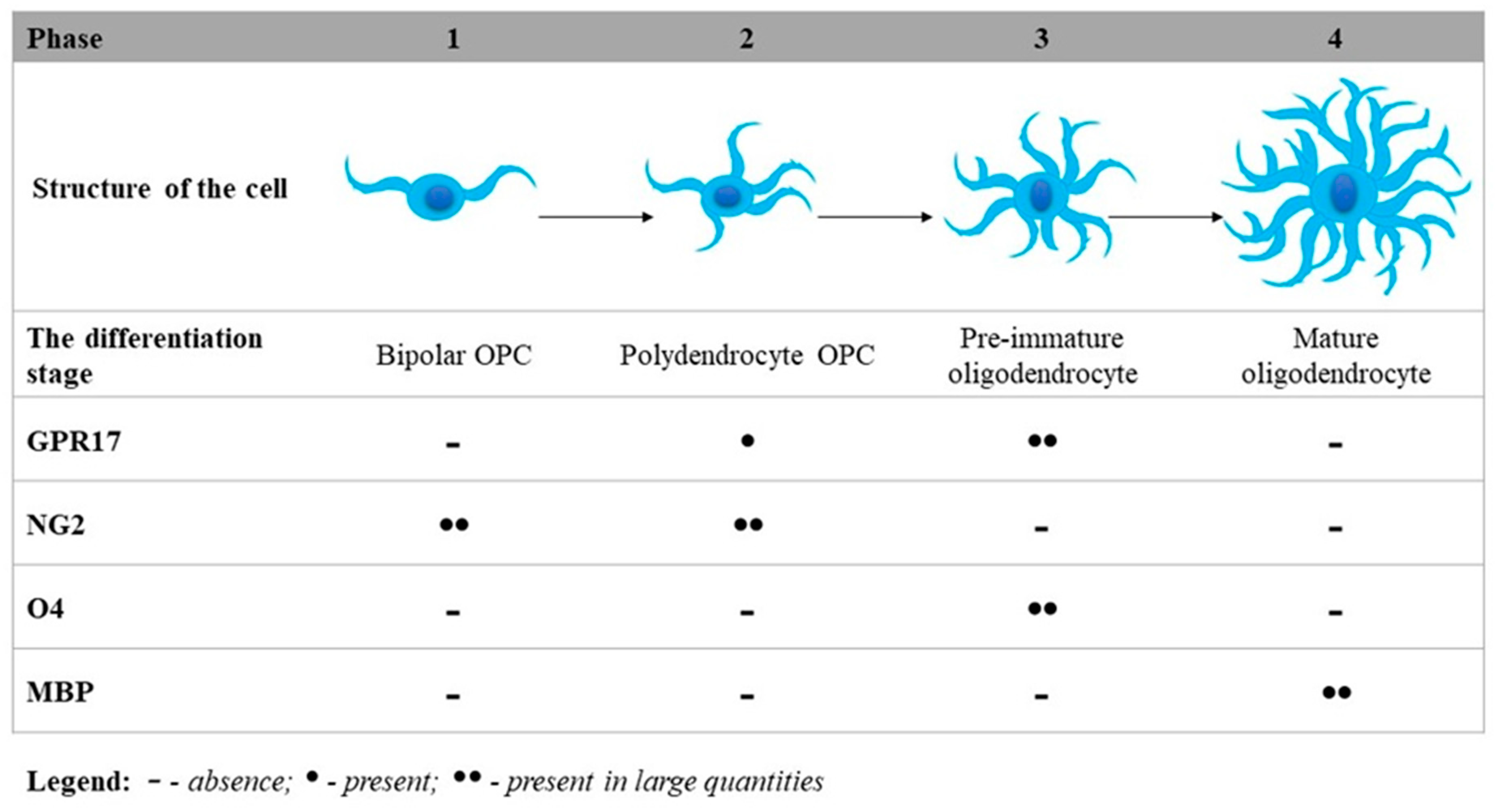
3. The Essential Role of GPR17 in Neurodegeneration
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gitler, A.D.; Dhillon, P.; Shorter, J. Neurodegenerative disease: Models, mechanisms, and a new hope. Dis. Models Mech. 2017, 10, 499–502. [Google Scholar] [CrossRef]
- Jennum, P.; Wanscher, B.; Frederiksen, J.; Kjellberg, J. The socioeconomic consequences of multiple sclerosis: A controlled national study. Eur. Neuropsychopharmacol. 2012, 22, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Raj, R.; Kaprio, J.; Korja, M.; Mikkonen, E.D.; Jousilahti, P.; Siironen, J. Risk of hospitalization with neurodegenerative disease after moderate-to-severe traumatic brain injury in the working-age population: A retrospective cohort study using the Finnish national health registries. PLoS Med. 2017, 14, e1002316. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, N.; Razavi, S.; Nikzad, E. Multiple Sclerosis: Pathogenesis, Symptoms, Diagnoses and Cell-Based Therapy. J. Cell Biol. 2017, 19, 1–10. [Google Scholar] [CrossRef]
- Honce, J.M. Gray matter Pathology in MS: Neuroimaging and Clinical Correlations. Mult. Scler. Int. 2013, 2013, 627870. [Google Scholar] [CrossRef]
- De Stefano, N.; Matthews, P.M.; Filippi, M.; Agosta, F.; De Luca, M.; Bartolozzi, M.L.; Guidi, L.; Ghezzi, A.; Montanari, E.; Cifelli, A.; et al. Evidence of early cortical atrophy in MS: Relevance to white matter changes and disability. Neurology 2003, 60, 1157–1162. [Google Scholar] [CrossRef]
- Chard, D.T.; Griffin, C.M.; Rashid, W.; Davies, G.R.; Altmann, D.R.; Kapoor, R.; Barker, G.J.; Thompson, A.J.; Miller, D.H. Progressive grey matter atrophy in clinically early relapsing-remitting multiple sclerosis. Mult. Scler. 2004, 10, 387–391. [Google Scholar] [CrossRef]
- Miller, E. Multiple sclerosis. Adv. Exp. Med. Biol. 2012, 724, 222–238. [Google Scholar] [CrossRef]
- Legroux, L.; Arbour, N. Multiple Sclerosis and T Lymphocytes: An Entangled Story. J. Neuroimmune Pharmacol. 2015, 10, 528–546. [Google Scholar] [CrossRef]
- Korn, T.; Mitsdoerffer, M.; Croxford, A.L.; Awasthi, A.; Dardalhon, V.A.; Galileos, G.; Vollmar, P.; Stritesky, G.L.; Kaplan, M.H.; Waisman, A.; et al. IL-6 controls Th17 immunity in vivo by inhibiting the conversion of conventional T cells into Foxp3+ regulatory T cells. Proc. Natl. Acad. Sci. USA 2008, 105, 18460–18465. [Google Scholar] [CrossRef]
- Sospedra, M.; Martin, R. Immunology of multiple sclerosis. Annu. Rev. Immunol. 2005, 23, 683–747. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.; Trapp, B.D. Relapsing and progressive forms of multiple sclerosis—Insights from pathology. Curr. Opin. Neurol. 2014, 27, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Senanayake, V.K.; Jin, W.; Mochizuki, A.; Chitou, B.; Goodenowe, D.B. Metabolic dysfunctions in multiple sclerosis: Implications as to causation, early detection, and treatment, a case control study. BMC Neurol. 2015, 15, 154. [Google Scholar] [CrossRef] [PubMed]
- Shah, P. Symptomatic management in multiple sclerosis. Ann. Indian Acad. Neurol. 2015, 18, S35–S42. [Google Scholar] [CrossRef] [PubMed]
- Cunnusamy, K.; Baughman, E.J.; Franco, J.; Ortega, S.B.; Sinha, S.; Chaudhary, P.; Greenberg, B.M.; Frohman, E.M.; Karandikar, N.J. Disease exacerbation of multiple sclerosis is characterized by loss of terminally differentiated autoregulatory CD8+ T cells. Clin. Immunol. 2014, 152, 115–126. [Google Scholar] [CrossRef]
- Wingerchuk, D.M.; Weinshenker, B.G. Disease modifying therapies for relapsing multiple sclerosis. BMJ 2016, 354, i3518. [Google Scholar] [CrossRef]
- Moss, B.P.; Cohen, J.A. The emergence of follow-on disease-modifying therapies for multiple sclerosis. Mult. Scler. J. 2019, 25, 1560–1565. [Google Scholar] [CrossRef]
- Ciccarelli, O.; Barkhof, F.; Bodini, B.; Stefano, N.D.; Golay, X.; Nicolay, K.; Pelletier, D.; Pouwels, P.J.; Smith, S.A.; Wheeler-Kingshott, C.A.; et al. Pathogenesis of multiple sclerosis: Insights from molecular and metabolic imaging. Lancet Neurol. 2014, 13, 807–822. [Google Scholar] [CrossRef]
- Dziedzic, A.; Bijak, M. Interactions between platelets and leukocytes in pathogenesis of multiple sclerosis. Adv. Clin. Exp. Med. 2018, 28, 277–285. [Google Scholar] [CrossRef]
- Al-Badri, G.; Castorina, A. Insights into the Role of Neuroinflammation in the Pathogenesis of Multiple Sclerosis. J. Funct. Morphol. Kinesiol. 2018, 3, 13. [Google Scholar] [CrossRef]
- Fujinami, R.S.; von Herrath, M.G.; Christen, U.; Whitton, J.L. Molecular Mimicry, Bystander Activation, or Viral Persistence: Infections and Autoimmune Disease. Clin. Microbiol. Rev. 2006, 19, 80–94. [Google Scholar] [CrossRef] [PubMed]
- Van Nierop, G.P.; van Luijn, M.M.; Michels, S.S.; Melief, M.J.; Janssen, M.; Langerak, A.W.; Ouwendijk, W.J.D.; Hintzen, R.Q.; Verjans, G.M.G.M. Phenotypic and functional characterization of T cells in white matter lesions of multiple sclerosis patients. Acta Neuropathol. 2017, 134, 383–401. [Google Scholar] [CrossRef] [PubMed]
- Peeters, L.M.; Vanheusden, M.; Somers, V.; Van Wijmeersch, B.; Stinissen, P.; Broux, B.; Hellings, N. Cytotoxic CD4+ T Cells Drive Multiple Sclerosis Progression. Front. Immunol. 2017, 8, 1160. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.M.; Lalor, S.J.; Sweeney, C.M.; Tubridy, N.; Mills, K.H.G. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin. Exp. Immunol. 2010, 162, 1–11. [Google Scholar] [CrossRef]
- Passos, G.R.D.; Sato, D.K.; Becker, J.; Fujihara, K. Th17 Cells Pathways in Multiple Sclerosis and Neuromyelitis Optica Spectrum Disorders: Pathophysiological and Therapeutic Implications. Mediators Inflamm. 2016, 2016, 5314541. [Google Scholar] [CrossRef]
- Arbelaez, C.A.; Glatigny, S.; Duhen, R.; Eberl, G.; Oukka, M.; Bettelli, E. IL-7/IL-7 Receptor Signaling Differentially Affects Effector CD4+T Cell Subsets Involved in Experimental Autoimmune Encephalomyelitis. J. Immunol. 2015, 195, 1974–1983. [Google Scholar] [CrossRef]
- Karussis, D. The diagnosis of multiple sclerosis and the various related demyelinating syndromes: A critical review. J. Autoimmun. 2014, 48–49, 134–142. [Google Scholar] [CrossRef]
- Wong, Y.Y.M.; de Mol, C.L.; van der Vuurst de Vries, R.M.; van Pelt, E.D.; Ketelslegers, I.A.; Catsman-Berrevoets, C.E.; Neuteboom, R.F.; Hintzen, R.Q. Real-world validation of the 2017 McDonald criteria for pediatric MS. Neurol. Neuroimmunol. Neuroinflamm. 2019, 6, e528. [Google Scholar] [CrossRef]
- Thompson, A.J.; Banwell, B.L.; Barkhof, F.; Carroll, W.M.; Coetzee, T.; Comi, G.; Correale, J.; Fazekas, F.; Filippi, M.; Freedman, M.S.; et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018, 17, 162–173. [Google Scholar] [CrossRef]
- Podbielska, M.; Banik, N.L.; Kurowska, E.; Hogan, E.L. Myelin Recovery in Multiple Sclerosis: The Challenge of Remyelination. Brain Sci. 2013, 3, 1282–1324. [Google Scholar] [CrossRef]
- Simons, M.; Nave, K.A. Oligodendrocytes: Myelination and Axonal Support. Cold Spring Harb. Perspect. Biol. 2015, 8, a020479. [Google Scholar] [CrossRef] [PubMed]
- Popescu, B.F.G.; Pirko, I.; Lucchinetti, C.F. Pathology of Multiple Sclerosis: Where Do We Stand? Continuum (Minneap Minn) 2013, 19, 901–921. [Google Scholar] [CrossRef] [PubMed]
- Lucchinetti, C.; Brück, W.; Parisi, J.; Scheithauer, B.; Rodriguez, M.; Lassmann, H. Heterogeneity of multiple sclerosis lesions: Implications for the pathogenesis of demyelination. Ann. Neurol. 2000, 47, 707–717. [Google Scholar] [CrossRef]
- Lassmann, H. Pathology and disease mechanisms in different stages of multiple sclerosis. J. Neurol. Sci. 2013, 333, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Harris, V.; Tuddenham, J.; Sadiq, S. Biomarkers of multiple sclerosis: Current findings. Degener. Neurol. Neuromuscul. Dis. 2017, 7, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Comabella, M.; Gandhi, R. Biomarkers in Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2019, 9, a029058. [Google Scholar] [CrossRef]
- Katsavos, S.; Anagnostouli, M. Biomarkers in Multiple Sclerosis: An Up-to-Date Overview. Mult. Scler. Int. 2013, 2013, 1–20. [Google Scholar] [CrossRef]
- Giovannoni, G. Multiple sclerosis cerebrospinal fluid biomarkers. Dis. Markers 2006, 22, 187–196. [Google Scholar] [CrossRef]
- Lewczuk, P.; Riederer, P.; O’Bryant, S.E.; Verbeek, M.M.; Dubois, B.; Visser, P.J.; Jellinger, K.A.; Engelborghs, S.; Ramirez, A.; Parnetti, L.; et al. Cerebrospinal fluid and blood biomarkers for neurodegenerative dementias: An update of the Consensus of the Task Force on Biological Markers in Psychiatry of the World Federation of Societies of Biological Psychiatry. World J. Biol. Psychiatry 2017, 19, 244–328. [Google Scholar] [CrossRef]
- Agrawal, M.; Biswas, A. Molecular diagnostics of neurodegenerative disorders. Front. Mol. Biosci. 2015, 2, 54. [Google Scholar] [CrossRef]
- Marinissen, M.J.; Gutkind, J.S. G-protein-coupled receptors and signaling networks: Emerging paradigms. Trends Pharmacol. Sci. 2001, 22, 368–376. [Google Scholar] [CrossRef]
- Fredriksson, R.; Lagerström, M.C.; Lundin, L.G.; Schiöth, H.B. The G-Protein-Coupled Receptors in the Human Genome Form Five Main Families. Phylogenetic Analysis, Paralogon Groups, and Fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.A.; Roth, B.L. Strategies to discover unexpected targets for drugs active at G protein–coupled receptors. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 117–144. [Google Scholar] [CrossRef] [PubMed]
- Katritch, V.; Cherezov, V.; Stevens, R.C. Diversity and modularity of G protein-coupled receptor structures. Trends Pharmacol. Sci. 2012, 33, 17–27. [Google Scholar] [CrossRef]
- Tuteja, N. Signaling through G protein coupled receptors. Plant. Signal. Behav. 2009, 4, 942–947. [Google Scholar] [CrossRef]
- Neves, S.R. G Protein Pathways. Science 2002, 296, 1636–1639. [Google Scholar] [CrossRef]
- Gupte, J.; Swaminath, G.; Danao, J.; Tian, H.; Li, Y.; Wu, X. Signaling property study of adhesion G-protein-coupled receptors. FEBS Lett. 2012, 586, 1214–1219. [Google Scholar] [CrossRef]
- Syrovatkina, V.; Alegre, K.O.; Dey, R.; Huang, X.Y. Regulation, Signaling, and Physiological Functions of G-Proteins. J. Mol. Biol. 2016, 428, 3850–3868. [Google Scholar] [CrossRef]
- Pleli, T.; Mondorf, A.; Ferreiros, N.; Thomas, D.; Dvorak, K.; Biondi, R.M.; Heringdorf, D.M.; Zeuzem, S.; Geisslinger, G.; Zimmermann, H.; et al. Activation of Adenylyl Cyclase Causes Stimulation of Adenosine Receptors. Cell Physiol. Biochem. 2018, 45, 2516–2528. [Google Scholar] [CrossRef]
- Redfern, C.H.; Degtyarev, M.Y.; Kwa, A.T.; Salomonis, N.; Cotte, N.; Nanevicz, T.; Fidelman, N.; Desai, K.; Vranizan, K.; Lee, E.K.; et al. Conditional expression of a Gi-coupled receptor causes ventricular conduction delay and a lethal cardiomyopathy. Proc. Natl. Acad. Sci. USA 2000, 97, 4826–4831. [Google Scholar] [CrossRef]
- Ho, M.K.C.; Yung, L.Y.; Chan, J.S.C.; Chan, J.H.P.; Wong, C.S.S.; Wong, Y.H. Gα14links a variety of Gi- and Gs-coupled receptors to the stimulation of phospholipase C. Br. J. Pharmacol. 2001, 132, 1431–1440. [Google Scholar] [CrossRef] [PubMed]
- McCloskey, D.T.; Turcato, S.; Wang, G.Y.; Turnbull, L.; Zhu, B.Q.; Bambino, T.; Nguyen, A.P.; Lovett, D.H.; Nissenson, R.A.; Karliner, J.S.; et al. Expression of a Gi-coupled receptor in the heart causes impaired Ca2+ handling, myofilament injury, and dilated cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H205–H212. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.C.Y.; Wong, Y.H. Role of G Protein-Coupled Receptors in the Regulation of Structural Plasticity and Cognitive Function. Molecules 2017, 22, 1239. [Google Scholar] [CrossRef] [PubMed]
- Billington, C.K.; Penn, R.B. Signaling and regulation of G protein-coupled receptors in airway smooth muscle. Respir. Res. 2003, 4, 2. [Google Scholar] [CrossRef]
- Peters, S.L.; Michel, M.C. cAMP-independent relaxation of smooth muscle cells via Gs-coupled receptors. Naunyn Schmiedebergs Arch. Pharmacol. 2003, 368, 329–330. [Google Scholar] [CrossRef]
- Billups, D.; Billups, B.; Challiss, R.A.J.; Nahorski, S.R. Modulation of Gq protein-cupled IP3 and Ca2+ signaling by the membrane potential. J. Neurosci. 2006, 26, 9983–9995. [Google Scholar] [CrossRef]
- Mizuno, N.; Itoh, H. Functions and Regulatory Mechanisms of Ga-Signaling Pathways. Neurosignals 2009, 17, 42–54. [Google Scholar] [CrossRef]
- Pelaia, G.; Renda, T.; Gallelli, L.; Vatrella, A.; Busceti, M.T.; Agati, S.; Caputi, M.; Cazzola, M.; Maselli, R.; Marsico, S.A. Molecular mechanisms underlying airway smooth muscle contraction and proliferation: Implications for asthma. Respir. Med. 2008, 102, 1173–1181. [Google Scholar] [CrossRef]
- Zhang, L.; Shi, G. Gq-Coupled Receptors in Autoimmunity. J. Immunol. Res. 2016, 2016, 3969023. [Google Scholar] [CrossRef]
- Moers, A.; Nieswandt, B.; Massberg, S.; Wettschureck, N.; Grüner, S.; Konrad, I.; Schulte, V.; Aktas, B.; Gratacap, M.P.; Simon, M.I.; et al. G13 is an essential mediator of platelet activation in hemostasis and thrombosis. Nat. Med. 2003, 9, 1418–1422. [Google Scholar] [CrossRef]
- Siehler, S. Regulation of RhoGEF proteins by G12/13-coupled receptors. Br. J. Pharmacol. 2009, 158, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Goulimari, P.; Knieling, H.; Engel, U.; Grosse, R. LARG and mDia1 link Galpha12/13 to cell polarity and microtubule dynamics. Mol. Biol. Cell. 2008, 19, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Dupré, D.J.; Robitaille, M.; Rebois, R.V.; Hébert, T.E. The Role of Gβγ Subunits in the Organization, Assembly, and Function of GPCR Signaling Complexes. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 31–56. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, D.M.; Rasmussen, S.G.; Kobilka, B.K. The structure and function of G-protein-coupled receptors. Nature 2009, 459, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.M.; Mai, T.L.; Chen, C.M. Visualizing the GPCR Network: Classification and Evolution. Sci. Rep. 2017, 7, 15495. [Google Scholar] [CrossRef] [PubMed]
- Schiöth, H.B.; Fredriksson, R. The GRAFS classification system of G-protein coupled receptors in comparative perspective. Gen. Comp. Endocrinol. 2005, 142, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Deupi, X. Relevance of rhodopsin studies for GPCR activation. Biochim. Biophys. Acta 2014, 1837, 674–682. [Google Scholar] [CrossRef]
- Joost, P.; Methner, A. Phylogenetic analysis of 277 human G-protein-coupled receptors as a tool for the prediction of orphan receptor ligands. Genome Biol. 2002, 3. [Google Scholar] [CrossRef]
- Yona, S.; Lin, H.H.; Dri, P.; Davies, J.Q.; Hayhoe, R.P.G.; Lewis, S.M.; Heinsbroek, S.E.; Brown, K.A.; Perretti, M.; Hamann, J.; et al. Ligation of the adhesion-GPCR EMR2 regulates human neutrophil function. FASEB J. 2008, 22, 741–751. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate Receptor Ion Channels: Structure, Regulation, and Function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef]
- Afroze, S.; Meng, F.; Jensen, K.; McDaniel, K.; Rahal, K.; Onori, P.; Gaudio, E.; Alpini, G.; Glaser, S.S. The physiological roles of secretin and its receptor. Ann. Transl. Med. 2013, 1, 29. [Google Scholar] [CrossRef] [PubMed]
- Patapoutia, A.; Reichardt, L.F. Roles of Wnt proteins in neural development and maintenance. Curr. Opin. Neurobiol. 2000, 10, 392–399. [Google Scholar] [CrossRef]
- Chandrashekar, J.; Mueller, K.L.; Hoon, M.A.; Adler, E.; Feng, L.; Guo, W.; Zuker, C.S.; Ryba, N.J. T2Rs function as bitter taste receptors. Cell 2000, 100, 703–711. [Google Scholar] [CrossRef]
- Raport, C.J.; Schweickart, V.L.; Chantry, D.; Eddy, R.L.; Shows, T.B.; Godiska, R.; Gray, P.W. New members of the chemokine receptor gene family. J. Leukoc. Biol. 1996, 59, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.K.; Nguyen, T.; Lynch, K.R.; Cheng, R.; Vanti, W.B.; Arkhitko, O.; Lewis, T.; Evans, J.F.; George, S.R.; O’Dowd, B.F. Discovery and mapping of ten novel G protein-coupled receptor genes. Gene 2001, 275, 83–91. [Google Scholar] [CrossRef]
- Lüttichau, H.R.; Lewis, I.C.; Gerstoft, J.; Schwartz, T.W. The herpesvirus 8-encoded chemokine vMIP-II, but not the poxvirus-encoded chemokine MC148, inhibits the CCR10 receptor. Eur. J. Immunol. 2001, 31, 1217–1220. [Google Scholar] [CrossRef]
- Ciana, P.; Fumagalli, M.; Trincavelli, M.L.; Verderio, C.; Rosa, P.; Lecca, D.; Ferrario, S.; Parravicini, C.; Capra, V.; Gelosa, P.; et al. The orphan receptor GPR17 identified as a new dual uracil nucleotides/cysteinyl-leukotrienes receptor. EMBO J. 2006, 25, 4615–4627. [Google Scholar] [CrossRef]
- Fumagalli, M.; Lecca, D.; Abbracchio, M.P. CNS remyelination as a novel reparative approach to neurodegenerative diseases: The roles of purinergic signaling and the P2Y-like receptor GPR17. Neuropharmacology 2016, 104, 82–93. [Google Scholar] [CrossRef]
- Khan, M.Z.; He, L. Neuro-psychopharmacological perspective of Orphan receptors of Rhodopsin (class A) family of G protein-coupled receptors. Psychopharmacology 2017, 234, 1181–1207. [Google Scholar] [CrossRef]
- Lecca, D.; Trincavelli, M.L.; Gelosa, P.; Sironi, L.; Ciana, P.; Fumagalli, M.; Villa, G.; Verderio, C.; Grumelli, C.; Guerrini, U.; et al. The Recently Identified P2Y-Like Receptor GPR17 Is a Sensor of Brain Damage and a New Target for Brain Repair. PLoS ONE 2008, 3, e3579. [Google Scholar] [CrossRef]
- Benned-Jensen, T.; Rosenkilde, M. Distinct expression and ligand-binding profiles of two constitutively active GPR17 splice variants. Br. J. Pharmacol. 2010, 159, 1092–1105. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Wang, H.; Li, C.; Song, S.; Fang, S.; Wei, E.; Shi, Q. GPR17 mediates ischemia-like neuronal injury via microglial activation. Int. J. Mol. Med. 2018, 42, 2750–2762. [Google Scholar] [CrossRef] [PubMed]
- Lecca, D.; Fumagalli, M.; Ceruti, S.; Abbracchio, M.P. Intertwining extracellular nucleotides and their receptors with Ca2+in determining adult neural stem cell survival, proliferation and final fate. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2016, 371, 20150433. [Google Scholar] [CrossRef] [PubMed]
- Ceruti, S.; Vigano, F.; Boda, E.; Ferrario, S.; Magni, G.; Boccazzi, M.; Rosa, P.; Buffo, A.; Abbracchio, M.P. Expression of the new P2Y-like receptor GPR17 during oligodendrocyte precursor cell maturation regulates sensitivity to ATP-induced death. Glia 2011, 59, 363–378. [Google Scholar] [CrossRef] [PubMed]
- Quillinan, N.; Herson, P.S.; Traystman, R.J. Neuropathophysiology of Brain Injury. Anesthesiol. Clin. 2016, 34, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Ettle, B.; Schlachetzki, J.C.M.; Winkler, J. Oligodendroglia and Myelin in Neurodegenerative Diseases: More Than Just Bystanders? Mol. Neurobiol. 2016, 53, 3046–3062. [Google Scholar] [CrossRef]
- Fields, R.D. Myelin—More than Insulation. Science 2014, 344, 264–266. [Google Scholar] [CrossRef]
- Mayoral, S.R.; Chan, J.R. The environment rules: Spatiotemporal regulation of oligodendrocyte differentiation. Curr. Opin. Neurobiol. 2016, 39, 47–52. [Google Scholar] [CrossRef]
- Boyd, J.G.; Gordon, T. Neurotrophic factors and their receptors in axonal regeneration and functional recovery after peripheral nerve injury. Mol. Neurobiol. 2003, 27, 277–324. [Google Scholar] [CrossRef]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte Chemoattractant Protein-1 (MCP-1): An Overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef]
- Zhang, J.M.; An, J. Cytokines, Inflammation, and Pain. Int. Anesthesiol. Clin. 2007, 45, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Franke, H.; Parravicini, C.; Lecca, D.; Zanier, E.R.; Heine, C.; Bremicker, K.; Fumagalli, M.; Rosa, P.; Longhi, L.; Stocchetti, N.; et al. Changes of the GPR17 receptor, a new target for neurorepair, in neurons and glial cells in patients with traumatic brain injury. Purinergic Signal. 2013, 9, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Simon, C.; Götz, M.; Dimou, L. Progenitors in the adult cerebral cortex: Cell cycle properties and regulation by physiological stimuli and injury. Glia 2011, 59, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Kucharova, K.; Stallcup, W.B. The NG2 proteoglycan promotes oligodendrocyte progenitor proliferation and developmental myelination. Neuroscience 2010, 166, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Stallcup, W.B. The NG2 proteoglycan: Past insights and future prospects. J. Neurocytol. 2002, 31, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M. NG2-expressing glial progenitor cells: An abundant and widespread population of cycling cells in the adult rat CNS. Mol. Cell. Neurosci. 2003, 24, 476–488. [Google Scholar] [CrossRef]
- Nishiyama, A.; Komitova, M.; Suzuki, R.; Zhu, X. Polydendrocytes (NG2 cells): Multifunctional cells with lineage plasticity. Nat. Rev. Neurosci. 2009, 10, 9–22. [Google Scholar] [CrossRef]
- Alavi, M.S.; Shamsizadeh, A.; Azhdari-Zarmehri, H.; Roohbakhsh, A. Orphan G protein-coupled receptors: The role in CNS disorders. Biomed. Pharmacother. 2018, 98, 222–232. [Google Scholar] [CrossRef]
- Ishii, K.; Toda, M.; Nakai, Y.; Asou, H.; Watanabe, M.; Nakamura, M.; Yato, Y.; Fujimura, Y.; Kawakami, Y.; Toyama, Y.; et al. Increase of oligodendrocyte progenitor cells after spinal cord injury. J. Neurosci. Res. 2001, 65, 500–507. [Google Scholar] [CrossRef]
- Miron, V.E.; Antel, J.P. Cells of the oligodendroglial lineage, myelination, and remyelination. Biochim. Biophys. Acta 2011, 1812, 184–193. [Google Scholar] [CrossRef]
- Hanafy, K.A.; Sloane, J.A. Regulation of remyelination in multiple sclerosis. FEBS Letters 2011, 585, 3821–3828. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; Bruck, W.; Lucchinetti, C.; Rodriguez, M. Remyelination in multiple sclerosis. Mult. Scler. 1997, 3, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Patrikios, P. Remyelination is extensive in a subset of multiple sclerosis patients. Brain 2006, 129, 3165–3172. [Google Scholar] [CrossRef] [PubMed]
- Bradl, M.; Lassmann, H. Oligodendrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, T.; Miron, V.; Cuo, Q.; Wegner, C.; Antel, J.; Brück, W. Differentiation block of oligodendroglial progenitorcells as a cause for remyelination failure in chronic multiple sclerosis. Brain 2008, 131, 1749–1758. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, M.; Daniele, S.; Lecca, D.; Lee, P.R.; Parravicini, C.; Fields, R.D.; Rosa, P.; Antonucci, F.; Verderio, C.; Trincavelli, M.L.; et al. Phenotypic Changes, Signaling Pathway, and Functional Correlates of GPR17-expressing Neural Precursor Cells during Oligodendrocyte Differentiation. J. Biol. Chem. 2011, 286, 10593–10604. [Google Scholar] [CrossRef] [PubMed]
- Jakovcevski, I. Oligodendrocyte development and the onset of myelination in the human fetal brain. Front. Neuroanat. 2009, 3, 5. [Google Scholar] [CrossRef]
- Satoh, J.; Kino, Y.; Yanaizu, M.; Tosaki, Y.; Sakai, K.; Ishida, T.; Saito, Y. Expression of GPR17, a regulator of oligodendrocyte differentiation and maturation, in Nasu-Hakola disease brains. Intractable Rare Dis. Res. 2017, 6, 50–54. [Google Scholar] [CrossRef]
- Lu, C.; Dong, L.; Zhou, H.; Li, Q.; Huang, G.; Bai, S.; Liao, L. G-Protein-Coupled Receptor Gpr17 Regulates Oligodendrocyte Differentiation in Response to Lysolecithin-Induced Demyelination. Sci. Rep. 2018, 8, 4502. [Google Scholar] [CrossRef]
- Stilund, M.; Glejstrup, M.C.; Petersen, T.; Møller, H.J.; Rasmussen, P.V.; Christensen, T. Biomarkers of inflammation and axonal degeneration/damage in patients with newly diagnosed multiple sclerosis: Contributions of the soluble CD163 CSF/serum ratio to a biomarker panel. PLoS ONE 2015, 10, e0119681. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, H.; Wang, S.; Koito, H.; Li, J.; Ye, F.; Hoang, J.; Escobar, S.S.; Gow, A.; Arnett, H.A.; et al. The oligodendrocyte-specific G protein–coupled receptor GPR17 is a cell-intrinsic timer of myelination. Nat. Neurosci. 2009, 12, 1398–1406. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Bercury, K.K.; Ahrendsen, J.T.; Macklin, W.B. Olig1 Function Is Required for Oligodendrocyte Differentiation in the Mouse Brain. J. Neurosci. 2015, 35, 4386–4402. [Google Scholar] [CrossRef] [PubMed]
- Othman, A.; Frim, D.M.; Polak, P.; Vujicic, S.; Arnason, B.G.W.; Boullerne, A.I. Olig1 is expressed in human oligodendrocytes during maturation and regeneration. Glia 2011, 59, 914–926. [Google Scholar] [CrossRef] [PubMed]
- Bjartmar, C.; Wujek, J.R.; Trapp, B.D. Axonal loss in the pathology of MS: Consequences for understanding the progressive phase of the disease. J. Neurol. Sci. 2003, 206, 165–171. [Google Scholar] [CrossRef]
- Zezula, J.; Casaccia-Bonnefil, P.; Ezhevsky, S.A.; Osterhout, D.J.; Levine, J.M.; Dowdy, S.F.; Chao, M.V.; Koff, A. p21cip1 is required for the differentiation of oligodendrocytes independently of cell cycle withdrawal. EMBO Rep. 2001, 2, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L. p27kip1 independently promotes neuronal differentiation and migration in the cerebral cortex. Genes Dev. 2006, 20, 1511–1524. [Google Scholar] [CrossRef]
- Wang, S.; Sdrulla, A.; Johnson, J.E.; Yokota, Y.; Barres, B.A. A role for the helix-loop-helix protein Id2 in the control of oligodendrocyte development. Neuron 2001, 29, 603–614. [Google Scholar] [CrossRef]
- Kondo, T.; Raff, M. The Id4 HLH protein and the timing of oligodendrocyte differentiation. EMBO J. 2000, 19, 1998–2007. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| G α-Subunits | Signal Transduction | Receptors | Effects on Human the Body | Ref. |
|---|---|---|---|---|
| Gαi | inhibition of AC; activation of phosphodiesterase; open K+ channels; closes Ca2+ channels | α2-adrenergic receptors; chemokine receptors; serotonin receptors; histamine receptors; dopamine receptors; rhodopsin; taste receptors; platelet’s receptors | smooth muscle contraction; depression of neuronal activity; vision; taste; maintains ionic balance; cell motility | [50,51,52,53] |
| Gαs | activation of AC | βa-adrenergic receptors; serotonin; dopamine receptors; histamine receptors; olfactory receptors | smooth muscle relaxation; stimulation of neuronal activity; smell; cell growth and motility; Ca2+ influx | [51,53,54,55] |
| Gαq/11 | inhibition of AC | α1-adrenergic receptors, endothelin receptors; vasopressin receptors; chemokine receptors | intracellular calcium mobilization; smooth muscle cell proliferation | [56,57,58,59] |
| Gα12/13 | activation of the Rho family of GTPases | - | platelet activation; neuronal activity; immune response (e.g., lymphocyte adhesion and migration, neutrophil chemokinesis and chemotaxis); cell polarity; cell shape; changes in the cytoskeleton | [60,61,62] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dziedzic, A.; Miller, E.; Saluk-Bijak, J.; Bijak, M. The GPR17 Receptor—A Promising Goal for Therapy and a Potential Marker of the Neurodegenerative Process in Multiple Sclerosis. Int. J. Mol. Sci. 2020, 21, 1852. https://doi.org/10.3390/ijms21051852
Dziedzic A, Miller E, Saluk-Bijak J, Bijak M. The GPR17 Receptor—A Promising Goal for Therapy and a Potential Marker of the Neurodegenerative Process in Multiple Sclerosis. International Journal of Molecular Sciences. 2020; 21(5):1852. https://doi.org/10.3390/ijms21051852
Chicago/Turabian StyleDziedzic, Angela, Elzbieta Miller, Joanna Saluk-Bijak, and Michal Bijak. 2020. "The GPR17 Receptor—A Promising Goal for Therapy and a Potential Marker of the Neurodegenerative Process in Multiple Sclerosis" International Journal of Molecular Sciences 21, no. 5: 1852. https://doi.org/10.3390/ijms21051852
APA StyleDziedzic, A., Miller, E., Saluk-Bijak, J., & Bijak, M. (2020). The GPR17 Receptor—A Promising Goal for Therapy and a Potential Marker of the Neurodegenerative Process in Multiple Sclerosis. International Journal of Molecular Sciences, 21(5), 1852. https://doi.org/10.3390/ijms21051852