Induction of Antitumor Immunity by Exosomes Isolated from Cryopreserved Cord Blood Monocyte-Derived Dendritic Cells

 ,
,
Abstract
1. Introduction
2. Results
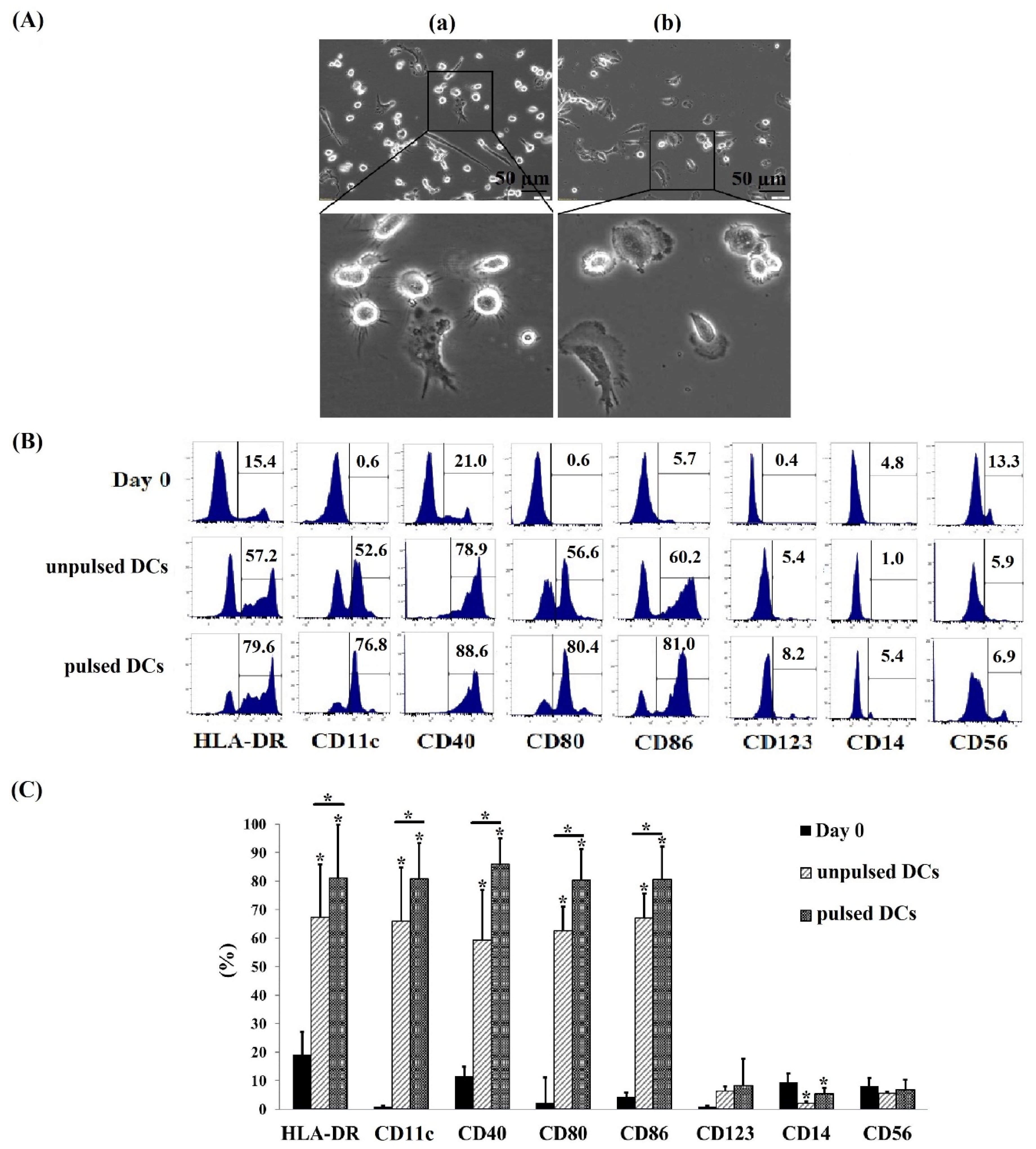
2.1. Successful Generation of Cryopreserved Umbilical Cord Blood Mononuclear Cell-Derived DCs (Cryo CBMDCs)
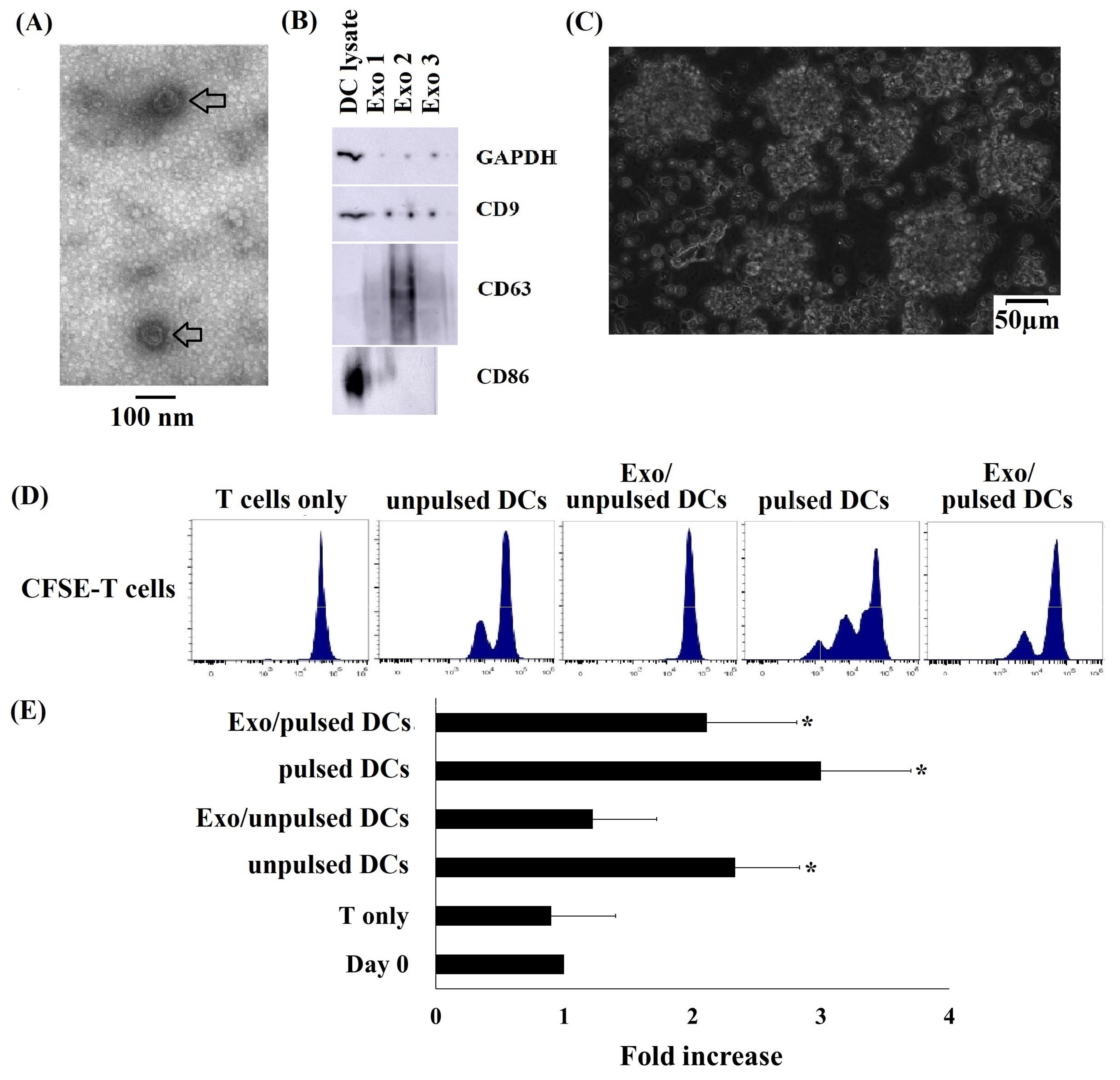
2.2. Typical Characteristics of DC-Derived Exosomes
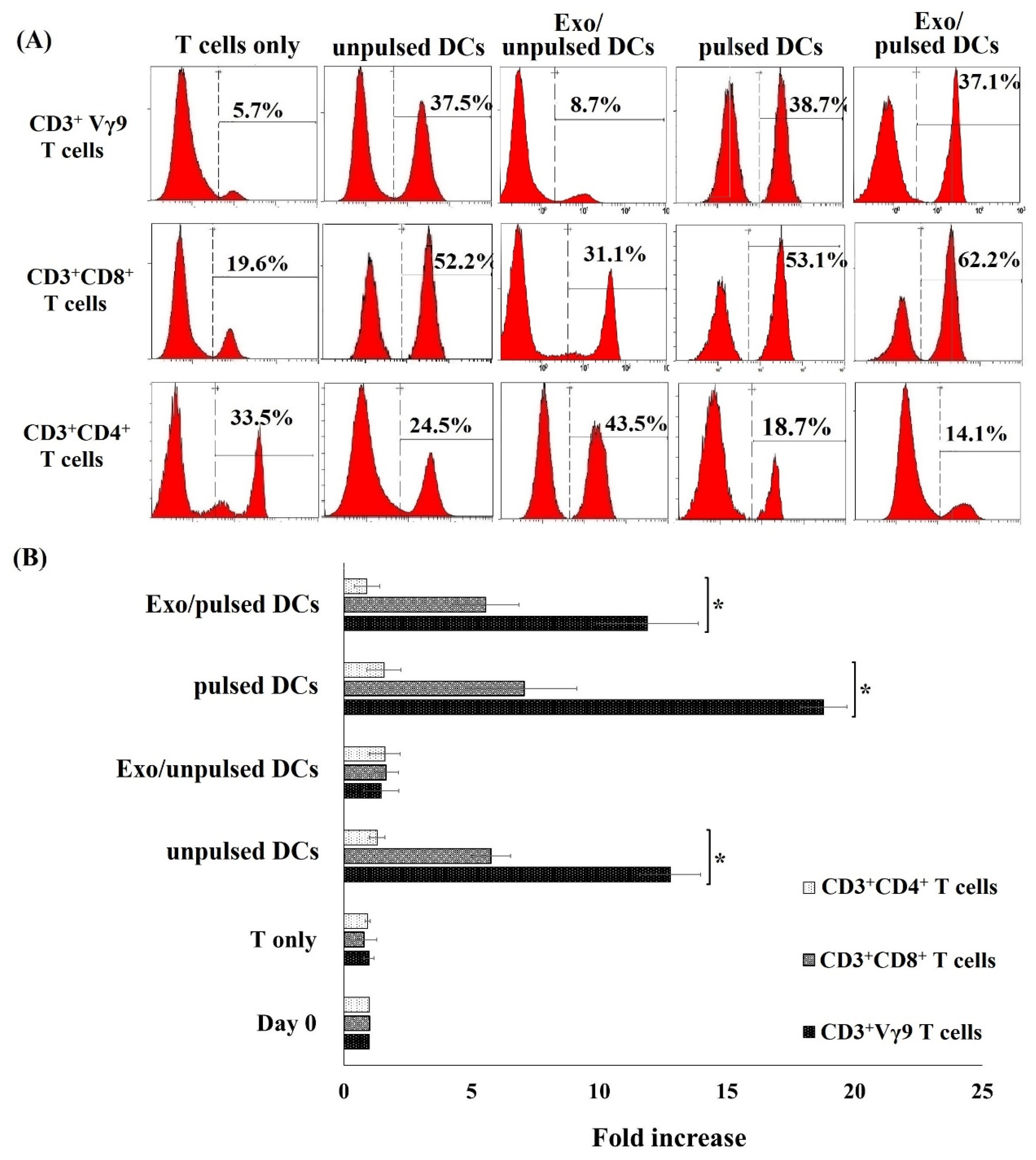
2.3. Cryo CBMDCs and their Exosomes Induced the Proliferation of Allogeneic T Cells
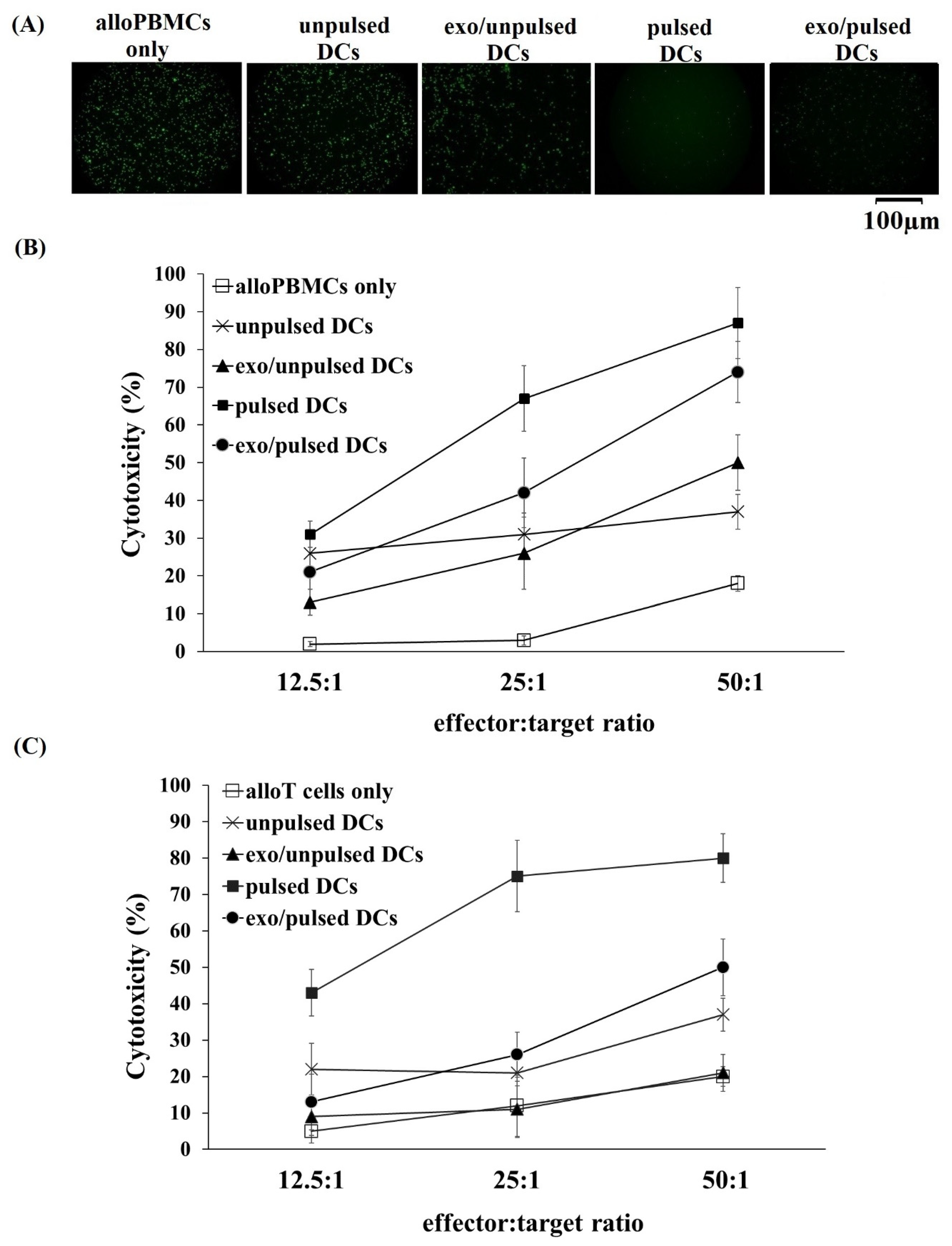
2.4. Greater Cytotoxic Activity of Allogeneic PBMCs and Allogeneic T Cells Primed with DCs and their Exosomes on A549 Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Thawing Cryopreserved CB Units and Mononuclear Cell Isolation
4.3. Generation of DCs
4.4. A549 Lung Carcinoma Cell Culture and Preparation of Tumor Cell Lysates
4.5. Exosome Isolation
4.6. Analysis of Exosome Morphology via Transmission Electron Microscopy and Western Blot Analysis
4.7. Allogeneic T Cell Purification and Proliferation Assay
4.8. Allogeneic T Cell Subset Proliferation Assay
4.9. Cytotoxicity Assay
4.10. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DCs | Dendritic cells |
| CB | Cord blood |
| MNCs | Mononuclear cells |
| CBMCs | Cord blood mononuclear cells |
| AutoDCs | Autologous dendritic cells |
| AlloDCs | Allogeneic dendritic cells |
| EVs | Extracellular vesicles |
| CSFE | Carboxyfluorescein succinimidyl ester |
| Calcein-AM | Calcein acetoxymethyl |
| FI | fluorescence intensity |
| HLA | human leukocyte antigen |
| NK cells | Natural killer cells |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| IL | Interleukin |
| TNFα | Tumor necrosis factor α |
| TEM | Transmission electron microscopy |
| FBS | fetal bovine serum |
| DMEM | Dulbecco’s modified Eagle’s medium |
| RPMI | Roswell Park Memorial Institute medium |
| PBS | phosphate-buffered saline |
References
- Saxena, M.; Bhardwaj, N. Re-emergence of dendritic cell vaccines for cancer treatment. Trends Cancer 2018, 4, 119–137. [Google Scholar] [CrossRef]
- van Willigen, W.W.; Bloemendal, M.; Gerritsen, W.R.; Schreibelt, G.; de Vries, I.J.M.; Bol, K.F. Dendritic cell cancer therapy: Vaccinating the right patient at the right time. Front. Immunol. 2018, 1, 2265. [Google Scholar] [CrossRef]
- Fabre, J.W. The allogeneic response and tumor immunity. Nat. Med. 2001, 7, 649–652. [Google Scholar] [CrossRef]
- Steinman, R.M. The dendritic cell system and its role in immunogenicity. Annu. Rev. Immunol. 1991, 9, 271–296. [Google Scholar] [CrossRef] [PubMed]
- de Gruijl, T.D.; van den Eertwegh, A.J.; Pinedo, H.M.; Scheper, R.J. Whole-cell cancer vaccination: From autologous to allogeneic tumor- and dendritic cell-based vaccines. Cancer Immunol. Immunother. 2008, 57, 1569–1577. [Google Scholar] [CrossRef] [PubMed]
- Mac Keon, S.; Ruiz, M.S.; Gazzaniga, S.; Wainstok, R. Dendritic cell-based vaccination in cancer: Therapeutic implications emerging from murine models. Front. Immunol. 2015, 6, 243. [Google Scholar] [CrossRef] [PubMed]
- Höltl, L.; Ramoner, R.; Zelle-Rieser, C.; Gander, H.; Putz, T.; Papesh, C.; Nussbaumer, W.; Falkensammer, C.; Bartsch, G.; Thurnher, M. Allogeneic dendritic cell vaccination against metastatic renal cell carcinoma with or without cyclophosphamide. Cancer Immunol. Immunother. 2005, 54, 663–670. [Google Scholar] [CrossRef]
- Hus, I.; Roliński, J.; Tabarkiewicz, J.; Wojas, K.; Bojarska-Junak, A.; Greiner, J.; Giannopoulos, K.; Dmoszyńska, A.; Schmitt, M. Allogeneic dendritic cells pulsed with tumor lysates or apoptotic bodies as immunotherapy for patients with early-stage B-cell chronic lymphocytic leukemia. Leukemia 2005, 19, 1621–1627. [Google Scholar] [CrossRef]
- Neves, A.R.; Ensina, L.F.; Anselmo, L.B.; Leite, K.R.; Buzaid, A.C.; Camara-Lopes, L.H.; Barbuto, J.A. Dendritic cells derived from metastatic cancer patients vaccinated with allogeneic dendritic cell-autologous tumor cell hybrids express more CD86 and induce higher levels of interferon-gamma in mixed lymphocyte reactions. Cancer Immunol. Immunother. 2005, 54, 61–66. [Google Scholar] [CrossRef]
- Tamir, A.; Basagila, E.; Kagahzian, A.; Jiao, L.; Jensen, S.; Nicholls, J.; Tate, P.; Stamp, G.; Farzaneh, F.; Harrison, P.; et al. Induction of tumor-specific T-cell responses by vaccination with tumor lysate-loaded dendritic cells in colorectal cancer patients with carcinoembryonic-antigen positive tumors. Cancer Immunol. Immunother. 2007, 56, 2003–2016. [Google Scholar] [CrossRef]
- Trefzer, U.; Weingart, G.; Chen, Y.; Herberth, G.; Adrian, K.; Winter, H.; Audring, H.; Guo, Y.; Sterry, W.; Walden, P. Hybrid cell vaccination for cancer immune therapy: First clinical trial with metastatic melanoma. Int. J. Cancer 2000, 85, 618–626. [Google Scholar] [CrossRef]
- Trefzer, U.; Herberth, G.; Wohlan, K.; Milling, A.; Thiemann, M.; Sherev, T.; Sparbier, K.; Sterry, W.; Walden, P. Vaccination with hybrids of tumor and dendritic cells induces tumor-specific T-cell and clinical responses in melanoma stage III and IV patients. Int. J. Cancer 2004, 110, 730–740. [Google Scholar] [CrossRef] [PubMed]
- Trefzer, U.; Herberth, G.; Wohlan, K.; Milling, A.; Thiemann, M.; Sharav, T.; Sparbier, K.; Sterry, W.; Walden, P. Tumour-dendritic hybrid cell vaccination for the treatment of patients with malignant melanoma: Immunological effects and clinical results. Vaccine 2005, 23, 2367–2373. [Google Scholar] [CrossRef] [PubMed]
- de Gruijl, T.; Santegoeds, S.; van Wetering, S.; Singh, S.K.; Hall, A.; van de Loosdrecht, A.A.; Kruisbeek, A. Allogeneic dendritic cell (DC) vaccination as an “off the shelf” treatment to prevent or delay relapse in elderly acute myeloid leukemia patients: Results of Phase I/IIa safety and feasibility study. J. Immunother Cancer. 2013, 1 (Suppl. 1), 205. [Google Scholar] [CrossRef]
- van de Loosdrecht, A.A.; van Wetering, S.; Santegoets, S.J.A.M.; Singh, S.K.; Eeltink, C.M.; den Hartog, Y.; Koppes, M.; Kaspers, J.; Ossenkoppele, G.J.; Kruisbeek, A.M.; et al. A novel allogeneic off-the-shelf dendritic cell vaccine for postremission treatment of elderly patients with acute myeloid leukemia. Cancer Immunol. Immunother. 2018, 67, 1505–1518. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Ji, Q.; Yang, Y.; Li, Q.; Wang, Z. Exosome: Function and role in cancer metastasis and drug resistance. Technol. Cancer Res. Treat. 2018, 17, 1533033818763450. [Google Scholar] [CrossRef]
- Barros, F.M.; Carneiro, F.; Machado, J.C.; Melo, S.A. Exosomes and immune response in cancer: Friends or foes? Front. Immunol. 2018, 9, 730. [Google Scholar] [CrossRef]
- Zitvogel, L.; Regnault, A.; Lozier, A.; Wolfers, J.; Flament, C.; Tenza, D.; Ricciardi-Castagnoli, P.; Raposo, G.; Amigorena, S. Eradication of established murine tumors using a novel cell-free vaccine: Dendritic cell-derived exosomes. Nat. Med. 1998, 4, 594–600. [Google Scholar] [CrossRef]
- Buschow, S.I.; Nolte-’t Hoen, E.N.; van Niel, G.; Pols, M.S.; ten Broeke, T.; Lauwen, M.; Ossendorp, F.; Melief, C.J.; Raposo, G.; Wubbolts, R.; et al. MHC II in dendritic cells is targeted to lysosomes or T cell-induced exosomes via distinct multivesicular body pathways. Traffic 2009, 10, 1528–1542. [Google Scholar] [CrossRef]
- Szaryńska, M.; Preis, K.; Zabul, P.; Kmieć, Z. Diversity of dendritic cells generated from umbilical cord or adult peripheral blood precursors. Cent. Eur. J. Immunol. 2018, 43, 306–313. [Google Scholar] [CrossRef]
- Théry, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006. [Google Scholar] [CrossRef] [PubMed]
- Wahlund, C.J.E.; Güclüler, G.; Hiltbrunner, S.; Veerman, R.E.; Näslund, T.I.; Gabrielsson, S. Exosomes from antigen-pulsed dendritic cells induce stronger antigen-specific immune responses than microvesicles in vivo. Sci. Rep. 2017, 7, 17095. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Dorval, T.; Chaput, N.; André, F.; Caby, M.P.; Novault, S.; Flament, C.; Leboulaire, C.; Borg, C.; Amigorena, S.; et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: Results of the first phase I clinical trial. J. Transl. Med. 2005, 3, 10. [Google Scholar] [CrossRef] [PubMed]
- Besse, B.; Charrier, M.; Lapierre, V.; Dansin, E.; Lantz, O.; Planchard, D.; Le Chevalier, T.; Livartoski, A.; Barlesi, F.; Laplanche, A.; et al. Dendritic cell-derived exosomes as maintenance immunotherapy after first line chemotherapy in NSCLC. Oncoimmunology 2016, 5, e1071008. [Google Scholar] [CrossRef]
- Morse, M.A.; Garst, J.; Osada, T.; Khan, S.; Hobeika, A.; Clay, T.M.; Valente, N.; Shreeniwas, R.; Sutton, M.A.; Delcayre, A.; et al. A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J. Transl. Med. 2005, 3, 9. [Google Scholar] [CrossRef]
- Pham, P.V.; Vu, B.T.; Pham, V.Q.; Le, P.M.; Le, H.T.; Phan, N.K. Production of dendritic cells and cytokine-induced killer cells from banked umbilical cord blood samples. Biomed. Res. 2015, 2, 402–408. [Google Scholar]
- Yi, H.J.; Lu, G.X. Adherent and non-adherent dendritic cells are equivalently qualified in GM-CSF, IL-4 and TNF-α culture system. Cell Immunol. 2012, 277, 44–48. [Google Scholar] [CrossRef]
- Aldahlawi, A.M. Modulation of dendritic cell immune functions by plant components. J. Microsc. Ultrastruct. 2016, 4, 55–62. [Google Scholar] [CrossRef]
- Delirezh, N.; Moazzeni, S.M.; Shokri, F.; Shokrgozar, M.A.; Atri, M.M.; Karbassian, H. In vitro analysis of T cell responses induced by breast tumor cell lysate pulsed with autologous dendritic cells. Adv. Biosci. Biotechnol. 2012, 3, 126–136. [Google Scholar] [CrossRef][Green Version]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Théry, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef]
- Yoshioka, Y.; Konishi, Y.; Kosaka, N.; Katsuda, T.; Kato, T.; Ochiya, T. Comparative marker analysis of extracellular vesicles in different human cancer types. J. Extracell. Vesicles 2013, 2, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ge, Q.; Palliser, D.; Eisen, H.N.; Chen, J. Homeostatic T cell proliferation in a T cell-dendritic cell coculture system. Proc. Natl. Acad. Sci. USA 2002, 99, 2983–2988. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1988, 392, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Delirezh, N.; Majedi, L.; Asri Rezaei, S.; Ranjkeshzadeh, H. Generation of mature monocyte-Derived dendritic cells in the presence of heparin and monocyte conditioned medium: Phenotypic and functional comparison. Iran. Biomed. J. 2011, 15, 79–84. [Google Scholar]
- Rainone, V.; Martelli, C.; Ottobrini, L.; Biasin, M.; Texido, G.; Degrassi, A.; Borelli, M.; Lucignani, G.; Trabattoni, D.; Clerici, M. Correction: Immunological characterization of whole tumor Lysate-loaded dendritic cells for cancer immunotherapy. PLoS ONE 2019, 11, e0151008. [Google Scholar] [CrossRef]
- Mittelbrunn, M.; Sanchez-Madrid, F. Intercellular communication: Diverse structures for exchange of genetic information. Nat. Rev. Mol. Cell Biol. 2012, 13, 328–335. [Google Scholar] [CrossRef]
- Munich, S.; Sobo-Vujanovic, A.; Buchser, W.J.; Beer-Stolz, D.; Vujanovic, N.L. Dendritic cell exosomes directly kill tumor cells and activate natural killer cells via TNF superfamily ligands. Oncoimmunology 2012, 1, 1074–1083. [Google Scholar] [CrossRef]
- Admyre, C.; Johansson, S.M.; Paulie, S.; Gabrielsson, S. Direct exosome stimulation of peripheral human T cells detected by ELISPOT. Eur. J. Immunol. 2006, 36, 1772–1781. [Google Scholar] [CrossRef]
- Aggarwal, R.; Lu, J.; Kanji, S.; Das, M.; Joseph, M.; Lutsberg, M.B.; Ray, A.; Pompili, V.J.; Shapiro, C.L.; Das, H. Human Vγ2Vδ2 T cells limit breast cancer growth by modeling cell survival-apoptosis-related molecules and microenvironment in tumors. Int. J. Cancer 2013, 133, 2133–2144. [Google Scholar] [CrossRef]
- Todaro, M.; D’Asaro, M.; Caccamo, N.; Iovino, F.; Francipane, M.G.; Meraviglia, S.; Orlando, V.; La Mendola, C.; Gulotta, G.; Salerno, A.; et al. Efficient killing of human colon cancer stem cells by gammadelta T lymphocytes. J. Immunol. 2009, 182, 7287–7296. [Google Scholar] [CrossRef]
- 41. Wang, L.; Kamath, A.; Das, H.; Li, L.; Bukowski, J.F. Antibacterial effect of human V gamma 2V delta 2 T cells in vivo. J. Clin. Investig. 2011, 108, 1349–1357. [Google Scholar] [CrossRef]
- Lo Presti, E.; Pizzolato, G.; Gulotta, E.; Cocorullo, G.; Gulotta, G.; Dieli, F.; Meraviglia, S. Current advances in γδ T cell-based tumor immunotherapy. Front. Immunol. 2017, 8, 1401. [Google Scholar] [CrossRef]
- Ryan, P.L.; Sumaria, N.; Holland, C.J.; Bradford, C.M.; Izotova, N.; Grandjean, C.L.; Jawad, A.S.; Bergmeier, L.A.; Pennington, D.J. Heterogeneous yet stable Vdelta2 (+) T-cell profiles define distinct cytotoxic effector potentials in healthy human individuals. Proc. Natl. Acad. Sci. USA 2016, 113, 14378–14383. [Google Scholar] [CrossRef]
- Fisher, J.P.; Heuijerjans, J.; Yan, M.; Gustafsson, K.; Anderson, J. Gammadelta T cells for cancer immunotherapy: A systematic review of clinical trials. Oncoimmunology 2014, 3, e27572. [Google Scholar] [CrossRef] [PubMed]
- Morita, C.T.; Mariuzza, R.A.; Brenner, M.B. Antigen recognition by human gamma delta T cells: Pattern recognition by the adaptive immune system. Springer Semin. Immunopathol. 2000, 22, 191–217. [Google Scholar] [CrossRef] [PubMed]
- Girardi, M.; Oppenheim, D.E.; Steele, C.R.; Lewis, J.M.; Glusac, E.; Filler, R.; Hobby, P.; Sutton, B.; Tigelaar, R.E.; Hayday, A.C. Regulation of cutaneous malignancy by gammadelta T cells. Science 2001, 294, 605–609. [Google Scholar] [CrossRef] [PubMed]
- Dworacki, G.; Cicinnati, V.R.; Beckebaum, S.; Pizzoferrato, E.; Hoffmann, T.K.; De Leo, A.B. Unpulsed dendritic cells induce broadly applicable anti-tumor immunity in mice. Cancer Biol. 2005, 4, 50–56. [Google Scholar] [CrossRef]
- Lo, J.; Xia, C.Q.; Peng, R.; Clare-Salzler, M.J. Immature dendritic cell therapy confers durable immune modulation in an antigen-dependent and antigen-independent manner in nonobese diabetic mice. J. Immunol. Res. 2018, 2018, 5463879. [Google Scholar] [CrossRef] [PubMed]
- Zelenay, S.; Reis e Sousa, C. Adaptive immunity after cell death. Trends Immunol. 2013, 34, 329–335. [Google Scholar] [CrossRef]
- Ahrens, S.; Zelenay, S.; Sancho, D.; Hanč, P.; Kjær, S.; Feest, C.; Fletcher, G.; Durkin, C.; Postigo, A.; Skehel, M.; et al. F-actin is an evolutionarily conserved damage-associated molecular pattern recognized by DNGR-1, a receptor for dead cells. Immunity 2012, 36, 635–645. [Google Scholar] [CrossRef]
- Viaud, S.; Terme, M.; Flament, C.; Taieb, J.; André, F.; Novault, S.; Escudier, B.; Robert, C.; Caillat-Zucman, S.; Tursz, T.; et al. Dendritic cell-derived exosomes promote natural killer cell activation and proliferation: A role for NKG2D ligands and IL-15Ralpha. PLoS ONE 2009, 4, e4942. [Google Scholar] [CrossRef] [PubMed]
- Simhadri, V.R.; Reiners, K.S.; Hansen, H.P.; Topolar, D.; Simhadri, V.L.; Nohroudi, K.; Kufer, T.A.; Engert, A.; Pogge von Strandmann, E. Dendritic cells release HLA-B-associated transcript-3 positive exosomes to regulate natural killer function. PLoS ONE 2008, 3, e3377. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Bevan, M.J. CD8+ T Cells: Foot soldiers of the immune system. Immunity 2011, 35, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Utsugi-Kobukai, S.; Fujimaki, H.; Hotta, C.; Nakazawa, M.; Minami, M. MHC class I-mediated exogenous antigen presentation by exosomes secreted from immature and mature bone marrow derived dendritic cells. Immunol. Lett. 2003, 89, 125–131. [Google Scholar] [CrossRef]
- Hao, S.; Bai, O.; Li, F.; Yuan, J.; Laferte, S.; Xiang, J. Mature dendritic cells pulsed with exosomes stimulate efficient cytotoxic T-lymphocyte responses and antitumour immunity. Immunology 2006, 120, 90–102. [Google Scholar] [CrossRef]
- Autenrieth, S.E.; Grimm, S.; Rittig, S.M.; Grünebach, F.; Gouttefangeas, C.; Bühring, H.J. Profiling of primary peripheral blood- and monocyte-derived dendritic cells using monoclonal antibodies from the HLDA10 Workshop in Wollongong, Australia. Clin. Transl. Immunol. 2015, 4, e50. [Google Scholar] [CrossRef]
- Crespo, I.; Paiva, A.; Couceiro, A.; Pimentel, P.; Orfão, A.; Regateiro, F. Immunophenotypic and fuctional characterization of cord blood dendritic cells. Stem Cells Dev. 2004, 13, 63–70. [Google Scholar] [CrossRef]
- van Nguyen, T.; Nguyen, P.H.; Chu, T.T.; Nguyen, T.D.; Than, U.T.T.; Bui, A.V.; Ngo, T.A.; Nguyen, L.T. Factors affecting human umbilical cord blood quality prior to cryopreservation: The importance of birth weight and gestational age. Stem Cells Transl. Med. 2019, 8 (Suppl. 1), S28. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CD3+Vγ9 T Cells | CD3+CD8+ T Cells | CD3+CD4+ T Cells | |
|---|---|---|---|
| Day 0 | 5.2 ± 1.7 | 25.8 ± 4.2 | 35.2 ± 7.4 |
| T cells only | 5.7 ± 2.5 | 22.4 ± 7.4 | 36.7 ± 7.8 |
| Unpulsed DCs | 31.2 ± 3.1 * | 55.3 ± 2.4 * | 20.5 ± 11.2 * |
| Exo/unpulsed DCs | 6.8 ± 1.2 | 30.4 ± 8.9 | 48.2 ± 8.6 |
| Pulsed DCs | 35.7 ± 7.4 * | 52.7 ± 5.1 * | 19.2 ± 5.8 * |
| Exo/pulsed DCs | 32.1 ± 5.8 * | 58.9 ± 4.6 * | 15.7 ± 2.5 * |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Than, U.T.T.; Le, H.T.; Hoang, D.H.; Nguyen, X.-H.; Pham, C.T.; Bui, K.T.V.; Bui, H.T.H.; Nguyen, P.V.; Nguyen, T.D.; Do, T.T.H.; et al. Induction of Antitumor Immunity by Exosomes Isolated from Cryopreserved Cord Blood Monocyte-Derived Dendritic Cells. Int. J. Mol. Sci. 2020, 21, 1834. https://doi.org/10.3390/ijms21051834
Than UTT, Le HT, Hoang DH, Nguyen X-H, Pham CT, Bui KTV, Bui HTH, Nguyen PV, Nguyen TD, Do TTH, et al. Induction of Antitumor Immunity by Exosomes Isolated from Cryopreserved Cord Blood Monocyte-Derived Dendritic Cells. International Journal of Molecular Sciences. 2020; 21(5):1834. https://doi.org/10.3390/ijms21051834
Chicago/Turabian StyleThan, Uyen Thi Trang, Huyen Thi Le, Diem Huong Hoang, Xuan-Hung Nguyen, Cuong Thi Pham, Khanh Thi Van Bui, Hue Thi Hong Bui, Phong Van Nguyen, Tu Dac Nguyen, Thu Thi Hoai Do, and et al. 2020. "Induction of Antitumor Immunity by Exosomes Isolated from Cryopreserved Cord Blood Monocyte-Derived Dendritic Cells" International Journal of Molecular Sciences 21, no. 5: 1834. https://doi.org/10.3390/ijms21051834
APA StyleThan, U. T. T., Le, H. T., Hoang, D. H., Nguyen, X.-H., Pham, C. T., Bui, K. T. V., Bui, H. T. H., Nguyen, P. V., Nguyen, T. D., Do, T. T. H., Chu, T. T., Bui, A. V., Nguyen, L. T., & Hoang, N. T. M. (2020). Induction of Antitumor Immunity by Exosomes Isolated from Cryopreserved Cord Blood Monocyte-Derived Dendritic Cells. International Journal of Molecular Sciences, 21(5), 1834. https://doi.org/10.3390/ijms21051834