Exploring Diverse-Ring Analogues on Combretastatin A4 (CA-4) Olefin as Microtubule-Targeting Agents



Abstract

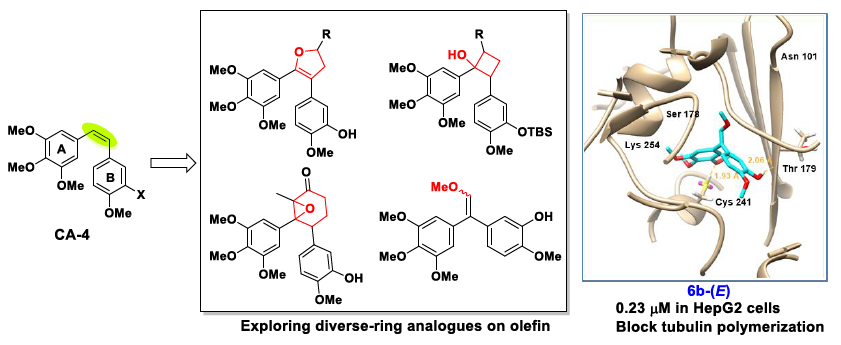
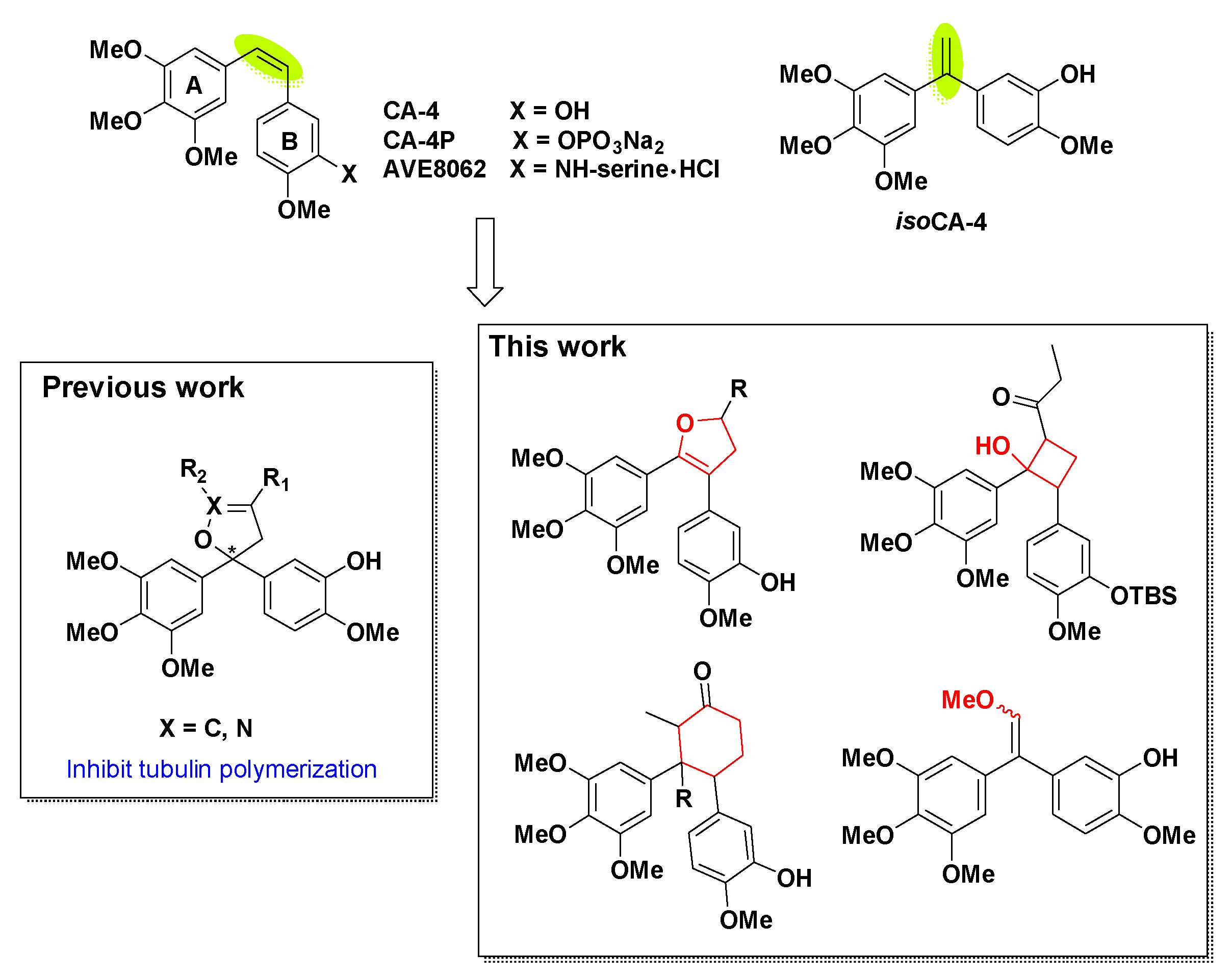
1. Introduction
2. Results and Discussion
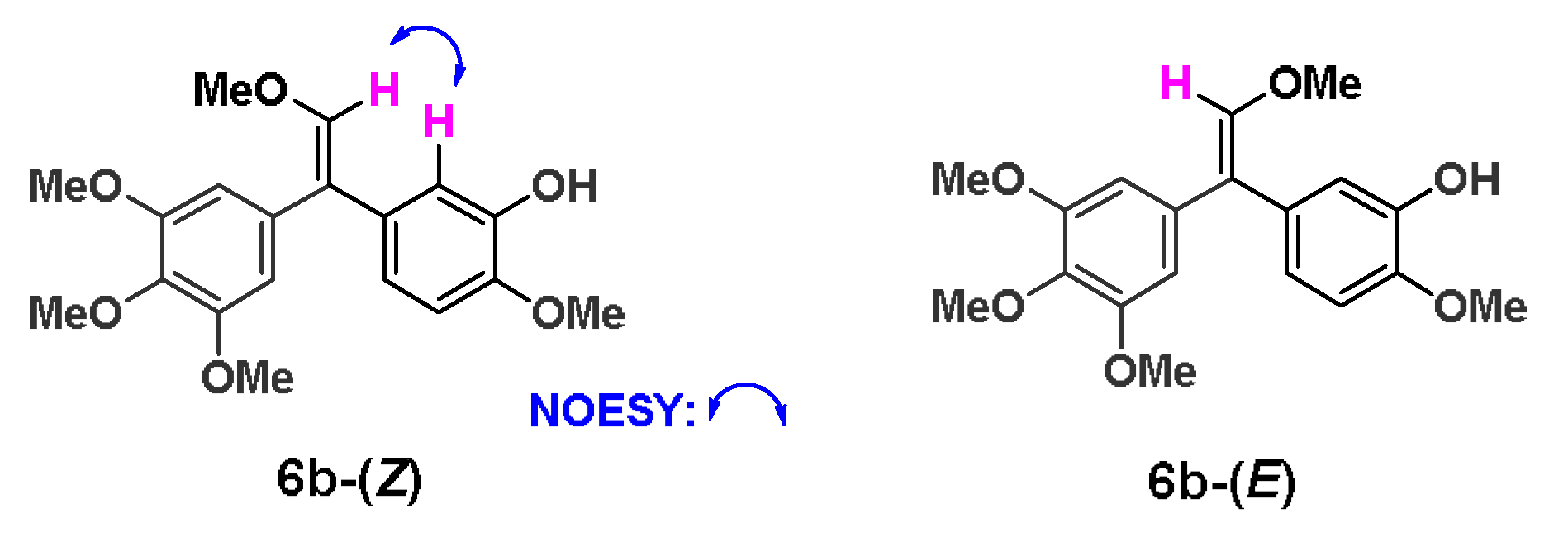
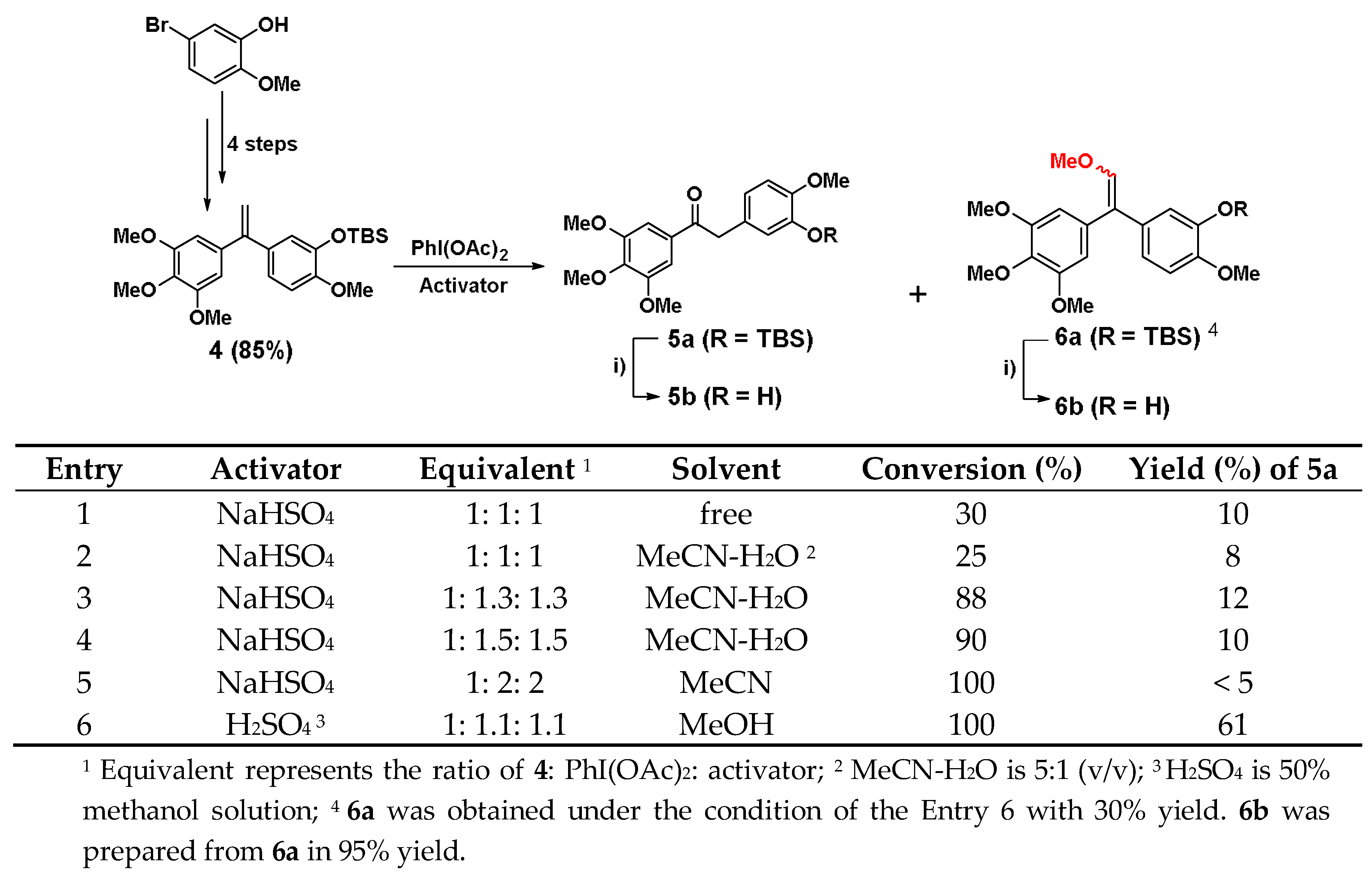
2.1. Chemistry
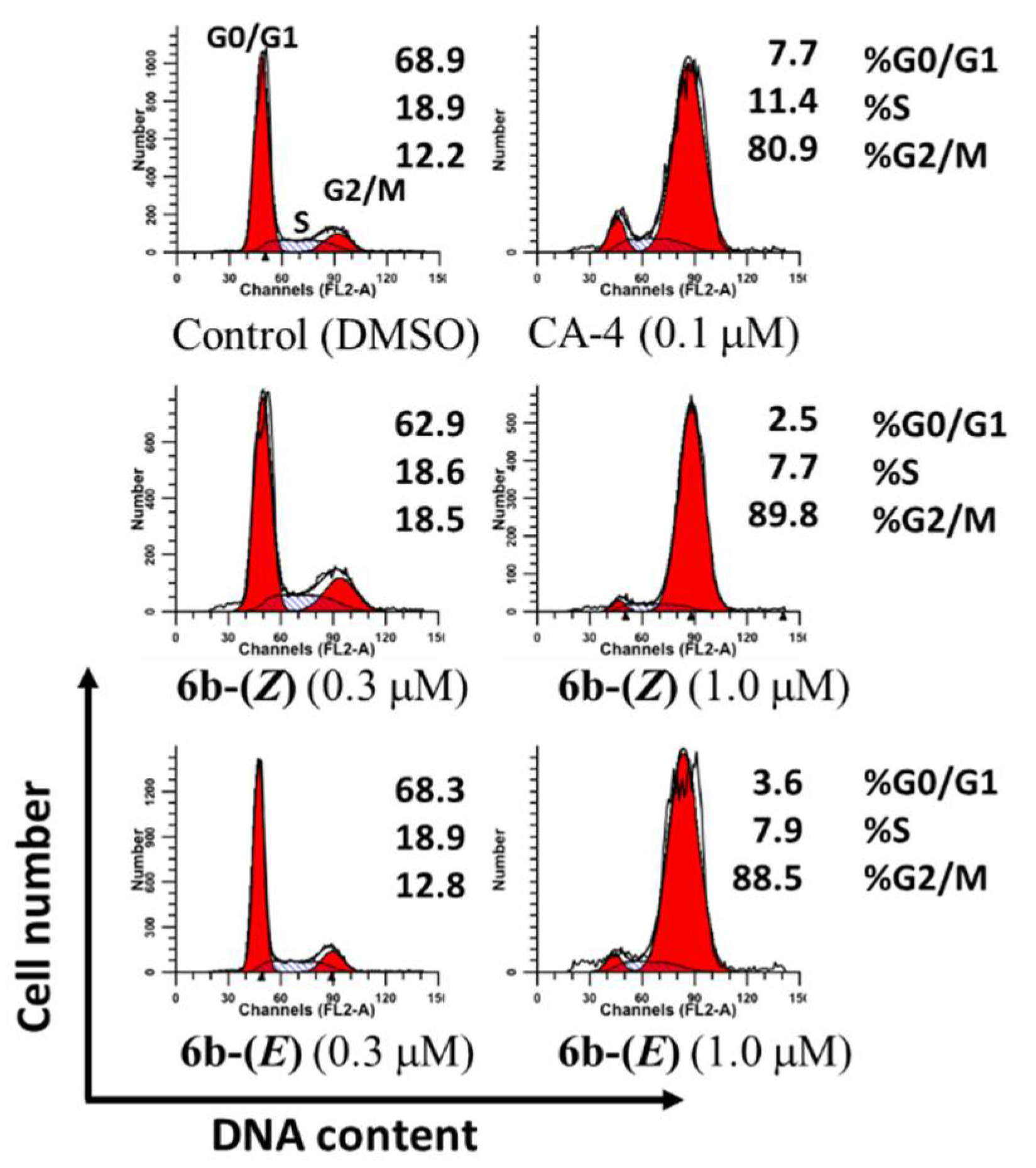
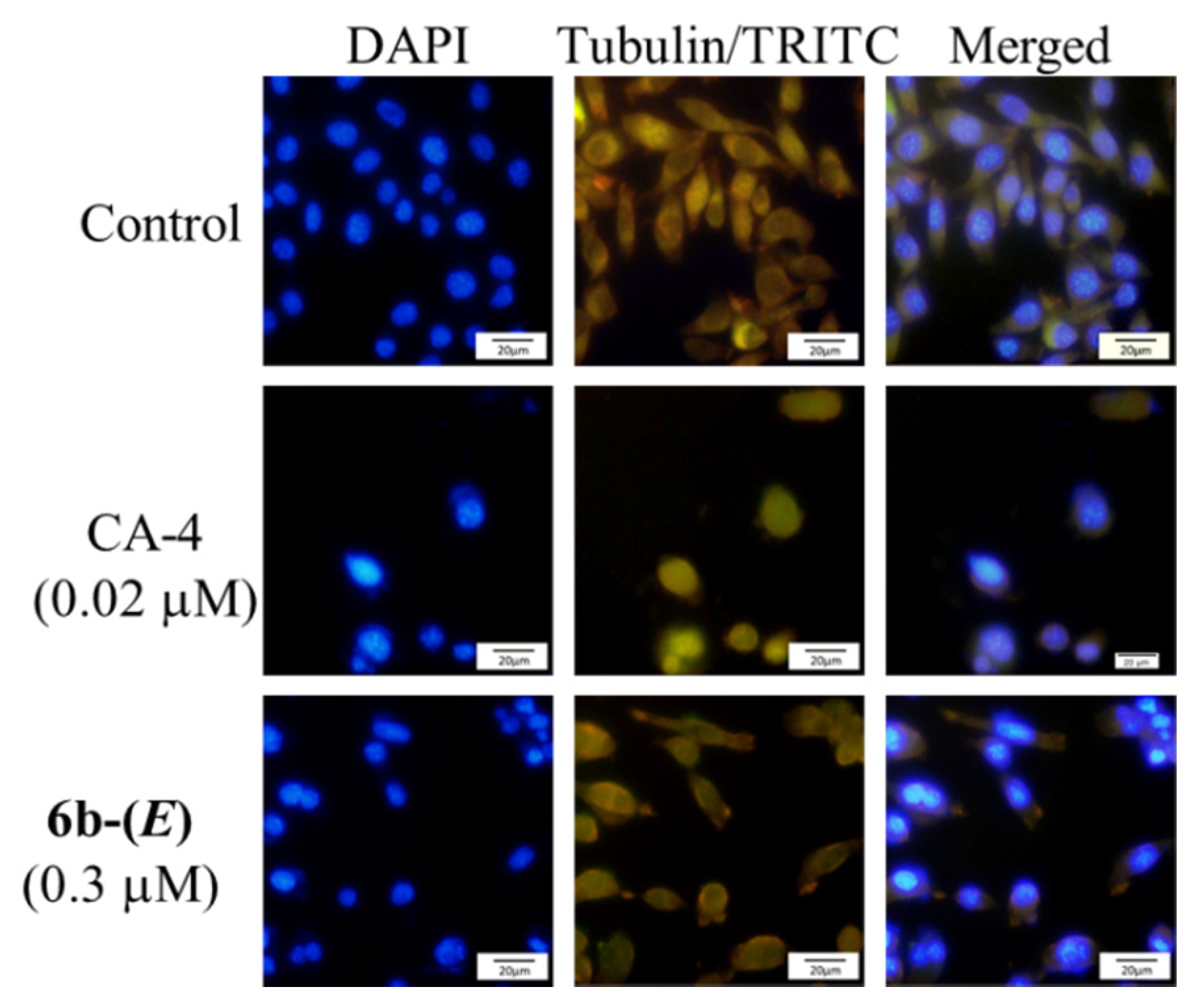
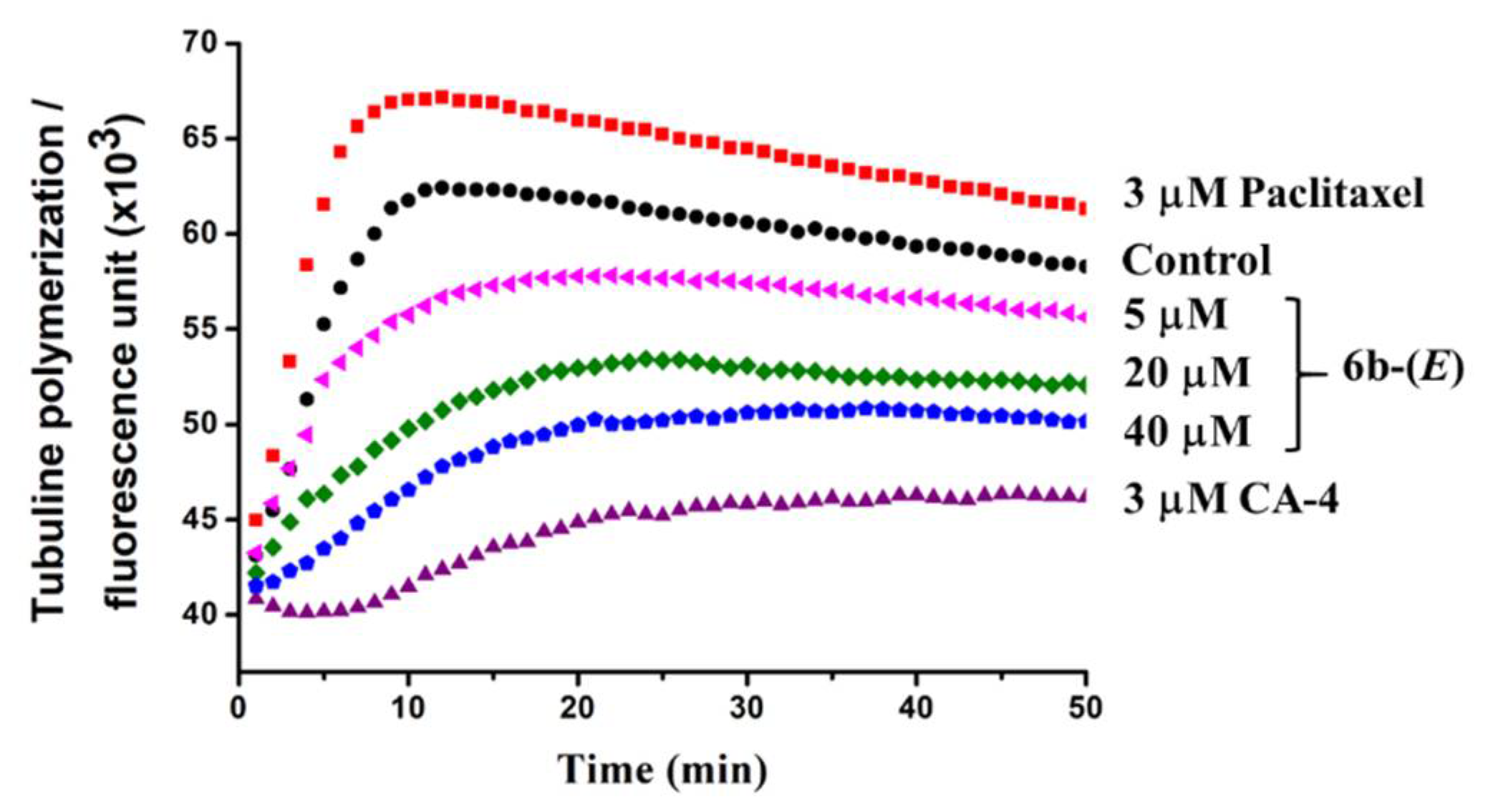
2.2. Biological Activity
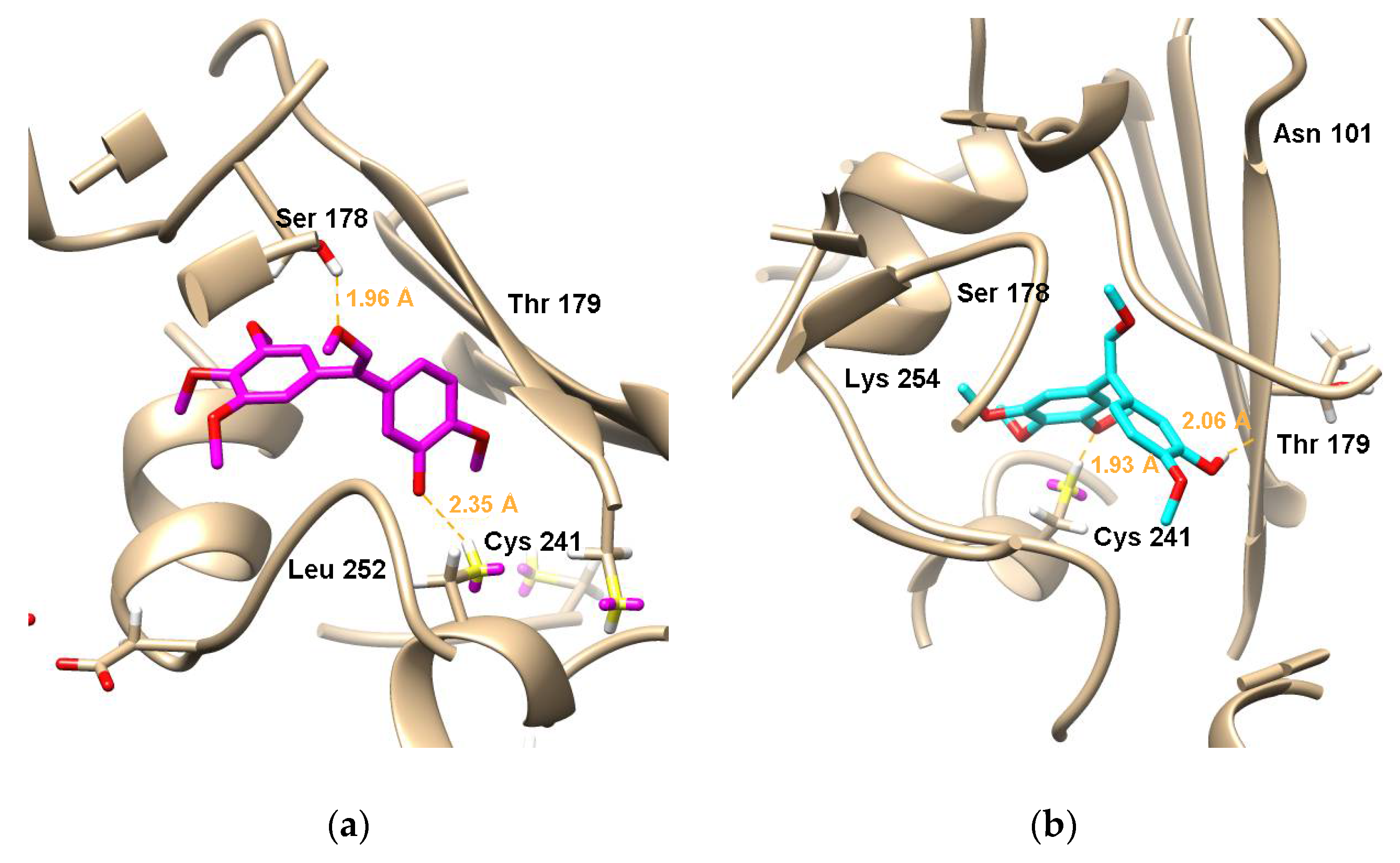
2.3. Molecular Docking
3. Materials and Methods
3.1. Chemistry
3.1.1. General
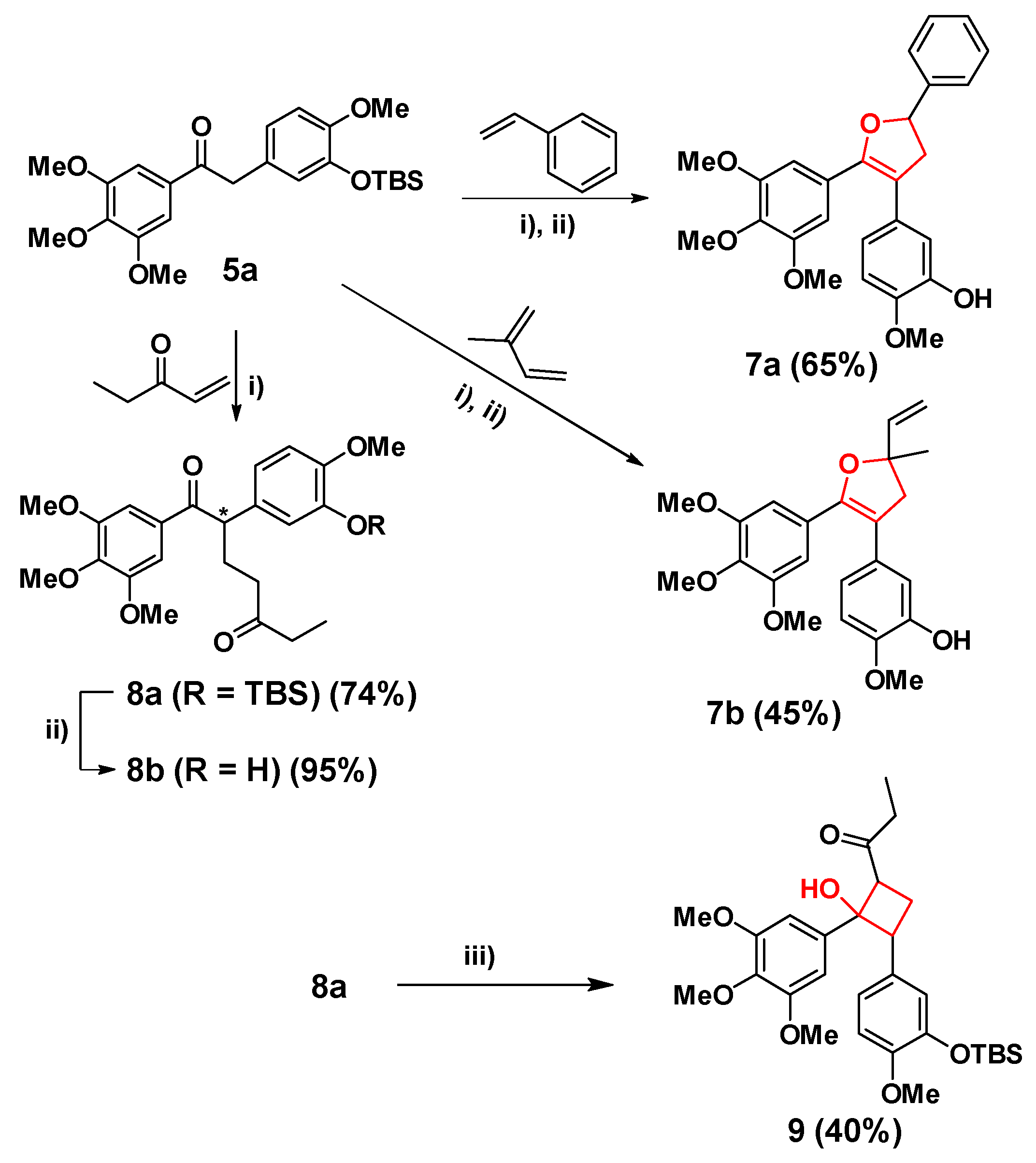
3.1.2. Synthesis of Diphenylethanone Analogue 5a, 5b and by-Products 6a, 6b
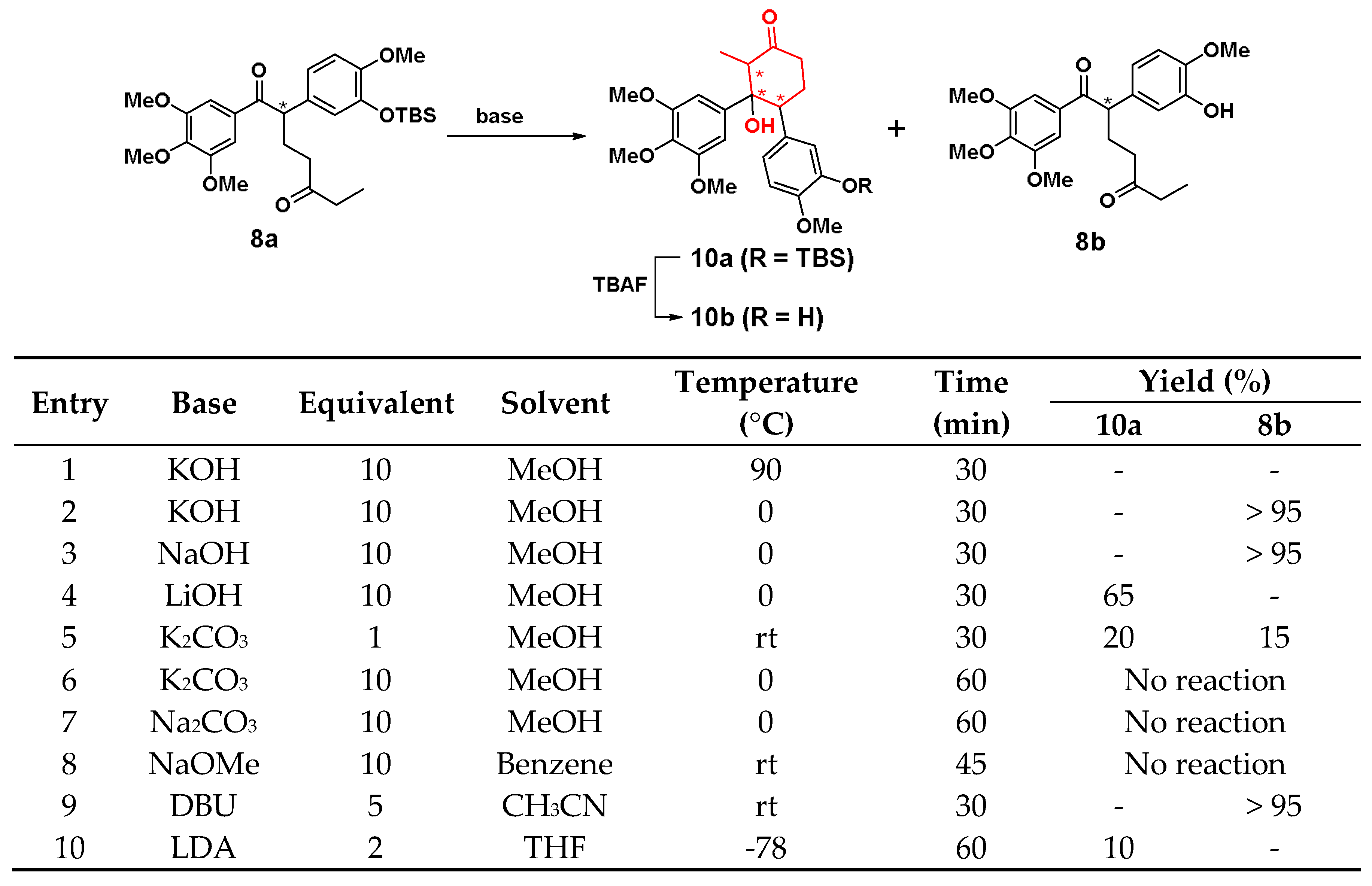
3.1.3. Synthesis of Cis-Locked Dihydrofuran Analogues 7a, 7b and α-Substituted Diphenylethanones 8a, 8b
3.1.4. Synthesis of Cyclobutane Analogue 9
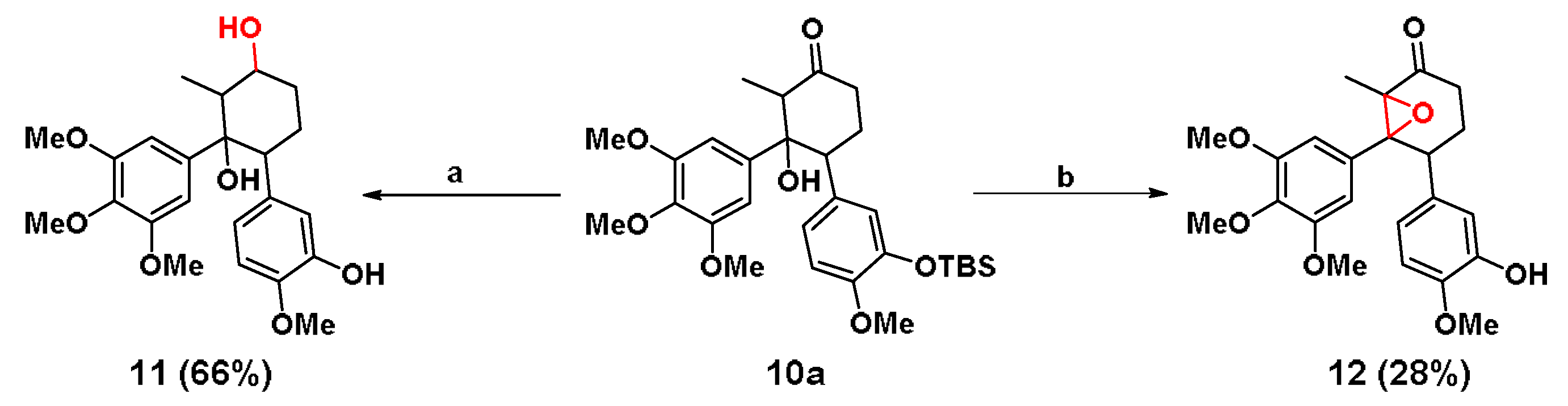
3.1.5. Synthesis of Cyclohexane Analogue 10a, 10b and 11, 12
3.2. Biological Activity Evaluation
3.2.1. Cell Culture
3.2.2. Cytotoxic Activity (SRB Assay)
3.2.3. Apoptosis-Inducing Activity (PI/Annexin V-FITC Assay)
3.2.4. Cell Cycle Distribution (PI Staining Assay)
3.2.5. Immunofluorescence Analysis (DAPI /α-Tubulin-TRITC Assay)
3.2.6. Tubulin Polymerization Assay
3.2.7. Statistics Analysis
3.3. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pettit, G.R.; Singh, S.B.; Hamel, E.; Lin, C.M.; Alberts, D.S.; Garciakendall, D. Isolation and structure of the strong cell growth and tubulin inhibitor combretastatin A-4. Experientia 1989, 45, 209–211. [Google Scholar] [CrossRef] [PubMed]
- Hamel, E. Antimitotic natural products and their interactions with tubulin. Med. Res. Rev. 1996, 16, 207–231. [Google Scholar] [CrossRef]
- Woods, J.A.; Hadfield, J.A.; Pettit, G.R.; Fox, B.W.; Mcgown, A.T. The Interaction with Tubulin of a Series of Stilbenes Based on Combretastatin a-4. Brit. J. Cancer 1995, 71, 705–711. [Google Scholar] [CrossRef]
- Young, S.L.; Chaplin, D.J. Combretastatin A4 phosphate: Background and current clinical status. Expert Opin. Investig. Drugs 2004, 13, 1171–1182. [Google Scholar] [CrossRef] [PubMed]
- Hori, K.; Saito, S. Microvascular mechanisms by which the combretastatin A-4 derivative AC7700 (AVE8062) induces tumour blood flow stasis. Br. J. Cancer 2003, 89, 1334–1344. [Google Scholar] [CrossRef] [PubMed]
- Available online: www.mateon.com (accessed on 15 February 2020).
- Lipeeva, A.V.; Shults, E.E.; Shakirov, M.M.; Pokrovsky, M.A.; Pokrovsky, A.G. Synthesis and Cytotoxic Activity of a New Group of Heterocyclic Analogues of the Combretastatins. Molecules 2014, 19, 7881–7900. [Google Scholar] [CrossRef]
- Jin, Y.H.; Qi, P.; Wang, Z.W.; Shen, Q.R.; Wang, J.; Zhang, W.G.; Song, H.R. 3D-QSAR Study of Combretastatin A-4 Analogs Based on Molecular Docking. Molecules 2011, 16, 6684–6700. [Google Scholar] [CrossRef]
- Piekus-Slomka, N.; Mikstacka, R.; Ronowicz, J.; Sobiak, S. Hybrid cis-stilbene Molecules: Novel Anticancer Agents. Int. J. Mol. Sci. 2019, 20, 1300. [Google Scholar] [CrossRef]
- Schmitt, F.; Gosch, L.C.; Dittmer, A.; Rothemund, M.; Mueller, T.; Schobert, R.; Biersack, B.; Volkamer, A.; Hopfner, M. Oxazole-Bridged Combretastatin A-4 Derivatives with Tethered Hydroxamic Acids: Structure-Activity Relations of New Inhibitors of HDAC and/or Tubulin Function. Int. J. Mol. Sci. 2019, 20, 383. [Google Scholar] [CrossRef]
- Tron, G.C.; Pirali, T.; Sorba, G.; Pagliai, F.; Busacca, S.; Genazzani, A.A. Medicinal Chemistry of Combretastatin A4: Present and Future Directions. J. Med. Chem. 2006, 49, 3033–3044. [Google Scholar] [CrossRef]
- Nam, N.H. Combretastatin A-4 analogues as antimitotic antitumor agents. Curr. Med. Chem. 2003, 10, 1697–1722. [Google Scholar] [CrossRef] [PubMed]
- Aprile, S.; Del Grosso, E.; Tron, G.C.; Grosa, G. In vitro metabolism study of combretastatin A-4 in rat and human liver microsomes. Drug Metab. Dispos. 2007, 35, 2252–2261. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Kajita, D.; Matsumoto, Y.; Hashimoto, Y. Design and synthesis of silicon-containing tubulin polymerization inhibitors: Replacement of the ethylene moiety of combretastatin A-4 with a silicon linker. Bioorg. Med. Chem. 2013, 21, 7381–7391. [Google Scholar] [CrossRef]
- Pang, Y.Q.; Yan, J.; An, B.J.; Huang, L.; Li, X.S. The synthesis and evaluation of new butadiene derivatives as tubulin polymerization inhibitors. Bioorg. Med. Chem. 2017, 25, 3059–3067. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Li, Y.; Sheng, C.; Lv, Z.; Dong, G.; Wang, T.; Liu, J.; Zhang, M.; Li, L.; Zhang, T.; et al. Design and synthesis of cyclopropylamide analogues of combretastatin-A4 as novel microtubule-stabilizing agents. J. Med. Chem. 2013, 56, 685–699. [Google Scholar] [CrossRef] [PubMed]
- Ty, N.; Pontikis, R.; Chabot, G.G.; Devillers, E.; Quentin, L.; Bourg, S.; Florent, J.C. Synthesis and biological evaluation of enantiomerically pure cyclopropyl analogues of combretastatin A4. Bioorg. Med. Chem. 2013, 21, 1357–1366. [Google Scholar] [CrossRef]
- Zhou, P.; Liu, Y.; Zhou, L.; Zhu, K.; Feng, K.; Zhang, H.; Liang, Y.; Jiang, H.; Luo, C.; Liu, M.; et al. Potent Antitumor Activities and Structure Basis of the Chiral beta-Lactam Bridged Analogue of Combretastatin A-4 Binding to Tubulin. J. Med. Chem. 2016, 59, 10329–10334. [Google Scholar] [CrossRef]
- Theeramunkong, S.; Caldarelli, A.; Massarotti, A.; Aprile, S.; Caprioglio, D.; Zaninetti, R.; Teruggi, A.; Pirali, T.; Grosa, G.; Tron, G.C.; et al. Regioselective Suzuki coupling of dihaloheteroaromatic compounds as a rapid strategy to synthesize potent rigid combretastatin analogues. J. Med. Chem. 2011, 54, 4977–4986. [Google Scholar] [CrossRef]
- Pirali, T.; Busacca, S.; Beltrami, L.; Imovilli, D.; Pagliai, F.; Miglio, G.; Massarotti, A.; Verotta, L.; Tron, G.C.; Sorba, G.; et al. Synthesis and cytotoxic evaluation of combretafurans, potential scaffolds for dual-action antitumoral agents. J. Med. Chem. 2006, 49, 5372–5376. [Google Scholar] [CrossRef]
- Assadieskandar, A.; Amini, M.; Ostad, S.N.; Riazi, G.H.; Cheraghi-Shavi, T.; Shafiei, B.; Shafiee, A. Design, synthesis, cytotoxic evaluation and tubulin inhibitory activity of 4-aryl-5-(3,4,5-trimethoxyphenyl)-2-alkylthio-1H-imidazole derivatives. Bioorg. Med. Chem. 2013, 21, 2703–2709. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, Q.; Bai, Z.; Sun, J.; Jiang, X.; Song, H.; Wu, Y.; Zhang, W. Synthesis and biological evaluation of 2,3-diarylthiophene analogues of combretastatin A-4. Med. Chem. Commun. 2015, 6, 971–976. [Google Scholar] [CrossRef]
- Simoni, D.; Grisolia, G.; Giannini, G.; Roberti, M.; Rondanin, R.; Piccagli, L.; Baruchello, R.; Rossi, M.; Romagnoli, R.; Invidiata, F.P.; et al. Heterocyclic and phenyl double-bond-locked combretastatin analogues possessing potent apoptosis-inducing activity in HL60 and in MDR cell lines. J. Med. Chem. 2005, 48, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Zhang, B.; Yang, H.K.; Li, Q.; Diao, P.C.; You, W.W.; Zhao, P.L. Design, synthesis, and biological evaluation of novel alkylsulfanyl-1,2,4-triazoles as cis-restricted combretastatin A-4 analogues. Eur. J. Med. Chem. 2017, 125, 1098–1106. [Google Scholar] [CrossRef] [PubMed]
- Subba Rao, A.V.; Swapna, K.; Shaik, S.P.; Lakshma Nayak, V.; Srinivasa Reddy, T.; Sunkari, S.; Shaik, T.B.; Bagul, C.; Kamal, A. Synthesis and biological evaluation of cis-restricted triazole/tetrazole mimics of combretastatin-benzothiazole hybrids as tubulin polymerization inhibitors and apoptosis inducers. Bioorg. Med. Chem. 2017, 25, 977–999. [Google Scholar] [CrossRef]
- Salehi, M.; Amini, M.; Ostad, S.N.; Riazi, G.H.; Assadieskandar, A.; Shafiei, B.; Shafiee, A. Synthesis, cytotoxic evaluation and molecular docking study of 2-alkylthio-4-(2,3,4-trimethoxyphenyl)-5-aryl-thiazoles as tubulin polymerization inhibitors. Bioorg. Med. Chem. 2013, 21, 7648–7654. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Yang, Z.; Liu, Y.; Ma, L.; Wu, Y.; He, L.; Shao, M.; Yu, K.; Wu, W.; Pu, Y.; et al. Synthesis and biological evaluation of diarylthiazole derivatives as antimitotic and antivascular agents with potent antitumor activity. Bioorg. Med. Chem. 2015, 23, 3337–3350. [Google Scholar] [CrossRef]
- Xu, Q.; Wang, Y.; Xu, J.; Sun, M.; Tian, H.; Zuo, D.; Guan, Q.; Bao, K.; Wu, Y.; Zhang, W. Synthesis and Bioevaluation of 3,6-Diaryl-[1,2,4]triazolo[4,3-b] Pyridazines as Antitubulin Agents. ACS Med. Chem. Lett. 2016, 7, 1202–1206. [Google Scholar] [CrossRef]
- Messaoudi, S.; Treguier, B.; Hamze, A.; Provot, O.; Peyrat, J.F.; De Losada, J.R.; Liu, J.M.; Bignon, J.; Wdzieczak-Bakala, J.; Thoret, S.; et al. Isocombretastatins a versus combretastatins a: The forgotten isoCA-4 isomer as a highly promising cytotoxic and antitubulin agent. J. Med. Chem. 2009, 52, 4538–4542. [Google Scholar] [CrossRef]
- Tréguier, B.; Hamze, A.; Provot, O.; Brion, J.-D.; Alami, M. Expeditious synthesis of 1,1-diarylethylenes related to isocombretastatin A-4 (isoCA-4) via palladium-catalyzed arylation of N-tosylhydrazones with aryl triflates. Tetrahedron Lett. 2009, 50, 6549–6552. [Google Scholar] [CrossRef]
- Stocker, V.; Ghinet, A.; Leman, M.; Rigo, B.; Millet, R.; Farce, A.; Desravines, D.; Dubois, J.; Waterlot, C.; Gautret, P. On the synthesis and biological properties of isocombretastatins: A case of ketone homologation during Wittig reaction attempts. Rsc Adv. 2013, 3, 3683–3696. [Google Scholar] [CrossRef]
- Aziz, J.; Brachet, E.; Hamze, A.; Peyrat, J.F.; Bernadat, G.; Morvan, E.; Bignon, J.; Wdzieczak-Bakala, J.; Desravines, D.; Dubois, J.; et al. Synthesis, biological evaluation, and structure-activity relationships of tri- and tetrasubstituted olefins related to isocombretastatin A-4 as new tubulin inhibitors. Org. Biomol. Chem. 2013, 11, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Rasolofonjatovo, E.; Provot, O.; Hamze, A.; Rodrigo, J.; Bignon, J.; Wdzieczak-Bakala, J.; Desravines, D.; Dubois, J.; Brion, J.D.; Alami, M. Conformationnally restricted naphthalene derivatives type isocombretastatin A-4 and isoerianin analogues: Synthesis, cytotoxicity and antitubulin activity. Eur. J. Med. Chem. 2012, 52, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Renko, D.; Provot, O.; Rasolofonjatovo, E.; Bignon, J.; Rodrigo, J.; Dubois, J.; Brion, J.D.; Hamze, A.; Alami, M. Rapid synthesis of 4-arylchromenes from ortho-substituted alkynols: A versatile access to restricted isocombretastatin A-4 analogues as antitumor agents. Eur. J. Med. Chem. 2015, 90, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Soussi, M.A.; Provot, O.; Bernadat, G.; Bignon, J.; Wdzieczak-Bakala, J.; Desravines, D.; Dubois, J.; Brion, J.D.; Messaoudi, S.; Alami, M. Discovery of azaisoerianin derivatives as potential antitumors agents. Eur. J. Med. Chem. 2014, 78, 178–189. [Google Scholar] [CrossRef]
- Rasolofonjatovo, E.; Provot, O.; Hamze, A.; Rodrigo, J.; Bignon, J.; Wdzieczak-Bakala, J.; Lenoir, C.; Desravines, D.; Dubois, J.; Brion, J.-D.; et al. Design, synthesis and anticancer properties of 5-arylbenzoxepins as conformationally restricted isocombretastatin A-4 analogs. Eur. J. Med. Chem. 2013, 62, 28–39. [Google Scholar] [CrossRef]
- Song, M.Y.; Cao, C.Y.; He, Q.R.; Dong, Q.M.; Li, D.; Tang, J.J.; Gao, J.M. Constructing novel dihydrofuran and dihydroisoxazole analogues of isocombretastatin-4 as tubulin polymerization inhibitors through [3+2] reactions. Bioorgan. Med. Chem. 2017, 25, 5290–5302. [Google Scholar] [CrossRef]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef]
- Simoni, D.; Romagnoli, R.; Baruchello, R.; Rondanin, R.; Rizzi, M.; Pavani, M.G.; Alloatti, D.; Giannini, G.; Marcellini, M.; Riccioni, T.; et al. Novel combretastatin analogues endowed with antitumor activity. J. Med. Chem. 2006, 49, 3143–3152. [Google Scholar] [CrossRef]
- Tang, J.J.; Fan, G.J.; Dai, F.; Ding, D.J.; Wang, Q.; Lu, D.L.; Li, R.R.; Li, X.Z.; Hu, L.M.; Jin, X.L.; et al. Finding more active antioxidants and cancer chemoprevention agents by elongating the conjugated links of resveratrol. Free Radic. Biol. Med. 2011, 50, 1447–1457. [Google Scholar] [CrossRef]
- Dong, S.; Tang, J.-J.; Zhang, C.-C.; Tian, J.-M.; Guo, J.-T.; Zhang, Q.; Li, H.; Gao, J.-M. Semisynthesis and in vitro cytotoxic evaluation of new analogues of 1-O-acetylbritannilactone, a sesquiterpene from Inula britannica. Eur. J. Med. Chem. 2014, 80, 71–82. [Google Scholar] [CrossRef]
- Tang, J.J.; Dong, S.; Han, Y.Y.; Lei, M.; Gao, J.M. Synthesis of 1-O-acetylbritannilactone analogues from Inula britannica and in vitro evaluation of their anticancer potential. Med. Chem. Comm. 2014, 5, 1584–1589. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd | IC50 (µM) 1 | Compd | IC50 (µM) |
|---|---|---|---|
| 5b | > 10 | 9 | > 10 |
| 6b-(Z) | 0.30 ± 0.05 | 10a | > 10 |
| 6b-(E) | 0.23 ± 0.07 | 10b | > 10 |
| 7a | > 10 | 11 | > 10 |
| 7b | > 10 | 12 | 3.67± 0.16 |
| 8a | > 10 | CA-4 | 0.013 ± 0.003 |
| 8b | > 10 | isoCA-4 | 0.017 ± 0.002 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, M.-Y.; He, Q.-R.; Wang, Y.-L.; Wang, H.-R.; Jiang, T.-C.; Tang, J.-J.; Gao, J.-M. Exploring Diverse-Ring Analogues on Combretastatin A4 (CA-4) Olefin as Microtubule-Targeting Agents. Int. J. Mol. Sci. 2020, 21, 1817. https://doi.org/10.3390/ijms21051817
Song M-Y, He Q-R, Wang Y-L, Wang H-R, Jiang T-C, Tang J-J, Gao J-M. Exploring Diverse-Ring Analogues on Combretastatin A4 (CA-4) Olefin as Microtubule-Targeting Agents. International Journal of Molecular Sciences. 2020; 21(5):1817. https://doi.org/10.3390/ijms21051817
Chicago/Turabian StyleSong, Ming-Yu, Qiu-Rui He, Yi-Lin Wang, Hao-Ran Wang, Tian-Cheng Jiang, Jiang-Jiang Tang, and Jin-Ming Gao. 2020. "Exploring Diverse-Ring Analogues on Combretastatin A4 (CA-4) Olefin as Microtubule-Targeting Agents" International Journal of Molecular Sciences 21, no. 5: 1817. https://doi.org/10.3390/ijms21051817
APA StyleSong, M.-Y., He, Q.-R., Wang, Y.-L., Wang, H.-R., Jiang, T.-C., Tang, J.-J., & Gao, J.-M. (2020). Exploring Diverse-Ring Analogues on Combretastatin A4 (CA-4) Olefin as Microtubule-Targeting Agents. International Journal of Molecular Sciences, 21(5), 1817. https://doi.org/10.3390/ijms21051817