1. Introduction
Neuronal synapses relay and transfer signals between cells and are key components of neural processing. The size of their sub-components (transmitter-filled vesicles, active zone, synaptic cleft, pre- and postsynaptic membranes) fall below the diffraction limits of visible light and thus conventional microscopy. Despite the advent of “superresolution” light microscopic techniques (e.g., direct stochastic optical reconstruction microscopy (dSTORM)), the possibility to visualize the biological membranes and the preservation of the cellular ultrastructure by transmission electron microscopy (TEM) remains unsurpassed [
1]. However, for an optimal imaging quality allowing the resolution of structures in the nanometer range, high requirements for tissue preservation have to be met. Aside from rare suitable brain biopsies or surgical specimens, histological studies of human central nervous system (CNS) diseases almost entirely rely on post mortem samples. Due to varying degrees of tissue degradation up until fixation, pathological changes can easily be obscured and achieving sufficient sample quality remains a challenging task [
2]. On the other hand, animal models allow perfect control over the modalities of sacrifice, perfusion, and tissue sampling to obtain the best ultrastructural quality. However, genetic animal models can only be obtained for diseases with known mutations that exclude disorders with complex modes of inheritance (e.g., essential tremor [
3]). Reprogramming of easily obtainable human cell types, such as fibroblasts, to either pluripotent stem cells [
4], followed by differentiation into neurons [
5] or to neural stem cells/neurons directly [
6,
7] allows the derivation of neurons from patients with a wide spectrum of diseases in vitro. Neurons in cell culture can readily be fixed and processed in ways suitable for TEM [
8]. We previously published a rather uncomplicated, straight forward protocol based on plasmid transfection that provides directly reprogrammed human induced neural stem cells (iNSC) [
9]. These cells can be cultured for many passages and differentiated into neurons or into astrocytes within one month. We, therefore, consider this protocol as a quite accessible way of obtaining neural cell types in vitro from healthy donors or patients. In this scientific study, we explored the ultrastructural preservation of neurons differentiated from iNSC under different culture conditions followed by fixation and TEM imaging of relevant neural structures. Furthermore, we compared different contrasting protocols in their ability to provide optimal visualization of the synaptic apparatus and other neuronal cell components.
2. Results
2.1. Differentiation of iNSC under Three Distinct Culture Conditions
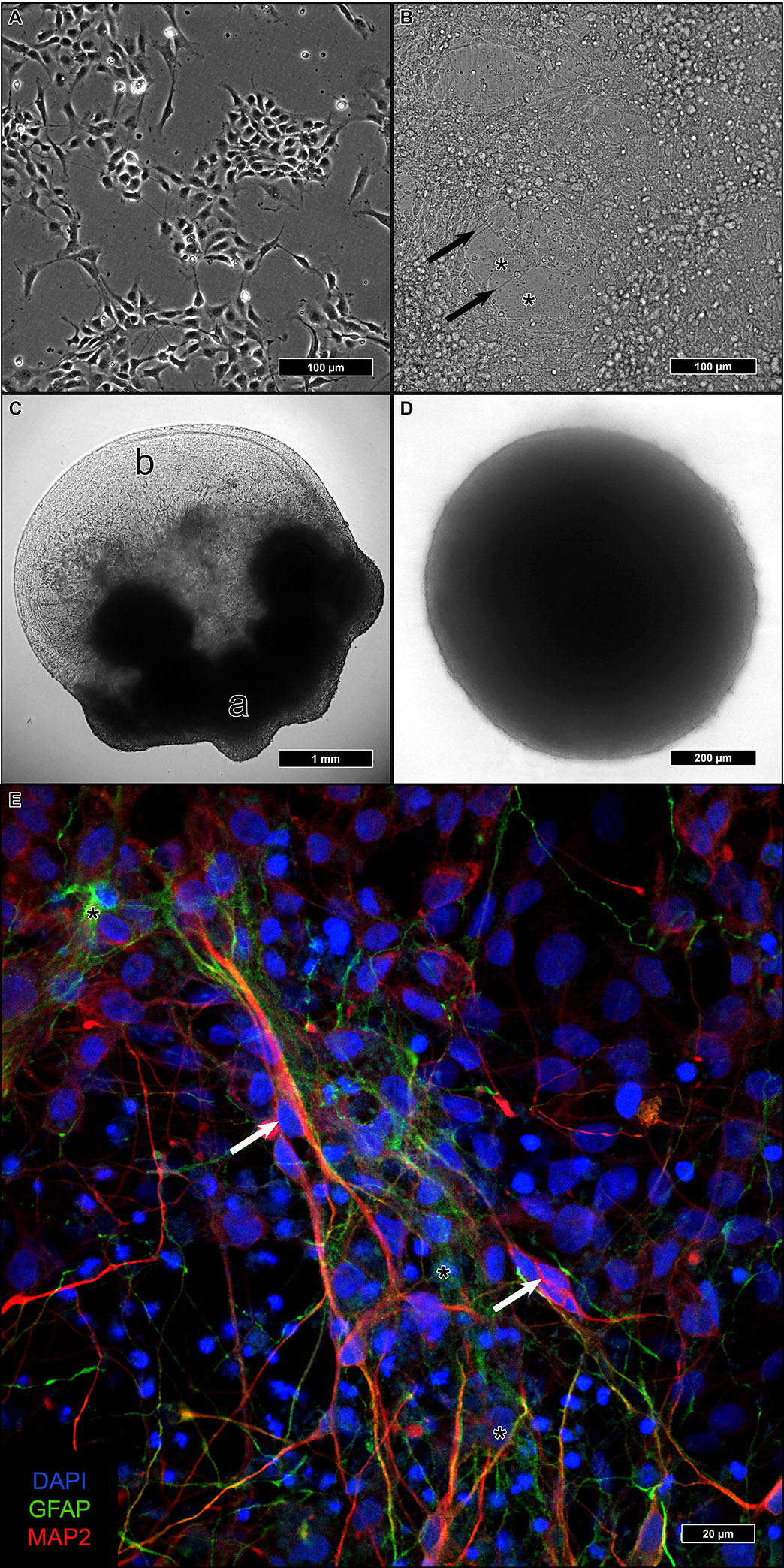
Differentiation of neural stem cells into adult neurons can be achieved under various culture conditions. We tested three of them to find out which one offered the best ultrastructural preservation for TEM.
Adherent differentiation on coated coverslips started from 2D-cultured iNSC (
Figure 1A). After reaching confluency, differentiation was initiated, and during the following four weeks, cells with a higher cytoplasma/nucleus ratio formed a basal layer with a dense network of neurite-sprouting cells on top (
Figure 1B). In a previous study, we identified the first type as astrocytes and the second as neurons [
9].
Aggregated iNSC in Matrigel remained at the embedding site and formed a dense fiber network spanning the entire droplet, which was several millimeters in size (
Figure 1C).
In u-shaped wells, iNSC aggregated within one to two weeks to a single compact sphere (
Figure 1D). After the addition of a differentiation medium, the size of the spheres remained constant. In contrast to the considerable size of the Matrigel droplets, individual neurospheres remained below 1 mm in diameter. No discernible changes in the spheres were visible upon differentiation.
After re-plating neurospheres on coated glass coverslips, attachment of the spheres and outgrowth of cells could be observed (
Figure 1E). Immunofluorescence could identify both microtubule-associated protein 2 (MAP2)-positive neurons as well as glial fibrillary acidic protein (GFAP)-positive astrocytes with a proportion of 5:1 (neurons:astrocytes).
2.2. Ultrastructural Preservation under Three Different Culture Conditions
INSC, differentiated under the described culture conditions, underwent fixation, contrastation, and embedding following standard protocols.
Cells on coverslips, despite remaining adherent during differentiation, tended to lift off during the preparation for TEM. When specimens were imaged by TEM, only processes containing intermediate filaments, and thus, most likely belonging to the astrocytic basal layer, remained (
Figure 2A,
Supplementary Figure S1). The ultrastructure was decently preserved, but no traces of neuronal cells (such as neurofilaments or synapses) could be found.
iNSC embedded and differentiated in Matrigel droplets exhibited inferior preservation of subcellular structures. Membranes and intracellular elements obviously had lost their integrity during sample processing for TEM and appeared fragmented (
Figure 2B).
The only culture condition that provided sufficient ultrastructural preservation for TEM analysis was the neurosphere culture. Neurospheres remained tightly packed during fixation and embedding. The high cell density inside the spheres allowed screening and visualization of a high number of features in a small area (
Figure 2C).
Since only neurospheres provided a sufficient ultrustructural quality after fixation and embedding, we settled on this differentiation method for further analyses.
2.3. Comparison of Three EM Preparation Protocols
Neuronal tissue staining for analysis of synaptic connections by electron microscopy requires optimal ultrastructural preservation in combination with strong deposition of heavy metal compounds into the biological membranes that outline neuronal processes, including axons and dendrites as well as synaptic vesicles. Besides combined primary fixation using a combination of glutaraldehyde and formaldehyde, several other parameters, including pH, osmolarity, and temperature of the washing buffer and primary fixative, are important for the success of ultrastructural preservation of neuronal tissues. In all three protocols, we used cacodylate buffer or phosphate buffer which have both been shown to be highly suitable for the preservation of neuronal tissue.
To enhance membrane contrast, standard staining protocols, including our standard-stain protocol, mostly used a combination of Osmium (Os) tetroxide (OsO4), and uranyl acetate (UA).
In the second protocol, we added the inorganic dye Ruthenium red (ammoniated ruthenium oxy-chloride) to OsO
4 to enhance the staining, as it has been shown that when used in combination the two compounds react to form ruthenium tetroxide, which reacts with several cellular components resulting in enhanced contrast of diverse tissues [
10,
11].
Our third protocol was based on a study published by Deerinck and colleagues (Deerinck et al., 2010), which was designed primarily to emphasize the contrast of cellular membranes for serial block-face electron microscopy. Our high contrast en-bloc staining protocol combined subsequent steps after primary aldehyde fixation, including ferrocyanide-reduced osmium tetroxide postfixation, thiocarbohydrazide-osmium liganding (OTO), and subsequent uranyl acetate and en bloc lead aspartate staining. As Ca2+ ions are known to enhance membrane preservation and staining, CaCl2 was included in a number of steps.
These three EM preparation protocols (standard stain, Ruthenium red stain, and high contrast en-bloc stain) were compared with respect to ultrastructural preservation and optimal visualization of biological membranes of the synaptic apparatus and other neuronal cell components.
All relevant subcellular structures were clearly discernible by all three protocols. However, unlike the neurospheres processed by the high contrast en-bloc stain, biological membranes were often not clearly visible in the neurospheres processed by the other two protocols (
Figure 3–panel 1). We noticed that the chromatin was weakly stained in the high contrast en-bloc stain-processed specimens, likely due to the presence of membrane enhancing reagents during sample preparation (
Figure 3–panel 1).
Detailed analysis of subcellular components in the differentially processed samples revealed that the continuity of nuclear envelope, and nuclear pores were preserved to a lesser extent in standard- or Ruthenium red stain-treated samples, whereas optimal preservation was achieved in the high contrast en-bloc stain-treated samples (
Figure 3–panel 2). Membranes were essentially parallel to each other and showed no breaks and nuclear pores were clearly visible. No artificial dilation of the intermembraneous space of the nuclear envelope (
Figure 3–panel 2), the rough endoplasmic reticulum (
Figure 3–panel 3), or the golgi apparatus (
Figure 3–panel 4) was seen in the high contrast en-bloc stain-treated samples compared to standard stain-treated samples. Dilation was seen to a lesser extent in Ruthenium red stain-treated samples (
Figure 3–panel 2, panel 3, panel 4). However, ribosomes of the outer nuclear membrane, rough endoplasmic reticulum as well as free ribosomes could only be seen clearly in standard- or Ruthenium red stain-treated samples (
Figure 3–panel 2, panel 3). Visualization of free ribosomes could be improved using post-staining of TEM sections with prolonged incubation with UAR (uranyl acetate replacement stain,
Supplementary Figure S2). Analysis of all samples processed according to the three different protocols revealed sufficient preservation of mitochondria with only mild shrinkage or swelling observed in the neurospheres (
Figure 3–panel 5). However, the double membranes and the cristae of mitochondria were only visible as continuous and undilated structures in the high contrast en-bloc stain-treated samples (
Figure 3–panel 5).
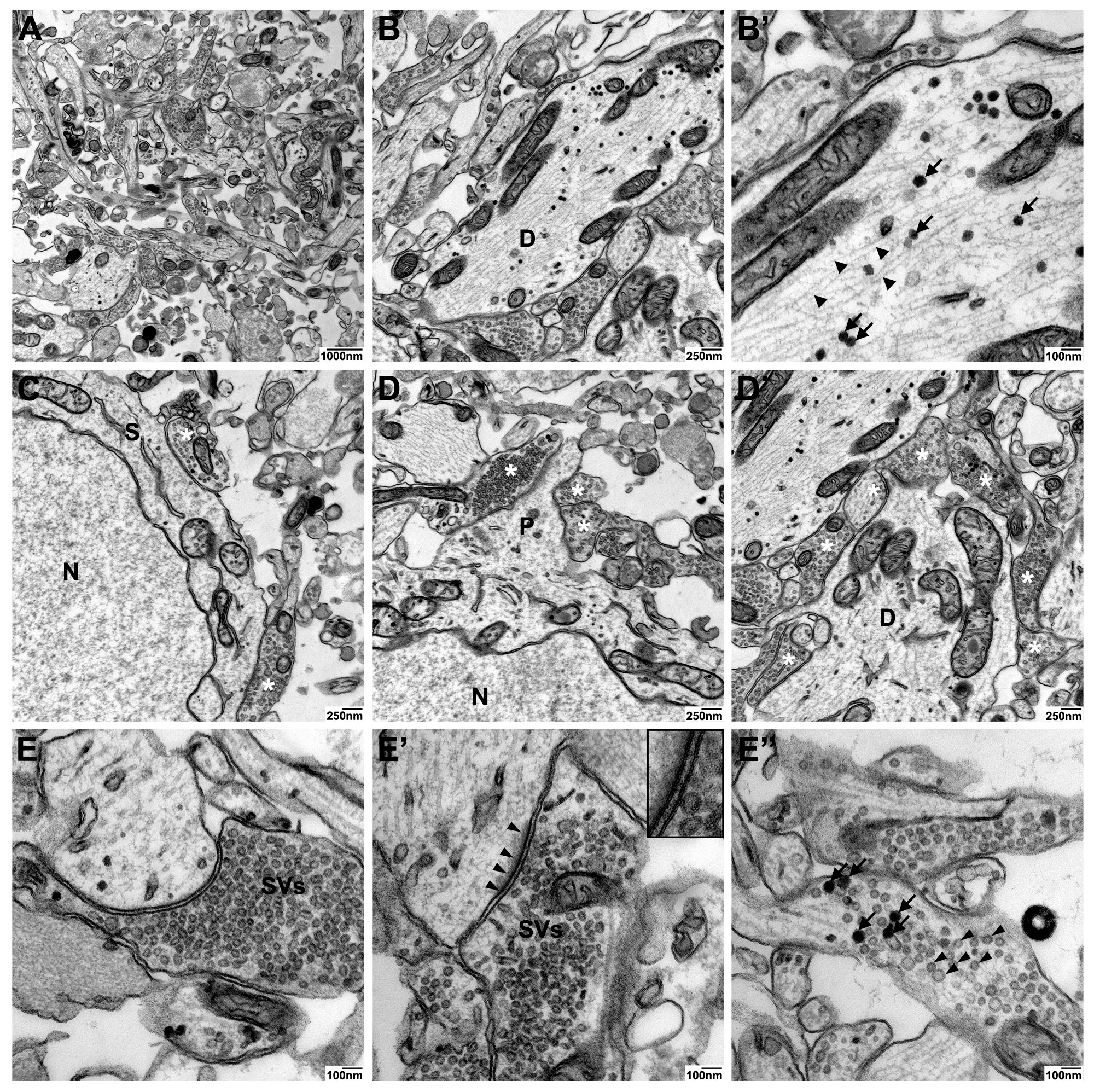
The particularly enhanced membrane contrast in high contrast en-bloc stain-treated neurospheres led to optimal preservation and discernability of axonal and dendritic processes in the neuropil (
Figure 4–panel 1, panel 2). In all three protocols, neurotubules were preserved, but neuronal membrane visualization was highly improved by high contrast en-bloc staining (
Figure 4–panel 2). In addition, this method offers high-quality ultrastructural preservation and excellent membrane staining of synaptic connections, although the postsynaptic density was stained less intensely (
Figure 5E’) compared to those processed with standard- or Ruthenium red stain (
Figure 4–panel 3). Since UA has been implied in labeling proteins of the postsynaptic density similar to heterochromatin, we speculate that the weaker staining of these structures in our high contrast en-bloc stain-processed neurospheres is a result of interference with other membrane enhancing reagents present during sample preparation.
Detailed analyses of neuronal and synaptic structures in high contrast en-bloc stain-processed neurospheres revealed high-quality ultrastructural preservation and excellent membrane staining of dendritic processes with neurofilaments, dense core vesicles as well as axosomatic and –dendritic synapses (
Figure 5A–D). A particular strength of the high contrast en-bloc stain was observed for visualizing subcomponents of the synaptic apparatus: The very clear membrane contrast made the pre- and postsynaptic membrane with the interjacent synaptic cleft easily discernible (
Figure 5E and E’). Different types of synaptic vesicles (clear vs. dense core), docked vesicles at the presynaptic membrance, or free vesicles of the resting pool (
Figure 5E–E’’) could be clearly visualized.
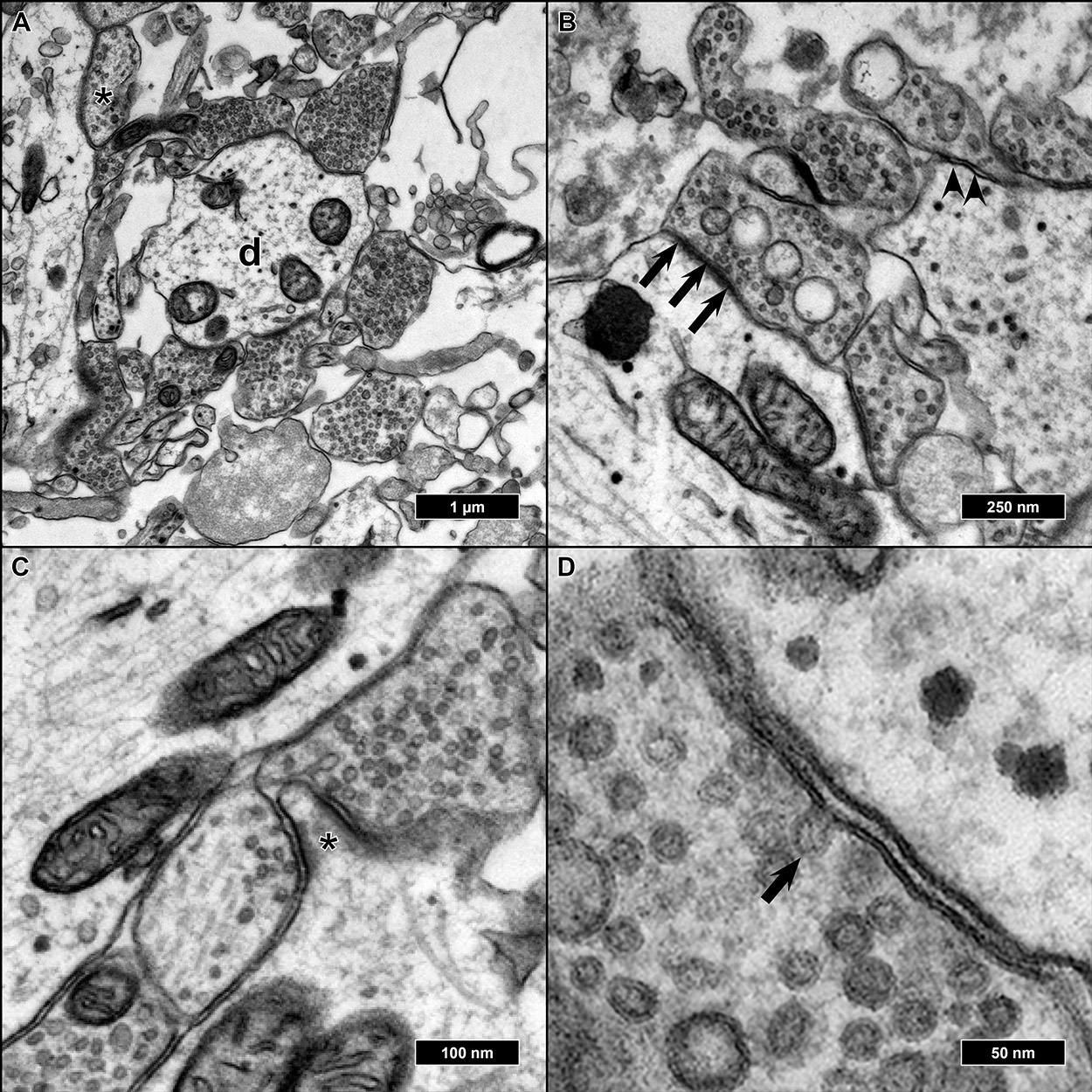
Features of advanced synaptic maturity could be observed in the analyzed samples: All synapses appeared as entirely or nearly filled with synaptic vesicles. We could never observe synapses with single or no vesicles inside (
Figure 6A). Of all synaptic contacts, 75% appeared as asymmetric synapses (postsynaptic membrane appearing thicker and more contrasted than the presynatic) (
Figure 6B). As already described, most synapses were either axo-dendritic or axo-somatic, sporadically synaptic contacts could also be found on protrusions from the main dendrite, presumably representing spines, but without smooth endoplasmatic reticulum present inside the protrusions (
Figure 6C). Occasionally, cells were fixed in the very moment of fusion between a synaptic vesicle and presynaptic membrane as a morphological correlate of synaptic transmission (
Figure 6D).
3. Discussion
For decades visualizing synaptic structures and studying their morphology has been a particular strength of the TEM. In the past, the general notion has been that synaptic transmission is highly conserved [
12] and thus was studied in a wider range of model organisms both invertebrate and vertebrate. However, the presence of distinctive features in the human neuromuscular synapse [
13] and striking differences in the postsynaptic human proteome in comparison to mouse [
14] challenge this notion. Therefore, there are obvious reasons to study synaptic neurotransmission in human neurons, but the aforementioned limitations of sufficiently preserved human CNS tissue challenge these endeavors. Reprogramming techniques can provide human neurons of healthy controls and patients with different kinds of diseases in vitro [
15]. After a sufficient time span of differentiation, a functional synaptic network is established [
9]. There have been studies in the past, which demonstrated the presence of synaptic connections between human neurons derived from reprogrammed stem cells by electron microscopy on a proof-of-principle basis [
8]. However, a systematic comparison of differentiation and TEM preparation protocols concerning optimal visualization of neuronal elements of these cells has not been performed.
The first (and until today, most employed) reprogramming paradigm is the reprogramming towards pluripotent stem cells [
4]. Most studies that had a more detailed look into the synapse and neuronal network formation of human neurons in vitro used this cell type [
16]. These cells and their pluripotency are maintained by complex and work-intensive protocols requiring almost daily media change and manual removal of spontaneously differentiated cells. For deriving mature neurons, multi-step protocols, combining neural induction, regional patterning, and terminal differentiation, have to be followed that can easily take several months [
15]. While this allows a certain enrichment of desired cell types, a 100% pure cell type is never achieved. Adherent differentiation on coated coverslips has been the standard approach leading to immature synapse formation after less than one week and spontaneous synaptic activity after roughly one month [
17]. However, a full maturity (e.g., formation of synapses on spines) is possibly not achieved. Neuralizing and differentiating pluripotent stem cells as 3D aggregates (organoids) results in a maturation over months and is more likely to result in a mature synaptic network [
18]. In our personal opinion, a culture technique requiring many months until specimens can be studied poses severe challenges to planning and performing experiments (especially if replicates are required).
The alternative is the generation of induced neurons (iN) from somatic cells: Overexpressing transcription factors associated with neuronal identity can reprogram non-neural cells to neurons [
19]. The time needed for the reprogramming process to be completed is of similar length as the time needed for neural differentiation from induced pluripotent stem cells (iPSC) [
20]. However, since iN are post-mitotic, the number derived is rather small and cannot be increased by proliferation. Thus, the reprogramming process has to be repeated for every set of new experiments.
A good compromise, in our opinion, is induced neural stem cells (iNSC) employed in this study: Derived from somatic cells by plasmid-based transfection, they are proliferative for at least 25 passages but cultivating them requires much less intervention and they are not prone to spontaneous differentiation [
9,
21]. Differentiation is simply initiated by a change in cell culture media and the addition of three recombinant growth factors and one small molecule. The timing until acquiring a certain level of maturity is not different from the other two methods described. The biggest disadvantage is that these cells are not responsive to patterning cues, meaning their regional identity cannot be altered. They exhibit a quite stable mix of neuronal subtypes (60% upper layer cortical layer, 20% GABAergic, 20% dopaminergic). Therefore, they might not be the first choice when it comes to obtaining a specific cell type, but their strength lies more in providing a good mixture of different neuronal subtypes in one dish.
Another important question in this context is the maturity and functionality of the synaptic network derived from reprogrammed stem cells. Neurons derived from iPSC might appear mature by morphology or specific protein expression after a couple of weeks in culture, yet the formation of functional networks usually takes more than a month, and still not all electrophysiological features associated with them might be present [
16]. The same seems to apply to synaptic contacts. Morphologically, synapses begin their existence as mere contacts between two neuronal membranes. Later synaptic vesicles fill the presynaptic bouton, the pre- and postsynaptic membrane becomes more and more defined (resulting in an increasing number of asymmetric synapses with a thicker postsynaptic membrane) and an active synapse capable of neurotransmitter release by fusion of the synaptic vesicles with the presynaptic membrane has come into existence [
22,
23]. Certain neurons form dendritic spines, highly dynamic structures for multiple synaptic contacts [
24]. The timing and sequence of synaptogenesis have already been studied in the human fetus decades ago, but only recently it has been acknowledged that neurons derived from human stem cells in vitro exhibit a different timing in synaptogenesis and certain features associated with synaptic maturity (e.g., spine formation) might be absent altogether [
17]. As usual, when dealing with stem cell-derived neurons, much is dependent on the individual protocol employed. The neurospheres from iNSC we employed in our study exhibited a particularly mature phenotype: The majority of synaptic boutons were densely filled with synaptic vesicles, and the majority exhibited an asymmetric morphology and due to the superior membrane contrast of the high contrast en-bloc stain, individual fusions between vesicles and the presynaptic membrane could be observed. We even observed membrane protrusions that could be dendritic spines, a feature that has only be observed in iPSC derived cerebral organoids after many months in culture [
25]. However, concerning this finding, uncertainty remained. We did not perform serial block-face scanning electron microscopy for three-dimensional reconstruction of the protrusions, which is the method of choice for unequivocally visualizing dendritic spines. Furthermore, we did not find a spine apparatus (smooth endoplasmatic reticulum inside the spine) in any protrusion. Not all spines contain a synaptic apparatus, but the presence of it is generally considered a sign of maturity [
26]. We would, therefore, consider the presence of possibly still immature spines with no signs of full maturity.
The iNSC employed in this study were cultured adherently on Matrigel. In a previous publication, we induced differentiation of iNSC into mature neurons on glass coverslips as it simplified staining and fluorescence microscopy, transfer to recording chambers, etc. [
9]. As we observed in the past, iNSC generated using our protocol differentiate into both neurons and astrocytes with the neurons exhibiting the tendency to aggregate on top of the astrocytes (
Figure 1B). Presumably, the numerous pipetting steps leading to repeated shear stress resulted in a detachment of the neuronal layer leaving only the astrocytic basal layer.
Aggregating iNSC into neurospheres and embedding them into Matrigel droplets followed a protocol similar to the formation of cerebral organoids [
18]. However, instead of forming a compact neuroepithelial layer with ventricle-like cavities, differentiating iNSC remained stationary and extended neurites throughout the aggregates. In contrast to iPSC-derived embryoid bodies that served as seeds for the cerebral organoids, our iNSC were different from early neuroepithelial precursors. We can only speculate that the comparatively large size of the aggregates of a few millimeters in conjunction with being mainly composed of a neurite fiber network, led to insufficient preservation for TEM.
Only the densely packed and rather small neurospheres reliably provided a sufficient ultrastructure. Neurospheres were generated by culturing iNSC under growth conditions until single spheres formed [
27,
28]. The growth of the spheres ceased after the induction of differentiation. Thus, the size of the spheres was mainly determined by the cell number seeded per well. This provided a good compromise between a small size allowing sufficient penetration of the fixatives and solutions during the embedding process and safe handling under the naked eye. Furthermore, the small size, yet a high density of cell bodies and processes, simplified screening during TEM analysis for relevant structures.
Besides the ultrastructural preservation, reliably identifying structures of interest by TEM requires high contrast with a sharp delineation. Neuronal structures that usually receive the most attention in TEM studies are either membrane-rich as synapses and mitochondria or filamentous, such as neurofilaments or neurotubuli. The standard- and Ruthenium red-stainings appeared “grainy”, and the contours of membranes were sometimes hard to discriminate from surrounding structures. A better contrast was achieved for structures rich in DNA or RNA (nucleus, ribosomes) or proteins (filaments and tubuli). The high contrast en-block stain was primarily designed for enhancing membrane contrast in mammalian tissue in serial block-face scanning electron microscopy (SBF-SEM) [
29]. Although developed for a different EM method, preserving and contrasting membranes proved useful for TEM as well. En-bloc contrasting protocols were optimized for better penetration of larger tissue samples and superseded the contrasting of ultra-thin cut sections on grids which is prone to non-specific deposition of the contrasting heavy metals [
30]. In contrast to the other two staining protocols tested here, CaCl
2 (which is known to improve the stability of lipid bilayers [
31]) was added to the fixative as well as to some of the contrasting steps. The increased contrast of membranes with this protocol was the result of a combination of different staining principles established in numerous individual studies: Ferrocyanide reduced osmium post-fixation in combination with cacodylate buffer, partially extracted the cytoplasmatic ground substance and mitochondrial matrix while strongly binding to membranes [
32]. Insufficient preservation of the lipid bilayer structure by the aforementioned method was compensated by a downstream osmium–thiocarbohydrazide–osmium (OTO) step [
33]. Using the classical contrasting agent uranyl–acetate (highly toxic and nowadays replaced by samarium and gadolinium [
34]) and lead aspartate in pre-embedding en-bloc staining of wet tissue overcame their tendency to form contaminating precipitates when applied post-embedding [
35,
36].
To our knowledge, this is the first systematic comparison of different protocols for the visualization of neurons derived from human reprogrammed cells by TEM and the first example of en-bloc staining techniques employed on three-dimensional cellular aggregates. Consequently, the paramount properties of en-bloc staining in the preservation and contrasting of neuronal membranes work for in vitro specimens as well.

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





