PARP Inhibition Increases the Reliance on ATR/CHK1 Checkpoint Signaling Leading to Synthetic Lethality—An Alternative Treatment Strategy for Epithelial Ovarian Cancer Cells Independent from HR Effectiveness

 , , , , and
, , , , and
Abstract
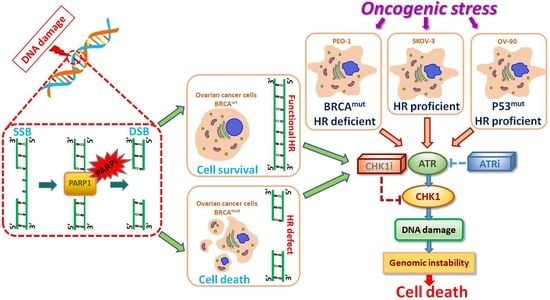
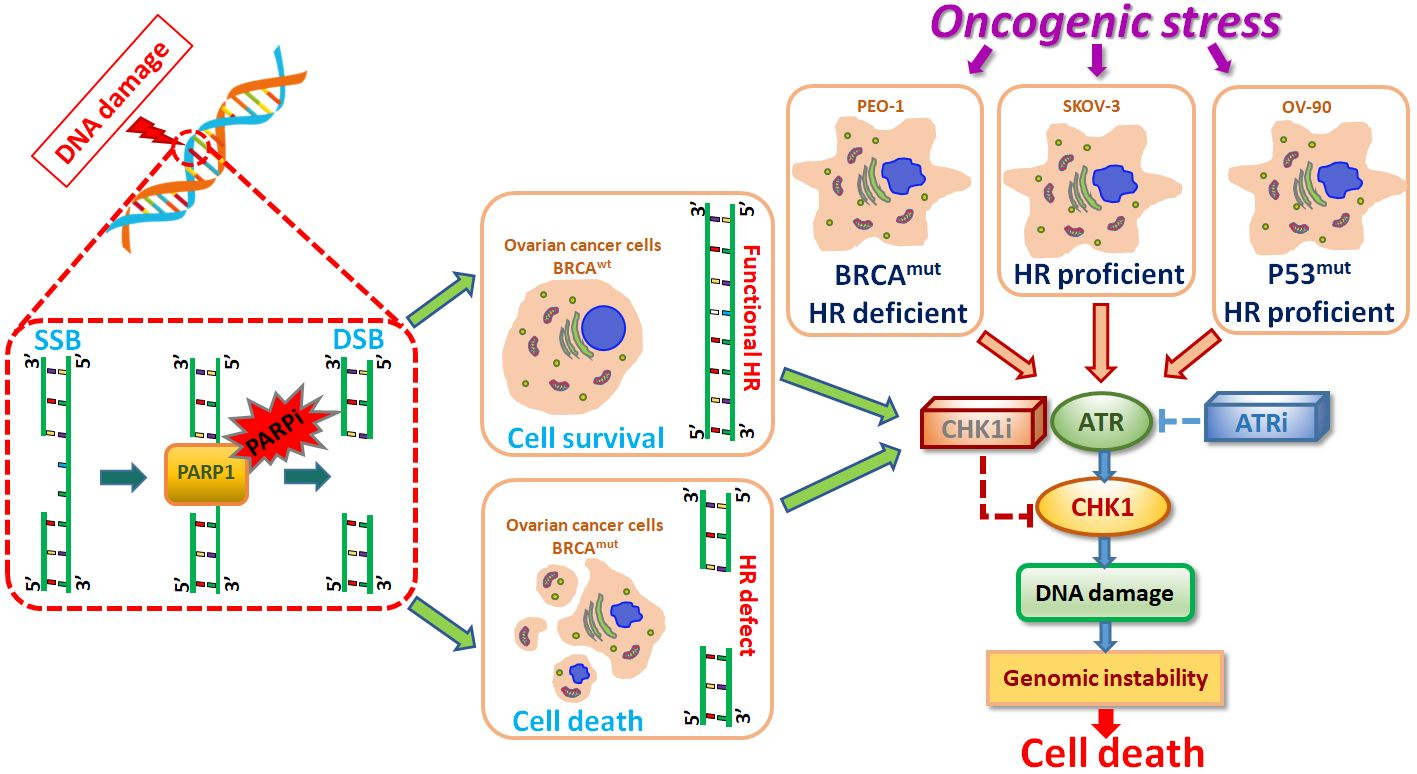{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. PARP Inhibition Is Not Sufficient to Kill Ovarian Cancer Cells, but Acts Synergistically with CHK1 or ATR Inhibition
2.2. PARPi in Combination with CHK1i or ATRi Increases DNA Damage in BRCAMUT and BRCAWT Ovarian Cells
2.3. Combined Treatment Induces Higher Levels of Apoptosis than Monotherapy in EOC Cells
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture and Drug Administration
4.3. MTT Assay
4.4. Clonogenic Assay
4.5. Morphological Assessment
4.6. Western Blot Analysis
4.7. Comet Assay
4.8. Caspase 3/7 Assay
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADP | Adenosine diphosphate |
| ATM | Ataxia telangiectasia mutated protein |
| ATR | Ataxia telangiectasia and Rad3-related protein |
| ATRi | Ataxia telangiectasia and Rad3-related protein inhibitor |
| BSA | Bovine serum albumin |
| CDI | Coefficient of drug interaction |
| CHK1 | Checkpoint kinase 1 |
| CHK1i | Checkpoint kinase 1 inhibitor |
| DAPI | 4,6-diamidino-2-phenylindole |
| DSB | Double-strand break |
| FANCI | Fanconi anemia complementation group I |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| HRR | Homologous recombination repair |
| MCM2 | DNA replication licensing factor |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide |
| PARP | Poly (ADP-ribose) polymerase |
| PARPi | Poly (ADP-ribose) polymerase inhibitor |
| PMSF | Phenylmethylsulfonyl fluoride |
| PVDF | Polyvinylidene difluoride |
| RPA | Replication protein A |
| SMARCAL1 | SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily A-like protein 1 |
| SSB | Single-strand break |
| ssDNA | single stranded DNA |
| WRN | Werner syndrome ATP-dependent helicase |
References
- He, S.; Zhang, C.; Liu, L.; Li, L.; Duan, Y.; Liu, J.; Shao, F. Efficacy and safety of PARP inhibitors as the maintenance therapy in ovarian cancer: A meta-analysis of nine randomized controlled trials. Biosci. Rep. 2020, 40. [Google Scholar] [CrossRef]
- Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [CrossRef] [PubMed]
- Buttarelli, M.; De Donato, M.; Raspaglio, G.; Babini, G.; Ciucci, A.; Martinelli, E.; Baccaro, P.; Pasciuto, T.; Fagotti, A.; Scambia, G.; et al. Clinical Value of lncRNA MEG3 in High-Grade Serous Ovarian Cancer. Cancers 2020, 12, 966. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Ceccaldi, R.; Shapiro, G.I.; D’Andrea, A.D. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov. 2015, 5, 1137–1154. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.C.; Hwang, S.; Kim, S.; Jung, S.G.; Park, H.; Joo, W.D.; Song, S.H.; Lee, C.; Kim, T.-H.; Kang, H.; et al. Clinical Impact of Somatic Variants in Homologous Recombination Repair-Related Genes in Ovarian High-Grade Serous Carcinoma. Cancer Res. Treat. 2020, 52, 634–644. [Google Scholar] [CrossRef] [PubMed]
- Martí, J.M.; Fernández-Cortés, M.; Serrano-Sáenz, S.; Zamudio-Martinez, E.; Delgado-Bellido, D.; Garcia-Diaz, A.; Oliver, F.J. The Multifactorial Role of PARP-1 in Tumor Microenvironment. Cancers 2020, 12, 739. [Google Scholar] [CrossRef]
- Bohrer, R.C.; Dicks, N.; Gutierrez, K.; Duggavathi, R.; Bordignon, V. Double-strand DNA breaks are mainly repaired by the homologous recombination pathway in early developing swine embryos. FASEB J. 2018, 32, 1818–1829. [Google Scholar] [CrossRef]
- Spriggs, D.R.; Longo, D.L. Progress in BRCA-Mutated Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2567–2568. [Google Scholar] [CrossRef]
- Sánchez, H.; Paul, M.W.; Grosbart, M.; Van Rossum-Fikkert, S.E.; Lebbink, J.H.G.; Kanaar, R.; Houtsmuller, A.B.; Wyman, C. Architectural plasticity of human BRCA2–RAD51 complexes in DNA break repair. Nucleic Acids Res. 2017, 45, 4507–4518. [Google Scholar] [CrossRef]
- Rajawat, J.; Shukla, N.; Mishra, D.P. Therapeutic Targeting of Poly(ADP-Ribose) Polymerase-1 (PARP1) in Cancer: Current Developments, Therapeutic Strategies, and Future Opportunities. Med. Res. Rev. 2017, 37, 1461–1491. [Google Scholar] [CrossRef]
- Tinker, A.V.; Gelmon, K. The role of PARP inhibitors in the treatment of ovarian carcinomas. Curr. Pharm. Des. 2012, 18, 3770–3774. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Ison, G.; McKee, A.E.; Zhang, H.; Tang, S.; Gwise, T.; Sridhara, R.; Lee, E.; Tzou, A.; Philip, R.; et al. FDA Approval Summary: Olaparib Monotherapy in Patients with Deleterious Germline BRCA-Mutated Advanced Ovarian Cancer Treated with Three or More Lines of Chemotherapy. Clin. Cancer Res. 2015, 21, 4257–4261. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef] [PubMed]
- Consortium, A.P.G. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef] [PubMed]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Drean, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat. Commun. 2018, 9, 1849. [Google Scholar] [CrossRef]
- Montemorano, L.; Lightfoot, M.; Bixel, K. Role of Olaparib as Maintenance Treatment for Ovarian Cancer: The Evidence to Date. OncoTargets Ther. 2019, 12, 11497–11506. [Google Scholar] [CrossRef]
- Engelke, C.G.; Parsels, L.A.; Qian, Y.; Zhang, Q.; Karnak, D.; Robertson, J.R.; Tanska, D.M.; Wei, D.; Davis, M.A.; Parsels, J.D.; et al. Sensitization of Pancreatic Cancer to Chemoradiation by the Chk1 Inhibitor MK8776. Clin. Cancer Res. 2013, 19, 4412–4421. [Google Scholar] [CrossRef]
- Gupta, D.; Lin, B.; Cowan, A.; Heinen, C.D. ATR-Chk1 activation mitigates replication stress caused by mismatch repair-dependent processing of DNA damage. Proc. Natl. Acad. Sci. USA 2018, 115, 1523–1528. [Google Scholar] [CrossRef]
- Menolfi, D.; Zha, S. ATM, ATR and DNA-PKcs kinases—The lessons from the mouse models: Inhibition ≠ deletion. Cell Biosci. 2020, 10. [Google Scholar] [CrossRef]
- Saldivar, J.C.; Cortez, D.; Cimprich, K.A. The essential kinase ATR: Ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 2017, 18, 622–636. [Google Scholar] [CrossRef]
- Zhang, X.; Lu, X.; Akhter, S.; Georgescu, M.-M.; Legerski, R.J. FANCI is a negative regulator of Akt activation. Cell Cycle 2016, 15, 1134–1143. [Google Scholar] [CrossRef] [PubMed]
- Yeom, G.; Kim, J.; Park, C.-J. Investigation of the core binding regions of human Werner syndrome and Fanconi anemia group J helicases on replication protein A. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef]
- Pugliese, G.M.; Salaris, F.; Palermo, V.; Marabitti, V.; Morina, N.; Rosa, A.; Franchitto, A.; Pichierri, P. Inducible SMARCAL1 knockdown in iPSC reveals a link between replication stress and altered expression of master differentiation genes. Dis. Models Mech. 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-J.; Min, A.; Im, S.-A.; Jang, H.; Lee, K.H.; Lau, A.; Lee, M.; Kim, S.; Yang, Y.; Kim, J.; et al. Anti-tumor activity of the ATR inhibitor AZD6738 in HER2 positive breast cancer cells. Int. J. Cancer 2017, 140, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Gamper, A.M.; Rofougaran, R.; Watkins, S.C.; Greenberger, J.S.; Beumer, J.H.; Bakkenist, C.J. ATR kinase activation in G1 phase facilitates the repair of ionizing radiation-induced DNA damage. Nucleic Acids Res. 2013, 41, 10334–10344. [Google Scholar] [CrossRef] [PubMed]
- Huntoon, C.J.; Flatten, K.S.; Wahner Hendrickson, A.E.; Huehls, A.M.; Sutor, S.L.; Kaufmann, S.H.; Karnitz, L.M. ATR Inhibition Broadly Sensitizes Ovarian Cancer Cells to Chemotherapy Independent of BRCA Status. Cancer Res. 2013, 73, 3683–3691. [Google Scholar] [CrossRef] [PubMed]
- Reaper, P.M.; Griffiths, M.R.; Long, J.M.; Charrier, J.-D.; MacCormick, S.; Charlton, P.A.; Golec, J.M.C.; Pollard, J.R. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat. Chem. Biol. 2011, 7, 428–430. [Google Scholar] [CrossRef]
- Kawahara, N.; Ogawa, K.; Nagayasu, M.; Kimura, M.; Sasaki, Y.; Kobayashi, H. Candidate synthetic lethality partners to PARP inhibitors in the treatment of ovarian clear cell cancer. Biomed. Rep. 2017, 7, 391–399. [Google Scholar] [CrossRef][Green Version]
- Ciardo, D.; Goldar, A.; Marheineke, K. On the Interplay of the DNA Replication Program and the Intra-S Phase Checkpoint Pathway. Genes 2019, 10, 94. [Google Scholar] [CrossRef]
- Rogalska, A.; Gajek, A.; Marczak, A. Epothilone B induces extrinsic pathway of apoptosis in human SKOV-3 ovarian cancer cells. Toxicol. Vitr. 2014, 28, 675–683. [Google Scholar] [CrossRef]
- Kampan, N.C.; Madondo, M.T.; McNally, O.M.; Quinn, M.; Plebanski, M. Paclitaxel and Its Evolving Role in the Management of Ovarian Cancer. BioMed Res. Int. 2015, 2015, 1–21. [Google Scholar] [CrossRef] [PubMed]
- De Donato, M.; Righino, B.; Filippetti, F.; Battaglia, A.; Petrillo, M.; Pirolli, D.; Scambia, G.; De Rosa, M.C.; Gallo, D. Identification and antitumor activity of a novel inhibitor of the NIMA-related kinase NEK6. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Lin, Z.P.; Zhu, Y.-L.; Lo, Y.-C.; Moscarelli, J.; Xiong, A.; Korayem, Y.; Huang, P.H.; Giri, S.; LoRusso, P.; et al. Combination of triapine, olaparib, and cediranib suppresses progression of BRCA-wild type and PARP inhibitor-resistant epithelial ovarian cancer. PLoS ONE 2018, 13. [Google Scholar] [CrossRef]
- Hjortkjær, M.; Malik Aagaard Jørgensen, M.; Waldstrøm, M.; Ørnskov, D.; Søgaard-Andersen, E.; Jakobsen, A.; Dahl-Steffensen, K. The clinical importance of BRCAness in a population-based cohort of Danish epithelial ovarian cancer. Int. J. Gynecol. Cancer 2019, 29, 166–173. [Google Scholar] [CrossRef]
- Baloch, T.; López-Ozuna, V.M.; Wang, Q.; Matanis, E.; Kessous, R.; Kogan, L.; Yasmeen, A.; Gotlieb, W.H. Sequential therapeutic targeting of ovarian Cancer harboring dysfunctional BRCA1. BMC Cancer 2019, 19. [Google Scholar] [CrossRef]
- Kim, W.; Zhao, F.; Wu, R.; Qin, S.; Nowsheen, S.; Huang, J.; Zhou, Q.; Chen, Y.; Deng, M.; Guo, G.; et al. ZFP161 regulates replication fork stability and maintenance of genomic stability by recruiting the ATR/ATRIP complex. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- George, E.; Kim, H.; Krepler, C.; Wenz, B.; Makvandi, M.; Tanyi, J.L.; Brown, E.; Zhang, R.; Brafford, P.; Jean, S.; et al. A patient-derived-xenograft platform to study BRCA-deficient ovarian cancers. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Kim, H.; George, E.; Ragland, R.L.; Rafail, S.; Zhang, R.; Krepler, C.; Morgan, M.A.; Herlyn, M.; Brown, E.J.; Simpkins, F. Targeting the ATR/CHK1 Axis with PARP Inhibition Results in Tumor Regression in BRCA-Mutant Ovarian Cancer Models. Clin. Cancer Res. 2017, 23, 3097–3108. [Google Scholar] [CrossRef]
- Kim, H.; Xu, H.; George, E.; Hallberg, D.; Kumar, S.; Jagannathan, V.; Medvedev, S.; Kinose, Y.; Devins, K.; Verma, P.; et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef]
- Burgess, B.T.; Anderson, A.M.; McCorkle, J.R.; Wu, J.; Ueland, F.R.; Kolesar, J.M. Olaparib Combined with an ATR or Chk1 Inhibitor as a Treatment Strategy for Acquired Olaparib-Resistant BRCA1 Mutant Ovarian Cells. Diagnostics 2020, 10, 121. [Google Scholar] [CrossRef]
- Jette, N.R.; Kumar, M.; Radhamani, S.; Arthur, G.; Goutam, S.; Yip, S.; Kolinsky, M.; Williams, G.J.; Bose, P.; Lees-Miller, S.P. ATM-Deficient Cancers Provide New Opportunities for Precision Oncology. Cancers 2020, 12, 687. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, R.; Falenta, K.; Wijnhoven, P.W.; Chabbert, C.; Stott, J.; Yates, J.; Lau, A.Y.; Young, L.A.; Hollingsworth, S.J. Abstract 337: The PARP inhibitor olaparib is synergistic with the ATR inhibitor AZD6738 in ATM deficient cancer cells. In Proceedings of the Molecular and Cellular Biology/Genetics, American Association for Cancer Research (AACR), Chicago, IL, USA, 14–18 April 2018; Volume 78, p. 337. [Google Scholar]
- Brill, E.; Yokoyama, T.; Nair, J.; Yu, M.; Ahn, Y.-R.; Lee, J.-M. Prexasertib, a cell cycle checkpoint kinases 1 and 2 inhibitor, increases in vitro toxicity of PARP inhibition by preventing Rad51 foci formation in BRCA wild type high-grade serous ovarian cancer. Oncotarget 2017, 8, 111026–111040. [Google Scholar] [CrossRef] [PubMed]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Parsels, L.A.; Karnak, D.; Parsels, J.D.; Zhang, Q.; Vélez-Padilla, J.; Reichert, Z.R.; Wahl, D.R.; Maybaum, J.; O’Connor, M.J.; Lawrence, T.S.; et al. PARP1 Trapping and DNA Replication Stress Enhance Radiosensitization with Combined WEE1 and PARP Inhibitors. Mol. Cancer Res. 2018, 16, 222–232. [Google Scholar] [CrossRef]
- Osoegawa, A.; Gills, J.J.; Kawabata, S.; Dennis, P.A. Rapamycin sensitizes cancer cells to growth inhibition by the PARP inhibitor olaparib. Oncotarget 2017, 8, 87044–87053. [Google Scholar] [CrossRef]
- Bianchi, A.; Lopez, S.; Altwerger, G.; Bellone, S.; Bonazzoli, E.; Zammataro, L.; Manzano, A.; Manara, P.; Perrone, E.; Zeybek, B.; et al. PARP-1 activity (PAR) determines the sensitivity of cervical cancer to olaparib. Gynecol. Oncol. 2019, 155, 144–150. [Google Scholar] [CrossRef]
- Parmar, K.; Kochupurakkal, B.S.; Lazaro, J.-B.; Wang, Z.C.; Palakurthi, S.; Kirschmeier, P.T.; Yang, C.; Sambel, L.A.; Färkkilä, A.; Reznichenko, E.; et al. The CHK1 Inhibitor Prexasertib Exhibits Monotherapy Activity in High-Grade Serous Ovarian Cancer Models and Sensitizes to PARP Inhibition. Clin. Cancer Res. 2019, 25, 6127–6140. [Google Scholar] [CrossRef]
- Szczepanska, J.; Poplawski, T.; Synowiec, E.; Pawlowska, E.; Chojnacki, C.J.; Chojnacki, J.; Blasiak, J. 2-Hydroxylethyl methacrylate (HEMA), a tooth restoration component, exerts its genotoxic effects in human gingival fibroblasts trough methacrylic acid, an immediate product of its degradation. Mol. Biol. Rep. 2011, 39, 1561–1574. [Google Scholar] [CrossRef]
- Angius, G.; Tomao, S.; Stati, V.; Vici, P.; Bianco, V.; Tomao, F. Prexasertib, a checkpoint kinase inhibitor: From preclinical data to clinical development. Cancer Chemother. Pharmacol. 2019, 85, 9–20. [Google Scholar] [CrossRef]
- Liu, Y.; Burness, M.L.; Martin-Trevino, R.; Guy, J.; Bai, S.; Harouaka, R.; Brooks, M.D.; Shang, L.; Fox, A.; Luther, T.K.; et al. RAD51 Mediates Resistance of Cancer Stem Cells to PARP Inhibition in Triple-Negative Breast Cancer. Clin. Cancer Res. 2017, 23, 514–522. [Google Scholar] [CrossRef]
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, R.A.; Chan, C.S. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2017, 25, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Karnitz, L.M.; Zou, L. Molecular Pathways: Targeting ATR in Cancer Therapy. Clin. Cancer Res. 2015, 21, 4780–4785. [Google Scholar] [CrossRef] [PubMed]
- Vance, S.; Liu, E.; Zhao, L.; Parsels, J.D.; Parsels, L.A.; Brown, J.L.; Maybaum, J.; Lawrence, T.S.; Morgan, M.A. Selective radiosensitization of p53 mutant pancreatic cancer cells by combined inhibition of Chk1 and PARP1. Cell Cycle 2014, 10, 4321–4329. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Liu, C.; Tao, Z.; Wang, M.; Jia, Y.; Sang, X.; Shen, L.; Xue, Y.; Jiang, K.; Luo, F.; et al. MYC status as a determinant of synergistic response to Olaparib and Palbociclib in ovarian cancer. EBioMedicine 2019, 43, 225–237. [Google Scholar] [CrossRef]
- Wang, D.; Li, C.; Zhang, Y.; Wang, M.; Jiang, N.; Xiang, L.; Li, T.; Roberts, T.M.; Zhao, J.J.; Cheng, H.; et al. Combined inhibition of PI3K and PARP is effective in the treatment of ovarian cancer cells with wild-type PIK3CA genes. Gynecol. Oncol. 2016, 142, 548–556. [Google Scholar] [CrossRef]
- Mitchell, C.; Park, M.; Eulitt, P.; Yang, C.; Yacoub, A.; Dent, P. Poly(ADP-Ribose) Polymerase 1 Modulates the Lethality of CHK1 Inhibitors in Carcinoma Cells. Mol. Pharmacol. 2010, 78, 909–917. [Google Scholar] [CrossRef]
- Zhou, J.; Yang, F.; Zhou, L.; Wang, J.-g.; Wen, P.; Luo, H.; Li, W.; Song, Z.; Sharman, E.H.; Bondy, S.C. Dietary melatonin attenuates age-related changes in morphology and in levels of key proteins in globus pallidus of mouse brain. Brain Res. 2014, 1546, 1–8. [Google Scholar] [CrossRef]
- Stingele, J.; Bellelli, R.; Alte, F.; Hewitt, G.; Sarek, G.; Maslen, S.L.; Tsutakawa, S.E.; Borg, A.; Kjær, S.; Tainer, J.A.; et al. Mechanism and Regulation of DNA-Protein Crosslink Repair by the DNA-Dependent Metalloprotease SPRTN. Mol. Cell 2016, 64, 688–703. [Google Scholar] [CrossRef]
- Yazinski, S.A.; Comaills, V.; Buisson, R.; Genois, M.M.; Nguyen, H.D.; Ho, C.K.; Todorova Kwan, T.; Morris, R.; Lauffer, S.; Nussenzweig, A.; et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 2017, 31, 318–332. [Google Scholar] [CrossRef]
- Koundrioukoff, S.; Carignon, S.; Techer, H.; Letessier, A.; Brison, O.; Debatisse, M. Stepwise activation of the ATR signaling pathway upon increasing replication stress impacts fragile site integrity. PLoS Genet. 2013, 9, e1003643. [Google Scholar] [CrossRef]
- Toledo, L.I.; Altmeyer, M.; Rask, M.B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef] [PubMed]
- Mengwasser, K.E.; Adeyemi, R.O.; Leng, Y.; Choi, M.Y.; Clairmont, C.; D’Andrea, A.D.; Elledge, S.J. Genetic Screens Reveal FEN1 and APEX2 as BRCA2 Synthetic Lethal Targets. Mol. Cell 2019, 73, 885–899.e6. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Acevedo, M.; Grace, L.; Teoh, D.; Whitaker, R.; Adams, D.J.; Jia, J.; Nixon, A.B.; Secord, A.A. Dasatinib (BMS-35482) potentiates the activity of gemcitabine and docetaxel in uterine leiomyosarcoma cell lines. Gynecol. Oncol. Res. Pract. 2014, 1. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gralewska, P.; Gajek, A.; Marczak, A.; Mikuła, M.; Ostrowski, J.; Śliwińska, A.; Rogalska, A. PARP Inhibition Increases the Reliance on ATR/CHK1 Checkpoint Signaling Leading to Synthetic Lethality—An Alternative Treatment Strategy for Epithelial Ovarian Cancer Cells Independent from HR Effectiveness. Int. J. Mol. Sci. 2020, 21, 9715. https://doi.org/10.3390/ijms21249715
Gralewska P, Gajek A, Marczak A, Mikuła M, Ostrowski J, Śliwińska A, Rogalska A. PARP Inhibition Increases the Reliance on ATR/CHK1 Checkpoint Signaling Leading to Synthetic Lethality—An Alternative Treatment Strategy for Epithelial Ovarian Cancer Cells Independent from HR Effectiveness. International Journal of Molecular Sciences. 2020; 21(24):9715. https://doi.org/10.3390/ijms21249715
Chicago/Turabian StyleGralewska, Patrycja, Arkadiusz Gajek, Agnieszka Marczak, Michał Mikuła, Jerzy Ostrowski, Agnieszka Śliwińska, and Aneta Rogalska. 2020. "PARP Inhibition Increases the Reliance on ATR/CHK1 Checkpoint Signaling Leading to Synthetic Lethality—An Alternative Treatment Strategy for Epithelial Ovarian Cancer Cells Independent from HR Effectiveness" International Journal of Molecular Sciences 21, no. 24: 9715. https://doi.org/10.3390/ijms21249715
APA StyleGralewska, P., Gajek, A., Marczak, A., Mikuła, M., Ostrowski, J., Śliwińska, A., & Rogalska, A. (2020). PARP Inhibition Increases the Reliance on ATR/CHK1 Checkpoint Signaling Leading to Synthetic Lethality—An Alternative Treatment Strategy for Epithelial Ovarian Cancer Cells Independent from HR Effectiveness. International Journal of Molecular Sciences, 21(24), 9715. https://doi.org/10.3390/ijms21249715