Targeting Fat Oxidation in Mouse Prostate Cancer Decreases Tumor Growth and Stimulates Anti-Cancer Immunity


 , ,
, , 
Abstract
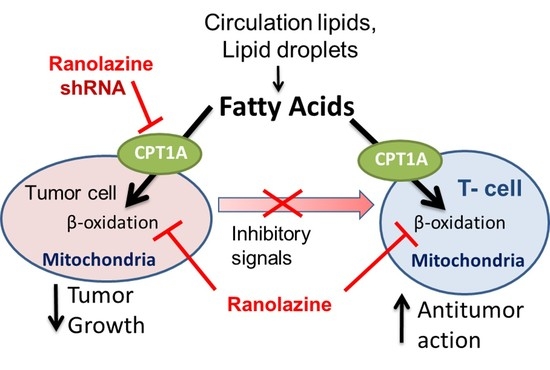
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Systemic Treatment with Ranolazine Decreases Tumor Growth in Immune Competent Mice
2.2. Treatment with Ranolazine Results in Changes in Immune Check Point Proteins (ICP) and Macrophages in the Tumors
2.3. Increased Content of CD8 T-Cells and Dendritic Cells in Drug-Treated Tumors
2.4. Increased Ex Vivo T-Cell Cytotoxic Activity of Drug-Treated Spleens
2.5. Mouse Prostate Cancer Cells Burn Less Lipid and Generate Less Acyl-Carnitines When Exposed to Ranolazine
2.6. Decreased PD1 Stain in T-Cells Incubated with TRAMPC1 Cells Deficient in Cpt1A Expression
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Reagents
4.2. Mouse Xenograft Generation with TRAMPC1 Cells
4.3. Tissue Harvesting and Flow Cytometry Assays
4.4. Multispectral Fluorescence Immunohistochemistry
4.5. Co-Culture Studies of TRAMPC1 Cells with Drug-Treated Derived Splenocytes
4.6. Lentiviral shRNA Transfections
4.7. Co-Culture Studies of Cpt1A-KD Cells with Splenocytes
4.8. Western Blot Analysis
4.9. Acyl Carnitine Analysis
4.10. Seahorse Metabolic Flux Analysis
4.11. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Rano | Ranolazine |
| KD | Knockdown |
| MFI | Mean Fluorescence Intensity |
| ICP | Immune Checkpoint Protein |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Nuhn, P.; De Bono, J.S.; Fizazi, K.; Freedland, S.J.; Grilli, M.; Kantoff, P.W.; Sonpavde, G.; Sternberg, C.N.; Yegnasubramanian, S.; Antonarakis, E.S. Update on Systemic Prostate Cancer Therapies: Management of Metastatic Castration-resistant Prostate Cancer in the Era of Precision Oncology. Eur. Urol. 2019, 75, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Srihari, S.; Kwong, R.; Tran, K.; Simpson, R.; Tattam, P.; Smith, E. Metabolic deregulation in prostate cancer. Mol. Omics 2018, 14, 320–329. [Google Scholar] [CrossRef]
- Laurent, V.; Guérard, A.; Mazerolles, C.; Le Gonidec, S.; Toulet, A.; Nieto, L.; Zaidi, F.; Majed, B.; Garandeau, D.; Socrier, Y.; et al. Periprostatic adipocytes act as a driving force for prostate cancer progression in obesity. Nat. Commun. 2016, 7, 10230. [Google Scholar] [CrossRef]
- Watt, M.J.; Clark, A.K.; Selth, L.A.; Haynes, V.R.; Lister, N.; Rebello, R.; Porter, L.H.; Niranjan, B.; Whitby, S.T.; Lo, J.; et al. Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Sci. Transl. Med. 2019, 11, eaau5758. [Google Scholar] [CrossRef]
- Wellberg, E.A.; Kabos, P.; Gillen, A.E.; Jacobsen, B.M.; Brechbuhl, H.M.; Johnson, S.J.; Rudolph, M.C.; Edgerton, S.M.; Thor, A.D.; Anderson, S.M.; et al. FGFR1 underlies obesity-associated progression of estrogen receptor-positive breast cancer after estrogen deprivation. JCI Insight 2018, 3, e120594. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer. Prostate Cancer Prostatic Dis. 2006, 9, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, I.R.; Rider, L.; Rodrigues, L.U.; Gijon, M.A.; Pac, C.T.; Romero, L.; Cimic, A.; Sirintrapun, S.J.; Glode, L.M.; Eckel, R.H.; et al. Lipid Catabolism via CPT1 as a Therapeutic Target for Prostate Cancer. Mol. Cancer Ther. 2014, 13, 2361–2371. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, R.N.; Karami-Tehrani, F.; Salami, S. Targeting prostate cancer cell metabolism: Impact of hexokinase and CPT-1 enzymes. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2015, 36, 2893–2905. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.; Stoykova, G.E.; Salzmann-Sullivan, M.; Dzieciatkowska, M.; Liebman, L.N.; Deep, G.; Schlaepfer, I.R. CPT1A Supports Castration-Resistant Prostate Cancer in Androgen-Deprived Conditions. Cells 2019, 8, 1115. [Google Scholar] [CrossRef]
- Schlaepfer, I.R.; Glode, L.M.; Hitz, C.A.; Pac, C.T.; Boyle, K.E.; Maroni, P.; Deep, G.; Agarwal, R.; Lucia, S.M.; Cramer, S.D.; et al. Inhibition of Lipid Oxidation Increases Glucose Metabolism and Enhances 2-Deoxy-2-[F]Fluoro-D-Glucose Uptake in Prostate Cancer Mouse Xenografts. Mol. Imaging Biol. 2015, 17, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Handy, C.E.; Antonarakis, E.S. Sipuleucel-T for the treatment of prostate cancer: Novel insights and future directions. Futur. Oncol. 2018, 14, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Massari, F.; Santoni, M.; Ciccarese, C.; Santini, D. The immunocheckpoints in modern oncology: The next 15 years. Expert. Opin. Biol. Ther. 2015, 15, 917–921. [Google Scholar] [CrossRef]
- Hussein, M.R.; Al-Assiri, M.; Musalam, A.O. Phenotypic characterization of the infiltrating immune cells in normal prostate, benign nodular prostatic hyperplasia and prostatic adenocarcinoma. Exp. Mol. Pathol. 2009, 86, 108–113. [Google Scholar] [CrossRef]
- Bettonville, M.; d’Aria, S.; Weatherly, K.; Porporato, P.E.; Zhang, J.; Bousbata, S.; Sonveaux, P.; Braun, M.Y. Long-term antigen exposure irreversibly modifies metabolic requirements for T cell function. eLife 2018, 7, e30938. [Google Scholar] [CrossRef]
- Buck, M.D.; O’Sullivan, D.; Pearce, E.L. T cell metabolism drives immunity. J. Exp. Med. 2015, 212, 1345–1360. [Google Scholar] [CrossRef]
- Carpenter, K.J.; Valfort, A.C.; Steinauer, N.; Chatterjee, A.; Abuirqeba, S.; Majidi, S.; Sengupta, M.; Di Paolo, R.J.; Shornick, L.P.; Zhang, J.; et al. LXR-inverse agonism stimulates immune-mediated tumor destruction by enhancing CD8 T-cell activity in triple negative breast cancer. Sci. Rep. 2019, 9, 19530. [Google Scholar] [CrossRef]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, I.R.; Joshi, M. CPT1A-mediated fat oxidation, mechanisms and therapeutic potential. Endocrinology 2020, 161, bqz046. [Google Scholar] [CrossRef] [PubMed]
- Bugan, I.; Kucuk, S.; Karagoz, Z.; Fraser, S.P.; Kaya, H.; Dodson, A.; Foster, C.S.; Altun, S.; Djamgoz, M.B.A. Anti-metastatic effect of ranolazine in an in vivo rat model of prostate cancer, and expression of voltage-gated sodium channel protein in human prostate. Prostate Cancer Prostatic Dis. 2019, 22, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Stoykova, G.E.; Schlaepfer, I.R. Lipid Metabolism and Endocrine Resistance in Prostate Cancer, and New Opportunities for Therapy. Int. J. Mol. Sci. 2019, 20, 2626. [Google Scholar] [CrossRef] [PubMed]
- Lionetti, V.; Stanley, W.C.; Recchia, F.A. Modulating fatty acid oxidation in heart failure. Cardiovasc. Res. 2011, 90, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Le, D.E.; Davis, C.M.; Wei, K.; Zhao, Y.; Cao, Z.; Nugent, M.; Scott, K.L.L.; Liu, L.; Nagarajan, S.; Alkayed, N.J.; et al. Ranolazine may exert its beneficial effects by increasing myocardial adenosine levels. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H189–H202. [Google Scholar] [CrossRef] [PubMed]
- Lisi, D.; Andrews, E.; Parry, C.; Hill, C.; Ombengi, D.; Ling, H. The Effect of Ranolazine on Glycemic Control: A Narrative Review to Define the Target Population. Cardiovasc. Drugs Ther. 2019, 33, 755–761. [Google Scholar] [CrossRef]
- Driffort, V.; Gillet, L.; Bon, E.; Marionneau-Lambot, S.; Oullier, T.; Joulin, V.; Collin, C.; Pagès, J.C.; Jourdan, M.L.; Chevalier, S.; et al. Ranolazine inhibits NaV1.5-mediated breast cancer cell invasiveness and lung colonization. Mol. Cancer 2014, 13, 264. [Google Scholar] [CrossRef]
- Flaig, T.W.; Salzmann-Sullivan, M.; Su, L.J.; Zhang, Z.; Joshi, M.; Gijon, M.A.; Kim, J.; Arcaroli, J.J.; Van Bokhoven, A.; Lucia, M.S.; et al. Lipid catabolism inhibition sensitizes prostate cancer cells to antiandrogen blockade. Oncotarget 2017, 8, 56051–56065. [Google Scholar] [CrossRef]
- Samudio, I.; Harmancey, R.; Fiegl, M.; Kantarjian, H.; Konopleva, M.; Korchin, B.; Kaluarachchi, K.; Bornmann, W.; Duvvuri, S.; Taegtmeyer, H.; et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J. Clin. Investig. 2010, 120, 142–156. [Google Scholar] [CrossRef]
- Klein Geltink, R.I.; O’Sullivan, D.; Corrado, M.; Bremser, A.; Buck, M.D.; Buescher, J.M.; Firat, E.; Zhu, X.; Niedermann, G.; Caputa, G.; et al. Mitochondrial Priming by CD28. Cell 2017, 171, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Foster, B.A.; Gingrich, J.R.; Kwon, E.D.; Madias, C.; Greenberg, N.M. Characterization of prostatic epithelial cell lines derived from transgenic adenocarcinoma of the mouse prostate (TRAMP) model. Cancer Res. 1997, 57, 3325–3330. [Google Scholar] [PubMed]
- Singh, K.B.; Kim, S.H.; Hahm, E.R.; Pore, S.K.; Jacobs, B.L.; Singh, S.V. Prostate cancer chemoprevention by sulforaphane in a preclinical mouse model is associated with inhibition of fatty acid metabolism. Carcinogenesis 2018, 39, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.V.; Warin, R.; Xiao, D.; Powolny, A.A.; Stan, S.D.; Arlotti, J.A.; Zeng, Y.; Hahm, E.R.; Marynowski, S.W.; Bommareddy, A.; et al. Sulforaphane inhibits prostate carcinogenesis and pulmonary metastasis in TRAMP mice in association with increased cytotoxicity of natural killer cells. Cancer Res. 2009, 69, 2117–2125. [Google Scholar] [CrossRef] [PubMed]
- Lang, J.; Capasso, A.; Jordan, K.R.; French, J.D.; Kar, A.; Bagby, S.M.; Barbee, J.; Yacob, B.W.; Head, L.S.; Tompkins, K.D.; et al. Development of an Adrenocortical Cancer Humanized Mouse Model to Characterize Anti-PD1 Effects on Tumor Microenvironment. J. Clin. Endocrinol. Metab. 2020, 105, dgz014. [Google Scholar] [CrossRef]
- Pollizzi, K.N.; Patel, C.H.; Sun, I.H.; Oh, M.H.; Waickman, A.T.; Wen, J.; Delgoffe, G.M.; Powell, J.D. mTORC1 and mTORC2 selectively regulate CD8⁺ T cell differentiation. J. Clin. Investig. 2015, 125, 2090–2108. [Google Scholar] [CrossRef]
- Wang, Z.; Aguilar, E.G.; Luna, J.I.; Dunai, C.; Khuat, L.T.; Le, C.T.; Mirsoian, A.; Minnar, C.M.; Stoffel, K.M.; Sturgill, I.R.; et al. Paradoxical effects of obesity on T cell function during tumor progression and PD-1 checkpoint blockade. Nat. Med. 2019, 25, 141–151. [Google Scholar] [CrossRef]
- Brown, Z.J.; Fu, Q.; Ma, C.; Kruhlak, M.; Zhang, H.; Luo, J.; Heinrich, B.; Yu, S.J.; Zhang, Q.; Wilson, A.; et al. Carnitine palmitoyltransferase gene upregulation by linoleic acid induces CD4(+) T cell apoptosis promoting HCC development. Cell Death Dis. 2018, 9, 620. [Google Scholar] [CrossRef]
- Bader, J.E.; Voss, K.; Rathmell, J.C. Targeting Metabolism to Improve the Tumor Microenvironment for Cancer Immunotherapy. Mol. Cell 2020, 78, 1019–1033. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, H.; Mao, C.; Sun, M.; Dominah, G.; Chen, L.; Zhuang, Z. Fatty acid oxidation contributes to IL-1β secretion in M2 macrophages and promotes macrophage-mediated tumor cell migration. Mol. Immunol. 2018, 94, 27–35. [Google Scholar] [CrossRef]
- Malinarich, F.; Duan, K.; Hamid, R.A.; Bijin, A.; Lin, W.X.; Poidinger, M.; Fairhurst, A.M.; Connolly, J.E. High mitochondrial respiration and glycolytic capacity represent a metabolic phenotype of human tolerogenic dendritic cells. J. Immunol. 2015, 194, 5174–5186. [Google Scholar] [CrossRef] [PubMed]
- Qorraj, M.; Bruns, H.; Böttcher, M.; Weigand, L.; Saul, D.; Mackensen, A.; Jitschin, R.; Mougiakakos, D. The PD-1/PD-L1 axis contributes to immune metabolic dysfunctions of monocytes in chronic lymphocytic leukemia. Leukemia 2017, 31, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Strauss, L.; Mahmoud, M.A.A.; Weaver, J.D.; Tijaro-Ovalle, N.M.; Christofides, A.; Wang, Q.; Pal, R.; Yuan, M.; Asara, J.; Patsoukis, N.; et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci. Immunol. 2020, 5, eaay1863. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Gladstone, M.; Shanley, C.; Goodrich, R.; Guth, A. A novel cancer immunotherapy utilizing autologous tumour tissue. Vox Sang. 2020, 115, 525–535. [Google Scholar] [CrossRef]
- Kivilompolo, M.; Öhrnberg, L.; Orešič, M.; Hyötyläinen, T. Rapid quantitative analysis of carnitine and acylcarnitines by ultra-high performance-hydrophilic interaction liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2013, 1292, 189–194. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guth, A.; Monk, E.; Agarwal, R.; Bergman, B.C.; Zemski-Berry, K.A.; Minic, A.; Jordan, K.; Schlaepfer, I.R. Targeting Fat Oxidation in Mouse Prostate Cancer Decreases Tumor Growth and Stimulates Anti-Cancer Immunity. Int. J. Mol. Sci. 2020, 21, 9660. https://doi.org/10.3390/ijms21249660
Guth A, Monk E, Agarwal R, Bergman BC, Zemski-Berry KA, Minic A, Jordan K, Schlaepfer IR. Targeting Fat Oxidation in Mouse Prostate Cancer Decreases Tumor Growth and Stimulates Anti-Cancer Immunity. International Journal of Molecular Sciences. 2020; 21(24):9660. https://doi.org/10.3390/ijms21249660
Chicago/Turabian StyleGuth, Amanda, Emily Monk, Rajesh Agarwal, Bryan C. Bergman, Karin A. Zemski-Berry, Angela Minic, Kimberly Jordan, and Isabel R. Schlaepfer. 2020. "Targeting Fat Oxidation in Mouse Prostate Cancer Decreases Tumor Growth and Stimulates Anti-Cancer Immunity" International Journal of Molecular Sciences 21, no. 24: 9660. https://doi.org/10.3390/ijms21249660
APA StyleGuth, A., Monk, E., Agarwal, R., Bergman, B. C., Zemski-Berry, K. A., Minic, A., Jordan, K., & Schlaepfer, I. R. (2020). Targeting Fat Oxidation in Mouse Prostate Cancer Decreases Tumor Growth and Stimulates Anti-Cancer Immunity. International Journal of Molecular Sciences, 21(24), 9660. https://doi.org/10.3390/ijms21249660