BET-Inhibitor I-BET762 and PARP-Inhibitor Talazoparib Synergy in Small Cell Lung Cancer Cells

 ,
,  , and
, and 
Abstract
1. Introduction
2. Results
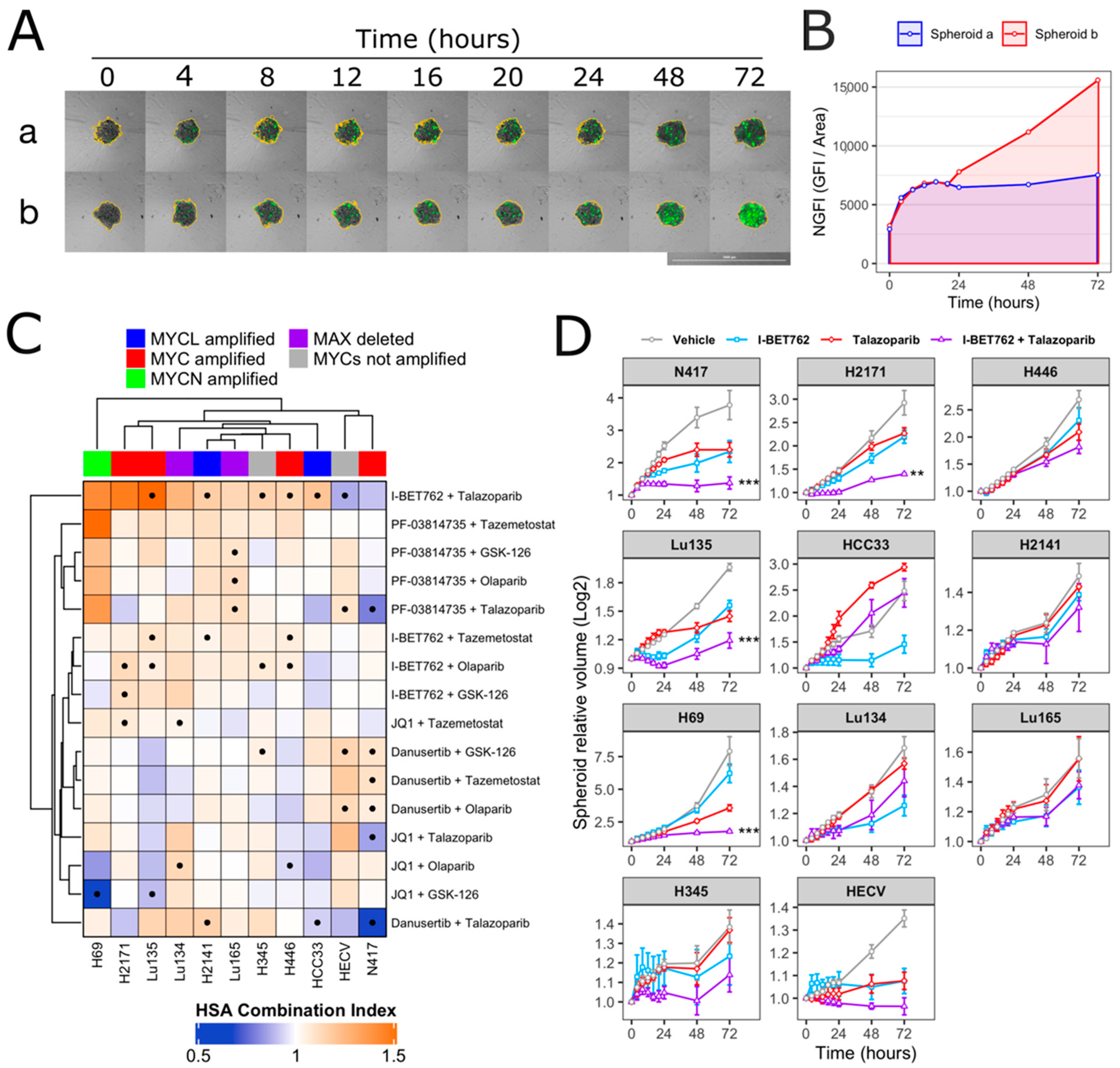
2.1. I-BET762 and Talazoparib Showed a Combinatorial Efficacy on MYCs-Amplified SCLC Cells
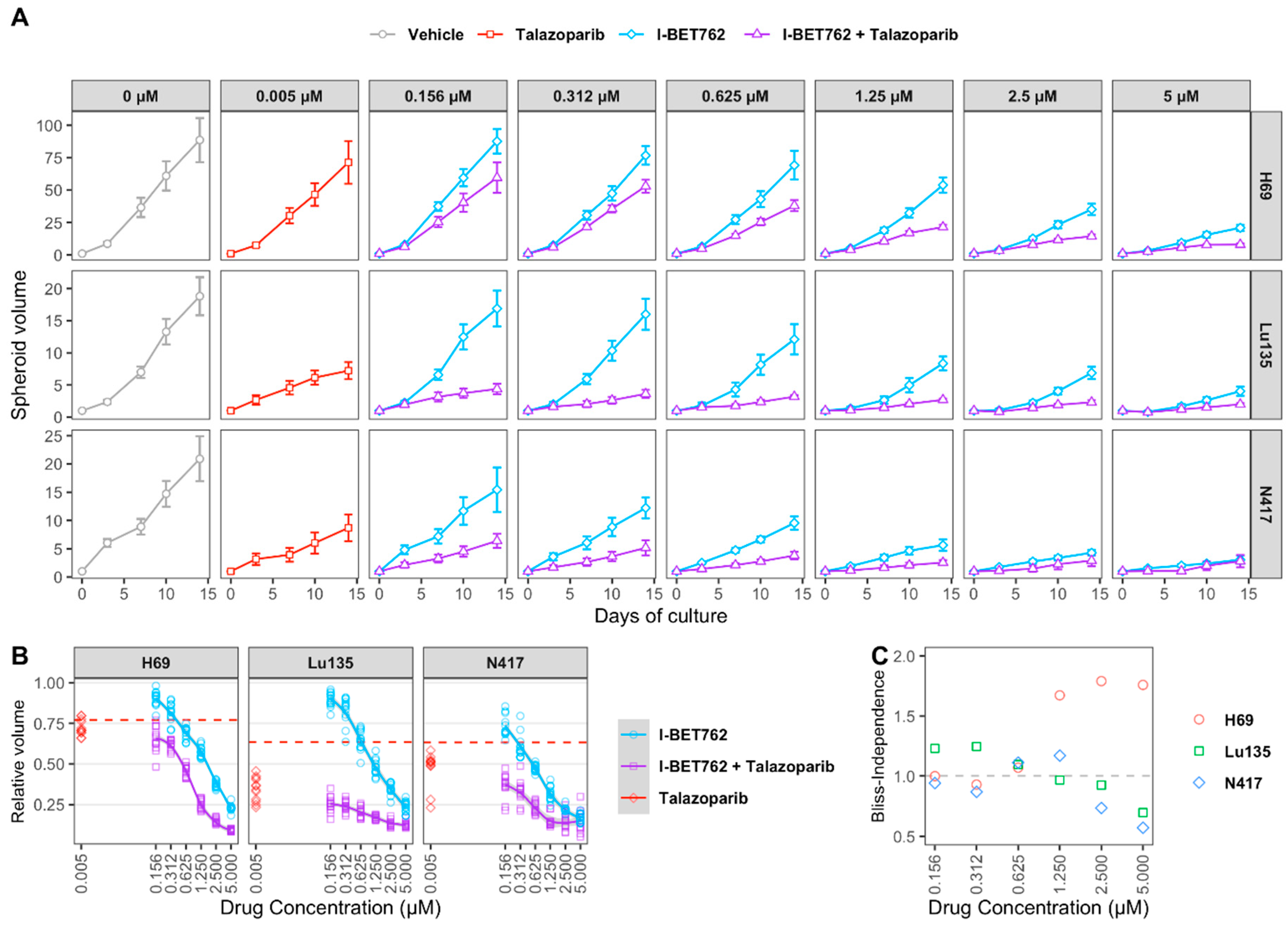
2.2. Combinatorial Treatment of I-BET762 and Talazoparib at Clinically Relevant Concentrations Showed Increased Efficacy on MYCs-Amplified SCLC Cell Growth
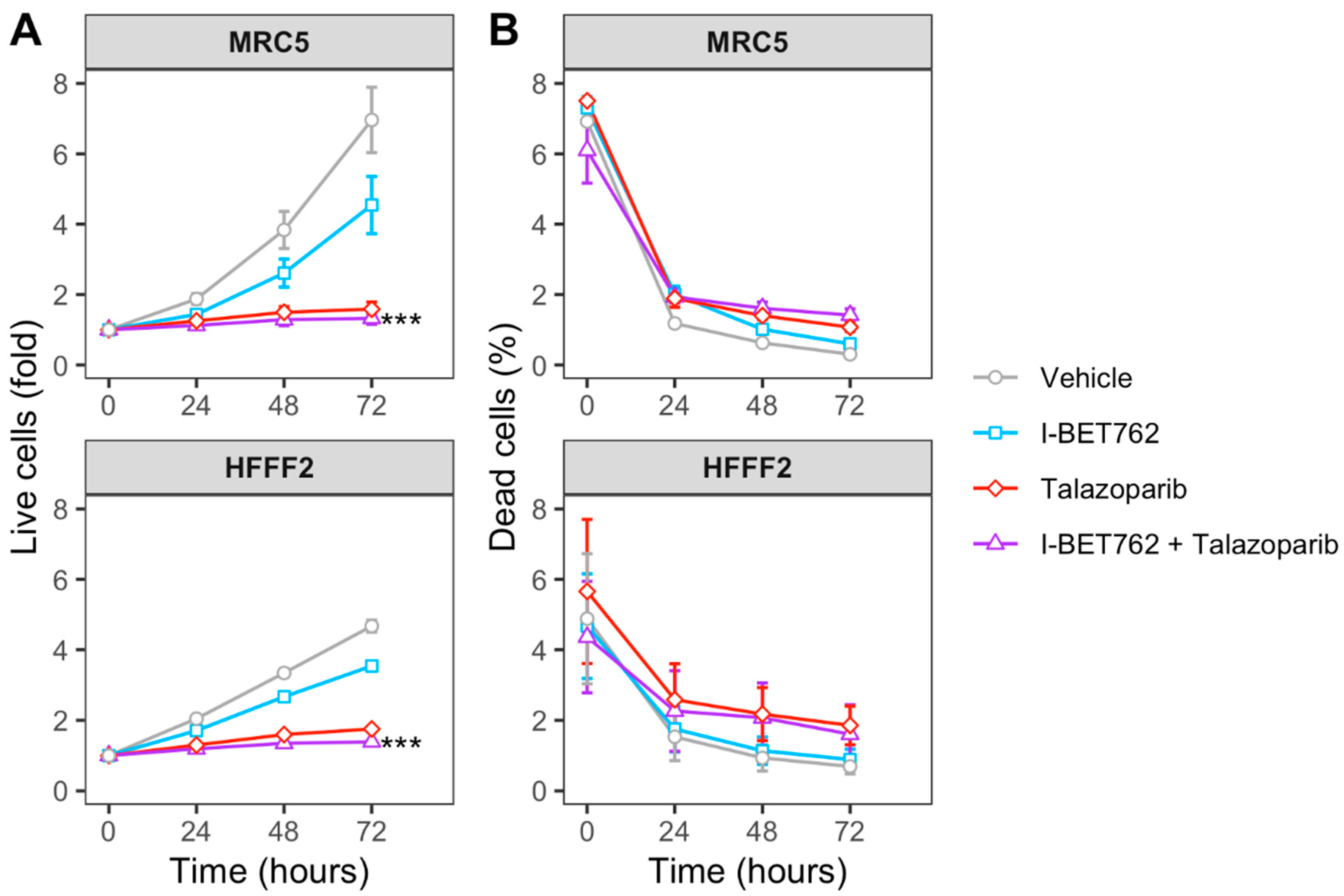
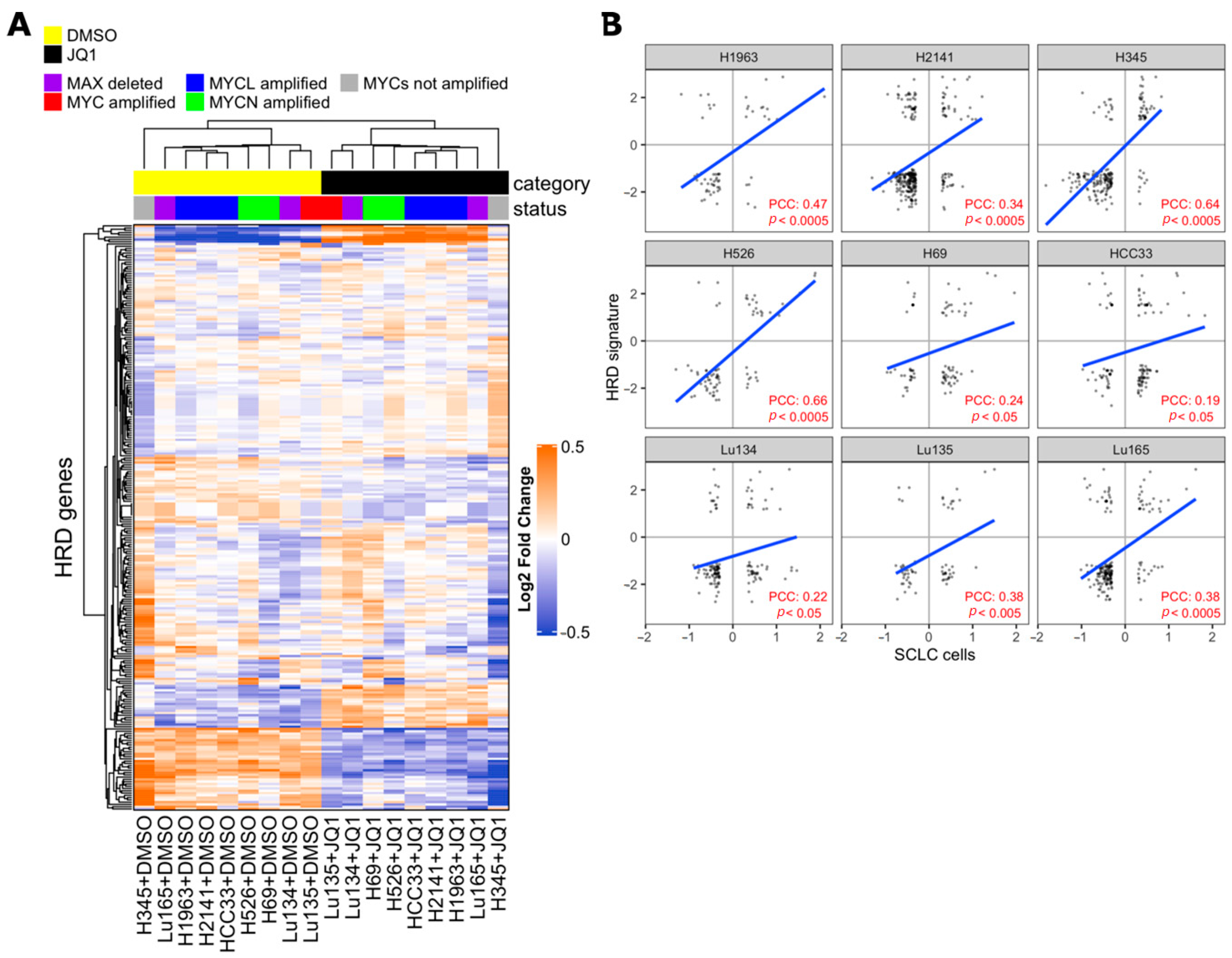
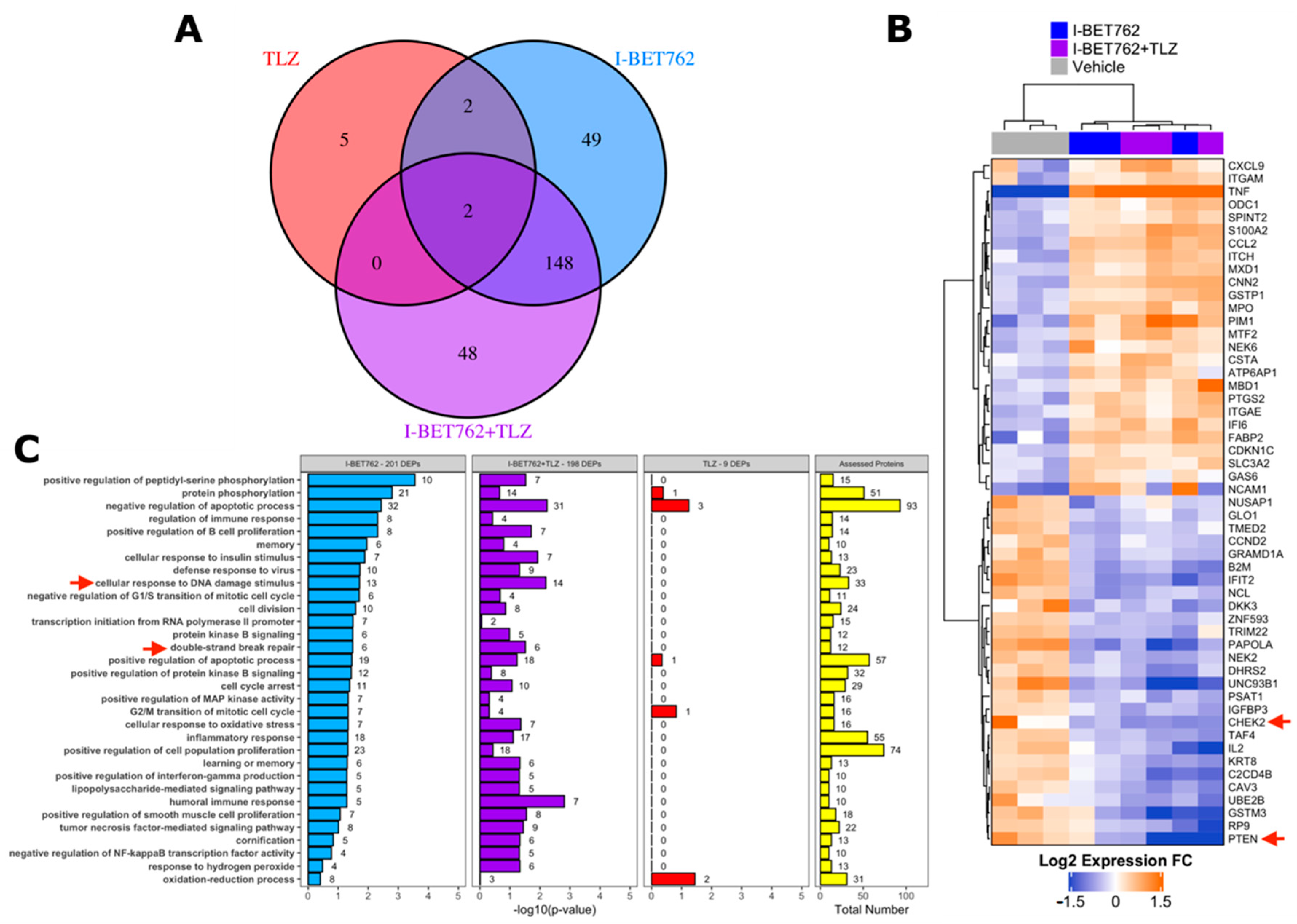
2.3. I-BET762 Treatment Induces Apoptosis and Down-Regulation of Double-Strand Break Repair Proteins
3. Discussion and Conclusions
4. Materials and Methods
4.1. Cell Culture
4.2. Kinetics of 3D Spheroid Cell Death Assay
4.3. Kinetics of Cell Proliferation Assay
4.4. Long-Term Tumor Spheroid Growth Assay
4.5. Cancer-Related Proteome and Phospho-Proteome Profiling
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef]
- Cooper, S.; Spiro, S.G. Small cell lung cancer: Treatment review. Respirology 2006, 11, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Sos, M.L.; Dietlein, F.; Peifer, M.; Schöttle, J.; Balke-Want, H.; Müller, C.; Koker, M.; Richters, A.; Heynck, S.; Malchers, F.; et al. A framework for identification of actionable cancer genome dependencies in small cell lung cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 17034–17039. [Google Scholar] [CrossRef]
- Iwakawa, R.; Takenaka, M.; Kohno, T.; Shimada, Y.; Totoki, Y.; Shibata, T.; Tsuta, K.; Nishikawa, R.; Noguchi, M.; Sato-Otsubo, A.; et al. Genome-wide identification of genes with amplification and/or fusion in small cell lung cancer. Genes Chromosomes Cancer 2013, 52, 802–816. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, F.P.; Tokgün, E.; Solé-Sánchez, S.; Giampaolo, S.; Tokgun, O.; Jauset, T.; Kohno, T.; Perucho, M.; Soucek, L.; Yokota, J. Growth suppression by MYC inhibition in small cell lung cancer cells with TP53 and RB1 inactivation. Oncotarget 2016, 7, 31014–31028. [Google Scholar] [CrossRef] [PubMed]
- Mertz, J.A.; Conery, A.R.; Bryant, B.M.; Sandy, P.; Balasubramanian, S.; Mele, D.A.; Bergeron, L.; Sims, R.J. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. USA 2011, 108, 16669–16674. [Google Scholar] [CrossRef] [PubMed]
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective Inhibition of Tumor Oncogenes by Disruption of Super-Enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef]
- Chapuy, B.; McKeown, M.R.; Lin, C.Y.; Monti, S.; Roemer, M.G.M.; Qi, J.; Rahl, P.B.; Sun, H.H.; Yeda, K.T.; Doench, J.G.; et al. Discovery and Characterization of Super-Enhancer-Associated Dependencies in Diffuse Large B Cell Lymphoma. Cancer Cell 2013, 24, 777–790. [Google Scholar] [CrossRef]
- Yang, Z.; Yik, J.H.N.; Chen, R.; He, N.; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 2005, 19, 535–545. [Google Scholar] [CrossRef]
- Shi, J.; Vakoc, C.R. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol. Cell 2014, 54, 728–736. [Google Scholar] [CrossRef]
- Fu, L.-L.; Tian, M.; Li, X.; Li, J.-J.; Huang, J.; Ouyang, L.; Zhang, Y.; Liu, B. Inhibition of BET bromodomains as a therapeutic strategy for cancer drug discovery. Oncotarget 2015, 6, 5501–5516. [Google Scholar] [CrossRef] [PubMed]
- Kato, F.; Fiorentino, F.P.; Alibés, A.; Perucho, M.; Sanchez-Cespedes, M.; Kohno, T.; Yokota, J. MYCL is a target of a BET bromodomain inhibitor, JQ1, on growth suppression efficacy in small cell lung cancer cells. Oncotarget 2016, 7, 77378–77388. [Google Scholar] [CrossRef] [PubMed]
- Lenhart, R.; Kirov, S.; Desilva, H.; Cao, J.; Lei, M.; Johnston, K.; Peterson, R.; Schweizer, L.; Purandare, A.; Ross-Macdonald, P.; et al. Sensitivity of Small Cell Lung Cancer to BET Inhibition Is Mediated by Regulation of ASCL1 Gene Expression. Mol. Cancer Ther. 2015, 14, 2167–2174. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Reinhart, R.A.; Monks, A.; Evans, D.; Morris, J.; Polley, E.; Teicher, B.A. Bromodomain and hedgehog pathway targets in small cell lung cancer. Cancer Lett. 2016, 371, 225–239. [Google Scholar] [CrossRef]
- Wang, H.; Hong, B.; Li, X.; Deng, K.; Li, H.; Lui, V.W.Y.; Lin, W. JQ1 synergizes with the Bcl-2 inhibitor ABT-263 against MYCN-amplified small cell lung cancer. Oncotarget 2017, 8, 86312–86324. [Google Scholar] [CrossRef][Green Version]
- Lam, L.T.; Lin, X.; Faivre, E.J.; Yang, Z.; Huang, X.; Wilcox, D.M.; Bellin, R.J.; Jin, S.; Tahir, S.K.; Mitten, M.; et al. Vulnerability of Small-Cell Lung Cancer to Apoptosis Induced by the Combination of BET Bromodomain Proteins and BCL2 Inhibitors. Mol. Cancer Ther. 2017, 16, 1511–1520. [Google Scholar] [CrossRef]
- Settleman, J. Cancer: Bet on drug resistance. Nature 2016, 529, 289–290. [Google Scholar] [CrossRef]
- Hook, K.E.; Garza, S.J.; Lira, M.E.; Ching, K.A.; Lee, N.V.; Cao, J.; Yuan, J.; Ye, J.; Ozeck, M.; Shi, S.T.; et al. An Integrated Genomic Approach to Identify Predictive Biomarkers of Response to the Aurora Kinase Inhibitor PF-03814735. Mol. Cancer Ther. 2012, 11, 710–719. [Google Scholar] [CrossRef]
- Brockmann, M.; Poon, E.; Berry, T.; Carstensen, A.; Deubzer, H.E.; Rycak, L.; Jamin, Y.; Thway, K.; Robinson, S.P.; Roels, F.; et al. Small Molecule Inhibitors of Aurora-A Induce Proteasomal Degradation of N-Myc in Childhood Neuroblastoma. Cancer Cell 2013, 24, 75–89. [Google Scholar] [CrossRef]
- Melichar, B.; Adenis, A.; Havel, L.; Lockhart, A.C.; Bennouna, J.; Schusterbauer, C.; Ullmann, C.D.; Zhang, B.; Benaim, E.; Dees, E.C. Phase (Ph) I/II study of investigational Aurora A kinase (AAK) inhibitor MLN8237 (alisertib): Updated ph II results in patients (pts) with small cell lung cancer (SCLC), non-SCLC (NSCLC), breast cancer (BrC), head and neck squamous cell carcinoma (HNSCC), and gastroesophageal cancer (GE). J. Clin. Oncol. 2013, 31 (Suppl. 15), 605. [Google Scholar]
- Byers, L.A.; Rudin, C.M. Small cell lung cancer: Where do we go from here? Cancer 2014, 121, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Melichar, B.; Adenis, A.; Lockhart, A.C.; Bennouna, J.; Dees, E.C.; Kayaleh, O.; Obermannova, R.; DeMichele, A.; Zatloukal, P.; Zhang, B.; et al. Safety and activity of alisertib, an investigational aurora kinase A inhibitor, in patients with breast cancer, small-cell lung cancer, non-small-cell lung cancer, head and neck squamous-cell carcinoma, and gastro-oesophageal adenocarcinoma: A five-arm phase 2 study. Lancet Oncol. 2015, 16, 395–405. [Google Scholar] [PubMed]
- Lopez, J.S.; Banerji, U. Combine and conquer: Challenges for targeted therapy combinations in early phase trials. Nat. Rev. Clin. Oncol. 2016, 14, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.W.; Loeb, L.A.; Salk, J.J. The influence of subclonal resistance mutations on targeted cancer therapy. Nat. Rev. Clin. Oncol. 2015, 13, 335–347. [Google Scholar] [CrossRef]
- Byers, L.A.; Wang, J.; Nilsson, M.B.; Fujimoto, J.; Saintigny, P.; Yordy, J.; Giri, U.; Peyton, M.; Fan, Y.H.; Diao, L.; et al. Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Discov. 2012, 2, 798–811. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Jain, P.G.; Patel, B.D. Medicinal chemistry approaches of poly ADP-Ribose polymerase 1 (PARP1) inhibitors as anticancer agents—A recent update. Eur. J. Med. Chem. 2019, 165, 198–215. [Google Scholar] [CrossRef]
- Sato, T.; Kaneda, A.; Tsuji, S.; Isagawa, T.; Yamamoto, S.; Fujita, T.; Yamanaka, R.; Tanaka, Y.; Nukiwa, T.; Marquez, V.E.; et al. PRC2 overexpression and PRC2-target gene repression relating to poorer prognosis in small cell lung cancer. Sci. Rep. 2013, 3, 1911. [Google Scholar] [CrossRef]
- Hubaux, R.; Thu, K.L.; Coe, B.P.; MacAulay, C.; Lam, S.; Lam, W.L. EZH2 Promotes E2F-Driven SCLC Tumorigenesis through Modulation of Apoptosis and Cell-Cycle Regulation. J. Thorac. Oncol. 2013, 8, 1102–1106. [Google Scholar] [CrossRef]
- Coe, B.P.; Thu, K.L.; Aviel-Ronen, S.; Vucic, E.A.; Gazdar, A.F.; Lam, S.; Tsao, M.-S.; Lam, W.L. Genomic Deregulation of the E2F/Rb Pathway Leads to Activation of the Oncogene EZH2 in Small Cell Lung Cancer. PLoS ONE 2013, 8, e71670. [Google Scholar] [CrossRef]
- Cardnell, R.J.; Feng, Y.; Diao, L.; Fan, Y.H.; Masrorpour, F.; Wang, J.; Shen, Y.; Mills, G.B.; Minna, J.D.; Heymach, J.V.; et al. Proteomic markers of DNA repair and PI3K pathway activation predict response to the PARP inhibitor BMN 673 in small cell lung cancer. Clin. Cancer Res. 2013, 19, 6322–6328. [Google Scholar] [CrossRef] [PubMed]
- Lok, B.H.; Gardner, E.E.; Schneeberger, V.E.; Ni, A.; Desmeules, P.; Rekhtman, N.; de Stanchina, E.; Teicher, B.A.; Riaz, N.; Powell, S.N.; et al. PARP Inhibitor Activity Correlates with SLFN11Expression and Demonstrates Synergy with Temozolomide in Small Cell Lung Cancer. Clin. Cancer Res. 2017, 23, 523–535. [Google Scholar] [CrossRef]
- Toyokawa, G.; Takada, K.; Tagawa, T.; Kinoshita, F.; Kozuma, Y.; Matsubara, T.; Haratake, N.; Takamori, S.; Akamine, T.; Hirai, F.; et al. Prevalence of Enhancer of Zeste Homolog 2 in Patients with Resected Small Cell Lung Cancer. Anticancer Res. 2018, 38, 3707–3711. [Google Scholar] [CrossRef] [PubMed]
- Poirier, J.T.; Gardner, E.E.; Connis, N.; Moreira, A.L.; de Stanchina, E.; Hann, C.L.; Rudin, C.M. DNA methylation in small cell lung cancer defines distinct disease subtypes and correlates with high expression of EZH2. Oncogene 2015, 34, 5869–5878. [Google Scholar] [CrossRef] [PubMed]
- Papavassiliou, K.A.; Papavassiliou, A.G. Bromodomains: Pockets with therapeutic potential. Trends Mol. Med. 2014, 20, 477–478. [Google Scholar] [CrossRef] [PubMed]
- Marchesi, I.; Bagella, L. Targeting Enhancer of Zeste Homolog 2 as a promising strategy for cancer treatment. World J. Clin. Oncol. 2016, 7, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.L.; Swisher, E.M.; Kaufmann, S.H. Poly (ADP-ribose) polymerase inhibitors: Recent advances and future development. J. Clin. Oncol. 2015, 33, 1397–1406. [Google Scholar] [CrossRef] [PubMed]
- Falchook, G.S.; Bastida, C.C.; Kurzrock, R. Aurora Kinase Inhibitors in Oncology Clinical Trials: Current State of the Progress. Semin. Oncol. 2015, 42, 832–848. [Google Scholar] [CrossRef]
- Selby, M.; Delosh, R.; Laudeman, J.; Ogle, C.; Reinhart, R.; Silvers, T.; Lawrence, S.; Kinders, R.; Parchment, R.; Teicher, B.A.; et al. 3D Models of the NCI60 Cell Lines for Screening Oncology Compounds. SLAS Discov. 2017, 22, 473–483. [Google Scholar] [CrossRef]
- Amati, B.; Littlewood, T.D.; Evan, G.I.; Land, H. The c-Myc protein induces cell cycle progression and apoptosis through dimerization with Max. EMBO J. 1993, 12, 5083–5087. [Google Scholar] [CrossRef]
- Romero, O.A.; Torres-Diz, M.; Pros, E.; Savola, S.; Gomez, A.; Moran, S.; Saez, C.; Iwakawa, R.; Villanueva, A.; Montuenga, L.M.; et al. MAX inactivation in small cell lung cancer disrupts MYC-SWI/SNF programs and is synthetic lethal with BRG1. Cancer Discov. 2014, 4, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Foucquier, J.; Guedj, M. Analysis of drug combinations: Current methodological landscape. Pharmacol. Res. Perspect. 2015, 3, e00149. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M. Talazoparib: First Global Approval. Drugs 2018, 78, 1939–1946. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Lu, L.-Y.; Yu, X. The role of BRCA1 in DNA damage response. Protein Cell 2010, 1, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Holloman, W.K. Unraveling the mechanism of BRCA2 in homologous recombination. Nat. Struct. Mol. Biol. 2011, 18, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T. The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, Y.; Shan, W.; Hu, Z.; Yuan, J.; Pi, J.; Wang, Y.; Fan, L.; Tang, Z.; Li, C.; et al. Repression of BET activity sensitizes homologous recombination-proficient cancers to PARP inhibition. Sci. Transl. Med. 2017, 9, eaal1645. [Google Scholar] [CrossRef]
- Karakashev, S.; Zhu, H.; Yokoyama, Y.; Zhao, B.; Fatkhutdinov, N.; Kossenkov, A.V.; Wilson, A.J.; Simpkins, F.; Speicher, D.; Khabele, D.; et al. BET Bromodomain Inhibition Synergizes with PARP Inhibitor in Epithelial Ovarian Cancer. Cell Rep. 2017, 21, 3398–3405. [Google Scholar] [CrossRef]
- Sun, C.; Yin, J.; Fang, Y.; Chen, J.; Jeong, K.J.; Chen, X.; Vellano, C.P.; Ju, Z.; Zhao, W.; Zhang, D.; et al. BRD4 Inhibition Is Synthetic Lethal with PARP Inhibitors through the Induction of Homologous Recombination Deficiency. Cancer Cell 2018, 33, 401–416.e8. [Google Scholar] [CrossRef]
- Wilson, A.J.; Stubbs, M.; Liu, P.; Ruggeri, B.; Khabele, D. The BET inhibitor INCB054329 reduces homologous recombination efficiency and augments PARP inhibitor activity in ovarian cancer. Gynecol. Oncol. 2018, 149, 575–584. [Google Scholar] [CrossRef]
- Mio, C.; Gerratana, L.; Bolis, M.; Caponnetto, F.; Zanello, A.; Barbina, M.; Di Loreto, C.; Garattini, E.; Damante, G.; Puglisi, F. BET proteins regulate homologous recombination-mediated DNA repair: BRCAness and implications for cancer therapy. Int. J. Cancer 2018, 144, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.L.; Fehling, S.C.; Garcia, P.L.; Gamblin, T.L.; Council, L.N.; van Waardenburg, R.C.A.M.; Yang, E.S.; Bradner, J.E.; Yoon, K.J. The BET inhibitor JQ1 attenuates double-strand break repair and sensitizes models of pancreatic ductal adenocarcinoma to PARP inhibitors. EBioMedicine 2019, 44, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Chun-Jen Lin, C.; Mo, W.; Dai, H.; Park, Y.-Y.; Kim, S.M.; Peng, Y.; Mo, Q.; Siwko, S.; Hu, R.; et al. Genome-wide transcriptome profiling of homologous recombination DNA repair. Nat. Commun. 2014, 5, 3361–3372. [Google Scholar] [CrossRef] [PubMed]
- Hoheisel, J.D.; Alhamdani, M.S.S.; Schröder, C. Affinity-based microarrays for proteomic analysis of cancer tissues. Proteom. Clin. Appl. 2013, 7, 8–15. [Google Scholar] [CrossRef]
- Bahassi, E.M.; Ovesen, J.L.; Riesenberg, A.L.; Bernstein, W.Z.; Hasty, P.E.; Stambrook, P.J. The checkpoint kinases Chk1 and Chk2 regulate the functional associations between hBRCA2 and Rad51 in response to DNA damage. Oncogene 2008, 27, 3977–3985. [Google Scholar] [CrossRef]
- McCabe, N.; Turner, N.C.; Lord, C.J.; Kluzek, K.; Białkowska, A.; Swift, S.; Giavara, S.; O’Connor, M.J.; Tutt, A.N.; Zdzienicka, M.Z.; et al. Deficiency in the Repair of DNA Damage by Homologous Recombination and Sensitivity to Poly(ADP-Ribose) Polymerase Inhibition. Cancer Res. 2006, 66, 8109–8115. [Google Scholar] [CrossRef]
- Höglund, A.; Strömvall, K.; Li, Y.; Forshell, L.P.; Nilsson, J.A. Chk2 deficiency in Myc overexpressing lymphoma cells elicits a synergistic lethal response in combination with PARP inhibition. Cell Cycle 2014, 10, 3598–3607. [Google Scholar] [CrossRef]
- Dedes, K.J.; Wetterskog, D.; Mendes Pereira, A.M.; Natrajan, R.; Lambros, M.B.; Geyer, F.C.; Vatcheva, R.; Savage, K.; Mackay, A.; Lord, C.J.; et al. PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Sci. Transl. Med. 2010, 2, 53ra75. [Google Scholar] [CrossRef]
- Mansour, W.Y.; Tennstedt, P.; Volquardsen, J.; Oing, C.; Kluth, M.; Hube-Magg, C.; Borgmann, K.; Simon, R.; Petersen, C.; Dikomey, E.; et al. Loss of PTEN-assisted G2/M checkpoint impedes homologous recombination repair and enhances radio-curability and PARP inhibitor treatment response in prostate cancer. Sci. Rep. 2018, 8, 3947–3959. [Google Scholar] [CrossRef]
- Mendes Pereira, A.M.; Martin, S.A.; Brough, R.; McCarthy, A.; Taylor, J.R.; Kim, J.S.; Waldman, T.; Lord, C.J.; Ashworth, A. Synthetic lethal targeting of PTENmutant cells with PARP inhibitors. EMBO Mol. Med. 2009, 1, 315–322. [Google Scholar] [CrossRef]
- Huang, M.; Miao, Z.-H.; Zhu, H.; Cai, Y.-J.; Lu, W.; Ding, J. Chk1 and Chk2 are differentially involved in homologous recombination repair and cell cycle arrest in response to DNA double-strand breaks induced by camptothecins. Mol. Cancer Ther. 2008, 7, 1440–1449. [Google Scholar] [CrossRef] [PubMed]
- Niedzwiedz, W.; Mosedale, G.; Johnson, M.; Ong, C.Y.; Pace, P.; Patel, K.J. The Fanconi anaemia gene FANCC promotes homologous recombination and error-prone DNA repair. Mol. Cell 2004, 15, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Hirano, S.; Yamamoto, K.; Ishiai, M.; Yamazoe, M.; Seki, M.; Matsushita, N.; Ohzeki, M.; Yamashita, Y.M.; Arakawa, H.; Buerstedde, J.-M.; et al. Functional relationships of FANCC to homologous recombination, translesion synthesis, and BLM. EMBO J. 2005, 24, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Tauchi, H.; Kobayashi, J.; Morishima, K.-I.; van Gent, D.C.; Shiraishi, T.; Verkaik, N.S.; vanHeems, D.; Ito, E.; Nakamura, A.; Sonoda, E.; et al. Nbs1 is essential for DNA repair by homologous recombination in higher vertebrate cells. Nature 2002, 420, 93–98. [Google Scholar] [CrossRef]
- Lewis, T.W.; Barthelemy, J.R.; Virts, E.L.; Kennedy, F.M.; Gadgil, R.Y.; Wiek, C.; Linka, R.M.; Zhang, F.; Andreassen, P.R.; Hanenberg, H.; et al. Deficiency of the Fanconi anemia E2 ubiqitin conjugase UBE2T only partially abrogates Alu-mediated recombination in a new model of homology dependent recombination. Nucleic Acids Res. 2019, 47, 3503–3520. [Google Scholar] [CrossRef]
- Chen, S.; Wang, C.; Sun, L.; Wang, D.-L.; Chen, L.; Huang, Z.; Yang, Q.; Gao, J.; Yang, X.-B.; Chang, J.-F.; et al. RAD6 Promotes Homologous Recombination Repair by Activating the Autophagy-Mediated Degradation of Heterochromatin Protein HP1. Mol. Cell. Biol. 2014, 35, 406–416. [Google Scholar] [CrossRef]
- Liu, C.; Wang, D.; Wu, J.; Keller, J.; Ma, T.; Yu, X. RNF168 forms a functional complex with RAD6 during the DNA damage response. J. Cell Sci. 2013, 126, 2042–2051. [Google Scholar] [CrossRef]
- Nakada, S.; Tai, I.; Panier, S.; Al-Hakim, A.; Iemura, S.-I.; Juang, Y.-C.; O’Donnell, L.; Kumakubo, A.; Munro, M.; Sicheri, F.; et al. Non-canonical inhibition of DNA damage-dependent ubiquitination by OTUB1. Nature 2010, 466, 941–946. [Google Scholar] [CrossRef]
- Swinney, D.C.; Anthony, J. How were new medicines discovered? Nat. Rev. Drug Discov. 2011, 10, 507–519. [Google Scholar] [CrossRef]
- Moffat, J.G.; Rudolph, J.; Bailey, D. Phenotypic screening in cancer drug discovery—Past, present and future. Nat. Rev. Drug Discov. 2014, 13, 588–602. [Google Scholar] [CrossRef]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretić, L.; Kong, G.; Leenders, F.; Lu, X.; Fernández-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Hargrave-Thomas, E.; Yu, B.; Reynisson, J. Serendipity in anticancer drug discovery. World J. Clin. Oncol. 2012, 3, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Eastman, A. Improving anticancer drug development begins with cell culture: Misinformation perpetrated by the misuse of cytotoxicity assays. Oncotarget 2017, 8, 8854–8866. [Google Scholar] [CrossRef] [PubMed]
- Obinu, A.; Rassu, G.; Corona, P.; Maestri, M.; Riva, F.; Miele, D.; Giunchedi, P.; Gavini, E. Poly (ethyl 2-cyanoacrylate) nanoparticles (PECA-NPs) as possible agents in tumor treatment. Colloids Surf. B Biointerfaces 2019, 177, 520–528. [Google Scholar] [CrossRef]
- Vinci, M.; Gowan, S.; Boxall, F.; Patterson, L.; Zimmermann, M.; Court, W.; Lomas, C.; Mendiola, M.; Hardisson, D.; Eccles, S.A. Advances in establishment and analysis of three-dimensional tumor spheroid-based functional assays for target validation and drug evaluation. BMC Biol. 2012, 10, 29. [Google Scholar] [CrossRef]
- Fiorentino, F.P.; Bagella, L.; Marchesi, I. A new parameter of growth inhibition for cell proliferation assays. J. Cell. Physiol. 2018, 233, 4106–4115. [Google Scholar] [CrossRef]
- Schröder, C.; Srinivasan, H.; Sill, M.; Linseisen, J.; Fellenberg, K.; Becker, N.; Nieters, A.; Hoheisel, J.D. Plasma protein analysis of patients with different B-cell lymphomas using high-content antibody microarrays. Proteom. Clin. Appl. 2013, 7, 802–812. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Name of Cell Line | Hystotype | MYC/MAX Gene Familiy Status |
|---|---|---|---|
| 1 | N417 | SCLC | MYC amplification |
| 2 | H2171 | SCLC | MYC amplification |
| 3 | H446 | SCLC | MYC amplification |
| 4 | Lu135 | SCLC | MYC amplification |
| 5 | H1963 | SCLC | MYCL amplification |
| 6 | H2141 | SCLC | MYCL amplification |
| 7 | H69 | SCLC | MYCN amplification |
| 8 | Lu134 | SCLC | MAX inactivation |
| 9 | Lu165 | SCLC | MAX inactivation |
| 10 | H345 | SCLC | - |
| 11 | HECV | Umbilical cord | - |
| No. | Compound Name | Aliases | Class | Status in Clinical Trials |
|---|---|---|---|---|
| 1 | JQ1 | JQ-1[+] | BET inhibitor | Not evaluated |
| 2 | GSK525762 | I-BET762 | BET inhibitor | Phase 2 |
| 3 | EPZ-6438 | Tazemetostat | EZH2 inhibitor | Phase 3 |
| 4 | GSK2816126 | GSK-126 | EZH2 inhibitor | Phase 1 terminated prior to the completion due to an unfavorable benefit risk profile. |
| 5 | AZD2281 | Olaparib | PARP-1 inhibitor | FDA approved |
| 6 | BMN-673 | Talazoparib | PARP-1 inhibitor | FDA approved |
| 7 | PF-03814735 | - | Aurora Kinase Inhibitor | Phase 1 |
| 8 | PHA-739358 | Danusertib | Aurora Kinase Inhibitor | Phase 2 |
| Cell line | Concentration of I-BET762 | Population Doubling Time (h) | |
|---|---|---|---|
| I-BET762 | Talazoparib 5 nM + I-BET762 | ||
| H69 | - | 52.2 ± 2.3 | 55 ± 2.8 |
| 0.156 µM | 52.2 ± 1.4 | 57.4 ± 2.9 | |
| 0.312 µM | 53.7 ± 1.1 | 58.7 ± 1.3 | |
| 0.625 µM | 55.2 ± 2 | 64.2 ± 1.9 | |
| 1.25 µM | 58.5 ± 1.7 | 76.1 ± 2.1 | |
| 2.5 µM | 65.7 ± 2.4 | 87.6 ± 3.4 | |
| 5 µM | 76.8 ± 2.7 | 112.6 ± 5.2 | |
| Lu135 | - | 80 ± 4.9 | 119.5 ± 10.7 |
| 0.156 µM | 83 ± 4.8 | 163.1 ± 28.6 | |
| 0.312 µM | 84.5 ± 4.8 | 186.9 ± 29.7 | |
| 0.625 µM | 94.4 ± 6.7 | 202.3 ± 14.7 | |
| 1.25 µM | 110.4 ± 6.6 | 236.7 ± 16.6 | |
| 2.5 µM | 122.1 ± 9.9 | 287.6 ± 46.3 | |
| 5 µM | 172.2 ± 27.8 | 344.1 ± 42.7 | |
| N417 | - | 77.3 ± 5.1 | 113.5 ± 24.2 |
| 0.156 µM | 86.5 ± 7 | 128.2 ± 15.1 | |
| 0.312 µM | 93.7 ± 5.5 | 150.2 ± 32.7 | |
| 0.625 µM | 103.8 ± 5.8 | 179.5 ± 26 | |
| 1.25 µM | 137 ± 13.2 | 259.7 ± 45 | |
| 2.5 µM | 161.4 ± 11.7 | 262.4 ± 111.8 | |
| 5 µM | 218 ± 35 | 262.9 ± 101.3 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fiorentino, F.P.; Marchesi, I.; Schröder, C.; Schmidt, R.; Yokota, J.; Bagella, L. BET-Inhibitor I-BET762 and PARP-Inhibitor Talazoparib Synergy in Small Cell Lung Cancer Cells. Int. J. Mol. Sci. 2020, 21, 9595. https://doi.org/10.3390/ijms21249595
Fiorentino FP, Marchesi I, Schröder C, Schmidt R, Yokota J, Bagella L. BET-Inhibitor I-BET762 and PARP-Inhibitor Talazoparib Synergy in Small Cell Lung Cancer Cells. International Journal of Molecular Sciences. 2020; 21(24):9595. https://doi.org/10.3390/ijms21249595
Chicago/Turabian StyleFiorentino, Francesco Paolo, Irene Marchesi, Christoph Schröder, Ronny Schmidt, Jun Yokota, and Luigi Bagella. 2020. "BET-Inhibitor I-BET762 and PARP-Inhibitor Talazoparib Synergy in Small Cell Lung Cancer Cells" International Journal of Molecular Sciences 21, no. 24: 9595. https://doi.org/10.3390/ijms21249595
APA StyleFiorentino, F. P., Marchesi, I., Schröder, C., Schmidt, R., Yokota, J., & Bagella, L. (2020). BET-Inhibitor I-BET762 and PARP-Inhibitor Talazoparib Synergy in Small Cell Lung Cancer Cells. International Journal of Molecular Sciences, 21(24), 9595. https://doi.org/10.3390/ijms21249595