Building Blocks to Design Liposomal Delivery Systems



Abstract
1. Introduction
- Lipid composition: The diversity and molar ratio of lipids present in the bilayer directly impact membrane fluidity, permeability, and surface charge, as well as the loading capacity of drugs.
- Drug loading and release: The nature of the encapsulated drug, which can be either hydrophilic or lipophilic. The inclusion of stimuli-sensitive lipids or other components allows for a triggered drug release under specific conditions.
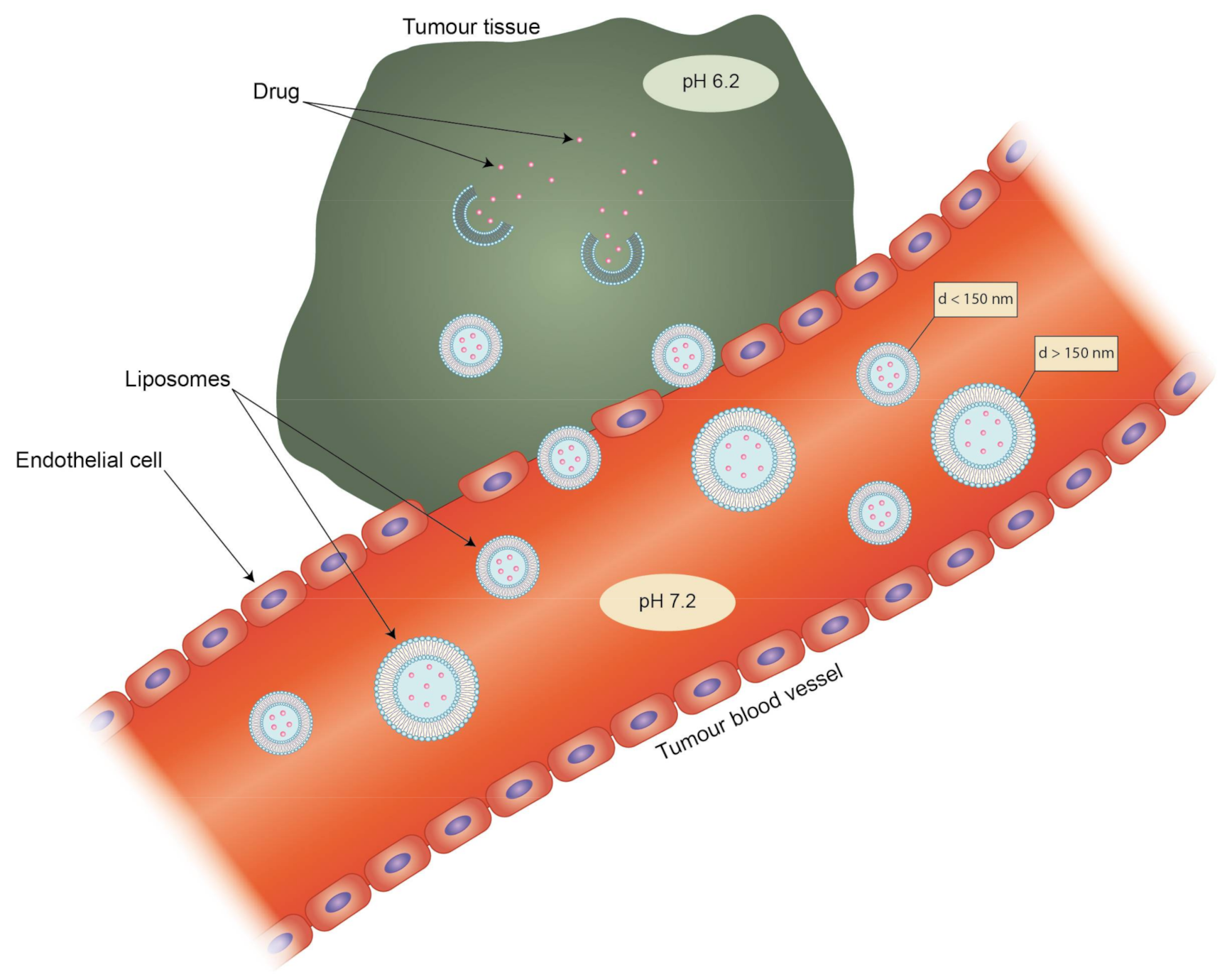
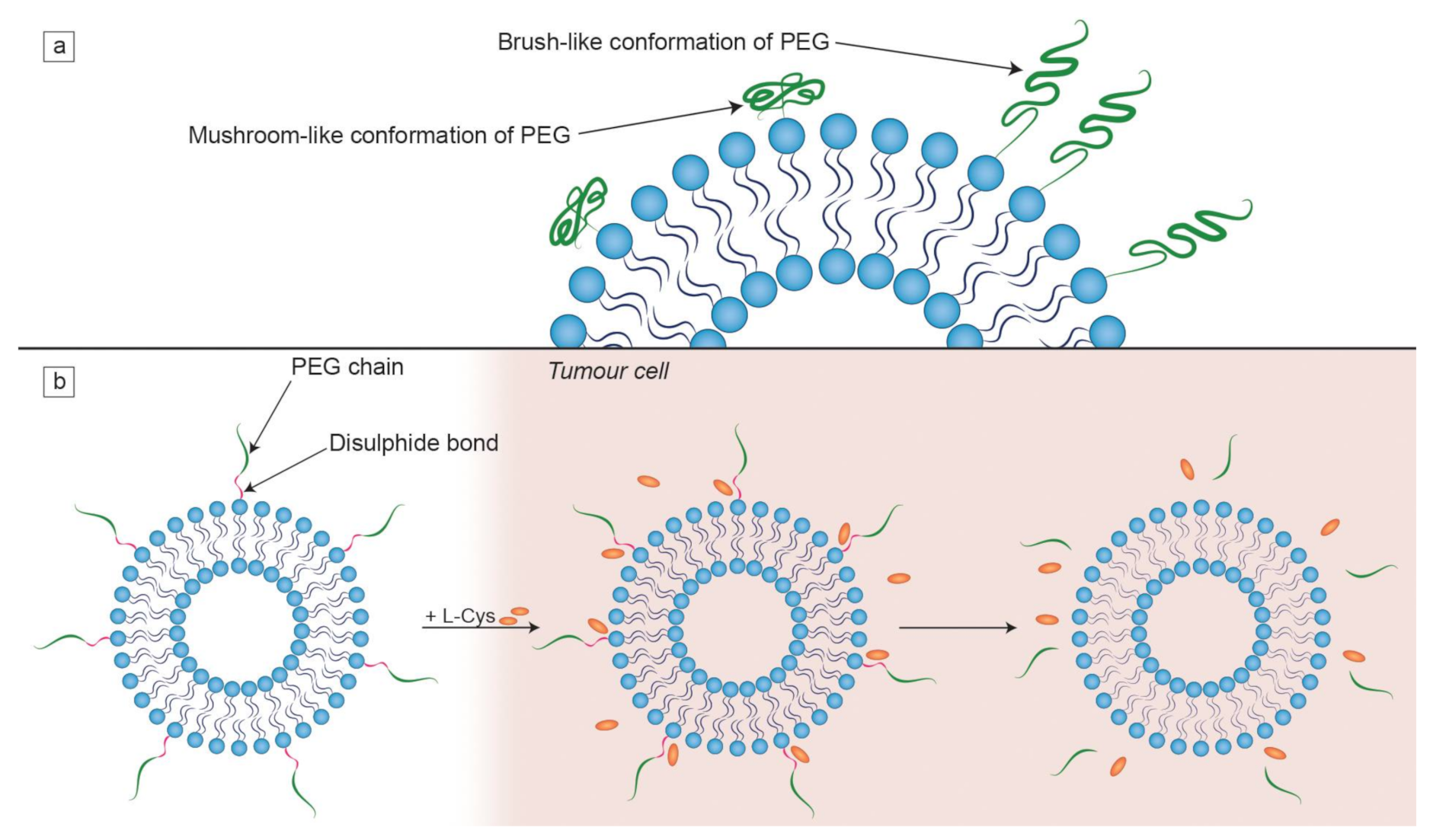
- Targeting methods: Active targeting by the attachment of ligands/molecules on the vesicle surface, which are preferentially (or exclusively) recognizable by target cells/tissues, and passive targeting through usage of the enhanced permeability and retention effect (EPR) effect. The vast majority of liposomal drug formulations contain “PEGylated lipids” (lipids with attached polyethylene glycol (PEG) chains) that affect the clearance of liposomes.
2. Lipid Composition
2.1. Phospholipids
2.2. Cholesterol
2.3. Charged Lipids
3. Drug Loading and Release
4. Targeting and Clearance
5. Individual Blocks That Make the Difference
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| MLV | multilamellar vesicles |
| SUV | small unilamellar vesicles |
| LUV | large unilamellar vesicles |
| SLN | solid lipid nanoparticles |
| NLC | nanostructured lipid carriers |
| EPR | enhanced permeability and retention effect |
| PEG | polyethylene glycol |
| POPC | 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine |
| DOPE | 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine |
| PE | phosphatidylethanolamine |
| DSPC | 1,2-distearoyl-sn-glycero-3-phosphocholine |
| LPC | lysophosphatidylcholine |
| Tm | main phase transition temperature |
| PC | phosphatidylcholine |
| DPPC | 1,2-dipalmitoyl-sn-glycero-3-phosphocholine |
| DMPC | 1,2-dimyristoyl-sn-glycero-3-phosphocholine |
| DOPC | 1,2-dioleoyl-sn-glycero-3-phosphocholine |
| Lo | liquid-ordered |
| Ld | liquid-disordered |
| RES | reticuloendothelial system |
| DMPG | 1,2-dimyristoyl-sn-glycero-3-phosphoglycerol |
| DMPS | 1,2-dimyristoyl-sn-glycero-3-phospho-L-serine |
| DPPG | 1,2-dipalmitoyl-sn-glycero-3-phosphorylglycerol |
| EMA | European Medicines Agency |
| FDA | Food and Drug Administration |
| HSPC | hydrogenated soya bean phosphatidylcholine |
| DSPG | 1,2-distearoyl-sn-glycero-3-phosphoglycerol |
| DEPC | 1,2-dierucoyl-sn-glycero-3-phosphocholine |
| DSPE-PEG(2000) | 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000] |
| EPC | acidic egg phosphatidylcholine |
| MPEG-2000-DSPE | 1,2-distearoyl-sn-glycero-3-phosphoethanolaMine-N-[biotinyl(polyethylene glycol)-2000] |
| DOPC | 1,2-dioleoyl-sn-glycero-3-phospho-L-serine |
| EPG | egg phosphatidylglycerol |
| DOTAP | 1,2-dioleoyl-3-trimethylammonium-propane |
| DOTMA | 2-di-O-octadecenyl-3-trimethylammonium propane |
| PEI | polyethylenimine |
| IgG | immunoglobulin G |
| BBB | blood–brain barrier |
| MPPC | 1-myristoyl-2-palmitoyl-sn-glycero-3-phosphocholine |
| MSPC | 1-myristoyl-2-stearoyl-sn-glycero-3-phosphocholine |
| MoSPC | monostearoyl-phosphatidylcholine |
| NIR | near-infrared |
| RFA | radiofrequency ablation |
| DSPE-PEG | PEGylated 1,2-distearoyl-sn-glycero-3-phosphorylethanolamine |
| AIA | adjuvant-induced arthritis |
| MPS | mononuclear phagocytic system |
| d | diameter |
| L-Cys | L-cysteine |
| hATTR | hereditary transthyretin-mediated |
| L-cl | liposome-coated lipoplex |
| HEPC | egg phosphatidylcholine |
| DC-Chol | 3b-(N-[dimethylaminoethane]carbamoyl)cholesterol) |
| HER2 | human epidermal growth factor receptor 2 |
| HSPC | hydrogenated soya bean phosphatidylcholine |
| EGFR | epidermal growth factor receptor |
| PKC | protein kinase C |
References
- Mozafari, M.R. Liposomes: An overview of manufacturing techniques. Cell. Mol. Biol. Lett. 2005, 10, 711–719. [Google Scholar]
- Gulati, M.; Grover, M.; Singh, S.; Singh, M. Lipophilic drug derivatives in liposomes. Int. J. Pharm. 1998, 165, 129–168. [Google Scholar] [CrossRef]
- Bozzuto, G.; Molinari, A. Liposomes as nanomedical devices. Int. J. Nanomed. 2015, 10, 975–999. [Google Scholar] [CrossRef] [PubMed]
- Elizondo, E.; Moreno, E.; Cabrera, I.; Córdoba, A.; Sala, S.; Veciana, J.; Ventosa, N. Liposomes and other vesicular systems: Structural characteristics, methods of preparation, and use in nanomedicine. In Progress in Molecular Biology and Translational Science; Villaverde, A., Ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2011; Volume 4, pp. 1–52. [Google Scholar]
- Danaei, M.; Dehghankhold, M.; Ataei, S.; Hasanzadeh Davarani, F.; Javanmard, R.; Dokhani, A.; Khorasani, S.; Mozafari, M.R. Impact of particle size and polydispersity index on the clinical applications of lipidic nanocarrier systems. Pharmaceutics 2018, 10, 1–17. [Google Scholar]
- Patil, Y.P.; Jadhav, S. Novel methods for liposome preparation. Chem. Phys. Lipids 2014, 177, 8–18. [Google Scholar] [CrossRef]
- Ruozi, B.; Belletti, D.; Tombesi, A.; Tosi, G.; Bondioli, L.; Forni, F.; Vandelli, M.A. AFM, ESEM, TEM, and CLSM in liposomal characterization: A comparative study. Int. J. Nanomed. 2011, 6, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Naseri, N.; Valizadeh, H.; Zakeri-Milani, P. Solid lipid nanoparticles and nanostructured lipid carriers: Structure preparation and application. Adv. Pharm. Bull. 2015, 5, 305–313. [Google Scholar] [CrossRef]
- Mennini, N.; Cirri, M.; Maestrelli, F.; Mura, P. Comparison of liposomal and NLC (nanostructured lipid carrier) formulations for improving the transdermal delivery of oxaprozin: Effect of cyclodextrin complexation. Int. J. Pharm. 2016, 515, 684–691. [Google Scholar] [CrossRef]
- Olechowski, F.; Müller, R.H.; Pyo, S.M. BergaCare SmartLipids: Commercial lipophilic active concentrates for improved performance of dermal products. Beilstein J. Nanotechnol. 2019, 10, 2152–2162. [Google Scholar] [CrossRef]
- Avasthi, P.; Marshall, W.F. Micelles and Nanoparticles for Ultrasonic Drug and Gene Delivery. Adv. Drug Deliv. Rev. 2008, 60, 1137–1152. [Google Scholar]
- Cullis, P.R.; Hope, M.J. Lipid Nanoparticle Systems for Enabling Gene Therapies. Mol. Ther. 2017, 25, 1467–1475. [Google Scholar] [CrossRef] [PubMed]
- Coimbra, M.; Isacchi, B.; Van Bloois, L.; Torano, J.S.; Ket, A.; Wu, X.; Broere, F.; Metselaar, J.M.; Rijcken, C.J.F.; Storm, G.; et al. Improving solubility and chemical stability of natural compounds for medicinal use by incorporation into liposomes. Int. J. Pharm. 2011, 416, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Hamill, R.J. Amphotericin B formulations: A comparative review of efficacy and toxicity. Drugs 2013, 73, 919–934. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K. Clinical usefulness of liposomal formulations in cancer therapy: Lessons from the experiences of doxorubicin. J. Pharm. Investig. 2019, 49, 203–214. [Google Scholar] [CrossRef]
- Larabi, M.; Pages, N.; Pons, F.; Appel, M.; Gulik, A.; Schalatter, J.; Bouvet, S.; Barratt, G. Study of the toxicity of a new lipid complex formulation of amphotericin B. J. Antimicrob. Chemother. 2004, 53, 81–88. [Google Scholar] [CrossRef]
- Adler-Moore, J.P.; Proffitt, R.T. Development, characterization, efficacy and mode of action of ambisome, a unilamellar liposomal formulation of amphotericin b. J. Liposome Res. 1993, 3, 429–450. [Google Scholar] [CrossRef]
- Patel, M.A.; Gadsden, J.C.; Nedeljkovic, S.S.; Bao, X.; Zeballos, J.L.; Yu, V.; Ayad, S.S.; Bendtsen, T.F. Brachial Plexus Block with Liposomal Bupivacaine for Shoulder Surgery Improves Analgesia and Reduces Opioid Consumption: Results from a Multicenter, Randomized, Double-Blind, Controlled Trial. Pain Med. 2020, 21, 387–400. [Google Scholar] [CrossRef]
- Richard, B.M.; Rickert, D.E.; Newton, P.E.; Ott, L.R.; Haan, D.; Brubaker, A.N.; Cole, P.I.; Ross, P.E.; Rebelatto, M.C.; Nelson, K.G. Safety Evaluation of EXPAREL (DepoFoam Bupivacaine) Administered by Repeated Subcutaneous Injection in Rabbits and Dogs: Species Comparison. J. Drug Deliv. 2011, 2011, 1–14. [Google Scholar] [CrossRef]
- Glantz, M.J.; Jaeckle, K.A.; Chamberlain, M.C.; Phuphanich, S.; Recht, L.; Swinnen, L.J.; Maria, B.; LaFollette, S.; Schumann, G.B.; Cole, B.F.; et al. A randomized controlled trial comparing intrathecal sustained-release cytarabine (DepoCyt) to intrathecal methotrexate in patients with neoplastic meningitis from solid tumors. Clin. Cancer Res. 1999, 5, 3394–3402. [Google Scholar]
- Forssen, E.A. The design and development of DaunoXome® for solid tumor targeting in vivo. Adv. Drug Deliv. Rev. 1997, 24, 133–150. [Google Scholar] [CrossRef]
- Martin, F.; Huang, A.; Uziely, B.; Kaufman, B.; Safra, T. Prolonged Circulation Time and Enhanced Accumulation in Malignant Exudates of Doxorubicin Encapsulated in Polyethylene-glycol Coated Liposomes. Cancer Res. 1994, 54, 987–992. [Google Scholar]
- Chao, T.C.; Wang, W.S.; Yen, C.C.; Chiou, T.J.; Liu, J.H.; Chen, P.M. A Dose-Escalating Pilot Study of Sterically Stabilized, Pegylated Liposomal Doxorubicin (Lipo-Dox®) in Patients with Metastatic Breast Cancer. Cancer Investig. 2003, 21, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Swenson, C.E.; Perkins, W.R.; Roberts, P.; Janoff, A.S. Liposome technology and the development of MyocetTM (liposomal doxorubicin citrate). Breast 2001, 10, 1–7. [Google Scholar] [CrossRef]
- Usonis, V.; Bakasénas, V.; Valentelis, R.; Katiliene, G.; Vidzeniene, D.; Herzog, C. Antibody titres after primary and booster vaccination of infants and young children with a virosomal hepatitis A vaccine (Epaxal®). Vaccine 2003, 21, 4588–4592. [Google Scholar] [CrossRef]
- Mischler, R.; Metcalfe, I.C. Inflexal®V a trivalent virosome subunit influenza vaccine: Production. Vaccine 2002, 20, 5–11. [Google Scholar] [CrossRef]
- Zhang, H. Onivyde for the therapy of multiple solid tumors. OncoTargets Ther. 2016, 9, 3001–3007. [Google Scholar] [CrossRef]
- Venkatakrishnan, K.; Liu, Y.; Noe, D.; Mertz, J.; Bargfrede, M.; Marbury, T.; Farbakhsh, K.; Oliva, C.; Milton, A. Pharmacokinetics and pharmacodynamics of liposomal mifamurtide in adult volunteers with mild or moderate hepatic impairment. Br. J. Clin. Pharmacol. 2014, 77, 998–1010. [Google Scholar] [CrossRef]
- Keck, S.; Glennon, C.; Ginsberg, B. DepoDur® extended-release epidural morphine: Reshaping postoperative care: What perioperative nurses need to know. Orthop. Nurs. 2007, 26, 86–93. [Google Scholar] [CrossRef]
- Bressler, N.M.; Bressler, S.B. Photodynamic Therapy with Verteporfin (Visudyne): Impact on Ophthalmology and Visual Sciences. Iovs 2000, 41, 624–628. [Google Scholar]
- Bedikian, A.Y.; Silverman, J.A.; Papadopoulos, N.E.; Kim, K.B.; Hagey, A.E.; Vardeleon, A.; Hwu, W.J.; Homsi, J.; Davies, M.; Hwu, P. Pharmacokinetics and safety of Marqibo (vincristine sulfate liposomes injection) in cancer patients with impaired liver function. J. Clin. Pharmacol. 2011, 51, 1205–1212. [Google Scholar] [CrossRef]
- Cullis, P.R.; De Kruijff, B. Lipid polymorphism and the functional roles of lipids in biological membranes. BBA Rev. Biomembr. 1979, 559, 399–420. [Google Scholar] [CrossRef]
- Mouritsen, O.G. Lipids, curvature, and nano-medicine. Eur. J. Lipid Sci. Technol. 2011, 113, 1174–1187. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.B.; Kennard, C.; Pedersen, S.L.; Jensen, K.J.; Uline, M.J.; Hatzakis, N.S.; Stamou, D. Membrane Curvature and Lipid Composition Synergize To Regulate N-Ras Anchor Recruitment. Biophys. J. 2017, 113, 1269–1279. [Google Scholar] [CrossRef] [PubMed]
- Lamichhane, N.; Udayakumar, T.S.; D’Souza, W.D.; Simone, C.B.; Raghavan, S.R.; Polf, J.; Mahmood, J. Liposomes: Clinical applications and potential for image-guided drug delivery. Molecules 2018, 23, 288. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Zhang, Q. Development of liposomal formulations: From concept to clinical investigations. Asian J. Pharm. Sci. 2013, 8, 81–87. [Google Scholar] [CrossRef]
- Blicher, A.; Wodzinska, K.; Fidorra, M.; Winterhalter, M.; Heimburg, T. The temperature dependence of lipid membrane permeability, its quantized nature, and the influence of anesthetics. Biophys. J. 2009, 96, 4581–4591. [Google Scholar] [CrossRef] [PubMed]
- Cruzeiro-Hansson, L.; Mouritsen, O.G. Passive ion permeability of lipid membranes modelled via lipid-domain interfacial area. BBA Biomembr. 1988, 944, 63–72. [Google Scholar] [CrossRef]
- Sercombe, L.; Veerati, T.; Moheimani, F.; Wu, S.Y.; Sood, A.K.; Hua, S. Advances and challenges of liposome assisted drug delivery. Front. Pharmacol. 2015, 6, 1–13. [Google Scholar] [CrossRef]
- Rawicz, W.; Olbrich, K.C.; Mcintosh, T.; Needham, D.; Evans, E. Effect of Chain Length and Unsaturation on Elasticity of Lipid Bilayers. Biophys J. 2000, 79, 1–12. [Google Scholar] [CrossRef]
- van Hoogevest, P.; Wendel, A. The use of natural and synthetic phospholipids as pharmaceutical excipients. Eur. J. Lipid Sci. Technol. 2014, 116, 1088–1107. [Google Scholar] [CrossRef]
- Yeh, M.K.; Chang, H.I. Clinical development of liposome-based drugs: Formulation, characterization, and therapeutic efficacy. Int. J. Nanomed. 2012, 7, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.C.; Crist, R.M.; Clogston, J.D.; McNeil, S.E. Zeta potential: A case study of cationic, anionic, and neutral liposomes. Anal. Bioanal. Chem. 2017, 409, 5779–5787. [Google Scholar] [CrossRef] [PubMed]
- Klasczyk, B.; Knecht, V.; Lipowsky, R.; Dimova, R. Interactions of alkali metal chlorides with phosphatidylcholine vesicles. Langmuir 2010, 26, 18951–18958. [Google Scholar] [CrossRef] [PubMed]
- Mombelli, E.; Morris, R.; Taylor, W.; Fraternali, F. Hydrogen-bonding propensities of sphingomyelin in solution and in a bilayer assembly: A molecular dynamics study. Biophys. J. 2003, 84, 1507–1517. [Google Scholar] [CrossRef]
- Steinbauer, B.; Mehnert, T.; Beyer, K. Hydration and lateral organization in phospholipid bilayers containing sphingomyelin: A 2H-NMR study. Biophys. J. 2003, 85, 1013–1024. [Google Scholar] [CrossRef][Green Version]
- Arsov, Z.; González-Ramírez, E.J.; Goñi, F.M.; Tristram-Nagle, S.; Nagle, J.F. Phase behavior of palmitoyl and egg sphingomyelin. Chem. Phys. Lipids 2018, 213, 102–110. [Google Scholar] [CrossRef]
- Taylor, K.M.G.; Morris, R.M. Thermal analysis of phase transition behaviour in liposomes. Thermochim. Acta 1995, 248, 289–301. [Google Scholar] [CrossRef]
- Çağdaş, M.; Sezer, A.D.; Bucak, S. Liposomes as Potential Drug Carrier Systems for Drug Delivery. In Application of Nanotechnology in Drug Delivery; IntechOpen: London, UK, 2014. [Google Scholar]
- Chang, H.-C.; Flanagan, D.R. Liposomal entrapment of suramin(II): Interaction of suramin with phospholipids of various chain lengths. J. Pharm. Sci. 1995, 84, 1078–1082. [Google Scholar] [CrossRef]
- Anderson, M.; Omri, A. The Effect of Different Lipid Components on the in Vitro Stability and Release Kinetics of Liposome Formulations. Drug Deliv. J. Deliv. Target. Ther. Agents 2004, 11, 33–39. [Google Scholar] [CrossRef]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal formulations in clinical use: An updated review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef]
- Ermilova, I.; Lyubartsev, A.P. Cholesterol in phospholipid bilayers: Positions and orientations inside membranes with different unsaturation degrees. Soft Matter 2019, 15, 78–93. [Google Scholar] [CrossRef] [PubMed]
- Scherfeld, D.; Kahya, N.; Schwille, P. Lipid Dynamics and Domain Formation in Model Membranes Composed of Ternary Mixtures of Unsaturated and Saturated Phosphatidylcholines and Cholesterol. Biophys. J. 2003, 85, 3758–3768. [Google Scholar] [CrossRef]
- Sinha, M.; Mishra, S.; Joshi, P.G. Liquid-ordered microdomains in lipid rafts and plasma membrane of U-87 MG cells: A time-resolved fluorescence study. Eur. Biophys. J. 2003, 32, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Sampaio, J.L. Membrane organization and lipid rafts. Cold Spring Harb. Perspect. Biol. 2011, 3, a004697. [Google Scholar] [CrossRef] [PubMed]
- Grzybek, M.; Kubiak, J.; Łach, A.; Przybyło, M.; Sikorski, A.F. A raft-associated species of phosphatidylethanolamine interacts with cholesterol comparably to sphingomyelin. A Langmuir-Blodgett monolayer study. PLoS ONE 2009, 4, e5053. [Google Scholar] [CrossRef]
- Demel, R.A.; De Kruyff, B. The function of sterols in membranes. BBA Rev. Biomembr. 1976, 457, 109–132. [Google Scholar] [CrossRef]
- Mannock, D.A.; Lewis, R.N.A.H.; McElhaney, R.N. Comparative calorimetric and spectroscopic studies of the effects of lanosterol and cholesterol on the thermotropic phase behavior and organization of dipalmitoylphosphatidylcholine bilayer membranes. Biophys. J. 2006, 91, 3327–3340. [Google Scholar] [CrossRef]
- Subczynski, W.K.; Pasenkiewicz-Gierula, M.; Widomska, J.; Mainali, L.; Raguz, M. High Cholesterol/Low Cholesterol: Effects in Biological Membranes: A Review. Cell Biochem. Biophys. 2017, 75, 369–385. [Google Scholar] [CrossRef]
- Magarkar, A.; Dhawan, V.; Kallinteri, P.; Viitala, T.; Elmowafy, M.; Róg, T.; Bunker, A. Cholesterol level affects surface charge of lipid membranes in saline solution. Sci. Rep. 2014, 4, 1–5. [Google Scholar] [CrossRef]
- Briuglia, M.L.; Rotella, C.; McFarlane, A.; Lamprou, D.A. Influence of cholesterol on liposome stability and on in vitro drug release. Drug Deliv. Transl. Res. 2015, 5, 231–242. [Google Scholar] [CrossRef]
- Abu Lila, A.S.; Ishida, T. Liposomal Delivery Systems: Design Optimization and Current Applications. Biol. Pharm. Bull. Pharm. Bull. 2016, 40, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Eloy, J.O.; Claro de Souza, M.; Petrilli, R.; Barcellos, J.P.A.; Lee, R.J.; Marchetti, J.M. Liposomes as carriers of hydrophilic small molecule drugs: Strategies to enhance encapsulation and delivery. Coll. Surf. B Biointerfaces 2014, 123, 345–363. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Yu, M.; Miao, Y.; He, S.; Dai, Z.; Song, W.; Liu, Y.; Song, S.; Ahmad, E.; Wang, D.; et al. Cholesterol-tuned liposomal membrane rigidity directs tumor penetration and anti-tumor effect. Acta Pharm. Sin. B 2019, 9, 858–870. [Google Scholar] [CrossRef] [PubMed]
- Matusewicz, L.; Podkalicka, J.; Sikorski, A.F. Immunoliposomes with simvastatin as a potential therapeutic in treatment of breast cancer cells overexpressing her2—An in vitro study. Cancers 2018, 10, 418. [Google Scholar] [CrossRef] [PubMed]
- Harashima, H.; Matsuo, H.; Kiwada, H. Identification of proteins mediating clearance of liposomes using a liver perfusion system. Adv. Drug Deliv. Rev. 1998, 32, 61–79. [Google Scholar] [CrossRef]
- Caracciolo, G.; Pozzi, D.; Capriotti, A.L.; Cavaliere, C.; Piovesana, S.; La Barbera, G.; Amici, A.; Laganà, A. The liposome-protein corona in mice and humans and its implications for in vivo delivery. J. Mater. Chem. B 2014, 2, 7419–7428. [Google Scholar] [CrossRef]
- Foteini, P.; Pippa, N.; Naziris, N.; Demetzos, C. Physicochemical study of the protein–liposome interactions: Influence of liposome composition and concentration on protein binding. J. Liposome Res. 2019, 29, 313–321. [Google Scholar] [CrossRef]
- Lauraeus, S.; Holopainen, J.M.; Taskinen, M.R.; Kinnunen, P.K.J. Aggregation of dimyristoylphosphatidylglycerol liposomes by human plasma low density lipoprotein. Biochim. Biophys. Acta Biomembr. 1998, 1373, 147–162. [Google Scholar] [CrossRef]
- Kerek, E.M.; Prenner, E.J. Inorganic cadmium affects the fluidity and size of phospholipid based liposomes. Biochim. Biophys. Acta Biomembr. 2016, 1858, 3169–3181. [Google Scholar] [CrossRef]
- Allen, T.M.; Cullis, P.R. Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef]
- Sawant, R.R.; Torchilin, V.P. Liposomes as’smart’ pharmaceutical nanocarriers. Soft Matter 2010, 6, 4026–4044. [Google Scholar] [CrossRef]
- Zhi, D.; Zhang, S.; Wang, B.; Zhao, Y.; Yang, B.; Yu, S. Transfection efficiency of cationic lipids with different hydrophobic domains in gene delivery. Bioconjug. Chem. 2010, 21, 563–577. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Bajaj, A. Advances in gene delivery through molecular design of cationic lipids. Chem. Commun. 2009, 4632–4656. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.J.; Liu, Y.H.; Zhang, J.; Zhang, Y.; Xia, Y.; Yu, X.Q. Cyclen-based cationic lipids with double hydrophobic tails for efficient gene delivery. Biomater. Sci. 2014, 2, 1460–1470. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Zhang, S.; Wang, B.; Cui, S.; Yan, J. Toxicity of cationic lipids and cationic polymers in gene delivery. J. Control. Release 2006, 114, 100–109. [Google Scholar] [CrossRef]
- Simões, S.; Filipe, A.; Faneca, H.; Mano, M.; Penacho, N.; Düzgünes, N.; de Lima, M.P. Cationic liposomes for gene delivery. Expert Opin. Drug Deliv. 2005, 2, 237–254. [Google Scholar] [CrossRef]
- Koltover, I.; Salditt, T.; Rädler, J.O.; Safinya, C.R. An inverted hexagonal phase of cationic liposome-DNA complexes related to DNA release and delivery. Science 1998, 281, 78–81. [Google Scholar] [CrossRef]
- Kolašinac, R.; Kleusch, C.; Braun, T.; Merkel, R.; Csiszár, A. Deciphering the functional composition of fusogenic liposomes. Int. J. Mol. Sci. 2018, 19, 346. [Google Scholar] [CrossRef]
- Yasuda, S.; Yoshida, H.; Nishikawa, M.; Takakura, Y. Comparison of the type of liposome involving cytokine production induced by non-cpG lipoplex in macrophages. Mol. Pharm. 2010, 7, 533–542. [Google Scholar] [CrossRef]
- Song, Y.K. Characterization of cationic liposome-mediated gene transfer in vivo by intravenous administration. Hum. Gene Ther. 1997, 8, 1585–1594. [Google Scholar] [CrossRef]
- Li, M.; Du, C.; Guo, N.; Teng, Y.; Meng, X.; Sun, H.; Li, S.; Yu, P.; Galons, H. Composition design and medical application of liposomes. Eur. J. Med. Chem. 2019, 164, 640–653. [Google Scholar] [CrossRef] [PubMed]
- Benjaminsen, R.V.; Mattebjerg, M.A.; Henriksen, J.R.; Moghimi, S.M.; Andresen, T.L. The possible “proton sponge” effect of polyethylenimine (PEI) does not include change in lysosomal pH. Mol. Ther. 2013, 21, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Rezaee, M.; Oskuee, R.K.; Nassirli, H.; Malaekeh-Nikouei, B. Progress in the development of lipopolyplexes as efficient non-viral gene delivery systems. J. Control. Release 2016, 236, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Levchenko, T.S.; Rammohan, R.; Lukyanov, A.N.; Whiteman, K.R.; Torchilin, V.P. Liposome clearance in mice: The effect of a separate and combined presence of surface charge and polymer coating. Int. J. Pharm. 2002, 240, 95–102. [Google Scholar] [CrossRef]
- Aggarwal, P.; Hall, J.B.; McLeland, C.B.; Dobrovolskaia, M.A.; McNeil, S.E. Nanoparticle interaction with plasma proteins as it relates to particle biodistribution, biocompatibility and therapeutic efficacy. Adv. Drug Deliv. Rev. 2009, 61, 428–437. [Google Scholar] [CrossRef]
- Campos-Martorell, M.; Cano-Sarabia, M.; Simats, A.; Hernández-Guillamon, M.; Rosell, A.; Maspoch, D.; Montaner, J. Charge effect of a liposomal delivery system encapsulating simvastatin to treat experimental ischemic stroke in rats. Int. J. Nanomed. 2016, 11, 3035–3048. [Google Scholar]
- Pauli, G.; Tang, W.L.; Li, S.D. Development and characterization of the solvent-assisted active loading technology (SALT) for liposomal loading of poorly water-soluble compounds. Pharmaceutics 2019, 11, 465. [Google Scholar] [CrossRef]
- Gubernator, J. Active methods of drug loading into liposomes: Recent strategies for stable drug entrapment and increased in vivo activity. Expert Opin. Drug Deliv. 2011, 8, 565–580. [Google Scholar] [CrossRef]
- Nounou, M.M.; El-Khordagui, L.K.; Khalafallah, N.A.; Khalil, S.A. In vitro release of hydrophilic and hydrophobic drugs from liposomal dispersions and gels. Acta Pharm. 2006, 56, 311–324. [Google Scholar]
- Bhardwaj, U.; Burgess, D.J. Physicochemical properties of extruded and non-extruded liposomes containing the hydrophobic drug dexamethasone. Int. J. Pharm. 2010, 388, 181–189. [Google Scholar] [CrossRef]
- Sawant, R.R.; Torchilin, V.P. Challenges in development of targeted liposomal therapeutics. AAPS J. 2012, 14, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yang, Y.; Zhang, F.C.; Wu, C.; Lü, W.L.; Mei, X.G. Epirubicin-encapsulated long-circulating thermosensitive liposome improves pharmacokinetics and antitumor therapeutic efficacy in animals. J. Liposome Res. 2011, 21, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Zangabad, P.S.; Mirkiani, S.; Shahsavari, S.; Masoudi, B.; Masroor, M.; Hamed, H.; Jafari, Z.; Taghipour, Y.D.; Hashemi, H.; Karimi, M.; et al. Stimulus-responsive liposomes as smart nanoplatforms for drug delivery applications. Nanotechnol. Rev. 2018, 7, 95–122. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Su, J.; Wu, K.; Ma, W.; Wang, B.; Li, M.; Sun, P.; Shen, Q.; Wang, Q.; Fan, Q. Multifunctional Thermosensitive Liposomes Based on Natural Phase-Change Material: Near-Infrared Light-Triggered Drug Release and Multimodal Imaging-Guided Cancer Combination Therapy. ACS Appl. Mater. Interfaces 2019, 11, 10540–10553. [Google Scholar] [CrossRef]
- Nguyen, V.D.; Min, H.K.; Kim, C.S.; Han, J.; Park, J.O.; Choi, E. Folate receptor-targeted liposomal nanocomplex for effective synergistic photothermal-chemotherapy of breast cancer in vivo. Coll. Surf. B Biointerfaces 2019, 173, 539–548. [Google Scholar] [CrossRef]
- Landon, C.D.; Park, J.Y.; Needham, D.; Dewhirst, M.W. Nanoscale drug delivery and hyperthermia: The materials design and preclinical and clinical testing of low temperature-sensitive liposomes used in combination with mild hyperthermia in the treatment of local cancer. Open Nanomed. J. 2011, 3, 38–64. [Google Scholar] [CrossRef]
- Poon, R.T.P.; Borys, N. Lyso-thermosensitive liposomal doxorubicin: A novel approach to enhance efficacy of thermal ablation of liver cancer. Expert Opin. Pharmacother. 2009, 10, 333–343. [Google Scholar] [CrossRef]
- Lencioni, R.; Cioni, D. RFA plus lyso-thermosensitive liposomal doxorubicin: In search of the optimal approach to cure intermediate-size hepatocellular carcinoma. Hepatic Oncol. 2016, 3, 193–200. [Google Scholar] [CrossRef]
- Celik, H.; Wakim, P.; Pritchard, W.F.; Castro, M.; Leonard, S.; Karanian, J.W.; Dewhirst, M.W.; Lencioni, R.; Wood, B.J. Radiofrequency Ablation Duration per Tumor Volume May Correlate with Overall Survival in Solitary Hepatocellular Carcinoma Patients Treated with Radiofrequency Ablation Plus Lyso-Thermosensitive Liposomal Doxorubicin. J. Vasc. Interv. Radiol. 2019, 30, 1908–1914. [Google Scholar] [CrossRef]
- Chen, K.J.; Liang, H.F.; Chen, H.L.; Wang, Y.; Cheng, P.Y.; Liu, H.L.; Xia, Y.; Sung, H.W. A thermoresponsive bubble-generating liposomal system for triggering localized extracellular drug delivery. ACS Nano 2013, 7, 438–446. [Google Scholar] [CrossRef]
- Chen, K.J.; Chaung, E.Y.; Wey, S.P.; Lin, K.J.; Cheng, F.; Lin, C.C.; Liu, H.L.; Tseng, H.W.; Liu, C.P.; Wei, M.C.; et al. Hyperthermia-mediated local drug delivery by a bubble-generating liposomal system for tumor-specific chemotherapy. ACS Nano 2014, 8, 5105–5115. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zou, T.; Dai, M.; He, X.Y.; Peng, N.; Wu, K.; Wang, X.Q.; Liao, C.Y.; Liu, Y. Doxorubicin loaded tumor-triggered targeting ammonium bicarbonate liposomes for tumor-specific drug delivery. Coll. Surf. B Biointerfaces 2019, 178, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Paliwal, S.R.; Paliwal, R.; Vyas, S.P. A review of mechanistic insight and application of pH-sensitive liposomes in drug delivery. Drug Deliv. 2015, 22, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Barattin, M.; Mattarei, A.; Balasso, A.; Paradisi, C.; Cantù, L.; Del Favero, E.; Viitala, T.; Mastrotto, F.; Caliceti, P.; Salmaso, S. PH-Controlled Liposomes for Enhanced Cell Penetration in Tumor Environment. ACS Appl. Mater. Interfaces 2018, 10, 17646–17661. [Google Scholar] [CrossRef]
- Li, J.; Wang, X.; Zhang, T.; Wang, C.; Huang, Z.; Luo, X.; Deng, Y. A review on phospholipids and their main applications in drug delivery systems. Asian J. Pharm. Sci. 2014, 10, 81–98. [Google Scholar] [CrossRef]
- Yin, X.; Chi, Y.; Guo, C.; Feng, S.; Liu, J.; Sun, K.; Wu, Z. Chitooligosaccharides Modified Reduction-Sensitive Liposomes: Enhanced Cytoplasmic Drug Delivery and Osteosarcomas-Tumor Inhibition in Animal Models. Pharm. Res. 2017, 34, 2172–2184. [Google Scholar] [CrossRef]
- Merino, M.; Zalba, S.; Garrido, M.J. Immunoliposomes in clinical oncology: State of the art and future perspectives. J. Control. Release 2018, 275, 162–176. [Google Scholar] [CrossRef]
- Jia, M.; Deng, C.; Luo, J.; Zhang, P.; Sun, X.; Zhang, Z.; Gong, T. A novel dexamethasone-loaded liposome alleviates rheumatoid arthritis in rats. Int. J. Pharm. 2018, 540, 57–64. [Google Scholar] [CrossRef]
- Bertrand, N.; Leroux, J.C. The journey of a drug-carrier in the body: An anatomo-physiological perspective. J. Control. Release 2012, 161, 152–163. [Google Scholar] [CrossRef]
- Cho, K.; Wang, X.; Nie, S.; Chen, Z.; Shin, D.M. Therapeutic nanoparticles for drug delivery in cancer. Clin. Cancer Res. 2008, 14, 1310–1316. [Google Scholar] [CrossRef]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Powell, D.; Chandra, S.; Dodson, K.; Shaheen, F.; Wiltz, K.; Ireland, S.; Syed, M.; Dash, S.; Wiese, T.; Mandal, T.; et al. Aptamer-functionalized hybrid nanoparticle for the treatment of breast cancer. Eur. J. Pharm. Biopharm. 2017, 114, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Lammers, T.; Hennink, W.E.; Storm, G. Tumour-targeted nanomedicines: Principles and practice. Br. J. Cancer 2008, 99, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.; Abu Lila, A.S.; Shimizu, T.; Alaaeldin, E.; Hussein, A.; Sarhan, H.A.; Szebeni, J.; Ishida, T. PEGylated liposomes: Immunological responses. Sci. Technol. Adv. Mater. 2019, 20, 710–724. [Google Scholar] [CrossRef] [PubMed]
- Nag, O.K.; Awasthi, V. Surface engineering of liposomes for stealth behavior. Pharmaceutics 2013, 5, 542–569. [Google Scholar] [CrossRef]
- Labouta, H.I.; Gomez-Garcia, M.J.; Sarsons, C.D.; Nguyen, T.; Kennard, J.; Ngo, W.; Terefe, K.; Iragorri, N.; Lai, P.; Rinker, K.D.; et al. Surface-grafted polyethylene glycol conformation impacts the transport of PEG-functionalized liposomes through a tumour extracellular matrix model. RSC Adv. 2018, 8, 7697–7708. [Google Scholar] [CrossRef]
- Barenholz, Y. Doxil®—The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Środa, K.; Rydlewski, J.; Langner, M.; Kozubek, A.; Grzybek, M.; Sikorski, A.F. Repeated injections of PEG-PE liposomes generate anti-PEG antibodies. Cell. Mol. Biol. Lett. 2005, 10, 37–47. [Google Scholar]
- Thai, T.T.H.; Pilkington, E.H.; Nguyen, D.H.; Lee, J.S.; Park, K.D.; Truong, N.P. The Importance of Poly (ethylene glycol) Alternatives for Overcoming PEG Immunogenicity in Drug. Polymers 2020, 12, 298. [Google Scholar] [CrossRef]
- Kuai, R.; Yuan, W.; Qin, Y.; Chen, H.; Tang, J.; Yuan, M.; Zhang, Z.; He, Q. Efficient delivery of payload into tumor cells in a controlled manner by TAT and thiolytic cleavable PEG Co-modified liposomes. Mol. Pharm. 2010, 7, 1816–1826. [Google Scholar] [CrossRef]
- Zhang, X.; Goel, V.; Robbie, G.J. Pharmacokinetics of Patisiran, the First Approved RNA Interference Therapy in Patients With Hereditary Transthyretin-Mediated Amyloidosis. J. Clin. Pharmacol. 2019, 60, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Toporkiewicz, M.; Meissner, J.; Matusewicz, L.; Czogalla, A.; Sikorski, A.F. Toward a magic or imaginary bullet? Ligands for drug targeting to cancer cells: Principles, hopes, and challenges. Int. J. Nanomed. 2015, 10, 1399–1414. [Google Scholar] [PubMed]
- Klibanov, A.L.; Maruyama, K.; Beckerleg, A.M.; Torchilin, V.P.; Huang, L. Activity of amphipathic poly(ethylene glycol) 5000 to prolong the circulation time of liposomes depends on the liposome size and is unfavorable for immunoliposome binding to target. BBA Biomembr. 1991, 1062, 142–148. [Google Scholar] [CrossRef]
- Maruyama, K.; Takizawa, T.; Yuda, T.; Kennel, S.J.; Huang, L.; Iwatsuru, M. Targetability of novel immunoliposomes modified with amphipathic poly(ethylene glycol) s conjugated at their distal terminals to monoclonal antibodies. BBA Biomembr. 1995, 1234, 74–80. [Google Scholar] [CrossRef]
- Bradley, A.J.; Devine, D.V.; Ansell, S.M.; Janzen, J.; Brooks, D.E. Inhibition of liposome-induced complement activation by incorporated poly(ethylene glycol)-lipids. Arch. Biochem. Biophys. 1998, 357, 185–194. [Google Scholar] [CrossRef]
- Chan, C.L.; Majzoub, R.N.; Shirazi, R.S.; Ewert, K.K.; Chen, Y.J.; Liang, K.S.; Safinya, C.R. Endosomal escape and transfection efficiency of PEGylated cationic liposome-DNA complexes prepared with an acid-labile PEG-lipid. Biomaterials 2012, 33, 4928–4935. [Google Scholar] [CrossRef]
- Kale, A.A.; Torchilin, V.P. Enhanced transfection of tumor cells in vivo using “Smart” pH-sensitive TAT-modified pegylated liposomes. J. Drug Target. 2007, 15, 538–545. [Google Scholar] [CrossRef]
- Fang, Y.; Xue, J.; Gao, S.; Lu, A.; Yang, D.; Jiang, H.; He, Y.; Shi, K. Cleavable PEGylation: A strategy for overcoming the “PEG dilemma” in efficient drug delivery. Drug Deliv. 2017, 24, 22–32. [Google Scholar] [CrossRef]
- Patel, J.D.; O’Carra, R.; Jones, J.; Woodward, J.G.; Mumper, R.J. Preparation and characterization of nickel nanoparticles for binding to his-tag proteins and antigens. Pharm. Res. 2007, 24, 343–352. [Google Scholar] [CrossRef]
- Koning, G.A.; Storm, G. Targeted drug delivery systems for the intracellular delivery of macromolecular drugs. Drug Discov. Today 2003, 8, 482–483. [Google Scholar] [CrossRef]
- Maruyama, K. PEG-Immunoliposome. Biosci. Rep. 2002, 22, 251–266. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Xu, C.; Zhao, X.; Lin, C.; Yang, X.; Xin, X.; Zhang, L.; Qin, C.; Han, X.; Yang, L.; et al. Nanoplatform Assembled from a CD44-Targeted Prodrug and Smart Liposomes for Dual Targeting of Tumor Microenvironment and Cancer Cells. ACS Nano 2018, 12, 1519–1536. [Google Scholar] [CrossRef] [PubMed]
- Lakkadwala, S.; Singh, J. Co-delivery of doxorubicin and erlotinib through liposomal nanoparticles for glioblastoma tumor regression using an in vitro brain tumor model. Coll. Surf. B Biointerfaces 2019, 173, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Wyrozumska, P.; Meissner, J.; Toporkiewicz, M.; Szarawarska, M.; Kuliczkowski, K.; Ugorski, M.; Walasek, M.A.; Sikorski, A.F. Liposome-coated lipoplex-based carrier for antisense oligonucleotides. Cancer Biol. Ther. 2015, 16, 66–76. [Google Scholar] [CrossRef]
- Meissner, J.M.; Toporkiewicz, M.; Czogalla, A.; Matusewicz, L.; Kuliczkowski, K.; Sikorski, A.F. Novel antisense therapeutics delivery systems: In vitro and in vivo studies of liposomes targeted with anti-CD20 antibody. J. Control. Release 2015, 220, 515–528. [Google Scholar] [CrossRef]
- Matusewicz, L.; Filip-Psurska, B.; Psurski, M.; Tabaczar, S.; Podkalicka, J.; Wietrzyk, J.; Ziółkowski, P.; Czogalla, A.; Sikorski, A.F. EGFR-targeted immunoliposomes as a selective delivery system of simvastatin, with potential use in treatment of triple-negative breast cancers. Int. J. Pharm. 2019, 569, 118605. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Product Name | Route of Administration | Lipid Composition (Molar Ratio 1) | Treatment | Ref. |
|---|---|---|---|---|---|
| Amphotericin B | Abelcet | Intravenous | DMPC, DMPG (7:3) | Systemic fungal infections | [16] |
| Ambisome | Intravenous | HSPC, DSPG, cholesterol (2:0.8:0.4) | Systemic fungal infections | [17] | |
| Bupivacaine | Exparel | Supraperiosteal Injection | DEPC, DPPG, cholesterol, tricaprylin | Postsurgical local analgesia | [18] |
| Nocita | Supraperiosteal Injection | DEPC, DPPG, cholesterol, tricaprylin | Postsurgical local analgesia (for dogs only) | [19] | |
| Cytarabine | Depocyt | Spinal | DOPC, DPPG, cholesterol, triolein (7:1:11:1) | Lymphomatous meningitis | [20] |
| Daunorubicin | DaunoXome | Intravenous | DSPC, cholesterol (2:1) | Kaposi’s sarcoma | [21] |
| Doxorubicin | Doxil/Caelyx 2 | Intravenous | HSPC, cholesterol, DSPE-PEG (2000) (56:39:5) | Kaposi’s sarcoma | [22] |
| Lipodox | Intravenous | DSPC, cholesterol, DSPE-PEG (2000) (56:39:5) | Kaposi’s sarcoma, ovarian/breast cancer | [23] | |
| Myocet liposomal 3 | Intravenous | EPC, cholesterol (55:45) | Metastatic breast cancer | [24] | |
| Inactivated hepatitis A virus | Epaxal | Intramuscular | DOPC, DOPE (75:25) | Hepatitis A | [25] |
| Inactivated hemagglutinin of influenza virus strains A and B | Inflexal V | Intramuscular | DOPC, DOPE (75:25) | Influenza | [26] |
| Irinotecan | Onivyde | Intravenous | DSPC, MPEG-2000-DSPE | metastatic adenocarcinoma of the pancreas | [27] |
| Mifamurtide | Mepact 2 | Intravenous | POPC, DOPS (7:3) | High-grade non-metastatic osteosarcoma | [28] |
| Morphine sulfate | DepoDur | Epidural | DOPC, DPPG, cholesterol, triolein (7:1:11:1) | Pain management | [29] |
| Verteporfin | Visudyne | Intravenous | DMPC, EPG (5:3) | Age-related macular degeneration, pathologic myopia, ocular histoplasmosis | [30] |
| Vincristine sulfate | Marqibo | Intravenous | Sphingomyelin, cholesterol (6:4) | Acute lymphoblastic leukaemia | [31] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Juszkiewicz, K.; Sikorski, A.F.; Czogalla, A. Building Blocks to Design Liposomal Delivery Systems. Int. J. Mol. Sci. 2020, 21, 9559. https://doi.org/10.3390/ijms21249559
Juszkiewicz K, Sikorski AF, Czogalla A. Building Blocks to Design Liposomal Delivery Systems. International Journal of Molecular Sciences. 2020; 21(24):9559. https://doi.org/10.3390/ijms21249559
Chicago/Turabian StyleJuszkiewicz, Katarzyna, Aleksander F. Sikorski, and Aleksander Czogalla. 2020. "Building Blocks to Design Liposomal Delivery Systems" International Journal of Molecular Sciences 21, no. 24: 9559. https://doi.org/10.3390/ijms21249559
APA StyleJuszkiewicz, K., Sikorski, A. F., & Czogalla, A. (2020). Building Blocks to Design Liposomal Delivery Systems. International Journal of Molecular Sciences, 21(24), 9559. https://doi.org/10.3390/ijms21249559