MiR-96-5p Induced by Palmitic Acid Suppresses the Myogenic Differentiation of C2C12 Myoblasts by Targeting FHL1



Abstract
1. Introduction
2. Results
2.1. PA Inhibited Myogenic Differentiation and FHL1 Expression but Induced miR-96-5p Expression
2.2. FHL1 Is Essential for Myogenic Differentiation
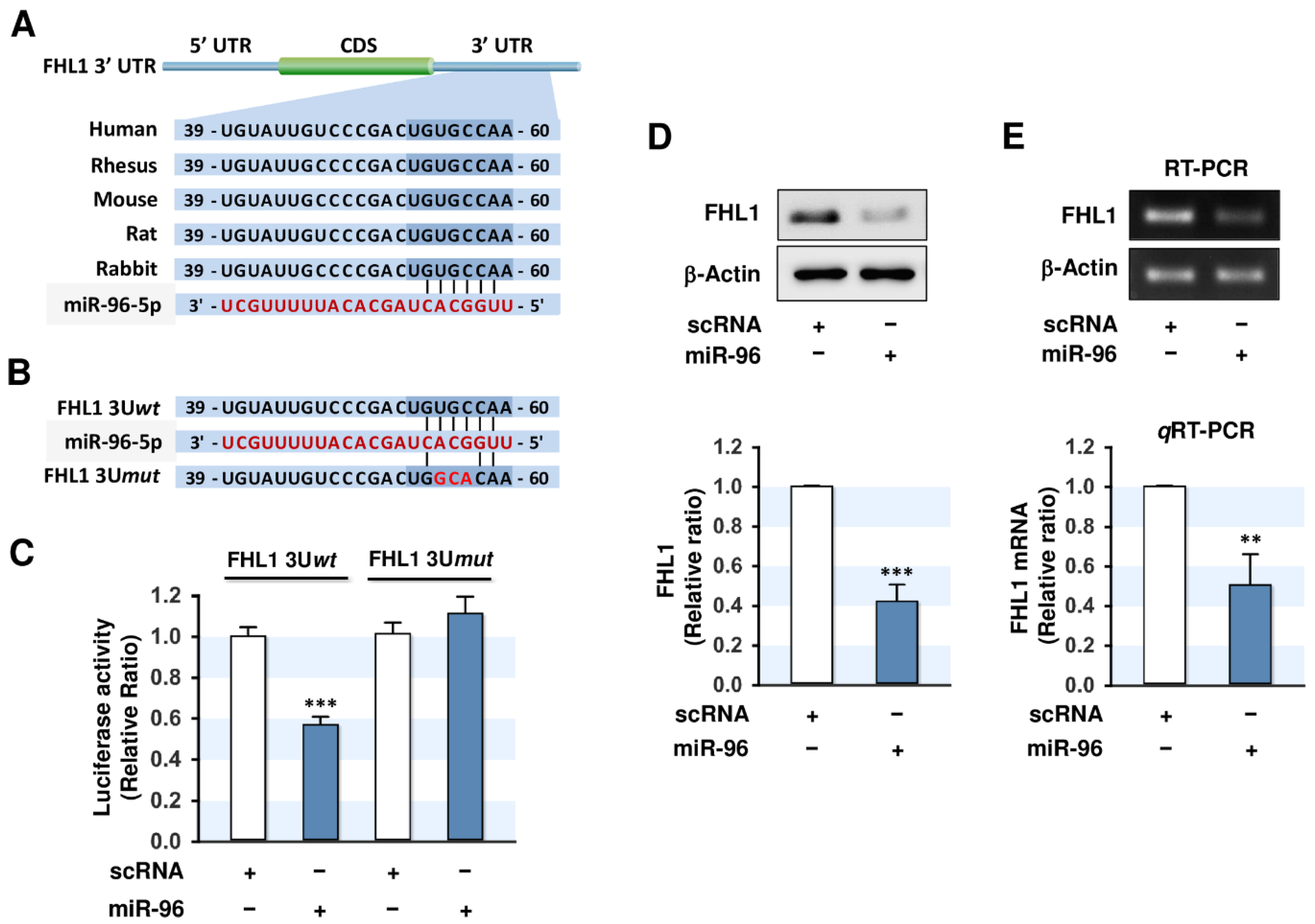
2.3. MiR-96-5p Directly Targeted the 3’UTR of FHL1
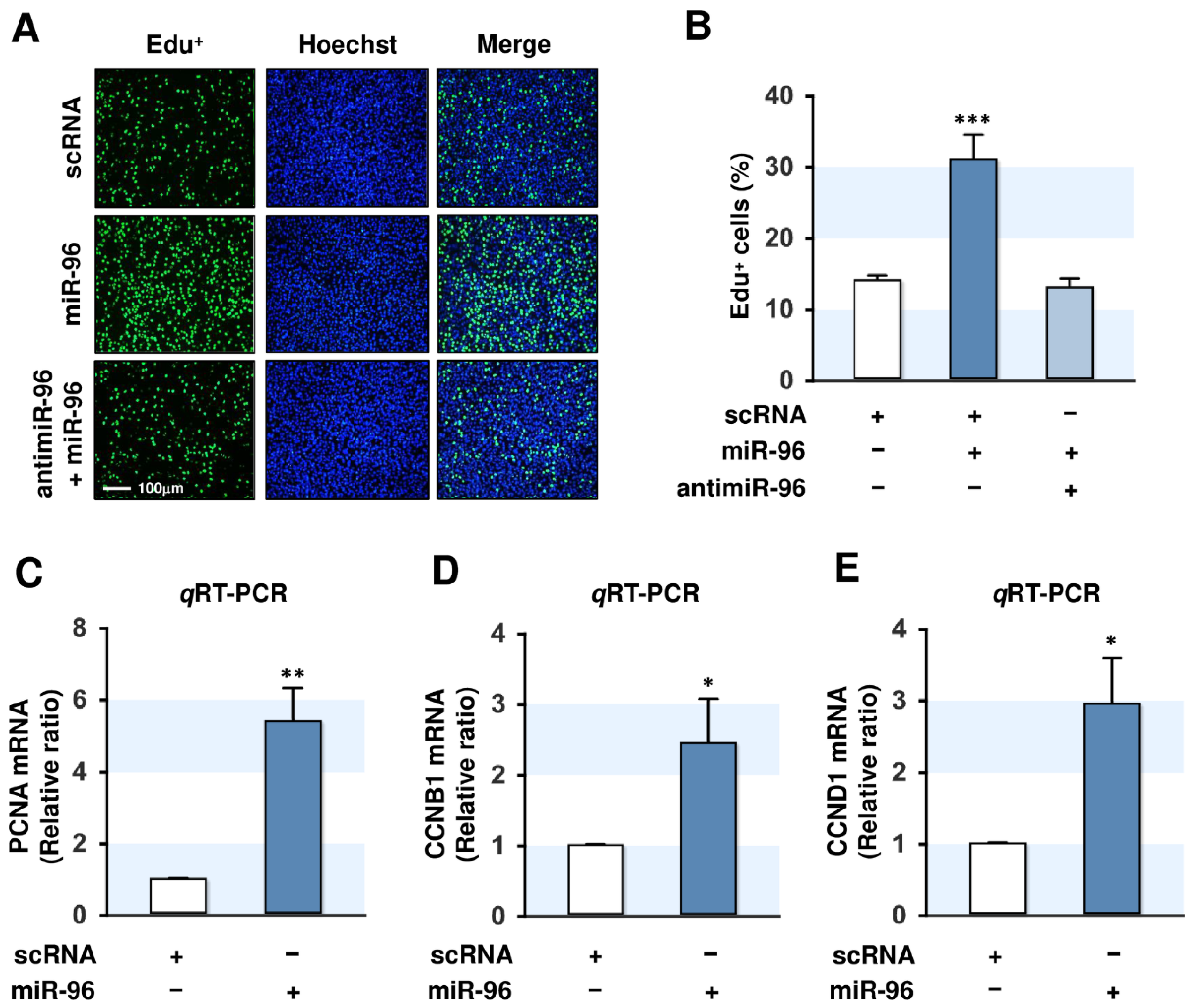
2.4. MiR-96-5p Increased the Proliferation of C2C12 Myoblasts
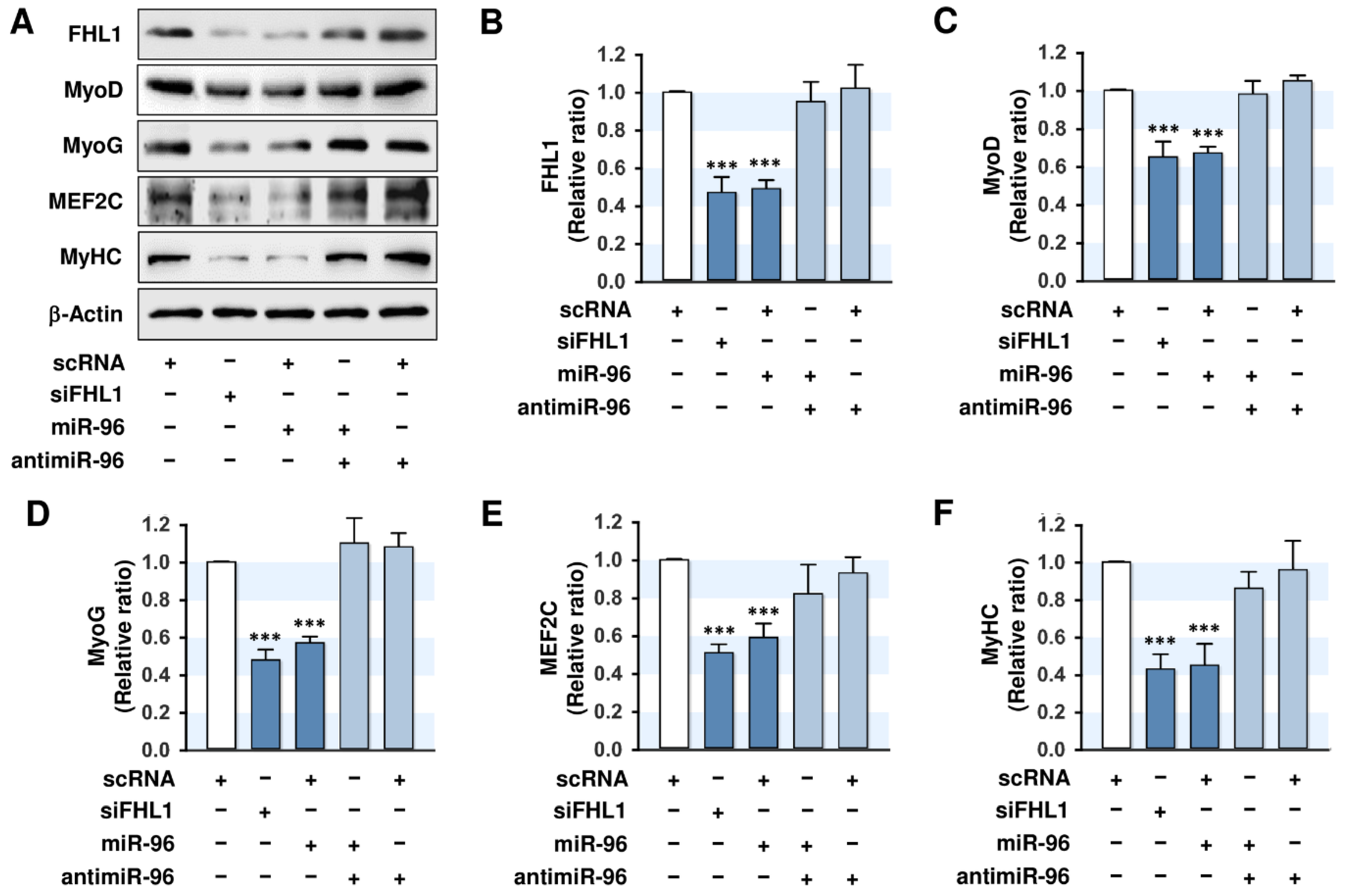
2.5. MiR-96-5p Inhibited the Expressions of Myogenic Factors
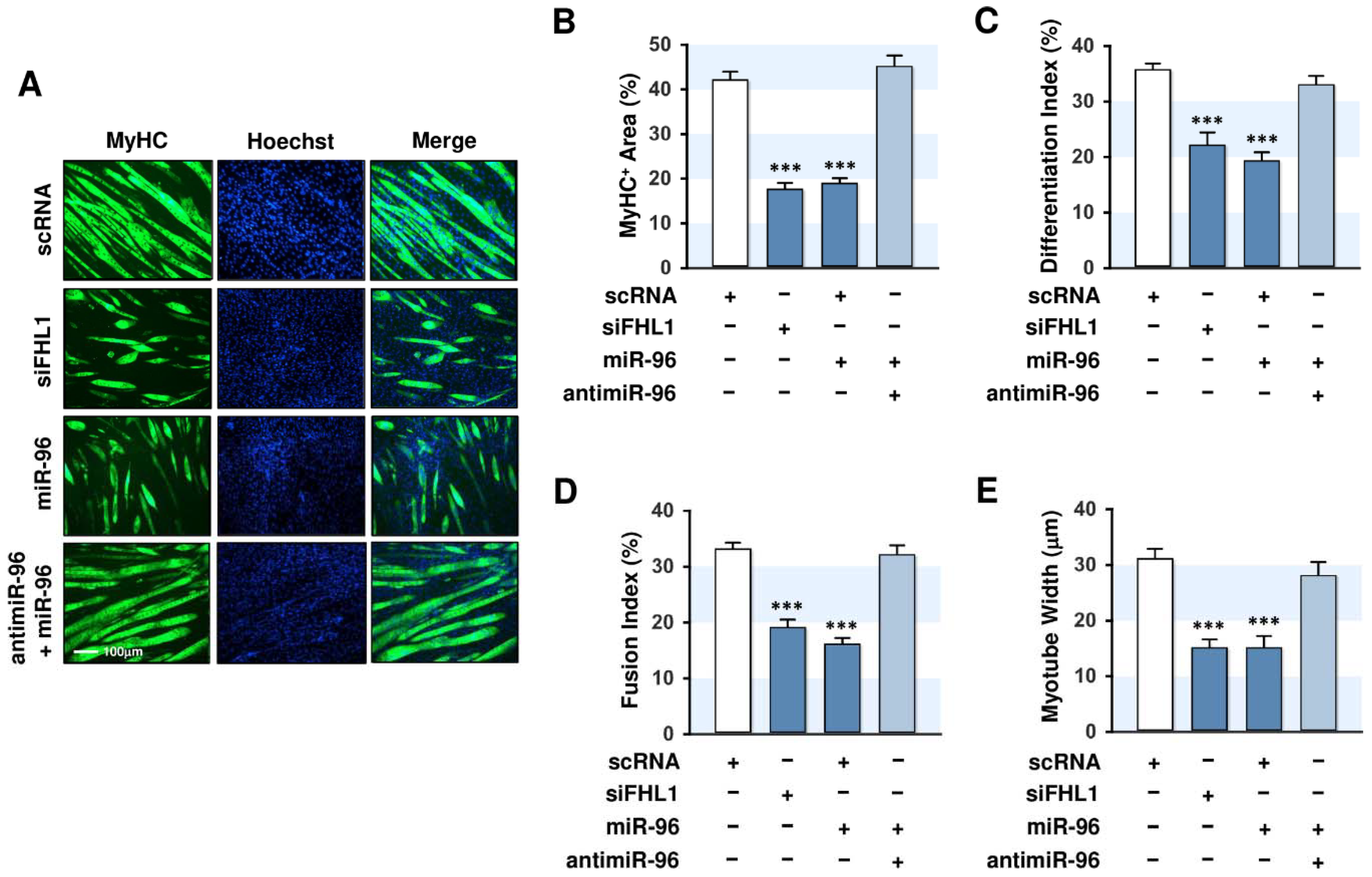
2.6. MiR-96-5p Inhibited the Myogenic Differentiation of C2C12 Myoblasts
3. Discussion
4. Materials and Methods
4.1. Cell Culture, Differentiation, and PA Treatment
4.2. Transfection of Oligonucleotides
4.3. RNA Preparation and Quantitative Reverse Transcription-Polymerase Chain Reaction (qRT-PCR)
4.4. Dual-Luciferase Reporter Assay
4.5. Immunoblot Analysis
4.6. Immunofluorescence Analysis
4.7. Ethynyl Deoxyuridine (EdU) Assay
4.8. Database and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dumont, N.A.; Bentzinger, C.F.; Sincennes, M.C.; Rudnicki, M.A. Satellite Cells and Skeletal Muscle Regeneration. Compr. Physiol. 2015, 5, 1027–1059. [Google Scholar] [PubMed]
- Chal, J.; Pourquie, O. Making muscle: Skeletal myogenesis in vivo and in vitro. Development 2017, 144, 2104–2122. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.F.; Hall, D.T.; Gallouzi, I.-E. The impact of mRNA turnover and translation on age-related muscle loss. Ageing Res. Rev. 2012, 11, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Benarroch, L.; Bonne, G.; Rivier, F.; Hamroun, D. The 2020 version of the gene table of neuromuscular disorders (nuclear genome). Neuromuscul. Disord. 2019, 29, 980–1018. [Google Scholar] [CrossRef]
- Asfour, H.A.; Allouh, M.Z.; Said, R.S. Myogenic regulatory factors: The orchestrators of myogenesis after 30 years of discovery. Exp. Biol. Med. 2018, 243, 118–128. [Google Scholar] [CrossRef]
- Akhmedov, D.; Berdeaux, R. The effects of obesity on skeletal muscle regeneration. Front. Physiol. 2013, 4, 371. [Google Scholar] [CrossRef]
- Teng, S.; Huang, P. The effect of type 2 diabetes mellitus and obesity on muscle progenitor cell function. Stem Cell Res. Ther. 2019, 10, 103. [Google Scholar] [CrossRef]
- Chang, Y.C.; Liu, H.W.; Chen, Y.T.; Chen, Y.A.; Chen, Y.J.; Chang, S.J. Resveratrol protects muscle cells against palmitate-induced cellular senescence and insulin resistance through ameliorating autophagic flux. J. Food Drug Anal. 2018, 26, 1066–1074. [Google Scholar] [CrossRef]
- Zhang, G.; Chen, X.; Lin, L.; Wen, C.; Rao, S. Effects of fatty acids on proliferation and differentiation of myoblast. Wei Sheng Yan Jiu 2012, 41, 883–888. [Google Scholar]
- Saini, A.; Sharples, A.P.; Al-Shanti, N.; Stewart, C.E. Omega-3 fatty acid EPA improves regenerative capacity of mouse skeletal muscle cells exposed to saturated fat and inflammation. Biogerontology 2017, 18, 109–129. [Google Scholar] [CrossRef]
- Xu, D.; Jiang, Z.; Sun, Z.; Wang, L.; Zhao, G.; Hassan, H.M.; Fan, S.; Zhou, W.; Han, S.; Zhang, L.; et al. Mitochondrial dysfunction and inhibition of myoblast differentiation in mice with high-fat-diet-induced pre-diabetes. J. Cell Physiol. 2019, 234, 7510–7523. [Google Scholar] [CrossRef] [PubMed]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Saliminejad, K.; Khorram Khorshid, H.R.; Soleymani Fard, S.; Ghaffari, S.H. An overview of microRNAs: Biology, functions, therapeutics, and analysis methods. J. Cell Physiol. 2019, 234, 5451–5465. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Chen, M.; Lian, D.; Li, Y.; Li, Y.; Wang, J.; Deng, S.; Yu, K.; Lian, Z. Non-Coding RNA Regulates the Myogenesis of Skeletal Muscle Satellite Cells, Injury Repair and Diseases. Cells 2019, 8, 988. [Google Scholar] [CrossRef]
- Mok, G.F.; Lozano-Velasco, E.; Munsterberg, A. microRNAs in skeletal muscle development. Semin. Cell Dev. Biol. 2017, 72, 67–76. [Google Scholar] [CrossRef]
- Ji, C.; Guo, X. The clinical potential of circulating microRNAs in obesity. Nat. Rev. Endocrinol. 2019, 15, 731–743. [Google Scholar] [CrossRef]
- Ortiz-Dosal, A.; Rodil-Garcia, P.; Salazar-Olivo, L.A. Circulating microRNAs in human obesity: A systematic review. Biomarkers 2019, 24, 499–509. [Google Scholar] [CrossRef]
- Kadrmas, J.L.; Beckerle, M.C. The LIM domain: From the cytoskeleton to the nucleus. Nat. Rev. Mol. Cell Biol. 2004, 5, 920–931. [Google Scholar] [CrossRef]
- Schessl, J.; Feldkirchner, S.; Kubny, C.; Schoser, B. Reducing body myopathy and other FHL1-related muscular disorders. Semin. Pediatr. Neurol. 2011, 18, 257–263. [Google Scholar] [CrossRef]
- Cowling, B.S.; McGrath, M.J.; Nguyen, M.A.; Cottle, D.L.; Kee, A.J.; Brown, S.; Schessl, J.; Zou, Y.; Joya, J.; Bonnemann, C.G.; et al. Identification of FHL1 as a regulator of skeletal muscle mass: Implications for human myopathy. J. Cell Biol. 2008, 183, 1033–1048. [Google Scholar] [CrossRef]
- Lee, J.Y.; Chien, I.C.; Lin, W.Y.; Wu, S.M.; Wei, B.H.; Lee, Y.E.; Lee, H.H. Fhl1 as a downstream target of Wnt signaling to promote myogenesis of C2C12 cells. Mol. Cell Biochem. 2012, 365, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Domenighetti, A.A.; Chu, P.H.; Wu, T.; Sheikh, F.; Gokhin, D.S.; Guo, L.T.; Cui, Z.; Peter, A.K.; Christodoulou, D.C.; Parfenov, M.G.; et al. Loss of FHL1 induces an age-dependent skeletal muscle myopathy associated with myofibrillar and intermyofibrillar disorganization in mice. Hum. Mol. Genet. 2014, 23, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Cong, Y.F.; Liu, B.; Miao, J.; Wang, L. Aberrant Protein Turn-Over Associated With Myofibrillar Disorganization in FHL1 Knockout Mice. Front. Genet. 2018, 9, 273. [Google Scholar] [CrossRef] [PubMed]
- Windpassinger, C.; Schoser, B.; Straub, V.; Hochmeister, S.; Noor, A.; Lohberger, B.; Farra, N.; Petek, E.; Schwarzbraun, T.; Ofner, L.; et al. An X-linked myopathy with postural muscle atrophy and generalized hypertrophy, termed XMPMA, is caused by mutations in FHL1. Am. J. Hum. Genet. 2008, 82, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zhu, Y.; Liu, X.; Zhang, W.; Liu, J.; Wu, S.; Xiao, J.; Yuan, Y.; Wang, Z. FHL1-related clinical, muscle MRI and genetic features in six Chinese patients with reducing body myopathy. J. Hum. Genet. 2019, 64, 919–926. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.H.; Raskind, W.H.; Parson, W.W.; Sonnen, J.A.; Vu, T.; Zheng, Y.; Matsushita, M.; Wolff, J.; Lipe, H.; Bird, T.D. A novel mutation in FHL1 in a family with X-linked scapuloperoneal myopathy: Phenotypic spectrum and structural study of FHL1 mutations. J. Neurol. Sci. 2010, 296, 22–29. [Google Scholar] [CrossRef]
- Gueneau, L.; Bertrand, A.T.; Jais, J.P.; Salih, M.A.; Stojkovic, T.; Wehnert, M.; Hoeltzenbein, M.; Spuler, S.; Saitoh, S.; Verschueren, A.; et al. Mutations of the FHL1 gene cause Emery-Dreifuss muscular dystrophy. Am. J. Hum. Genet. 2009, 85, 338–353. [Google Scholar] [CrossRef]
- Bruyere, C.; Versaevel, M.; Mohammed, D.; Alaimo, L.; Luciano, M.; Vercruysse, E.; Gabriele, S. Actomyosin contractility scales with myoblast elongation and enhances differentiation through YAP nuclear export. Sci. Rep. 2019, 9, 15565. [Google Scholar] [CrossRef]
- Han, S.; Cui, C.; He, H.; Shen, X.; Chen, Y.; Wang, Y.; Li, D.; Zhu, Q.; Yin, H. FHL1 regulates myoblast differentiation and autophagy through its interaction with LC3. J. Cell Physiol. 2020, 235, 4667–4678. [Google Scholar] [CrossRef]
- Dambal, S.; Shah, M.; Mihelich, B.; Nonn, L. The microRNA-183 cluster: The family that plays together stays together. Nucleic Acids Res. 2015, 43, 7173–7188. [Google Scholar] [CrossRef]
- Li, G.; Luna, C.; Qiu, J.; Epstein, D.L.; Gonzalez, P. Alterations in microRNA expression in stress-induced cellular senescence. Mech. Ageing Dev. 2009, 130, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Dai, T.; Xiong, H.; Zhao, X.; Chen, X.; Yu, C.; Li, J.; Wang, X.; Song, L. Unregulated miR-96 induces cell proliferation in human breast cancer by downregulating transcriptional factor FOXO3a. PLoS ONE 2010, 5, e15797. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, N.; Leidinger, P.; Becker, K.; Backes, C.; Fehlmann, T.; Pallasch, C.; Rheinheimer, S.; Meder, B.; Stähler, C.; Meese, E.; et al. Distribution of miRNA expression across human tissues. Nucleic Acids Res. 2016, 44, 3865–3877. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; He, X.; Chang, Y.; Xu, C.; Jiang, X.; Sun, S.; Lin, J. Inhibition of miR-96 expression reduces cell proliferation and clonogenicity of HepG2 hepatoma cells. Oncol. Rep. 2013, 29, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Zheng, D.; Hu, P.; Zeng, Z.; Li, M.; Tucker, L.; Monahan, R.; Resnick, M.B.; Liu, M.; Ramratnam, B. Glycogen synthase kinase 3 beta inhibits microRNA-183-96-182 cluster via the beta-Catenin/TCF/LEF-1 pathway in gastric cancer cells. Nucleic Acids Res. 2014, 42, 2988–2998. [Google Scholar] [CrossRef]
- Ress, A.L.; Stiegelbauer, V.; Winter, E.; Schwarzenbacher, D.; Kiesslich, T.; Lax, S.; Jahn, S.; Deutsch, A.; Bauernhofer, T.; Ling, H.; et al. MiR-96-5p influences cellular growth and is associated with poor survival in colorectal cancer patients. Mol. Carcinog. 2015, 54, 1442–1450. [Google Scholar] [CrossRef]
- Zhang, Q.; Ren, W.; Huang, B.; Yi, L.; Zhu, H. MicroRNA-183/182/96 cooperatively regulates the proliferation of colon cancer cells. Mol. Med. Rep. 2015, 12, 668–674. [Google Scholar] [CrossRef]
- Wei, X.; Zhang, H. Four and a half LIM domains protein 1 can be as a double-edged sword in cancer progression. Cancer Biol. Med. 2020, 17, 270–281. [Google Scholar]
- Ding, L.; Wang, Z.; Yan, J.; Yang, X.; Liu, A.; Qiu, W.; Zhu, J.; Han, J.; Zhang, H.; Lin, J.; et al. Human four-and-a-half LIM family members suppress tumor cell growth through a TGF-beta-like signaling pathway. J. Clin. Investig. 2009, 119, 349–361. [Google Scholar]
- Zhang, F.; Feng, F.; Yang, P.; Li, Z.; You, J.; Xie, W.; Gao, X.; Yang, J. Four-and-a-half-LIM protein 1 down-regulates estrogen receptor alpha activity through repression of AKT phosphorylation in human breast cancer cell. Int. J. Biochem. Cell Biol. 2012, 44, 320–326. [Google Scholar] [CrossRef]
- Niu, C.; Liang, C.; Guo, J.; Cheng, L.; Zhang, H.; Qin, X.; Zhang, Q.; Ding, L.; Yuan, B.; Xu, X.; et al. Downregulation and growth inhibitory role of FHL1 in lung cancer. Int. J. Cancer 2012, 130, 2549–2556. [Google Scholar] [CrossRef] [PubMed]
- Asada, K.; Ando, T.; Niwa, T.; Nanjo, S.; Watanabe, N.; Okochi-Takada, E.; Yoshida, T.; Miyamoto, K.; Enomoto, S.; Ichinose, M.; et al. FHL1 on chromosome X is a single-hit gastrointestinal tumor-suppressor gene and contributes to the formation of an epigenetic field defect. Oncogene 2013, 32, 2140–2149. [Google Scholar] [CrossRef] [PubMed]
- Jurikova, M.; Danihel, L.; Polak, S.; Varga, I. Ki67, PCNA, and MCM proteins: Markers of proliferation in the diagnosis of breast cancer. Acta Histochem. 2016, 118, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Petrachkova, T.; Wortinger, L.A.; Bard, A.J.; Singh, J.; Warga, R.M.; Kane, D.A. Lack of Cyclin B1 in zebrafish causes lengthening of G2 and M phases. Dev. Biol. 2019, 451, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Belso, N.; Guban, B.; Manczinger, M.; Kormos, B.; Bebes, A.; Nemeth, I.; Vereb, Z.; Szell, M.; Kemeny, L.; Bata-Csorgo, Z. Differential role of D cyclins in the regulation of cell cycle by influencing Ki67 expression in HaCaT cells. Exp. Cell Res. 2019, 374, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Moon, Y.A.; Liang, G.; Xie, X.; Frank-Kamenetsky, M.; Fitzgerald, K.; Koteliansky, V.; Brown, M.S.; Goldstein, J.L.; Horton, J.D. The Scap/SREBP pathway is essential for developing diabetic fatty liver and carbohydrate-induced hypertriglyceridemia in animals. Cell Metab. 2012, 15, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Jeon, T.I.; Esquejo, R.M.; Roqueta-Rivera, M.; Phelan, P.E.; Moon, Y.A.; Govindarajan, S.S.; Esau, C.C.; Osborne, T.F. An SREBP-responsive microRNA operon contributes to a regulatory loop for intracellular lipid homeostasis. Cell Metab. 2013, 18, 51–61. [Google Scholar] [CrossRef]
- Shao, X.; Wang, M.; Wei, X.; Deng, S.; Fu, N.; Peng, Q.; Jiang, Y.; Ye, L.; Xie, J.; Lin, Y. Peroxisome Proliferator-Activated Receptor-gamma: Master Regulator of Adipogenesis and Obesity. Curr. Stem Cell Res. Ther. 2016, 11, 282–289. [Google Scholar] [CrossRef]
- John, E.; Wienecke-Baldacchino, A.; Liivrand, M.; Heinaniemi, M.; Carlberg, C.; Sinkkonen, L. Dataset integration identifies transcriptional regulation of microRNA genes by PPARgamma in differentiating mouse 3T3-L1 adipocytes. Nucleic Acids Res. 2012, 40, 4446–4460. [Google Scholar] [CrossRef]
- Lefterova, M.I.; Zhang, Y.; Steger, D.J.; Schupp, M.; Schug, J.; Cristancho, A.; Feng, D.; Zhuo, D.; Stoeckert, C.J., Jr.; Liu, X.S.; et al. PPARgamma and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes Dev. 2008, 22, 2941–2952. [Google Scholar] [CrossRef]
- Um, M.Y.; Moon, M.K.; Ahn, J.; Youl Ha, T. Coumarin attenuates hepatic steatosis by down-regulating lipogenic gene expression in mice fed a high-fat diet. Br. J. Nutr. 2013, 109, 1590–1597. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhou, J.; Mei, S.; Wu, D.; Mu, Z.; Chen, B.; Xie, Y.; Ye, Y.; Liu, J. Circulating exosomal microRNA-96 promotes cell proliferation, migration and drug resistance by targeting LMO7. J. Cell Mol. Med. 2017, 21, 1228–1236. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Liu, Y.; Zhang, J.; Bian, Z.; Yao, S.; Fei, B.; Zhou, L.; Yin, Y.; Huang, Z. A panel of serum exosomal microRNAs as predictive markers for chemoresistance in advanced colorectal cancer. Cancer Chemother. Pharmacol. 2019, 84, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.M.; Jeong, H.J.; Park, S.W.; Lee, W. Obesity-induced miR-15b is linked causally to the development of insulin resistance through the repression of the insulin receptor in hepatocytes. Mol. Nutr. Food Res. 2015, 59, 2303–2314. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.S.; Park, S.Y.; Ma, D.; Zhang, J.; Lee, W. The induction of microRNA targeting IRS-1 is involved in the development of insulin resistance under conditions of mitochondrial dysfunction in hepatocytes. PLoS ONE 2011, 6, e17343. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) Mouse primer lists for qRT-PCR and RT-PCR | |||||
| Gene | Primer Sequence (5′-3′) | Product Size | Annealing Temp. (°C) | Cycle | |
| miR-96 | F.P | TTTGGCACTAGCACATTTTTGCT | 90 | 55 | 40 (qRT-PCR) 30 (RT-PCR) |
| R.P | AGCAAAAATGTGCTAGTGCCAAA | ||||
| miRNA universal Primer | R.P | miScript universal primer (Qiagen) | |||
| U6 | F.P | CTCGCTTCGGCAGCACA | 94 | ||
| R.P | AACGCTTCACGAATTTGCGT | ||||
| FHL1 | F.P | CTGAAGTGCTTTGACAAGTTC | 102 | 58 | |
| R.P | GTGCCAGTAGCGATTCTTAT | ||||
| GAPDH | F.P | AACATCAAATGGGGTGAGGCC | 252 | 58 | |
| R.P | GTTGTCATGGATGACCTTGGC | ||||
| CCND1 | F.P | ACCAATCTCCTCAACGACCG | 228 | 58 | |
| R.P | ACGGAAGGGAAGAGAAGGG | ||||
| CCNB1 | F.P | GAGCTATCCTCATTGACTGG | 125 | 58 | |
| R.P | CATCTTCTTGGGCACACAAC | ||||
| PCNA | F.P | GAACCTGCAGAGCATGGACTC | 201 | 58 | |
| R.P | GGTGTCTGCATTATCTTCAGCCC | ||||
| (B) Primer lists for the cloning of FHL1 3’UTRs | |||||
| 3’UTRs | Primer Sequence (5′-3′) | Product Size | Annealing Temperature | Cycle | |
| wild-type FHL1 3’UTR | F.P | ATCTGGCCAACAAGCGCT | 303 | 58 | 35 |
| R.P | AATTGCAGCCGGACAGAAA | ||||
| mutant FHL1 3’UTR | F.P | ATCTGGCCAACAAGCGCT | 65 | ||
| R.P | CTTTTTGCGTCAGTCGGGA | ||||
| F.P | TCCCGACTGACGCAAAAAG | 257 | |||
| R.P | AAATTGCAGCCGGACAGAAA | ||||
| Antibody | Manufacturer | Cat. No. |
|---|---|---|
| FHL1 | Santa Cruz Biotechnology, Dallas, TX, USA | sc-374246 |
| MyHC | DSHB, Iowa city, IA, USA | MF20 |
| MyoD | Santa Cruz Biotechnology, Dallas, TX, USA | sc-377460 |
| MyoG | Santa Cruz Biotechnology, Dallas, TX, USA | sc-12732 |
| MEF2C | Thermo Fisher Scientific, Waltham, MA, USA | PA5-28247 |
| β-actin | Sigma-Aldrich Chemicals, St. Louis, MO, USA | A2066 |
| Antibodies HRP-linked anti-rabbit IgG | Cell Signaling Technology, Danvers, MA, USA | #7074 |
| Goat anti-mouse(H + L) | Thermo Fisher Scientific, Waltham, MA, USA | #32430 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, M.T.; Min, K.-H.; Lee, W. MiR-96-5p Induced by Palmitic Acid Suppresses the Myogenic Differentiation of C2C12 Myoblasts by Targeting FHL1. Int. J. Mol. Sci. 2020, 21, 9445. https://doi.org/10.3390/ijms21249445
Nguyen MT, Min K-H, Lee W. MiR-96-5p Induced by Palmitic Acid Suppresses the Myogenic Differentiation of C2C12 Myoblasts by Targeting FHL1. International Journal of Molecular Sciences. 2020; 21(24):9445. https://doi.org/10.3390/ijms21249445
Chicago/Turabian StyleNguyen, Mai Thi, Kyung-Ho Min, and Wan Lee. 2020. "MiR-96-5p Induced by Palmitic Acid Suppresses the Myogenic Differentiation of C2C12 Myoblasts by Targeting FHL1" International Journal of Molecular Sciences 21, no. 24: 9445. https://doi.org/10.3390/ijms21249445
APA StyleNguyen, M. T., Min, K.-H., & Lee, W. (2020). MiR-96-5p Induced by Palmitic Acid Suppresses the Myogenic Differentiation of C2C12 Myoblasts by Targeting FHL1. International Journal of Molecular Sciences, 21(24), 9445. https://doi.org/10.3390/ijms21249445