Bevacizumab Augments the Antitumor Efficacy of Infigratinib in Hepatocellular Carcinoma

 ,
, 
Abstract
1. Introduction
2. Results
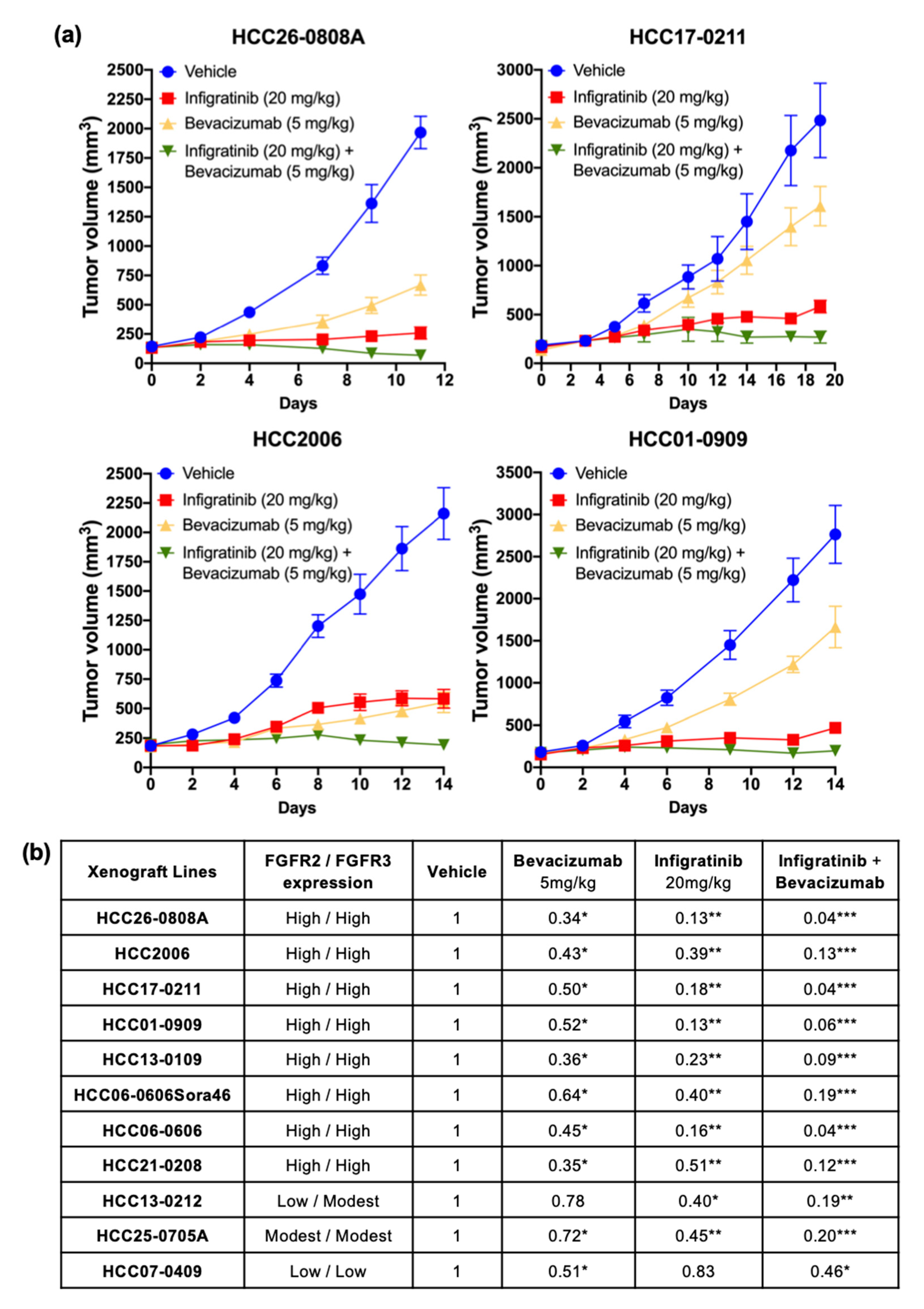
2.1. Infigratinib/Bevacizumab Demonstrates a Synergistic Effect that Inhibits Tumor Growth in High FGFR-Expressing HCC PDX Models
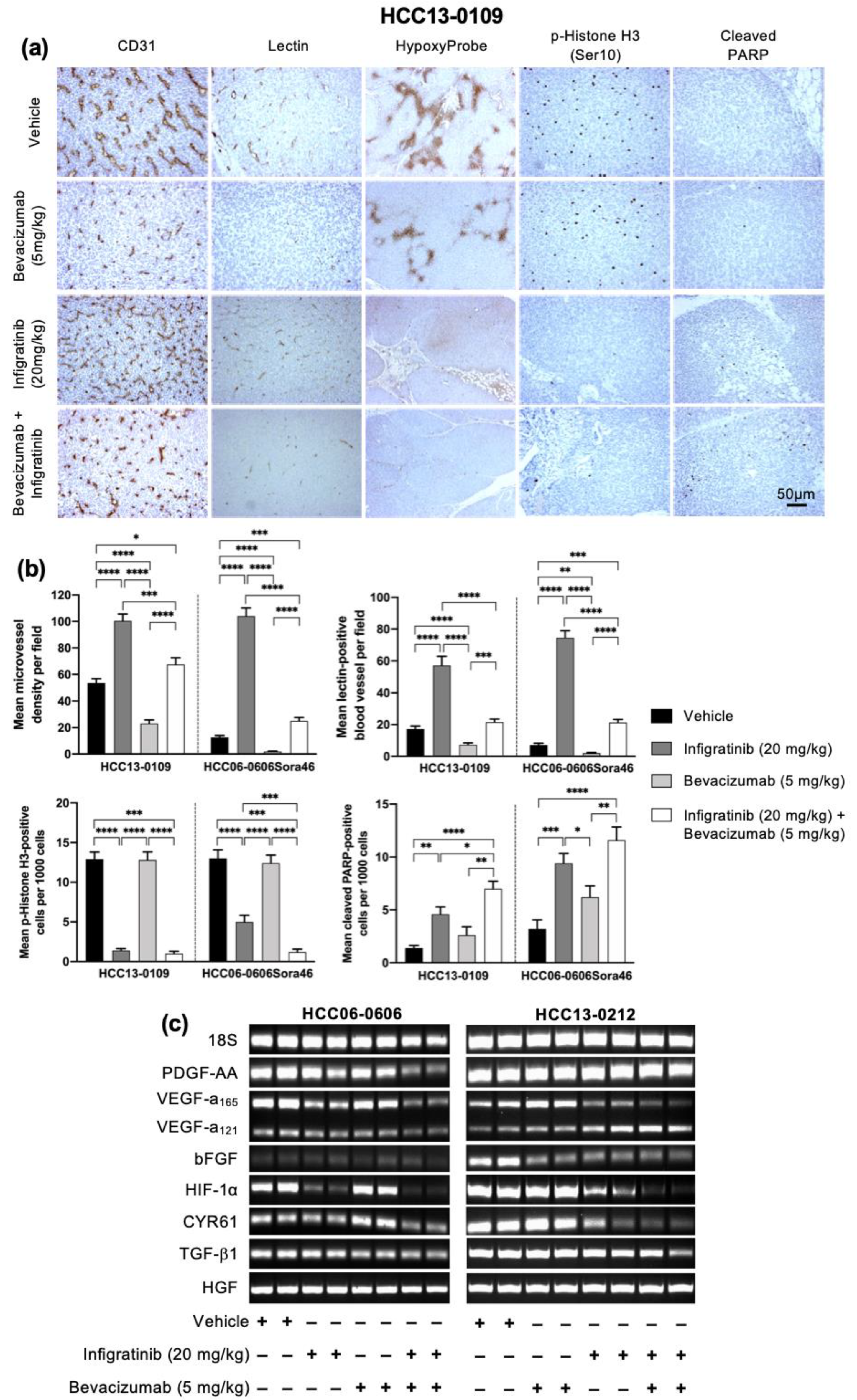
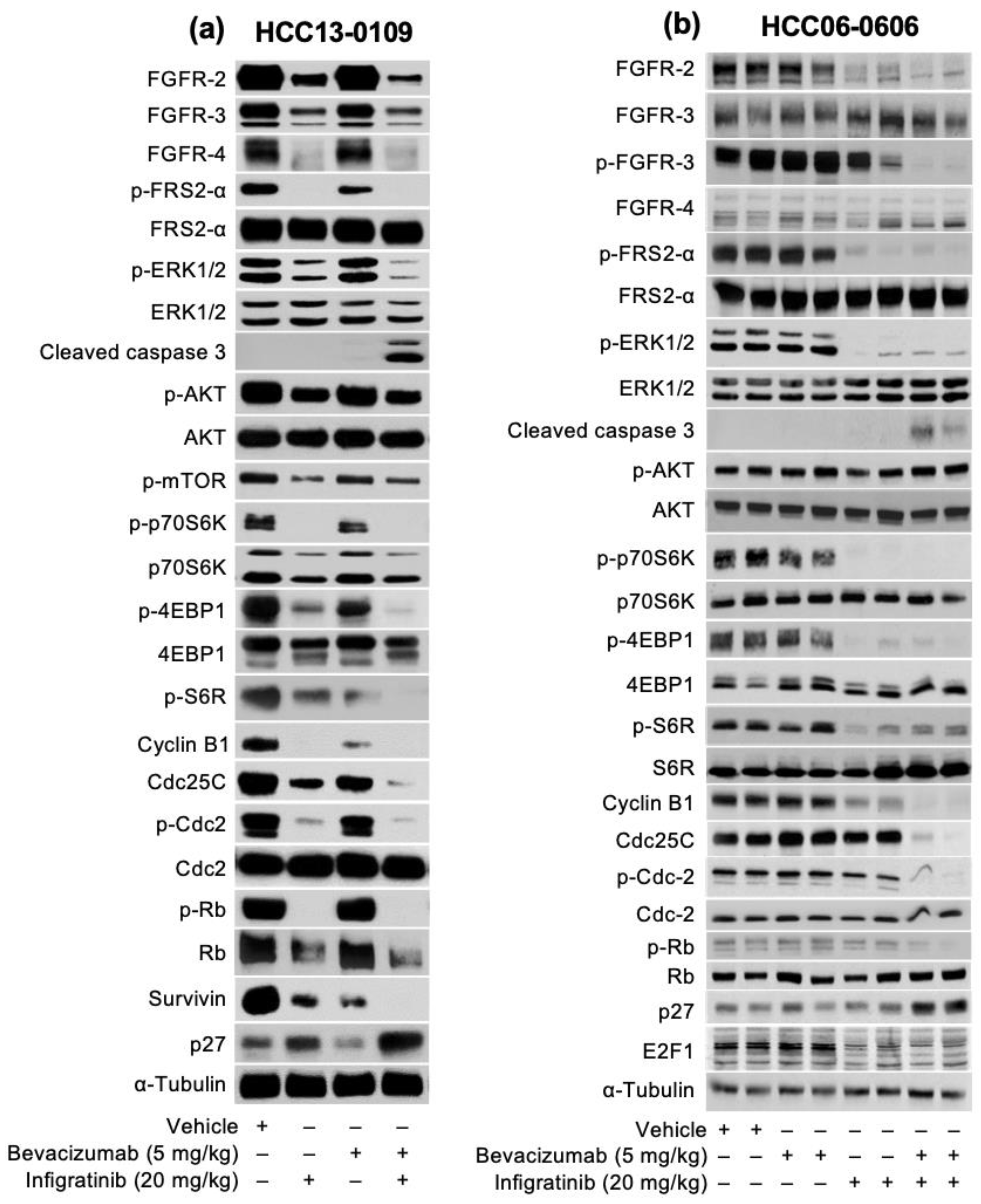
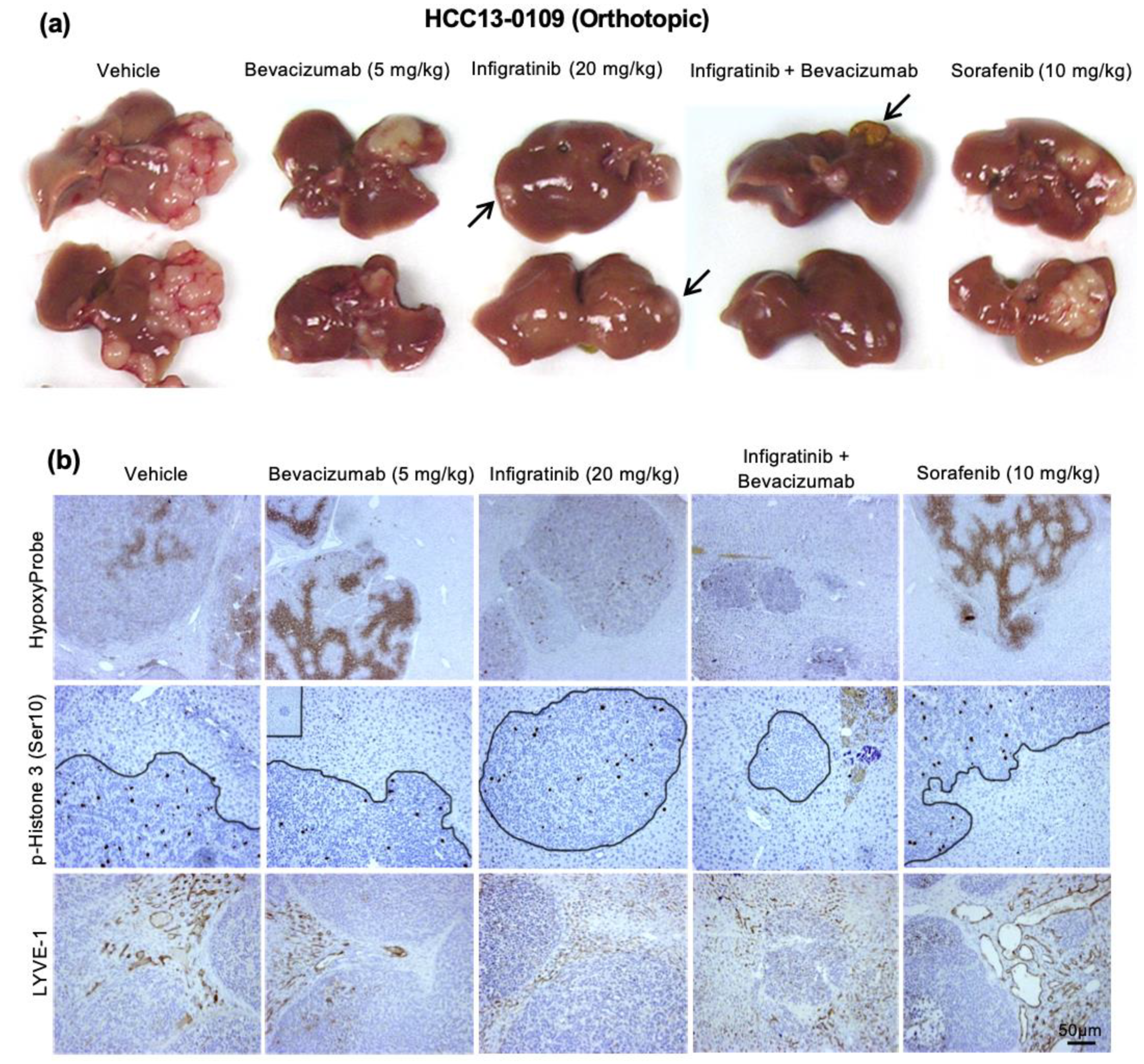
2.2. Infigratinib Acts Synergistically with Bevacizumab to Inhibit Tumor Growth, Cell Invasion, Hypoxia, and LYVE-1+ Peritumoral Lymphatic Vessels in an HCC13-0109 Orthotopic Model
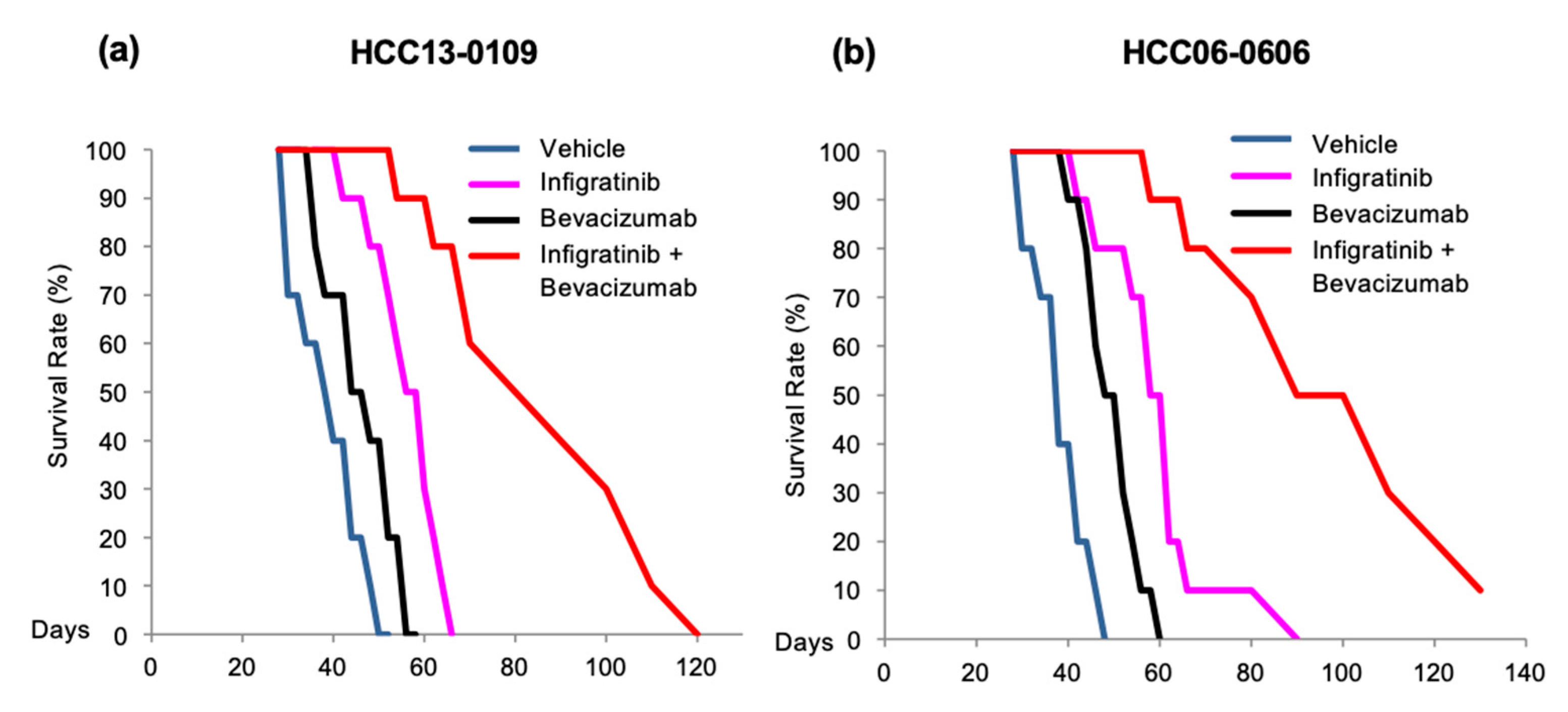
2.3. Infigratinib/Bevacizumab Prolongs the Survival of Mice Bearing HCC Orthotopic Tumors
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Xenograft Models
4.3. Development of Sorafenib-Resistant HCC Model
4.4. Drug Treatment and Data Collection
4.5. Vessel Perfusion Study
4.6. Immunohistochemistry
4.7. Western Blot Analysis
4.8. Study of Angiogenic Rescue Program

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Forward (5′–3′) | Reverse (5′–3′) | |
|---|---|---|
| GAPDH | TCTCCTCTGACTTCAACAGCGACAC | TGTTGCTGTAGCCAAATTCGTTGTC |
| PDGF-AA | CACGGGGTCCATGCCACTAAGCAT | ATCCGGATTCAGGCTTGTGGTCGC |
| VEGF | CGAAGTGGTGAAGTTCATGGATG | TTCTGTATCAGTCTTTCCTGGTGAG |
| bFGF | TACAACTTCAAGCAGAAGAG | CGACTCTTAGCAGACATTGG |
| HIF-1α | ACAGCAGCCAGACGATCATGC | ACCACGTACTGCTGGCAAAGC |
| CYR61 | TAAGGTCTGCGCCAAGCAGCTCAA | CGGCGCCATCAATACATGTGCACT |
| TGF-β1 | CAGAAATACAGCAACAATTCCTGG | TTGCAGTGTGTTATCCGTGCTGTC |
| HGF | GATTCTTTCACCCAGGCATC | TTTCCTTTGTCCCTCTGCAT |
4.9. Study of the Liver, Kidney and Hematological Injury-Related Parameters during the In Vivo Administration of Infigratinib/Bevacizumab
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| bFGF | Basic Fibroblast Growth Factor |
| CYR61 | Cysteine-rich Angiogenic Inducer 61 |
| FGF | Fibroblast Growth Factor |
| FGFR | Fibroblast Growth Factor Receptor |
| HCC | Hepatocellular Carcinoma |
| HGF | Hepatocyte Growth Factor |
| HIF-1α | Hypoxia-inducible Factor 1α |
| IP | Intraperitoneal |
| OS | Overall Survival |
| PARP | Poly (ADP-ribose) polymerase |
| PDGF-AA | Platelet-derived Growth Factor with 2A subunits |
| PVDF | Polyvinylidene fluoride |
| SCID | Severe combined immunodeficient |
| TGF | Transforming Growth Factor |
| VEGF | Vascular Endothelial Growth Factor |
| VEGFR | Vascular Endothelial Growth Factor Receptor |
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- EASL-EORTC clinical practice guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2012, 56, 908–943. [CrossRef] [PubMed]
- Nagasue, N.; Kohno, H.; Chang, Y.C.; Taniura, H.; Yamanoi, A.; Uchida, M.; Kimoto, T.; Takemoto, Y.; Nakamura, T.; Yukaya, H. Liver resection for hepatocellular carcinoma. Results of 229 consecutive patients during 11 years. Ann. Surg. 1993, 217, 375–384. [Google Scholar] [CrossRef]
- Altekruse, S.F.; McGlynn, K.A.; Reichman, M.E. Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 1485–1491. [Google Scholar] [CrossRef]
- Cheng, A.-L.; Kang, Y.-K.; Chen, Z.; Tsao, C.-J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.-S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.-W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef]
- Asada, N.; Tanaka, Y.; Hayashido, Y.; Toratani, S.; Kan, M.; Kitamoto, M.; Nakanishi, T.; Kajiyama, G.; Chayama, K.; Okamoto, T. Expression of fibroblast growth factor receptor genes in human hepatoma-derived cell lines. In Vitro Cell. Dev. Biol. Anim. 2003, 39, 321–328. [Google Scholar] [CrossRef]
- Ogasawara, S.; Yano, H.; Iemura, A.; Hisaka, T.; Kojiro, M. Expressions of basic fibroblast growth factor and its receptors and their relationship to proliferation of human hepatocellular carcinoma cell lines. Hepatology 1996, 24, 198–205. [Google Scholar] [CrossRef]
- Hu, Z.; Evarts, R.P.; Fujio, K.; Omori, N.; Omori, M.; Marsden, E.R.; Thorgeirsson, S.S. Expression of transforming growth factor alpha/epidermal growth factor receptor, hepatocyte growth factor/c-met and acidic fibroblast growth factor/fibroblast growth factor receptors during hepatocarcinogenesis. Carcinogenesis 1996, 17, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Yu, C.; Jin, C.; Kobayashi, M.; Bowles, C.A.; Wang, F.; McKeehan, W.L. Ectopic activity of fibroblast growth factor receptor 1 in hepatocytes accelerates hepatocarcinogenesis by driving proliferation and vascular endothelial growth factor-induced angiogenesis. Cancer Res. 2006, 66, 1481–1490. [Google Scholar] [CrossRef] [PubMed]
- Gu, Q.; Zhang, B.; Sun, H.; Xu, Q.; Tan, Y.; Wang, G.; Luo, Q.; Xu, W.; Yang, S.; Li, J.; et al. Genomic characterization of a large panel of patient-derived hepatocellular carcinoma xenograft tumor models for preclinical development. Oncotarget 2015, 6, 20160–20176. [Google Scholar] [CrossRef] [PubMed]
- Harimoto, N.; Taguchi, K.; Shirabe, K.; Adachi, E.; Sakaguchi, Y.; Toh, Y.; Okamura, T.; Kayashima, H.; Taketomi, A.; Maehara, Y. The significance of fibroblast growth factor receptor 2 expression in differentiation of hepatocellular carcinoma. Oncology 2010, 78, 361–368. [Google Scholar] [CrossRef]
- Paur, J.; Nika, L.; Maier, C.; Moscu-Gregor, A.; Kostka, J.; Huber, D.; Mohr, T.; Heffeter, P.; Schrottmaier, W.C.; Kappel, S.; et al. Fibroblast growth factor receptor 3 isoforms: Novel therapeutic targets for hepatocellular carcinoma? Hepatology 2015, 62, 1767–1778. [Google Scholar] [CrossRef]
- Moscatelli, D.; Joseph-Silverstein, J.; Presta, M.; Rifkin, D.B. Multiple forms of an angiogenesis factor: Basic fibroblast growth factor. Biochimie 1988, 70, 83–87. [Google Scholar] [CrossRef]
- Hsu, P.I.; Chow, N.H.; Lai, K.H.; Yang, H.B.; Chan, S.H.; Lin, X.Z.; Cheng, J.S.; Huang, J.S.; Ger, L.P.; Huang, S.M.; et al. Implications of serum basic fibroblast growth factor levels in chronic liver diseases and hepatocellular carcinoma. Anticancer Res. 1997, 17, 2803–2809. [Google Scholar]
- Mise, M.; Arii, S.; Higashituji, H.; Furutani, M.; Niwano, M.; Harada, T.; Ishigami, S.; Toda, Y.; Nakayama, H.; Fukumoto, M.; et al. Clinical significance of vascular endothelial growth factor and basic fibroblast growth factor gene expression in liver tumor. Hepatology 1996, 23, 455–464. [Google Scholar] [CrossRef]
- Beenken, A.; Mohammadi, M. The FGF family: Biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 2009, 8, 235–253. [Google Scholar] [CrossRef]
- Lieu, C.; Heymach, J.; Overman, M.; Tran, H.; Kopetz, S. Beyond VEGF: Inhibition of the fibroblast growth factor pathway and antiangiogenesis. Clin. Cancer Res. 2011, 17, 6130–6139. [Google Scholar] [CrossRef]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Haugsten, E.M.; Wiedlocha, A.; Olsnes, S.; Wesche, J. Roles of fibroblast growth factor receptors in carcinogenesis. Mol. Cancer Res. 2010, 8, 1439–1452. [Google Scholar] [CrossRef] [PubMed]
- Huynh, H.; Lee, L.Y.; Goh, K.Y.; Ong, R.; Hao, H.-X.; Huang, A.; Wang, Y.; Graus Porta, D.; Chow, P.; Chung, A. Infigratinib Mediates Vascular Normalization, Impairs Metastasis, and Improves Chemotherapy in Hepatocellular Carcinoma. Hepatology 2019, 69, 943–958. [Google Scholar] [CrossRef] [PubMed]
- Guagnano, V.; Furet, P.; Spanka, C.; Bordas, V.; Le Douget, M.; Stamm, C.; Brueggen, J.; Jensen, M.R.; Schnell, C.; Schmid, H.; et al. Discovery of 3-(2,6-dichloro-3,5-dimethoxy-phenyl)-1-{6-[4-(4-ethyl-piperazin-1-yl)-phenylamino]-pyrimidin-4-yl}-1-methyl-urea (NVP-BGJ398), a potent and selective inhibitor of the fibroblast growth factor receptor family of receptor tyrosine kinase. J. Med. Chem. 2011, 54, 7066–7083. [Google Scholar] [CrossRef] [PubMed]
- Guagnano, V.; Kauffmann, A.; Wohrle, S.; Stamm, C.; Ito, M.; Barys, L.; Pornon, A.; Yao, Y.; Li, F.; Zhang, Y.; et al. FGFR genetic alterations predict for sensitivity to NVP-BGJ398, a selective pan-FGFR inhibitor. Cancer Discov. 2012, 2, 1118–1133. [Google Scholar] [CrossRef] [PubMed]
- Semela, D.; Dufour, J.-F. Angiogenesis and hepatocellular carcinoma. J. Hepatol. 2004, 41, 864–880. [Google Scholar] [CrossRef] [PubMed]
- Arbiser, J.L.; Weiss, S.W.; Arbiser, Z.K.; Bravo, F.; Govindajaran, B.; Caceres-Rios, H.; Cotsonis, G.; Recavarren, S.; Swerlick, R.A.; Cohen, C. Differential expression of active mitogen-activated protein kinase in cutaneous endothelial neoplasms: Implications for biologic behavior and response to therapy. J. Am. Acad. Dermatol. 2001, 44, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Dempke, W.C.M.; Heinemann, V. Resistance to EGF-R (erbB-1) and VEGF-R modulating agents. Eur. J. Cancer 2009, 45, 1117–1128. [Google Scholar] [CrossRef]
- Bergers, G.; Hanahan, D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 2008, 8, 592–603. [Google Scholar] [CrossRef]
- Yoshiji, H.; Kuriyama, S.; Yoshii, J.; Ikenaka, Y.; Noguchi, R.; Hicklin, D.J.; Huber, J.; Nakatani, T.; Tsujinoue, H.; Yanase, K.; et al. Synergistic effect of basic fibroblast growth factor and vascular endothelial growth factor in murine hepatocellular carcinoma. Hepatology 2002, 35, 834–842. [Google Scholar] [CrossRef]
- Kano, M.R.; Morishita, Y.; Iwata, C.; Iwasaka, S.; Watabe, T.; Ouchi, Y.; Miyazono, K.; Miyazawa, K. VEGF-A and FGF-2 synergistically promote neoangiogenesis through enhancement of endogenous PDGF-B-PDGFRbeta signaling. J. Cell Sci. 2005, 118, 3759–3768. [Google Scholar] [CrossRef] [PubMed]
- Huynh, H.; Soo, K.C.; Chow, P.K.H.; Panasci, L.; Tran, E. Xenografts of human hepatocellular carcinoma: A useful model for testing drugs. Clin. Cancer Res. 2006, 12, 4306–4314. [Google Scholar] [CrossRef] [PubMed]
- Nogova, L.; Sequist, L.V.; Perez Garcia, J.M.; Andre, F.; Delord, J.-P.; Hidalgo, M.; Schellens, J.H.M.; Cassier, P.A.; Camidge, D.R.; Schuler, M.; et al. Evaluation of BGJ398, a Fibroblast Growth Factor Receptor 1-3 Kinase Inhibitor, in Patients With Advanced Solid Tumors Harboring Genetic Alterations in Fibroblast Growth Factor Receptors: Results of a Global Phase I, Dose-Escalation and Dose-Expansion St. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.; Lowery, M.; Shroff, R.T.; Weiss, K.H.; Springfeld, C.; Borad, M.J.; Ramanathan, R.K.; Goyal, L.; Sadeghi, S.; Macarulla, T.; et al. Phase II Study of BGJ398 in Patients With FGFR-Altered Advanced Cholangiocarcinoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Höpfner, M.; Schuppan, D.; Scherübl, H. Growth factor receptors and related signalling pathways as targets for novel treatment strategies of hepatocellular cancer. World J. Gastroenterol. 2008, 14, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X. Development of sorafenib and other molecularly targeted agents in hepatocellular carcinoma. Cancer 2008, 112, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Pàez-Ribes, M.; Allen, E.; Hudock, J.; Takeda, T.; Okuyama, H.; Viñals, F.; Inoue, M.; Bergers, G.; Hanahan, D.; Casanovas, O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009, 15, 220–231. [Google Scholar] [CrossRef]
- Ebos, J.M.L.; Lee, C.R.; Cruz-Munoz, W.; Bjarnason, G.A.; Christensen, J.G.; Kerbel, R.S. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 2009, 15, 232–239. [Google Scholar] [CrossRef]
- Huynh, H.; Ngo, V.C.; Koong, H.N.; Poon, D.; Choo, S.P.; Toh, H.C.; Thng, C.H.; Chow, P.; Ong, H.S.; Chung, A.; et al. AZD6244 enhances the anti-tumor activity of sorafenib in ectopic and orthotopic models of human hepatocellular carcinoma (HCC). J. Hepatol. 2010, 52, 79–87. [Google Scholar] [CrossRef]
- Saad, R.S.; Lindner, J.L.; Liu, Y.; Silverman, J.F. Lymphatic vessel density as prognostic marker in esophageal adenocarcinoma. Am. J. Clin. Pathol. 2009, 131, 92–98. [Google Scholar] [CrossRef]
- Brundler, M.-A.; Harrison, J.A.; de Saussure, B.; de Perrot, M.; Pepper, M.S. Lymphatic vessel density in the neoplastic progression of Barrett’s oesophagus to adenocarcinoma. J. Clin. Pathol. 2006, 59, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Renyi-Vamos, F.; Tovari, J.; Fillinger, J.; Timar, J.; Paku, S.; Kenessey, I.; Ostoros, G.; Agocs, L.; Soltesz, I.; Dome, B. Lymphangiogenesis correlates with lymph node metastasis, prognosis, and angiogenic phenotype in human non-small cell lung cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 7344–7353. [Google Scholar] [CrossRef] [PubMed]
- Koukourakis, M.I.; Giatromanolaki, A.; Sivridis, E.; Simopoulos, C.; Gatter, K.C.; Harris, A.L.; Jackson, D.G. LYVE-1 immunohistochemical assessment of lymphangiogenesis in endometrial and lung cancer. J. Clin. Pathol. 2005, 58, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Padera, T.P.; Kadambi, A.; di Tomaso, E.; Carreira, C.M.; Brown, E.B.; Boucher, Y.; Choi, N.C.; Mathisen, D.; Wain, J.; Mark, E.J.; et al. Lymphatic metastasis in the absence of functional intratumor lymphatics. Science 2002, 296, 1883–1886. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, I.; Van Laere, S.J.; Van den Eynden, G.G.; Benoy, I.; van Dam, P.; Colpaert, C.G.; Fox, S.B.; Turley, H.; Harris, A.L.; Van Marck, E.A.; et al. Increased angiogenesis and lymphangiogenesis in inflammatory versus noninflammatory breast cancer by real-time reverse transcriptase-PCR gene expression quantification. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 7965–7971. [Google Scholar] [CrossRef] [PubMed]
- Kerbel, R.S.; Yu, J.; Tran, J.; Man, S.; Viloria-Petit, A.; Klement, G.; Coomber, B.L.; Rak, J. Possible mechanisms of acquired resistance to anti-angiogenic drugs: Implications for the use of combination therapy approaches. Cancer Metastasis Rev. 2001, 20, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Sweeney, C.J.; Sledge, G.W.J. The Snark is a Boojum: The continuing problem of drug resistance in the antiangiogenic era. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2003, 14, 20–28. [Google Scholar] [CrossRef]
- Miller, K.D.; Sweeney, C.J.; Sledge, G.W.J. Can tumor angiogenesis be inhibited without resistance? EXS 2005, 95–112. [Google Scholar]
- Maione, P.; Gridelli, C.; Troiani, T.; Ciardiello, F. Combining targeted therapies and drugs with multiple targets in the treatment of NSCLC. Oncologist 2006, 11, 274–284. [Google Scholar] [CrossRef]
- Casanovas, O.; Hicklin, D.J.; Bergers, G.; Hanahan, D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 2005, 8, 299–309. [Google Scholar] [CrossRef]
- Krneta, J.; Kroll, J.; Alves, F.; Prahst, C.; Sananbenesi, F.; Dullin, C.; Kimmina, S.; Phillips, D.J.; Augustin, H.G. Dissociation of angiogenesis and tumorigenesis in follistatin- and activin-expressing tumors. Cancer Res. 2006, 66, 5686–5695. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.G.; Heijn, M.; di Tomaso, E.; Griffon-Etienne, G.; Ancukiewicz, M.; Koike, C.; Park, K.R.; Ferrara, N.; Jain, R.K.; Suit, H.D.; et al. Anti-Vascular endothelial growth factor treatment augments tumor radiation response under normoxic or hypoxic conditions. Cancer Res. 2000, 60, 5565–5570. [Google Scholar] [PubMed]
- Jain, R.K. Tumor angiogenesis and accessibility: Role of vascular endothelial growth factor. Semin. Oncol. 2002, 29, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Antiangiogenic therapy for cancer: Current and emerging concepts. Oncology (Williston Park). 2005, 19, 7–16. [Google Scholar]
- Huynh, H.; Nguyen, T.T.T.; Chow, K.-H.P.; Tan, P.H.; Soo, K.C.; Tran, E. Over-expression of the mitogen-activated protein kinase (MAPK) kinase (MEK)-MAPK in hepatocellular carcinoma: Its role in tumor progression and apoptosis. BMC Gastroenterol. 2003, 3, 19. [Google Scholar] [CrossRef] [PubMed]
- Wiesenauer, C.A.; Yip-Schneider, M.T.; Wang, Y.; Schmidt, C.M. Multiple anticancer effects of blocking MEK-ERK signaling in hepatocellular carcinoma. J. Am. Coll. Surg. 2004, 198, 410–421. [Google Scholar] [CrossRef]
- Huynh, H.; Chow, K.H.P.; Soo, K.C.; Toh, H.C.; Choo, S.P.; Foo, K.F.; Poon, D.; Ngo, V.C.; Tran, E. RAD001 (everolimus) inhibits tumour growth in xenograft models of human hepatocellular carcinoma. J. Cell. Mol. Med. 2009, 13, 1371–1380. [Google Scholar] [CrossRef]
- Tille, J.C.; Wood, J.; Mandriota, S.J.; Schnell, C.; Ferrari, S.; Mestan, J.; Zhu, Z.; Witte, L.; Pepper, M.S. Vascular endothelial growth factor (VEGF) receptor-2 antagonists inhibit VEGF- and basic fibroblast growth factor-induced angiogenesis in vivo and in vitro. J. Pharmacol. Exp. Ther. 2001, 299, 1073–1085. [Google Scholar]
- Allen, E.; Walters, I.B.; Hanahan, D. Brivanib, a dual FGF/VEGF inhibitor, is active both first and second line against mouse pancreatic neuroendocrine tumors developing adaptive/evasive resistance to VEGF inhibition. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 5299–5310. [Google Scholar] [CrossRef]
- Dieci, M.V.; Arnedos, M.; Andre, F.; Soria, J.C. Fibroblast growth factor receptor inhibitors as a cancer treatment: From a biologic rationale to medical perspectives. Cancer Discov. 2013, 3, 264–279. [Google Scholar] [CrossRef]
- Guide for the Care and Use of Laboratory Animals, 8th ed.; National Academies Press: Washington, DC, USA, 2011; ISBN 9780309154000.





| PDX Line | FGFR1 | FGFR2 | FGFR3 |
|---|---|---|---|
| HCC13-0109 | Low | High | High |
| HCC01-0909 | Low | High | High |
| HCC06-0606 | Low | High | High |
| HCC21-0208 | Low | High | High |
| HCC26-0808A | Low | High | High |
| HCC17-0211 | Low | High | High |
| HCC2006 | Low | High | Modest |
| HCC13-0212 | Low | Low | Modest |
| HCC25-0705A | Low | Modest | Modest |
| HCC06-0606Sor64 | Low | High | High |
| HCC07-0409 | Low | Low | Low |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le, T.B.U.; Vu, T.C.; Ho, R.Z.W.; Prawira, A.; Wang, L.; Goh, B.C.; Huynh, H. Bevacizumab Augments the Antitumor Efficacy of Infigratinib in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2020, 21, 9405. https://doi.org/10.3390/ijms21249405
Le TBU, Vu TC, Ho RZW, Prawira A, Wang L, Goh BC, Huynh H. Bevacizumab Augments the Antitumor Efficacy of Infigratinib in Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2020; 21(24):9405. https://doi.org/10.3390/ijms21249405
Chicago/Turabian StyleLe, Thi Bich Uyen, Thanh Chung Vu, Rebecca Zhi Wen Ho, Aldo Prawira, Lingzhi Wang, Boon Cher Goh, and Hung Huynh. 2020. "Bevacizumab Augments the Antitumor Efficacy of Infigratinib in Hepatocellular Carcinoma" International Journal of Molecular Sciences 21, no. 24: 9405. https://doi.org/10.3390/ijms21249405
APA StyleLe, T. B. U., Vu, T. C., Ho, R. Z. W., Prawira, A., Wang, L., Goh, B. C., & Huynh, H. (2020). Bevacizumab Augments the Antitumor Efficacy of Infigratinib in Hepatocellular Carcinoma. International Journal of Molecular Sciences, 21(24), 9405. https://doi.org/10.3390/ijms21249405