Abstract
The development of new therapeutic approaches to diseases relies on the identification of key molecular targets involved in amplifying disease processes. One such molecule is thioredoxin-interacting protein (TXNIP), also designated thioredoxin-binding protein-2 (TBP-2), a member of the α-arrestin family of proteins and a central regulator of glucose and lipid metabolism, involved in diabetes-associated vascular endothelial dysfunction and inflammation. TXNIP sequesters reduced thioredoxin (TRX), inhibiting its function, resulting in increased oxidative stress. Many different cellular stress factors regulate TXNIP expression, including high glucose, endoplasmic reticulum stress, free radicals, hypoxia, nitric oxide, insulin, and adenosine-containing molecules. TXNIP is also directly involved in inflammatory activation through its interaction with the nucleotide-binding domain, leucine-rich-containing family, and pyrin domain-containing-3 (NLRP3) inflammasome complex. Neurodegenerative diseases such as Alzheimer’s disease have significant pathologies associated with increased oxidative stress, inflammation, and vascular dysfunctions. In addition, as dysfunctions in glucose and cellular metabolism have been associated with such brain diseases, a role for TXNIP in neurodegeneration has actively been investigated. In this review, we will focus on the current state of the understanding of possible normal and pathological functions of TXNIP in the central nervous system from studies of in vitro neural cells and the brains of humans and experimental animals with reference to other studies. As TXNIP can be expressed by neurons, microglia, astrocytes, and endothelial cells, a complex pattern of regulation and function in the brain is suggested. We will examine data suggesting TXNIP as a therapeutic target for neurodegenerative diseases where further research is needed.
1. Introduction
Oxidative stress resulting from an imbalance in cellular redox can occur due to the production of excessive levels of reactive oxygen species (ROS) and/or a deficit of cellular antioxidant systems. Supplementation with various classes of antioxidants has been widely investigated for neurodegenerative diseases (for recent reviews, see [1,2]). Excess levels of ROS can occur from inflammatory responses by innate immune cells (e.g., neutrophils, monocytes/macrophages/microglia) due to infections or chronic autoimmune responses [2], deficiencies in mitochondrial oxidative phosphorylation pathways [3,4], and environmental factors such as UV light, toxic chemicals, or heavy metals. Oxidative stress is linked to many different chronic human diseases, particularly those with inflammatory and/or metabolic components (e.g., diabetes and diabetes-related retinopathy [5], stroke [6], cancers [7], vascular diseases, and neurodegenerative diseases [8,9]). Developing effective therapies for these diseases requires the identification of key pathological targets responsible for amplifying disease processes.
Thioredoxin-interacting protein (TXNIP) has emerged as a key pathological regulator of diseases, particularly those associated with glucose and lipid abnormalities and inflammation. TXNIP has been identified in disease mechanisms involved in various cancers [10,11,12], diabetes mellitus [13], cardiovascular disease [14], renal disease [15], and retinal disease [16], among others. Many of these diseases associated with TXNIP can arise from vascular complications of diabetes [17]. The involvement of TXNIP in diabetes, pancreatic beta-cell death, and glucose metabolism has been the focus of several recent review articles [14,17,18]. TXNIP was identified (and named) due to its interaction and inhibition of the key antioxidant proteins thioredoxin-1 (TRX1) and thioredoxin-2 (TRX2) [19,20,21], but more recent findings identified properties involved in cellular metabolism and transcription regulation [22], apoptosis and cell death [13], inflammation [23], and tumor suppression [11,24] that might not be directly related to the modulation of TRX and oxidative stress.
In this review, we will consider the known features of TXNIP expression with a particular focus on human and animal brains and different types of neural cells. Particular consideration will be on how TXNIP and the TRX system may be involved in the pathological features of Alzheimer’s disease (AD), a chronic neurodegenerative disease of elderly people, which have been associated with abnormalities in cellular glucose metabolism, mitochondrial dysfunction, inflammation, and oxidative stress, among others [25]. TXNIP might be an important hub coordinator of different pathological processes in this disease, but there has been only one published study on its expression and distribution in human AD brains, and further studies are needed [26]. We will also consider the evidence for the pathological contribution of diabetes and diabetes-induced oxidative stress to Alzheimer’s disease (AD) as part of this review [27], and how it can be linked to TXNIP.
2. Biochemistry of TXNIP: Overview
The gene for TXNIP is located on the human chromosome 1q21.1 and is transcribed to four messenger RNA (mRNA) splice variants [28]. TXNIP is a member of the α-arrestin protein family and has two arrestin-like domains: one is a PxxP sequence and the other is a PPxY sequence. One mRNA for TXNIP codes for 336 amino acids for a polypeptide of approximately 37.4 kDa, whereas another codes for 391 amino acids for a polypeptide of approximately 43.7 kDa. The two other splice variants have not been associated with proteins. Reported molecular weights of TXNIP vary depending on the expressing cell type, but the major protein band(s) detected by SDS-gel electrophoresis is/are 50–55 kDa. This would suggest a certain amount of post-translational modification occurring. Protein bands of approximately 37 kDa have been observed on immunoblots with certain antibodies and cell types, but 50–55 kDa polypeptides appear to be the major form. TXNIP was identified initially by several investigators as vitamin D3-upregulated protein-1 (VDUP1) in different cell types [19,20,21]. Interestingly, other studies have not confirmed TXNIP induction by vitamin D3 in additional cell types, and noticeably, there is a lack of a vitamin D3-responsive element in the TXNIP gene promoter [29,30]. It has been suggested that vitamin D3 stabilizes cellular TXNIP protein rather than inducing expression [29].
2.1. TXNIP and Thioredoxin System
Earlier studies provided the first link between VDUP1/TXNIP and oxidative stress by demonstrating binding to reduced, but not oxidized, thioredoxin (TRX) [19,20,21]. TXNIP can bind two cysteine residues (Cys32 and Cys35) present in the active catalytic site of TRX. The binding of oxidized TXNIP to reduced TRX involves the formation of disulfide bonds between TXNIP-Cys247 and TRX-Cys32 [31]. A mutation in TXNIP-Cys247 is sufficient to remove its ability to sequester TRX activity [31]. Although classified as belonging to the α-arrestin family, these cysteine residues are unique to TXNIP. TXNIP can bind and inactivate both TRX1 and TRX2 [19,32]. The TRX cellular disulfide oxidoreductase system is a highly conserved system found from prokaryotes to plants to mammals [33,34,35]. TRX plays a central role in protecting cells from oxidative stress. Oxidized TRX is reduced by NADPH through a reaction catalyzed by thioredoxin reductase complex (TRX-R). Reduced TRX can then directly reduce disulfides in target proteins [33]. The main form of TRX is TRX1, which is located primarily in the cytosol but can be translocated to the plasma membrane and/or nucleus, particularly under inflammatory conditions [36,37]. Closely related in structure, TRX2 is specific to mitochondria [38,39]. Immunohistochemistry and in situ hybridization mRNA studies have shown that TRX was highly expressed in mammalian brains, particularly in subsets of neurons in areas of high metabolic activity and oxidative burden [40,41,42]. TRX mRNA expression was highest in the piliform cortex, dentate gyrus, CA3/CA4 region of the hippocampus, locus coeruleus, and nucleus of the hypothalamus and solitary tract [41]. Trx2 mRNA and protein were highly expressed in rat brains in neurons in the olfactory bulb, frontal cortex, hippocampus, some hypothalamic and thalamic nuclei, cerebellum, and numerous brainstem nuclei [42].
The significance of the TRX system for maintaining cellular health was demonstrated in transgenic mice overexpressing forms of TRX. Increased expression of human TRX in mice resulted in resistance to oxidative stress and increased lifespan [43]. Other studies have shown that TRX overexpression in transgenic mice was protective of transient ischemic brain damage [44]. This effect was also observed by intravenous administration of TRX protein [45]. These examples of the beneficial outcomes of increased TRX are used to introduce the widely appreciated pathological consequences of reduced TRX activity. The first study linking TRX to AD showed a significant reduction in TRX protein levels in most brain regions in AD cases compared to nondemented controls with accompanying increased TRX-R activity. This study also demonstrated the protective effect of TRX when added to cultures of neurons treated with toxic doses of amyloid beta (Aβ) peptide [46]. A further study showed that TRX1 levels were increased significantly in the cerebrospinal fluid (CSF) and plasma of AD cases compared to mild cognitive impairment (MCI) cases [47]. This suggested that TRX1 was being secreted from damaged neurons in AD brains. A noticeable alteration in the patterns of TRX1 and TRX2 cellular immunoreactivity in hippocampal neurons of AD cases was also observed.
2.2. TXNIP Interacting Proteins
TXNIP has been shown to interact with other proteins besides TRX, including importin-α1 [48], human ecdysoneless (hEcd) [49], and NOD-, LRR-, and pyrin domain-containing protein-3 (NLRP3), a component of the inflammasome complex [23,50]. These interactions relate to additional identified properties of TXNIP. The significance of the interaction of TXNIP with inflammasome components and enhancement of inflammation will be considered.
3. Regulation of TXNIP Expression
Many different cellular stress factors positively or negatively regulate TXNIP expression. These include UV light, heat shock [51], hypoxia, ROS, nitric oxide [52], nicotinamide adenine dinucleotide [53], ATP, glutamine, nicotine, vascular endothelial growth factor, basic fibroblast growth factor [54], transforming growth factor β [55], estradiol [56], calcium channel blockers [57], activators of advanced glycation endoproduct receptor (RAGE) [58], insulin, and glucose [59]. Activation of the TXNIP promoter, which contains a carbohydrate response element (ChoRE), is regulated by transcription factors MondoA:Max-like protein X (MLx), nuclear factor Y (NF-Y), and the carbohydrate response element-binding protein (ChREBP) [45]. Transcription factors forkhead box O1 (FOXO1) and FOXO3a can bind to the TXNIP promoter and by competing with ChREBP can downregulate TXNIP transcription [46,47]. Activation of AMP protein kinase (AMPK) can also lead to inhibition of TXNIP mRNA transcription [48].
The mechanism whereby TXNIP expression is induced through the activation of the receptor for advanced glycation endproducts (RAGE) by the RAGE ligand S100b is of relevance for th consideration of AD as RAGE is a receptor for Aβ and is involved in Aβ toxicity and AD pathogenesis [58]. It was demonstrated in Schwann cells in vitro and injured sciatic nerve in vivo that binding of S100b to RAGE induced TXNIP with TXNIP being involved in the downstream activation of p38 mitogen-activated protein kinase (MAPK), cAMP response element-binding protein (CREB), and nuclear factor κB (NFκB). RAGE silencing blocked the induction of TXNIP, whereas the silencing of TXNIP inhibited the activation of these signaling pathways, preventing RAGE-induced fibronectin and IL1β synthesis and Schwann cell migration [58]. Induction of TXNIP by RAGE ligands in retinal endothelial cells through the activation of p38 MAPK and NFκB also led to enhanced expression of inflammation-associated genes, including cyclooxygenase 2 (Cox2), vascular endothelial growth factor (VEGFA), and intercellular adhesion molecule-1 (ICAM1). Expression of these genes was reduced when TXNIP expression was inhibited, and enhanced with TXNIP overexpression [60].
3.1. Cell Types
TXNIP expression has been studied in a range of different cell types derived from different tissues. Expression of TXNIP is not tissue-specific, with TXNIP RNA being detected across all tissues and many different cell types. The highest expression levels were demonstrated in the lungs, thyroid glands, adipose tissue, skeletal muscle, and blood mononuclear cells (https://www.proteinatlas.org/ENSG00000265972-TXNIP/summary/rna). By comparison, expression in neural tissue was relatively low, with the highest levels in the retina. The highest levels of constitutive expression of TXNIP mRNA were in muscle-derived cell lines (https://www.proteinatlas.org/ENSG00000265972-TXNIP/summary/rna).
A common feature of all expressing cell types has been the induction of TXNIP by different forms of cellular stress. Increased levels of palmitate and other lipids that led to cellular endoplasmic reticulum (ER) stress [61,62], increased levels of N-methyl D-aspartate (NMDA) resulting in NMDA receptor activation, and increased cellular calcium [63], increased ischemia/hypoxia, and increased glucose, resulting in enhanced oxidative stress, which all induced TXNIP expression. Increased levels of ROS from dysfunctional mitochondria can also increase TXNIP expression [32].
The promoter for VDUP1/TXNIP contains many different cis-regulatory sequences, but the presence of the heat shock factor element (HSE) sequence appeared critical for controlling cellular responses to certain stresses [51]. Using HEK-293 human embryonic kidney-derived Bosc cells, TXNIP was upregulated when cells were grown at high density or with serum deprivation, but this effect was abolished if the HSE sequence was deleted from the promoter. Overexpression of heat shock factor (HSF) in these cells upregulated the expression of TXNIP [51]. Many different stress factors have been shown to induce TXNIP expression in different cell types. One common factor can be ER stress. In an experimental cell model using INS-1 rat islet-derived cells, the induction of unfolded protein response (UPR) due to thapsigargin-induced ER stress resulted in the rapid induction of TXNIP expression along with the UPR sensor molecules Inositol-requiring transmembrane kinase/endoribonuclease 1α (IRE1α) and protein kinase RNA-like ER kinase (PERK). In cells deficient in IRE1α and PERK, an ER stress-induced increase in TXNIP did not occur. Activated IRE1α increased TXNIP mRNA stability by reducing levels of a TXNIP-destabilizing microRNA, miR-17. As a result of elevated TXNIP protein expression, NLRP3 becomes activated, resulting in the formation of caspase-1 and bioactive IL-1β [50]. This mechanism is believed to be a key event in diabetes, as excess TXNIP leads to pancreatic cell death, whereas knockdown of TXNIP prevented ER stress-induced cell death [64]. It has been previously shown that excess glucose-induced TXNIP expression by activating the transcription factor carbohydrate response element-binding protein (ChREBP) causes binding to an element in the TXNIP promoter and increasing transcription [65]. Activation of ChREBP and translocation to the nucleus also occurred as a result of ER stress [64]. The described mechanism for the ER stress-induced unfolded protein response (UPR) could have direct relevance for neurodegenerative diseases such as AD and Parkinson’s disease (PD) where accumulations of toxic Aβ or α-synuclein, primarily produced by neurons, can induce UPR [66,67]. Other transcription factors can positively or negatively affect the expression of TXNIP. Overexpression of transcription factor Ets-1, which is activated by inflammatory/oxidative stress associated extracellular regulated kinase (ERK1/2), resulted in increased TXNIP in Min6 β-cells and mouse islet cells. Knockdown of Ets1 reduced the expression of TXNIP. Overexpression of Ets and TXNIP negatively affected cellular proliferation and glucose regulatory functions [68].
3.1.1. Peripheral Cells
Many studies of TXNIP cellular expression in peripheral cells have focused on pancreatic islet cells in terms of effects on glucose uptake and metabolism, insulin secretion, and cell death. These studies have been extensively reviewed for their significance in understanding mechanisms of diabetes (for recent reviews, see [69,70]). For this review focusing on brain diseases with inflammatory components, we will consider TXNIP expression in macrophages and its consequences on inflammation. Increasing TXNIP expression in human blood-derived macrophages with a peroxisome proliferator-activated receptor-gamma (PPAR-γ) agonist resulted in enhanced levels of activated caspase-3, indicative of inflammasome activation, and also increased apoptosis [71]. In contrast, stimulating macrophages with a PPAR-alpha agonist resulted in increased TRX and reduced the expression of TXNIP, reduced caspase-3 activation, and reduced numbers of apoptotic cells [72]. Induction of TXNIP in RAW264 murine macrophage cells with the sugar D-allose or by plasmid-mediated overexpression inhibited the differentiation of osteoclasts by inhibiting the transcriptional activity of activator protein 1, NFκB, and nuclear factor of activated T cells (NFAT) [73]. Earlier studies have identified D-allose to have significant antiproliferative properties in several types of cancer cells [74,75,76] through the activation of the transcription factor MondoA:Mlx and the induction of TXNIP expression [77].
TXNIP deficiency due to treatment with lipopolysaccharide (LPS) used as a model of endotoxic shock can exacerbate inflammation. Macrophages from TXNIP-deficient mice produced increased amounts of nitric oxide in response to LPS treatment, and in vivo, TXNIP-deficient mice were rescued from endotoxic-shock-induced death by a nitric oxide synthase inhibitor. The mechanism is due to the LPS inhibition of MondoA:Mlx activation preventing the induction of TXNIP expression [78,79]. In contrast to LPS, chemicals that induce endoplasmic reticulum (ER) stress in macrophages, including tunicamycin, produced enhanced inflammation through increased production of ROS causing TXNIP induction NFκB activation and inflammasome/caspase-1 activation resulting in enhanced IL-1β secretion [80].
3.1.2. Neurons
The first study describing TXNIP function in neural cells employed cerebellar granular neurons, which underwent apoptosis when culture conditions were switched from high potassium depolarizing conditions to physiological potassium conditions. TXNIP expression was significantly enhanced in neurons under this and other apoptosis-inducing conditions. TXNIP induction was inhibited by increased calcium influx through voltage-dependent calcium channels [57]. Glutamate neurotoxicity was shown to be dependent on TXNIP inflammasome activation. Treatment of mice hippocampal slice cultures or SH-SY5Y cells with toxic doses of glutamate resulted in increased ROS, ER stress, increased TXNIP expression, and NLRP3 inflammasome activation, resulting in cell toxicity [81]. Treatment with the antioxidant curcumin was protective by the activation of AMPK and reducing the activation of IRE1α and PERK, resulting in the decreased expression of TXNIP, reduced NLRP3 inflammasome activation, and reduced cell death. Curcumin increased AMPK activity, whereas knockdown of AMPKα with specific siRNA abrogated its inhibitory effects. The protective effects of curcumin on inhibiting phosphorylated (p)-IRE1α, p-PERK, and NLRP3 expression in the hippocampus CA1 region was also demonstrated in vivo in rats subjected to hypoxia/ischemia insults [81]. A recent study that characterized the gene expression profiling of SH-SY5Y-derived neurons stably overexpressing the mitochondria-localized antioxidant protein mitochondrial ferritin (FtMt) demonstrated significantly reduced constitutive levels of expression of TXNIP mRNA. As increased FtMt will reduce ROS produced by mitochondria, this finding demonstrated a link between mitochondria and TXNIP expression in neurons [82]. Over-expression of FtMt and reduced TXNIP was protective for these cells.
3.1.3. Microglia
The interaction of ROS/oxidative stress and inflammasome activation can be demonstrated in microglia, the major mediator of innate inflammatory responses in the brain. Microglia treated with thrombin, a factor involved in brain injury after infarct or hemorrhage, expressed increased amounts of ROS and TXNIP, causing inflammasome activation and apoptosis. Treatment with N-acetyl cysteine, a free-radical scavenger, prevented TXNIP induction, resulting in cell death [83]. Resveratrol, a polyphenol with antioxidant, sirtuin, and PPAR-γ-activating properties, inhibited Aβ-induced inflammatory activation and proliferation of microglia by inhibiting the activation of TXNIP/TRX/NLRP3 inflammasome pathway [84]. As a model of chronic stress, treatment of primary mouse microglia with the stress hormone corticosterone increased TXNIP expression, TXNIP/NLRP3 interactions, and IL-1β levels, indicating inflammatory activation [85]. Knocking-down TXNIP expression significantly inhibited corticosterone-induced inflammatory activation. Similar findings were shown in mouse brains of corticosterone-treated animals [85].
A recent RNA sequencing profiling study of microglia purified from AD model mice identified the decreased expression of TXNIP between microglia considered homeostatic and microglia activated to a stage 1 disease-associated microglia (DAM) [86,87]. Similarly, a number of homeostatic microglial genes (P2ry12, P2ry13, Tmem119, Cxcr1) were also downregulated between these groups. DAM is a newly defined phenotype describing activated plaque-associated microglia with the function to reduce the amplification of inflammation.
3.1.4. Astrocytes
Astrocytes, the major support cells of the central nervous system providing growth factors, metabolic support, and detoxifying potentially damaging molecules (e.g., glutamate), can become activated by inflammatory cytokines and contribute to neurodegenerative processes by increased ROS and nitric oxide production. In a rat model of obesity, high-fat diet (HFD)-fed rats, both wild-type and spontaneously hypertensive rats, had evidence of abnormal capillary formation and degeneration along with the enhanced expression of TXNIP, NFκB, and inflammatory cytokines IL-1β and TNFα [88]. Increased expression of TXNIP was detected in astrocytes, retinal endothelial cells, and some microglia. Increased interaction of TXNIP and NLRP3 was also demonstrated in brain tissues from HFD rats. In a model of chronic mild stress/depression, deletion of the mitochondrial uncoupling protein-2 resulted in enhanced oxidative stress, enhanced TXNIP expression, and NLRP3 inflammasome activation in astrocytes [89]. In a rat model of subarachnoid hemorrhage, the peak induction of TXNIP correlated with the peak of cell death. Inhibition of TXNIP expression and the use of ER stress inhibitors resulted in an insignificant reduction of cell death. TXNIP expression was detected in astrocytes, microglia, and neurons in this model [90]. The role of astrocyte TXNIP in cellular lactate and glucose energy metabolism was demonstrated [91]. Activation of AMPK in astrocytes inhibited TXNIP by its phosphorylation and by promoting its degradation and inhibition of transcription. Activation of AMPK resulted in improved levels of glut-1 transporter expression in astrocytes. In AMPK astrocyte-specific deletion mice, glucose and lactate metabolism was impaired by high TXNIP expression resulting in an energy imbalance in neurons and, consequently, and cell death [91].
3.1.5. Endothelial Cells
TXNIP is involved in vascular dysfunction due to damage to vessel endothelial cells, the major cause of complications resulting from diabetes [92]. A number of studies have shown common mechanisms that high glucose and high levels of oxidized low-density lipoproteins (oxLDL) result in oxidative stress and inflammation through the enhanced expression of TXNIP and inflammasome activation. As an example, treatment of endothelial cells with high glucose and oxLDL combined with rosmarinic acid, an anti-inflammatory and antioxidant, inhibited TXNIP expression, inflammasome activation, and endothelial dysfunction through the inhibition of p38 MAPK and FOXO1 signaling. Similar results were obtained with ROS scavengers and p38 MAPK and FOXO1 inhibitors [93]. TXNIP knockout mice were shown to be resistant to the retinal endothelial dysfunction that occurred with a HFD in wild-type mice. The TXNIP gene deletion mice did not have the breakdown of the blood–retinal endothelial barrier that occurred in 18-week HFD-fed wild-type mice [94]. Overexpression of TXNIP in human aortic endothelial cells induced by high glucose or the overexpression of ChREBP directly caused apoptosis and impaired NO function. In vivo, type-1 diabetes modeled in rats caused increased TXNIP in peripheral blood along with decreased NO and VEGF, increased ROS, and increased inflammation-associated vascular cell adhesion molecule 1 (VCAM-1) [95].
3.1.6. Retinal Pigment Epithelial Cells
Retinal pigment epithelial (RPE) cells are the major support cells for the photoreceptor neurons in the eye. These cells transport nutrients, ions, and water, protect against photooxidation, reisomerization of all-trans-retinal into 11-cis-retinal, crucial for the visual cycle, phagocytosis of damaged photoreceptor constituents, and secrete essential factors for the structural integrity of the retina, for example pigment epithelial-derived factor (PEDF). These cells are damaged by the high-glucose/oxidative stress consequences of diabetes [96], and their dysfunction is involved in the development of diabetic retinopathy [97]. High-glucose-induced TXNIP in RPE cells resulted in mitochondrial depolarization, mitochondrial fragmentation, and shuttling to lysosomes. TXNIP overexpressing RPE cells showed significant changes in lysosome structure and autophagy. These high-glucose effects were inhibited if TXNIP expression was suppressed [98]. Surprisingly, another study demonstrated that the downregulation of TXNIP by oxidative stress in the RPE cell line ARPE-19 resulted in reduced cellular proliferation. The consequences of downregulation of TXNIP were increased autophagic flux, and increased p53 activation resulting in AMPK activation. The consequences of these were reduced blood–retinal barrier integrity [99].
3.1.7. Brain Cancer Cells
Overexpression of TXNIP has been correlated with inhibition of tumor cell suppression in multiple studies. In addition, alterations in the expression of all proteins of the TRX system, including TRX, TRX reductase (TRXR), and TXNIP, in gliomas and glioblastomas were associated with differing clinical outcomes. High cytoplasmic TRXR and TRX expression in patients with high-grade gliomas was associated with poor outcomes, whereas in medulloblastoma, high expression of cytoplasmic TRXR, TRX, and nuclear TRX was associated with worse prognosis [100]. In similar studies of malignant glioma, higher TXNIP expression levels were associated with extended patient survival time [24]. In vitro analysis in U251 cells showed increased growth, migration, and invasion with downregulated TXNIP expression compared with their non-transfected counterparts. Although not studied in brain cancer cells, it was shown in thyroid cancer cells that TXNIP overexpression resulted in decreased glucose uptake and cell division. In a mouse model, transplanted TXNIP-overexpressing cells had reduced metastatic potential [101]. A similar feature was observed in prostate cancer cells. Overexpression of TXNIP in a prostate cancer cell line blocked cell proliferation and glucose uptake. Cell division became halted in the G0/G1 phase, which promoted cell apoptosis [11]. This study also showed in a large series of human prostate benign hyperplasia and cancer samples that there was significantly less TXNIP expression in the cancer samples. Levels of TXNIP in these samples negatively correlated with the expression of glucose transporter (glut1)-1 mRNA [11].
4. TXNIP, Hyperglycemia, and Oxidative Stress—NLRP3 Inflammasome Complex
Presentations of research data on TXNIP in relation to disease have frequently considered TXNIP/TRX interactions and the resulting oxidative stress as a separate mechanism from its role in the NLRP3 inflammasome activation, but these two features are interrelated. Although associated with enhanced inflammation, NLRP3 inflammasome activation occurs in all brain cell types, including neurons, astrocytes, and endothelial cells, not just microglia [81,102]. If one considers the events occurring under hyperglycemic conditions, excess glucose leads to increased levels of ROS by inducing the overproduction of NADH and increased mitochondrial-derived ROS that inhibits GAPDH, the enzyme that removes excess cellular glucose [103]. However, this inhibition that activates alternative glucose metabolic pathways leads to further ROS production. Excess ROS directly induces the expression of TXNIP and the inflammation-associated transcription factor NFκB. Excess TXNIP will further exacerbate oxidative stress by binding TRX and, in cooperation with NFκB, bind and activate the inflammasome complex. The inflammasome complex consists of an association of NLRP3, apoptosis-associated speck-like protein containing a caspase-binding domain (ASC) and pro-caspase-1 [104,105]. In the presence of excess ROS, TXNIP interaction with TRX can be reversed, which then allows released TXNIP to bind and activate NLRP3 inflammasome, resulting in enhanced inflammation. Activation of NLRP3-inflammasome promotes the formation of activated caspase-1, which processes interleukin (IL)-1β, IL-18, and IL-33 into bioactive forms [106], and also induces cell death through pyroptosis [107]. An earlier study also demonstrated that inflammasome activators such as uric acid crystals induced the dissociation of TXNIP from TRX in the presence of ROS, allowing it to bind and activate the NLRP3 complex and enhance caspase activation [50].
5. TXNIP Expression in the Brain
Studies focusing on the cellular distribution of TXNIP in brain cells are limited. Detailed immunochemical characterization of TXNIP/VDUP1 by comparing its distribution in Drosophila and rat brains showed relatively conserved patterns of expression. The antibodies employed in this study identified this protein in both of these species. Constitutive expression in subsets of neurons and astrocytes were identified using double immunohistochemistry staining with appropriate markers. This study did not report the constitutive microglial expression of TXNIP but observed nuclear immunoreactivity under hyperglycemic conditions [108]. A further study employed immunohistochemistry to compare the neuroanatomical distribution of different TRXs, TRRs, glutathione/glutaredoxin, peroxiredoxins, and TXNIP in rat brains. Expression of these antioxidants and related molecules was highest in brain regions susceptible to damage in conditions of hypoxia/ischemia including the cerebellum, cortex, hippocampus, substantia nigra, striatum, and spinal cord. Weak expression of TXNIP was detected in subsets of neurons, not glia, in these brain regions, except the spinal cord, and with the highest expression in the retina. The pattern of expression of TRX1 and TXNIP showed extensive overlap [109]. A direct link between metabolic activity and TXNIP was demonstrated in mice with the expression of TXNIP in medial hypothalamic neurons, which increased under conditions of nutrient excess and obesity [110]. A further study using directed TXNIP gene deletion and overexpression in Agrp hypothalamic neurons showed that the overexpression of TXNIP led to reduced energy expenditure and activity, resulting in obesity and fat accumulations, whereas the deletion of TXNIP in Agrp neurons had a reverse effect, with increased energy metabolism and activity [111]. Another study showed strong induction of TXNIP in the hypothalamus of mice in a state of lowered energy metabolism, though increased TXNIP expression was detected in ependymal-lining cells, not neurons, in the hypothalamus. Increased TXNIP expression was also detected in the liver and adipose tissues of these animals [112]. These studies highlighted physiological functions for TXNIP in regulating energy metabolism.
5.1. TXNIP in Acute Neurological Conditions
The role of TXNIP in ischemic stroke disease using animal models has been examined. Wild-type, TXNIP knockout, and wild-type mice treated with the TXNIP inhibitor resveratrol were subjected to middle cerebral artery occlusion (MCAO) to model ischemic stroke. These studies showed TXNIP knockout, and resveratrol-treated mice had an approximately 40% decrease in infarct size and significantly improved neurological scores. This improvement correlated with reduced oxidative stress and reduced evidence of NLRP3 inflammasome activation [113]. A similar model using rats subjected to MCAO with reperfusion was significantly protected by treatment with umbelliferone, an antioxidant. This protection occurred as treatment increased the expression of PPAR-γ, which resulted in reduced NLRP3 inflammasome activation due to the inhibition of TXNIP expression [114]. A recent study utilized neonatal rat pups subjected to common carotid artery ligand and hypoxia to induce ischemic injury [113]. Animals treated with a PPAR-β/δ agonist had a significantly reduced infarct area and improved neurological features. This study showed that PPAR-β/δ activation induced the expression of miRNA miR17-5p, which reduced TXNIP levels and the activation of Apoptosis signal-regulating kinase 1 (ASK1) and p38 MAPK [115]. Promoting TXNIP activation in vivo using a TXNIP CRISPR activation plasmid reversed the protective effects, as did the administration of a PPAR-β/δ antagonist. The consequence of PPAR-β/δ activation by GW0742 was decreased TXNIP, NLRP3, IL-6, and TNF-α and reduced numbers of activated microglia [116]. Similar results from PPAR activation were observed in vitro using PC12 neuronal-like cells subjected to oxygen/glucose deprivation (OGD) [116].
A recent study employing a rat model for cerebral venous sinus thrombosis demonstrated that neuronal pyroptosis was caused by endoplasmic reticulum stress and oxidative stress that activated the TXNIP/peroxynitrite/NLRP3 inflammasome pathway. Immunohistochemical staining revealed that increased TXNIP was primarily in neurons and also in subsets of microglia and astrocytes [117]. TXNIP immunoreactivity is colocalized with p-IREα1 and p-PERK, mainly in neurons. Time-course studies showed maximal induction of TXNIP occurred three days after surgery and then expression declined. The same time course of induction was observed for NLRP3, whose expression was in neurons and microglia. Direct interaction of TXNIP and NLRP3 at three days was demonstrated by immunoprecipitation and immunoblot analysis, producing results indicative of inflammasome activation [117], and at three days, IL-1β levels were also at their highest. A significant feature of this study was the decline of TXNIP and NLRP3 levels after reaching maximal levels. This suggests that the time for the increased expression of these proteins will be restricted and limited to ongoing active sites of pathology.
5.2. TXNIP in Neurodegenerative Diseases (Animal and Cellular Models)
TXNIP has been implicated in neurodegenerative diseases, but there have only been a limited number of studies on this topic. Figure 1 illustrates the potential interacting features of TXNIP in the progression of AD. Similar processes could occur in Parkinson’s disease (PD). This is a hypothetical model as further human-focused studies of brain expression of TXNIP are needed. Overexpression of α-synuclein, the protein implicated in the pathogenesis of PD, in transgenic mice or transfected HEK cells, produced increased levels of TXNIP [118]. In this study, it was also shown that the overexpression of TXNIP resulted in the enhanced accumulation of α-synuclein. This resulted in increased accumulation of the autophagy proteins LC3-II and p62, indicating excess TXNIP caused a blockade of autophagic flux. TXNIP impaired lysosomal function by downregulating the expression of the lysosomal membrane protein ATP13A2. Overexpression of TXNIP by the injection of a viral transduction vector into mice substantia nigra pars compacta resulted in a 38% decrease in the numbers of tyrosine-hydroxylase-positive neurons [118]. This was the first study linking TXNIP to PD pathogenesis, but there have been no studies on human brain tissues. The only study to date examining TXNIP expression in human AD and control brains observed an increased expression of TXNIP mRNA and protein in AD cases and increased TXNIP immunoreactivity of cells associated with Aβ plaques and phosphorylated tau (p-Tau)-positive structures. The identities of TXNIP-immunopositive cells were not confirmed but the authors implied that they were microglia due to their morphology as other components of the NLRP3 inflammasome complex and IL-1β levels were increased [26]. An experimental study using APP/PS1 plaque-developing transgenic mice demonstrated that TXNIP protein levels were significantly elevated whereas TRX levels were not in these mice at 9 and 12 months of age when Aβ deposits had developed compared to wild-type mice. The treatment of murine primary cortical neurons or HT22-derived hippocampal neurons with oligomeric Aβ (1-42) produced a significantly increased expression of TXNIP but not TRX [119]. Inhibiting TXNIP expression in Aβ-treated cells resulted in reduced amounts of oxidative modifications of cellular proteins. Wild-type mice treated with a regime of chronic unpredictable stress over 28 days had increased levels of TXNIP, but not TRX, protein in the frontal cortex and hippocampus. The consequence of this was evidenced by increased levels of cysteine oxidative modifications, such as increased protein sulfenylation and nitrosylation and increased amounts of phosphorylated (activated) ASK1 [120]. Another study showed increased levels of TXNIP and decreased levels of TRX in the hippocampus of the female 5xFAD line of amyloid plaque-developing mice. Using the SH-SY5Y neuroblastoma cell line, this study showed that estradiol could protect from TXNIP induction and cell toxicity induced by in vitro Aβ (1-42) treatment. The basis of this study was the analysis of human gene expression profiling data that showed significantly higher levels of TXNIP mRNA in samples from female AD compared to male AD subjects [121]. The effect of estradiol modulation of TXNIP upregulation was through the activation of AMPK. Using the 3xTg-AD mouse model, significantly enhanced TXNIP was detected in the cortex but coincident with significantly reduced expression in the spleen compared to wild-type mice. The mean spleen weight of 3xTg-AD mice at 17 months was three times that of wild-type mice, whereas levels of spleen TXNIP in 3xTg-AD mice was only approximately 5% of wild-type levels. The enhanced cerebrum TXNIP expression was associated with increased histone acetylation, increased transcription factor CCCTC-binding factor (CTCF) binding, and TXNIP promoter hypomethylation, whereas the reduced spleen TXNIP expression was associated with increased histone methylation, reduced CTCF binding, and TXNIP promoter hypermethylation. Increased cortical expression of TXNIP was detectable at seven months before increased levels of p-Tau and inflammatory mediators became significant. It was concluded that the interaction of the brain and peripheral TXNIP contributed to cognitive decline, but identifying mechanisms will require further study [122]. The relationship of TXNIP and enhanced tau phosphorylation was investigated in 5xFAD mice and SH-SY5Y neuroblastoma cells. Treatment of SH-SY5Y cells with Aβ (1-42) resulted in TXNIP overexpression, followed by enhanced oxidative stress, p38 MAPK, and increased tau phosphorylation at Ser202/Thr205. Knockdown of TXNIP significantly reduced TXNIP expression and tau phosphorylation in SH-SY5Y cells, whereas the treatment of 5xFAD mice with the voltage-dependent calcium channel blocker verapamil had similar effects [123]. A consensus on how TXNIP could be involved in AD-type degenerative processes is still lacking.
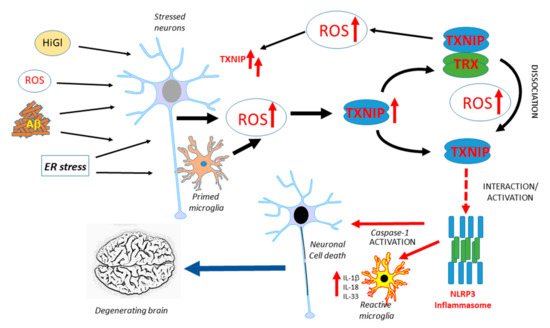
Figure 1.
Overview of features of TXNIP expression and function that might contribute to AD pathogenesis. Multiple factors have been identified to be possible initiators of AD-related neurotoxicity, all of which have been shown to induce TXNIP. The potential interactions leading to enhanced inflammation and neurodegeneration are illustrated. ROS: reactive oxygen species, HiGl: High glucose, ER: endoplasmic reticulum, TXNIP: thioredoxin-interacting protein, TRX: thioredoxin, IL: interleukin.
A separate series of studies identified features of NLRP3 inflammasome activation related to enhanced inflammation associated with AD pathology in human AD brains and mouse models [124,125,126,127]. These studies did not examine the involvement of TXNIP in the features of inflammasome activation, but it can be deduced based on other studies that it is involved in the observed pathological changes. It was shown that there was increased caspase-1 activation in AD and MCI human brains. In mice with genetic deficiencies of NLRP3, microglia showed a smaller inflammatory response and increased phagocytosis of amyloid beta [124]. It was shown in AD plaque-developing mice that injection of brain-derived ASC specks produced upon inflammasome activation promoted the seeding of Aβ plaques in brains [128].
Alterations in the expression of TXNIP have also been identified in blood cells from patients with multiple sclerosis (MS), a degenerative disease of white matter associated with autoimmune responses to myelin. In patients with newly diagnosed MS, for those receiving interferon or other immunosuppressive therapies, TXNIP mRNA expression was decreased whereas TRX1 mRNA was significantly increased. This suggested a disturbance in oxidative stress as part of the disease pathology [129].
5.3. Linking Diabetes, TXNIP, and Alzheimer’s Disease: Hypothesis
The role of TXNIP in inducing pancreatic cell death and causing type-1 diabetes appears to depend on properties not directly or exclusively related to its binding of TRX. A mutant form of TXNIP that does not bind TRX was shown to maintain glucose uptake inhibition properties, which was a property of the α-arrestin domain [130]. Subsequent studies identified that TXNIP inhibited the expression of glucose transporter-1 (Glut-1) and promoted endocytosis of the protein from the plasma membrane [131]. Activation of AMPK under conditions of energy stress phosphorylated TXNIP and promoted its degradation, preventing interactions with Glut-1 on the plasma membrane and restoring cellular glucose uptake and metabolism [131]. TXNIP also inhibits glucose uptake due to inhibiting glycolysis and oxidative phosphorylation. Cells lacking TXNIP showed increased glucose uptake [131]. How these features might be involved in AD is unclear but can be suggested based on glucose hypometabolism, a well-established feature of AD [132]. Glut-1 is highly expressed in the brain, particularly by cerebrovascular endothelial cells and astrocytes, whereas Glut-3 is the major neuronal transporter of glucose [133].
Figure 2 illustrates the potential involvement of peripheral TXNIP in modulating cerebral glucose metabolism in the brain and AD, contributing to neurodegeneration. As several studies have shown that the neuronal expression of TXNIP is more commonly observed under stress/injury conditions, further experimental studies to determine if TXNIP overexpression affects Glut-3 expression and localization are needed. The concept of how AD neurodegeneration could be linked to diabetes, deficits in insulin signaling and glucose metabolism, and endothelial dysfunction should be considered. The term type-3 diabetes has been used to describe the lack of response of neurons to insulin and insulin-like growth factor signaling and the consequent deficits in glucose metabolism [134]. There have been epidemiological data suggesting patients with insulin-resistant type-2 diabetes are at greater risk of developing AD-type dementia and that antidiabetic medication was effective at reducing the risk of AD [135], though these findings have not been extensively confirmed. Examining the neuropathological records from an autopsy series, there was no significant correlation between type-2 diabetes and the severity of plaque and tangle load in brain regions affected by AD [136], with the exception of subjects with an apolipoprotein E4 allele. It was confirmed in a more recent study that type-2 diabetes did not correlate with the amyloid plaque or neurofibrillary tangle load, but it was commented on that both diseases are associated at early stages with a significant cerebrovascular disease that can affect the development of AD pathology [27]. Much interest in this topic has been on whether patients treated with the insulin sensitizer metformin have a reduced risk of dementia [137,138,139]. Metformin, which activates AMPK leading to inhibition of TXNIP expression, has significant activity in inhibiting the activation of the NLRP3 inflammasome complex [140,141,142]. Metformin also inhibited the interaction of TXNIP and NLRP3 [143]. Further studies are needed on human brains from cases with diabetes to determine whether there is increased expression of TXNIP and whether this is correlated with AD or severity of AD pathology.
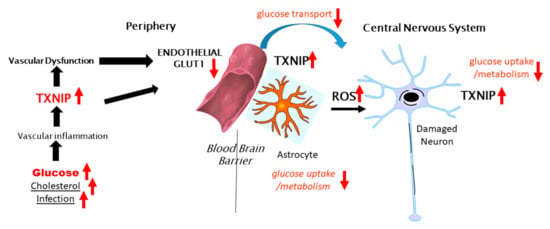
Figure 2.
Potential peripheral–CNS interactions involving TXNIP in glucose metabolism in the brain.
6. Physiological Consequences of Loss of TXNIP
6.1. Experimental Animals
A number of studies have reported the phenotypes of mice constructed to have total TXNIP gene deletion. The consequences of this have been both beneficial and detrimental depending on the disease model. In an initial report, the cause of hyperlipidemia in a mutant mouse strain (Hcb-19) was a mutation in the Txnip gene resulting in reduced expression. These mutant mice had hypertriglyceridemia, hypercholesterolemia, elevated plasma apolipoprotein B, and increased secretion of triglyceride-rich lipoproteins with increased ketone body synthesis [144]. In a TXNIP gene-deleted mouse model, they developed significantly increased fatty liver, with high levels of triacylglycerol, cholesterol ester, and total cholesterol, with higher-serum non-esterified fatty acids. Elevated fatty acid synthesis was the primary cause of liver lipogenesis in TXNIP knockout mice [145]. Gene deletion of TXNIP also resulted in impaired immunity with reduced numbers of natural killer cells and the impaired dendritic cell maturation of T cells [146,147,148]. TXNIP gene deletion can lead to lethality under fasting conditions due to a switch in metabolism to hyperlipidemia and hypoglycemia. Under these conditions, mice experienced enhanced glucose-induced insulin sensitivity and secretion and an increased expression of PPAR-target genes [149]. TXNIP gene-deletion mice fed a high-fat diet developed significantly greater fat deposits and body mass but with increased insulin sensitivity. This study correlated the loss of TXNIP with the augmented activation of PPAR-γ [150]. These effects on enhanced hyperlipidemia due to TXNIP gene deletion were detrimental to the affected organisms, but beneficial outcomes upon reversing the consequences of diabetes/hyperglycemia were also observed. Knockdown of TXNIP in vitro protected mesangial cells grown under high-glucose conditions from apoptosis and ASK1 and p38 MAP kinase signaling [151,152]. In wild-type and TXNIP knockout mice rendered diabetic with streptozotocin, the TXNIP knockout mice showed significant protection from diabetic nephropathy. The knockout diabetic mice did not show increased thickening of the basal membrane or increased glomerular TGF-β1, collagen IV, and fibrosis was seen in the wild-type mice [153]. Loss of TXNIP provided significant protection in mice fed a HFD, showing significant protection from retinal degeneration and microvascular dysfunction due to reduced activation by the retinal endothelial cells of TXNIP/NLRP3 inflammasome [94]. TXNIP gene-deletion mice have been shown in general to be protected from the vascular consequences of high-fat/obesity or high-sugar diets compared to wild-type mice. In a model that studied the consequence of HFD and TXNIP gene deletion, it was shown that wild-type mice had significantly impaired blood flow and vascular density due to impaired angiogenesis compared to the TXNIP knockout mice. In this model, there was significantly higher IL-1β, increased numbers of infiltrating macrophages, and increased vascular endothelial growth factor (VEGF) expression and VEGF receptor activation in wild-type mice only. The enhanced vascular inflammation was due to the high-fat diet activating the TXNIP/NLRP3 inflammasome, which did not occur in TXNIP knockout mice [154].
6.2. Human Subjects
From the above-described experimental studies, the loss of TXNIP in mice has been beneficial in reversing the consequences of hyperglycemia but with a detrimental effect on liver fatty acid metabolism. These animals were constructed with Txnip gene deletions, but the loss of TXNIP has been identified in rare genetic cases in human subjects. The published case report covers children of 4, 9, and 13 years of age [155]. This showed that the loss of TXNIP is nonlethal, but the affected subjects show lactic acidosis, low-serum methionine, and impaired oxidative phosphorylation in response to glucose and pyruvate [155]. There are no reports as yet if such individuals are resistant to diabetes or neurodegenerative diseases. These studies demonstrate that the pharmacological lowering of TXNIP levels is unlikely to be associated with significant side effects.
Table 1 lists the most significant studies related to the effects of TXNIP expression in the brain or brain-derived cells. This is a subjective list based on the theme of this article and is proposed to illustrate the different features of TXNIP in interactions with the nervous system.

Table 1.
Summary of major studies of TXNIP in the brain or brain-derived cells.
7. Modulating TXNIP Expression
A number of different classes of agents can modulate TXNIP expression, including PPAR-γ agonists, which include classes of drugs with insulin-sensitizing properties used for treating diabetes [157]. There have been conflicting results on the interactions of PPAR-γ and TXNIP. One study using human kidney proximal tubule cells showed PPAR-γ agonists attenuated high-glucose-mediated TXNIP expression [158], whereas another using human macrophages showed that the PPAR-γ agonist GW929 enhanced TXNIP expression [71]. Using a rat insulinoma cell line INS-1E to represent pancreatic beta cells, it was shown that hyperglycemia activated TXNIP expression and inhibited the activation of AMPK. Activation of AMPK with metformin or aminoimidazole-carboxamide ribonucleotide (AICAR) reduced TXNIP expression, an effect also observed when cells were treated with the lipid palmitate [142]. AMPK activation inhibited glucose-stimulated ChREBP nuclear entry and binding to the TXNIP promoter, thereby inhibiting TXNIP mRNA expression. These investigators also demonstrated that the addition of insulin to high-glucose-treated INS-1E cells reduced TXNIP expression and prevented high-glucose-mediated apoptosis. Treatment of cells with nitric oxide (NO) stimulated insulin secretion and reduced the expression of TXNIP; this effect was reversed when cells were treated with a nitric oxide synthase inhibitor [159]. A recent study showed that insulin-like growth factor-1 (IGF-1) negatively regulated TXNIP expression in vitro and in vivo. Furthermore, this study demonstrated that oxidative stress and glucose-induced TXNIP expression could be reversed by the administration of IGF-1 [160]. The calcium channel blocker verapamil, which is in widespread clinical use for hypertension, has been demonstrated to significantly inhibit TXNIP expression with therapeutic benefits. In an animal model of diabetic cardiomyopathy, three weeks of treatment with verapamil had a significant therapeutic benefit, which correlated with the reduced expression of TXNIP in cardiomyocytes [161]. The mechanism of action of verapamil appeared to be mediated by the enhanced activation of the transcription repressor nuclear factor Y (NPY) [162]. As mentioned above, verapamil was also effective in inhibiting tau phosphorylation in vivo in 5xFAD AD model mice, inhibiting Aβ-induced tau phosphorylation in vitro, and inhibiting the expression of TXNIP and the activation of p38 MAPK [123].
In a recent study, we identified a novel fluorinated derivative of curcumin that was highly effective in reducing stress-induced TXNIP expression. Cellular models examined used the human retinal pigment epithelial cell line ARPE-19, which have high constitutive levels of TXNIP expression, and macrophages derived from the human THP-1 monocyte cell line, which have lower constitutive expression. In this study, the effectiveness of fluorinated curcumin derivative Shiga Y6 compared to its non-fluorinated derivative Shiga Y5 in reducing TXNIP protein and mRNA expression under constitutive, high-glucose, endoplasmic reticulum stress, and inflammatory activation was demonstrated [163]. This compound was also effective in inducing TRX protein and mRNA levels in these cellular models. These studies demonstrate that derivatized curcumin molecules were more effective than curcumin alone in lowering TXNIP expression. Other studies have shown the effectiveness of curcumin in vitro and in vivo in inhibiting TXNIP expression but at higher doses than we used for testing the fluorinated curcumin compound [163,164]. The effect of curcumin on TXNIP expression appears to be through the activation of AMPK [81,165].
8. Summary
When considering the involvement of TXNIP in human acute neurological and chronic neurodegenerative diseases, there is mostly circumstantial evidence from animal and in vitro experiments about how it might accelerate pathology. It is clear there is a need for further detailed studies. With regards to AD, there have been many studies on microglia and inflammation in AD and studies showing NLRP3 inflammasome activation in AD brains without the consideration of TXNIP ([124]). TXNIP is essential for driving NLRP3 inflammasome activation and the formation of activated caspase-1 [50]. Preventing the interaction of TXNIP and NLRP3 will have significant effects. This can be done by inhibiting its expression, inhibiting the stress stimuli that causes increased expression, or manipulating the intermediate signaling pathways. As mentioned, a number of different classes of chemicals have been identified to inhibit TXNIP expression, which might have therapeutic potential for a number of diseases. The major issue for agents aimed at brain cells is the penetration of the blood–brain barrier. An issue for treating elderly patients with neurodegenerative diseases is whether inhibiting a particular target could exacerbate age-associated cancers. As decreased levels of TXNIP correlate with the increased growth of different types of cancer cells, this issue needs to be considered [166].
Understanding this protein with multiple properties related to sequestering TRX1 in the cytosol, such as sequestering TRX2 in mitochondria, modulating the expression or stability of key glucose transporters, acting as a nuclear transcription factor, and activating inflammasome, has been complicated. Its normal physiological function has not been elucidated. Some of the properties are independent of the enhancement of oxidative stress due to sequestering TRX. The interaction of oxidative stress with the activation of other properties, for example transcription factor activation, requires further study.
From this review of the literature, there is clearly a need for further studies of TXNIP expression in normal, aged, and disease-affected human brains. Only one study provided a limited description of the cellular and neuroanatomical distribution of TXNIP in human brains in AD [26]. Without this knowledge, identifying sites of TXNIP activity in relation to neuropathology, identifying the neuroanatomical distribution and cell types mediating TXNIP activity in particular, will hinder progress on developing TXNIP as a therapeutic target for brain diseases.
Funding
The work in the laboratory (D.G.W.) has been supported by grant 19K07843 from the Japan Society for the Promotion of Science.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ramli, N.Z.; Yahaya, M.F.; Tooyama, I.; Damanhuri, H.A. A Mechanistic Evaluation of Antioxidant Nutraceuticals on Their Potential against Age-Associated Neurodegenerative Diseases. Antioxidants 2020, 9, 1019. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef] [PubMed]
- Pradeepkiran, J.A.; Reddy, P.H. Defective mitophagy in Alzheimer’s disease. Ageing Res. Rev. 2020, 64, 101191. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Jo, M.G.; Kim, S.Y.; Chung, C.G.; Lee, S.B. Dietary Antioxidants and the Mitochondrial Quality Control: Their Potential Roles in Parkinson’s Disease Treatment. Antioxidants 2020, 9, 1056. [Google Scholar] [CrossRef] [PubMed]
- López-Contreras, A.K.; Martínez-Ruiz, M.G.; Olvera-Montaño, C.; Robles-Rivera, R.R.; Arévalo-Simental, D.E.; Castellanos-González, J.A.; Hernández-Chávez, A.; Huerta-Olvera, S.G.; Cardona-Muñoz, E.G.; Rodríguez-Carrizalez, A.D. Importance of the Use of Oxidative Stress Biomarkers and Inflammatory Profile in Aqueous and Vitreous Humor in Diabetic Retinopathy. Antioxidants 2020, 9, 891. [Google Scholar] [CrossRef]
- Tian, Y.; Su, Y.; Ye, Q.; Chen, L.; Yuan, F.; Wang, Z. Silencing of TXNIP Alleviated Oxidative Stress Injury by Regulating MAPK-Nrf2 Axis in Ischemic Stroke. Neurochem. Res. 2020, 45, 428–436. [Google Scholar] [CrossRef]
- Su, C.; Shi, A.; Cao, G.; Tao, T.; Chen, R.; Hu, Z.; Shen, Z.; Tao, H.; Cao, B.; Hu, D.; et al. Fenofibrate suppressed proliferation and migration of human neuroblastoma cells via oxidative stress dependent of TXNIP upregulation. Biochem. Biophys. Res. Commun. 2015, 460, 983–988. [Google Scholar] [CrossRef]
- Pereira, J.D.; Fraga, V.G.; Santos, A.L.M.; Carvalho, M.D.G.; Caramelli, P.; Gomes, K.B. Alzheimer’s disease and type 2 diabetes mellitus: A systematic review of proteomic studies. J. Neurochem. 2020, 1–24. [Google Scholar] [CrossRef]
- Saleem, U.; Sabir, S.; Niazi, S.G.; Nadeem, M.; Ahmad, B. Role of Oxidative Stress and Antioxidant Defense Biomarkers in Neurodegenerative Diseases. Crit. Rev. Eukaryot. Gene Expr. 2020, 30, 311–322. [Google Scholar] [CrossRef]
- Nishizawa, K.; Nishiyama, H.; Matsui, Y.; Kobayashi, T.; Saito, R.; Kotani, H.; Masutani, H.; Oishi, S.; Toda, Y.; Fujii, N.; et al. Thioredoxin-interacting protein suppresses bladder carcinogenesis. Carcinogenesis 2011, 32, 1459–1466. [Google Scholar] [CrossRef]
- Xie, M.; Xie, R.; Xie, S.; Wu, Y.; Wang, W.; Li, X.; Xu, Y.; Liu, B.; Zhou, Y.; Wang, T.; et al. Thioredoxin interacting protein (TXNIP) acts as a tumor suppressor in human prostate cancer. Cell Biol. Int. 2020, 44, 2094–2106. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, S.F.; Miele, M.E.; Hatta, N.; Takata, M.; Paquette-Straub, C.; Freedman, L.P.; Welch, D.R. Melanoma metastasis suppression by chromosome 6: Evidence for a pathway regulated by CRSP3 and TXNIP. Cancer Res. 2003, 63, 432–440. [Google Scholar] [PubMed]
- Chen, J.; Saxena, G.; Mungrue, I.N.; Lusis, A.J.; Shalev, A. Thioredoxin-interacting protein: A critical link between glucose toxicity and beta-cell apoptosis. Diabetes 2008, 57, 938–944. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.-R.; Chan, W.P.A.; Nguyen, T.H.; Liu, S.; Procter, N.E.K.; Ngo, D.T.; Sverdlov, A.L.; Chirkov, Y.Y.; Horowitz, J.D. Thioredoxin-Interacting Protein: Pathophysiology and Emerging Pharmacotherapeutics in Cardiovascular Disease and Diabetes. Cardiovasc. Drugs Ther. 2014, 28, 347–360. [Google Scholar] [CrossRef]
- Du, C.; Wu, M.; Liu, H.; Ren, Y.; Du, Y.; Wu, H.; Wei, J.; Liu, C.; Yao, F.; Wang, H.; et al. Thioredoxin-interacting protein regulates lipid metabolism via Akt/mTOR pathway in diabetic kidney disease. Int. J. Biochem. Cell Biol. 2016, 79, 1–13. [Google Scholar] [CrossRef]
- Devi, T.S.; Hosoya, K.-I.; Terasaki, T.; Singh, L.P. Critical role of TXNIP in oxidative stress, DNA damage and retinal pericyte apoptosis under high glucose: Implications for diabetic retinopathy. Exp. Cell Res. 2013, 319, 1001–1012. [Google Scholar] [CrossRef]
- Hu, J.; Yu, Y. The Function of Thioredoxin-Binding Protein-2 (TBP-2) in Different Diseases. Oxidative Med. Cell. Longev. 2018, 2018, 4582130. [Google Scholar] [CrossRef]
- Alhawiti, N.M.; Al Mahri, S.; Aziz, M.A.; Malik, S.S.; Mohammad, S. TXNIP in Metabolic Regulation: Physiological Role and Therapeutic Outlook. Curr. Drug Targets 2017, 18, 1095–1103. [Google Scholar] [CrossRef]
- Nishiyama, A.; Matsui, M.; Iwata, S.; Hirota, K.; Masutani, H.; Nakamura, H.; Takagi, Y.; Sono, H.; Gon, Y.; Yodoi, J. Identification of Thioredoxin-binding Protein-2/Vitamin D(3)Up-regulated Protein 1 as a Negative Regulator of Thioredoxin Function and Expression. J. Biol. Chem. 1999, 274, 21645–21650. [Google Scholar] [CrossRef]
- Junn, E.; Han, S.H.; Im, J.Y.; Yang, Y.; Cho, E.W.; Um, H.D.; Kim, D.K.; Lee, K.W.; Han, P.L.; Rhee, S.G.; et al. Vitamin D3 Up-Regulated Protein 1 Mediates Oxidative Stress Via Suppressing the Thioredoxin Function. J. Immunol. 2000, 164, 6287–6295. [Google Scholar] [CrossRef]
- Yamanaka, H.; Maehira, F.; Oshiro, M.; Asato, T.; Yanagawa, Y.; Takei, H.; Nakashima, Y. A Possible Interaction of Thioredoxin with VDUP1 in HeLa Cells Detected in a Yeast Two-Hybrid System. Biochem. Biophys. Res. Commun. 2000, 271, 796–800. [Google Scholar] [CrossRef] [PubMed]
- Farrell, M.R.; Rogers, L.K.; Liu, Y.; Welty, S.E.; Tipple, T.E. Thioredoxin-interacting protein inhibits hypoxia-inducible factor transcriptional activity. Free Radic. Biol. Med. 2010, 49, 1361–1367. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Zhou, R.; Tschopp, J. The NLRP3 Inflammasome: A Sensor for Metabolic Danger? Science 2010, 327, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Gao, J.; Wang, X.; Wen, W.; Yang, H.; Tian, Y.; Liu, N.; Wang, Z.; Liu, H.; Zhang, Y.; et al. A novel indication of thioredoxin-interacting protein as a tumor suppressor gene in malignant glioma. Oncol. Lett. 2017, 14, 2053–2058. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.Y.; Roy, J.; Fung, M.L.; Heng, B.C.; Zhang, C.; Lim, L.W. Relationships between Mitochondrial Dysfunction and Neurotransmission Failure in Alzheimer’s Disease. Aging Dis. 2020, 11, 1291–1316. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ismael, S.; Nasoohi, S.; Sakata, K.; Liao, F.-F.; McDonald, M.P.; Ishrat, T. Thioredoxin-Interacting Protein (TXNIP) Associated NLRP3 Inflammasome Activation in Human Alzheimer’s Disease Brain. J. Alzheimers Dis. 2019, 68, 255–265. [Google Scholar] [CrossRef]
- Gąsecka, A.; Siwik, D.; Gajewska, M.; Jaguszewski, M.J.; Mazurek, T.; Filipiak, K.J.; Postuła, M.; Eyileten, C. Early Biomarkers of Neurodegenerative and Neurovascular Disorders in Diabetes. J. Clin. Med. 2020, 9, 2807. [Google Scholar] [CrossRef]
- Vionnet, N.; Hani, E.H.; Dupont, S.; Gallina, S.; Francke, S.; Dotte, S.; De Matos, F.; Durand, E.; Leprêtre, F.; Lecoeur, C.; et al. Genomewide Search for Type 2 Diabetes–Susceptibility Genes in French Whites: Evidence for a Novel Susceptibility Locus for Early-Onset Diabetes on Chromosome 3q27-qter and Independent Replication of a Type 2–Diabetes Locus on Chromosome 1q21–q24. Am. J. Hum. Genet. 2000, 67, 1470–1480. [Google Scholar] [CrossRef]
- Abu El Maaty, M.A.; Almouhanna, F.; Wölfl, S. Expression of TXNIP in Cancer Cells and Regulation by 1,25(OH)2D3: Is It Really the Vitamin D3 Upregulated Protein? Int. J. Mol. Sci. 2018, 19, 796. [Google Scholar] [CrossRef]
- Ludwig, D.L.; Kotanides, H.; Le, T.; Chavkin, D.; Bohlen, P.; Witte, L. Cloning, genetic characterization, and chromosomal mapping of the mouse VDUP1 gene. Gene 2001, 269, 103–112. [Google Scholar] [CrossRef]
- Patwari, P.; Higgins, L.J.; Chutkow, W.A.; Yoshioka, J.; Lee, R.T. The interaction of thioredoxin with Txnip. Evidence for formation of a mixed disulfide by disulfide exchange. J. Biol. Chem. 2006, 281, 21884–21891. [Google Scholar] [CrossRef] [PubMed]
- Saxena, G.; Chen, J.; Shalev, A. Intracellular Shuttling and Mitochondrial Function of Thioredoxin-interacting Protein. J. Biol. Chem. 2010, 285, 3997–4005. [Google Scholar] [CrossRef]
- Luthman, M.; Holmgren, A. Rat liver thioredoxin and thioredoxin reductase: Purification and characterization. Biochemistry 1982, 21, 6628–6633. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, A.; Fagerstedt, M. The in vivo distribution of oxidized and reduced thioredoxin in Escherichia coli. J. Biol. Chem. 1982, 257, 6926–6930. [Google Scholar]
- Berstermann, A.; Vogt, K.; Follmann, H. Plant Seeds Contain Several Thioredoxins of Regular Size. Eur. J. Biochem. 1983, 131, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Chen, X.; Sun, J.; Li, Q.; Lian, X.; Li, S.; Wang, Y.; Tian, L. Inhibition of nuclear thioredoxin aggregation attenuates PM(2.5)-induced NF-κB activation and pro-inflammatory responses. Free Radic. Biol. Med. 2019, 130, 206–214. [Google Scholar] [CrossRef]
- Obikane, H.; Abiko, Y.; Ueno, H.; Kusumi, Y.; Esumi, M.; Mitsumata, M. Effect of endothelial cell proliferation on atherogenesis: A role of p21(Sdi/Cip/Waf1) in monocyte adhesion to endothelial cells. Atherosclerosis 2010, 212, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Bodenstein-Lang, J.; Buch, A.; Follmann, H. Animal and plant mitochondria contain specific thioredoxins. FEBS Lett. 1989, 258, 22–26. [Google Scholar] [CrossRef]
- Bodenstein, J.; Follmann, H. Characterization of Two Thioredoxins in Pig Heart Including a New Mitochondrial Protein. Zeitschrift für Naturforschung C 1991, 46, 270–279. [Google Scholar] [CrossRef]
- Rozell, B.; Hansson, H.A.; Luthman, M.; Holmgren, A. Immunohistochemical localization of thioredoxin and thioredoxin reductase in adult rats. Eur. J. Cell Biol. 1985, 38, 79–86. [Google Scholar] [PubMed]
- Lippoldt, A.; Padilla, C.A.; Gerst, H.; Andbjer, B.; Richter, E.; Holmgren, A.; Fuxe, K. Localization of thioredoxin in the rat brain and functional implications. J. Neurosci. 1995, 15, 6747–6756. [Google Scholar] [CrossRef] [PubMed]
- Rybnikova, E.; Damdimopoulos, A.E.; Gustafsson, J.Å.; Spyrou, G.; Pelto-Huikko, M. Expression of novel antioxidant thioredoxin-2 in the rat brain. Eur. J. Neurosci. 2000, 12, 1669–1678. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, A.; Hamuro, J.; Nakamura, H.; Kondo, N.; Hirabayashi, Y.; Ishizaki-Koizumi, S.; Hirakawa, T.; Inoue, T.; Yodoi, J. Overexpression of Human Thioredoxin in Transgenic Mice Controls Oxidative Stress and Life Span. Antioxid. Redox Signal. 2002, 4, 693–696. [Google Scholar] [CrossRef] [PubMed]
- Takagi, Y.; Mitsui, A.; Nishiyama, A.; Nozaki, K.; Sono, H.; Gon, Y.; Hashimoto, N.; Yodoi, J. Overexpression of thioredoxin in transgenic mice attenuates focal ischemic brain damage. Proc. Natl. Acad. Sci. USA 1999, 96, 4131–4136. [Google Scholar] [CrossRef] [PubMed]
- Hattori, I.; Takagi, Y.; Nakamura, H.; Nozaki, K.; Bai, J.; Kondo, N.; Sugino, T.; Nishimura, M.; Hashimoto, N.; Yodoi, J. Intravenous Administration of Thioredoxin Decreases Brain Damage Following Transient Focal Cerebral Ischemia in Mice. Antioxid. Redox Signal. 2004, 6, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Xie, C.; Gabbita, S.P.; Markesbery, W.R. Decreased thioredoxin and increased thioredoxin reductase levels in alzheimer’s disease brain. Free Radic. Biol. Med. 2000, 28, 418–427. [Google Scholar] [CrossRef]
- Arodin, L.; Lamparter, H.; Karlsson, H.; Nennesmo, I.; Björnstedt, M.; Schröder, J.; Fernandes, A.P. Alteration of Thioredoxin and Glutaredoxin in the Progression of Alzheimer’s Disease. J. Alzheimers Dis. 2014, 39, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Nishinaka, Y.; Masutani, H.; Oka, S.-I.; Matsuo, Y.; Yamaguchi, Y.; Nishio, K.; Ishii, Y.; Yodoi, J. Importin α1 (Rch1) Mediates Nuclear Translocation of Thioredoxin-binding Protein-2/Vitamin D(3)-up-regulated Protein 1. J. Biol. Chem. 2004, 279, 37559–37565. [Google Scholar] [CrossRef]
- Suh, H.-W.; Yun, S.; Song, H.; Jung, H.; Park, Y.-J.; Kim, T.-D.; Yoon, S.R.; Choi, I. TXNIP interacts with hEcd to increase p53 stability and activity. Biochem. Biophys. Res. Commun. 2013, 438, 264–269. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- Kim, K.-Y.; Shin, S.M.; Kim, J.K.; Paik, S.G.; Yang, Y.; Choi, I. Heat shock factor regulates VDUP1 gene expression. Biochem. Biophys. Res. Commun. 2004, 315, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Schulze, P.C.; Liu, H.; Choe, E.; Yoshioka, J.; Shalev, A.; Bloch, K.D.; Lee, R.T. Nitric Oxide–Dependent Suppression of Thioredoxin-Interacting Protein Expression Enhances Thioredoxin Activity. Arter. Thromb. Vasc. Biol. 2006, 26, 2666–2672. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.-X.; Goh, S.-R.; Dai, R.-P.; Luo, Y. Adenosine-Containing Molecules Amplify Glucose Signaling and Enhance Txnip Expression. Mol. Endocrinol. 2009, 23, 932–942. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ng, M.K.C.; Wu, J.; Chang, E.; Wang, B.; Katzenberg-Clark, R.; Ishii-Watabe, A.; Cooke, J.P. A Central Role for Nicotinic Cholinergic Regulation of Growth Factor–Induced Endothelial Cell Migration. Arter. Thromb. Vasc. Biol. 2007, 27, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Masaki, S.; Masutani, H.; Yoshihara, E.; Yodoi, J. Deficiency of Thioredoxin Binding Protein-2 (TBP-2) Enhances TGF-β Signaling and Promotes Epithelial to Mesenchymal Transition. PLoS ONE 2012, 7, e39900. [Google Scholar] [CrossRef] [PubMed]
- DeRoo, B.J.; Hewitt, S.C.; Peddada, S.D.; Korach, K.S. Estradiol Regulates the Thioredoxin Antioxidant System in the Mouse Uterus. Endocrinology 2004, 145, 5485–5492. [Google Scholar] [CrossRef]
- Saitoh, T.; Tanaka, S.; Koike, T. Rapid induction and Ca(2+) influx-mediated suppression of vitamin D3 up-regulated protein 1 (VDUP1) mRNA in cerebellar granule neurons undergoing apoptosis. J. Neurochem. 2001, 78, 1267–1276. [Google Scholar] [CrossRef]
- Sbai, O.; Devi, T.S.; Melone, M.A.B.; Féron, F.; Khrestchatisky, M.; Singh, L.P.; Perrone, L. RAGE-TXNIP axis is required for S100B-promoted Schwann cell migration, fibronectin expression and cytokine secretion. J. Cell Sci. 2010, 123, 4332–4339. [Google Scholar] [CrossRef]
- Parikh, H.; Carlsson, E.; Chutkow, W.A.; Johansson, L.E.; Storgaard, H.; Poulsen, P.; Saxena, R.; Ladd, C.; Schulze, P.C.; Mazzini, M.J.; et al. TXNIP Regulates Peripheral Glucose Metabolism in Humans. PLoS Med. 2007, 4, e158. [Google Scholar] [CrossRef]
- Perrone, L.; Devi, T.S.; Hosoya, K.-I.; Terasaki, T.; Singh, L.P. Thioredoxin interacting protein (TXNIP) induces inflammation through chromatin modification in retinal capillary endothelial cells under diabetic conditions. J. Cell. Physiol. 2009, 221, 262–272. [Google Scholar] [CrossRef]
- Chen, J.; Fontes, G.; Saxena, G.; Poitout, V.; Shalev, A. Lack of TXNIP protects against mitochondria-mediated apoptosis but not against fatty acid-induced ER stress-mediated beta-cell death. Diabetes 2010, 59, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Mandala, A.; Das, N.; Bhattacharjee, S.; Mukherjee, B.; Mukhopadhyay, S.; Roy, S.S. Thioredoxin interacting protein mediates lipid-induced impairment of glucose uptake in skeletal muscle. Biochem. Biophys. Res. Commun. 2016, 479, 933–939. [Google Scholar] [CrossRef]
- Martel, M.-A.; Soriano, F.X.; Baxter, P.; Rickman, C.; Duncan, R.R.; Wyllie, D.J.A.; Hardingham, G.E. Inhibiting pro-death NMDA receptor signaling dependent on the NR2 PDZ ligand may not affect synaptic function or synaptic NMDA receptor signaling to gene expression. Channels 2009, 3, 12–15. [Google Scholar] [CrossRef]
- Lerner, A.G.; Upton, J.-P.; Praveen, P.V.K.; Ghosh, R.; Nakagawa, Y.; Igbaria, A.; Shen, S.; Nguyen, V.; Backes, B.J.; Heiman, M.; et al. IRE1α Induces Thioredoxin-Interacting Protein to Activate the NLRP3 Inflammasome and Promote Programmed Cell Death under Irremediable ER Stress. Cell Metab. 2012, 16, 250–264. [Google Scholar] [CrossRef]
- Yu, F.-X.; Luo, Y. Tandem ChoRE and CCAAT Motifs and Associated Factors Regulate Txnip Expression in Response to Glucose or Adenosine-Containing Molecules. PLoS ONE 2009, 4, e8397. [Google Scholar] [CrossRef] [PubMed]
- Heman-Ackah, S.M.; Manzano, R.; Hoozemans, J.J.M.; Scheper, W.; Flynn, R.; Haerty, W.; Cowley, S.A.; Bassett, A.R.; Wood, M.J.A. Alpha-synuclein induces the unfolded protein response in Parkinson’s disease SNCA triplication iPSC-derived neurons. Hum. Mol. Genet. 2017, 26, 4441–4450. [Google Scholar] [CrossRef] [PubMed]
- Poirier, Y.; Grimm, A.; Schmitt, K.; Eckert, A. Link between the unfolded protein response and dysregulation of mitochondrial bioenergetics in Alzheimer’s disease. Cell. Mol. Life Sci. 2019, 76, 1419–1431. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; He, F.; Hu, L.; Hai, L.; Huang, M.; Xu, Z.; Zhang, J.; Zhou, Z.; Liu, F.; Dai, Y.-S. Transcription Factor Ets1 Regulates Expression of Thioredoxin-Interacting Protein and Inhibits Insulin Secretion in Pancreatic β-Cells. PLoS ONE 2014, 9, e99049. [Google Scholar] [CrossRef]
- Ling, C. Epigenetic regulation of insulin action and secretion – role in the pathogenesis of type 2 diabetes. J. Intern. Med. 2020, 288, 158–167. [Google Scholar] [CrossRef]
- Wondafrash, D.Z.; Nire’A, A.T.; Tafere, G.G.; Desta, D.M.; Asmerom, D.; Zewdie, K.A. Thioredoxin-Interacting Protein as a Novel Potential Therapeutic Target in Diabetes Mellitus and Its Underlying Complications. Diabetes Metab. Syndr. Obesity Targets Ther. 2020, 13, 43–51. [Google Scholar] [CrossRef]
- Billiet, L.; Furman, C.; Larigauderie, G.; Copin, C.; Page, S.; Fruchart, J.; Brand, K.; Rouis, M. Enhanced VDUP-1 gene expression by PPARgamma agonist induces apoptosis in human macrophage. J. Cell. Physiol. 2008, 214, 183–191. [Google Scholar] [CrossRef]
- Billiet, L.; Furman, C.; Cuaz-Pérolin, C.; Paumelle, R.; Raymondjean, M.; Simmet, T.; Rouis, M. Thioredoxin-1 and its natural inhibitor, vitamin D3 up-regulated protein 1, are differentially regulated by PPARalpha in human macrophages. J. Mol. Biol. 2008, 384, 564–576. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, F.; Takata, M.; Kamitori, K.; Nonaka, M.; Dong, Y.; Sui, L.; Tokuda, M. Rare sugar D-allose induces specific up-regulation of TXNIP and subsequent G1 cell cycle arrest in hepatocellular carcinoma cells by stabilization of p27kip1. Int. J. Oncol. 2008, 32, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, F.; Kamitori, K.; Sanada, K.; Horii, M.; Dong, Y.; Sui, L.; Tokuda, M. Rare sugar d-allose enhances anti-tumor effect of 5-fluorouracil on the human hepatocellular carcinoma cell line HuH-7. J. Biosci. Bioeng. 2008, 106, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Hoshikawa, H.; Mori, T.; Mori, N. In vitro and in vivo effects of D-allose: Up-regulation of thioredoxin-interacting protein in head and neck cancer cells. Ann. Otol. Rhinol. Laryngol. 2010, 119, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Saito, M.; Tsukamoto, I.; Yamaguchi, F.; Sui, L.; Kamitori, K.; Dong, Y.; Uehara, E.; Konishi, R.; Janjua, N.; et al. Analysis of the inhibitory mechanism of d-allose on MOLT-4F leukemia cell proliferation. J. Biosci. Bioeng. 2009, 107, 562–568. [Google Scholar] [CrossRef]
- Stoltzman, C.A.; Kaadige, M.R.; Peterson, C.W.; Ayer, D.E. MondoA senses non-glucose sugars: Regulation of thioredoxin-interacting protein (TXNIP) and the hexose transport curb. J. Biol. Chem. 2011, 286, 38027–38034. [Google Scholar] [CrossRef]
- Park, Y.-J.; Yoon, S.-J.; Suh, H.-W.; Kim, D.O.; Park, J.-R.; Jung, H.; Kim, T.-D.; Yoon, S.R.; Min, J.-K.; Na, H.-J.; et al. TXNIP Deficiency Exacerbates Endotoxic Shock via the Induction of Excessive Nitric Oxide Synthesis. PLoS Pathog. 2013, 9, e1003646. [Google Scholar] [CrossRef]
- Kanari, Y.; Sato, Y.; Aoyama, S.; Muta, T. Thioredoxin-Interacting Protein Gene Expression via MondoA Is Rapidly and Transiently Suppressed during Inflammatory Responses. PLoS ONE 2013, 8, e59026. [Google Scholar] [CrossRef]
- Kim, S.; Joe, Y.; Jeong, S.O.; Zheng, M.; Back, S.H.; Park, S.W.; Ryter, S.W.; Chung, H.T. Endoplasmic reticulum stress is sufficient for the induction of IL-1β production via activation of the NF-κB and inflammasome pathways. Innate Immun. 2014, 20, 799–815. [Google Scholar] [CrossRef]
- Li, Y.; Li, J.; Li, S.; Li, Y.; Wang, X.; Liu, B.; Fu, Q.; Ma, S. Curcumin attenuates glutamate neurotoxicity in the hippocampus by suppression of ER stress-associated TXNIP/NLRP3 inflammasome activation in a manner dependent on AMPK. Toxicol. Appl. Pharmacol. 2015, 286, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Mendsaikhan, A.; Takeuchi, S.; Walker, D.G.; Tooyama, I. Differences in Gene Expression Profiles and Phenotypes of Differentiated SH-SY5Y Neurons Stably Overexpressing Mitochondrial Ferritin. Front. Mol. Neurosci. 2018, 11, 470. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Zuo, D.; Yu, L.; Zhang, L.; Tang, J.; Cui, C.; Bao, L.; Zan, K.; Zhang, Z.; Yang, X.; et al. ROS/TXNIP pathway contributes to thrombin induced NLRP3 inflammasome activation and cell apoptosis in microglia. Biochem. Biophys. Res. Commun. 2017, 485, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Zhang, L. Resveratrol Suppresses Aβ-Induced Microglial Activation Through the TXNIP/TRX/NLRP3 Signaling Pathway. DNA Cell Biol. 2019, 38, 874–879. [Google Scholar] [CrossRef] [PubMed]
- Bharti, V.; Tan, H.; Zhou, H.; Wang, J.-F. Txnip mediates glucocorticoid-activated NLRP3 inflammatory signaling in mouse microglia. Neurochem. Int. 2019, 131, 104564. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 2018, 173, 1073–1081. [Google Scholar] [CrossRef]
- Mohamed, I.N.; Hafez, S.S.; Fairaq, A.; Ergul, A.; Imig, J.D.; El-Remessy, A.B. Thioredoxin-interacting protein is required for endothelial NLRP3 inflammasome activation and cell death in a rat model of high-fat diet. Diabetologia 2014, 57, 413–423. [Google Scholar] [CrossRef]
- Du, R.-H.; Wu, F.-F.; Lu, M.; Shu, X.-D.; Ding, J.-H.; Wu, G.; Hu, G. Uncoupling protein 2 modulation of the NLRP3 inflammasome in astrocytes and its implications in depression. Redox Biol. 2016, 9, 178–187. [Google Scholar] [CrossRef]
- Zhao, Q.; Che, X.; Zhang, H.; Fan, P.; Tan, G.; Liu, L.; Jiang, D.; Zhao, J.; Xiang, X.; Liang, Y.; et al. Thioredoxin-interacting protein links endoplasmic reticulum stress to inflammatory brain injury and apoptosis after subarachnoid haemorrhage. J. Neuroinflammation 2017, 14, 1–15. [Google Scholar] [CrossRef]
- Muraleedharan, R.; Gawali, M.V.; Tiwari, D.; Sukumaran, A.; Oatman, N.; Anderson, J.; Nardini, D.; Bhuiyan, M.A.N.; Tkáč, I.; Ward, A.L.; et al. AMPK-Regulated Astrocytic Lactate Shuttle Plays a Non-Cell-Autonomous Role in Neuronal Survival. Cell Rep. 2020, 32, 108092. [Google Scholar] [CrossRef] [PubMed]
- Dunn, L.L.; Buckle, A.M.; Cooke, J.P.; Ng, M.K.C. The Emerging Role of the Thioredoxin System in Angiogenesis. Arter. Thromb. Vasc. Biol. 2010, 30, 2089–2098. [Google Scholar] [CrossRef] [PubMed]
- Nyandwi, J.B.; Ko, Y.S.; Jin, H.; Yun, S.P.; Park, S.W.; Kim, H.J. Rosmarinic acid inhibits oxLDL-induced inflammasome activation under high-glucose conditions through downregulating the p38-FOXO1-TXNIP pathway. Biochem. Pharmacol. 2020, 182, 114246. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, I.N.; Sheibani, N.; El-Remessy, A.B. Deletion of Thioredoxin-Interacting Protein (TXNIP) Abrogates High Fat Diet-Induced Retinal Leukostasis, Barrier Dysfunction and Microvascular Degeneration in a Mouse Obesity Model. Int. J. Mol. Sci. 2020, 21, 3983. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kover, K.L.; Heruth, D.P.; Watkins, D.J.; Guo, Y.; Moore, W.V.; He, L.G.; Zang, M.; Clements, M.A.; Yan, Y. Thioredoxin-interacting protein promotes high-glucose-induced macrovascular endothelial dysfunction. Biochem. Biophys. Res. Commun. 2017, 493, 291–297. [Google Scholar] [CrossRef]
- Simó, R.; Villarroel, M.; Corraliza, L.; Hernández, C.; García-Ramírez, M. The Retinal Pigment Epithelium: Something More than a Constituent of the Blood-Retinal Barrier—Implications for the Pathogenesis of Diabetic Retinopathy. J. Biomed. Biotechnol. 2010, 2010, 190724. [Google Scholar] [CrossRef]
- Singh, L.P. Thioredoxin Interacting Protein (TXNIP) and Pathogenesis of Diabetic Retinopathy. J. Clin. Exp. Ophthalmol. 2013, 4. [Google Scholar] [CrossRef]
- Devi, T.S.; Yumnamcha, T.; Yao, F.; Somayajulu, M.; Kowluru, R.A.; Singh, L.P. TXNIP mediates high glucose-induced mitophagic flux and lysosome enlargement in human retinal pigment epithelial cells. Biol. Open 2019, 8, bio038521. [Google Scholar] [CrossRef]
- Cho, M.J.; Yoon, S.-J.; Kim, W.; Park, J.; Lee, J.; Park, J.-G.; Cho, Y.-L.; Kim, J.H.; Jang, H.; Park, Y.-J.; et al. Oxidative stress-mediated TXNIP loss causes RPE dysfunction. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Yao, A.; Storr, S.J.; Al-Hadyan, K.; Rahman, R.; Smith, S.; Grundy, R.; Paine, S.; Martin, S.G. Thioredoxin System Protein Expression Is Associated with Poor Clinical Outcome in Adult and Paediatric Gliomas and Medulloblastomas. Mol. Neurobiol. 2020, 57, 2889–2901. [Google Scholar] [CrossRef]
- Morrison, J.A.; Pike, L.A.; Sams, S.B.; Sharma, V.; Zhou, Q.; Severson, J.J.; Tan, A.C.; Wood, W.M.; Haugen, B.R. Thioredoxin interacting protein (TXNIP) is a novel tumor suppressor in thyroid cancer. Mol. Cancer 2014, 13, 62. [Google Scholar] [CrossRef] [PubMed]
- Koka, S.; Xia, M.; Chen, Y.; Bhat, O.M.; Yuan, X.; Boini, K.M.; Li, P.-L. Endothelial NLRP3 inflammasome activation and arterial neointima formation associated with acid sphingomyelinase during hypercholesterolemia. Redox Biol. 2017, 13, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.-J. Pathogenesis of Chronic Hyperglycemia: From Reductive Stress to Oxidative Stress. J. Diabetes Res. 2014, 2014, 137919. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.-W.; Wu, J.; Zhang, Z.; Datta, P.K.; Ibrahimi, I.M.; Taniguchi, S.; Sagara, J.; Fernandes-Alnemri, T.; Alnemri, E.S. Cryopyrin and pyrin activate caspase-1, but not NF-κB, via ASC oligomerization. Cell Death Differ. 2006, 13, 236–249. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; He, F.-F.; Tang, H.; Lei, C.-T.; Chen, S.; Meng, X.-F.; Su, H.; Zhang, C. NADPH Oxidase-Induced NALP3 Inflammasome Activation Is Driven by Thioredoxin-Interacting Protein Which Contributes to Podocyte Injury in Hyperglycemia. J. Diabetes Res. 2015, 2015, 504761. [Google Scholar] [CrossRef]
- Li, H.; Willingham, S.B.; Ting, J.P.-Y.; Re, F. Cutting Edge: Inflammasome Activation by Alum and Alum’s Adjuvant Effect Are Mediated by NLRP3. J. Immunol. 2008, 181, 17–21. [Google Scholar] [CrossRef]
- Luo, B.; Huang, F.; Liu, Y.; Liang, Y.; Wei, Z.; Ke, H.; Zeng, Z.; Huang, W.; He, Y. NLRP3 Inflammasome as a Molecular Marker in Diabetic Cardiomyopathy. Front. Physiol. 2017, 8, 519. [Google Scholar] [CrossRef]
- Levendusky, M.C.; Basle, J.; Chang, S.; Mandalaywala, N.V.; Voigt, J.M.; Dearborn, R.E.J. Expression and regulation of vitamin D3 upregulated protein 1 (VDUP1) is conserved in mammalian and insect brain. J. Comp. Neurol. 2009, 517, 581–600. [Google Scholar] [CrossRef]
- Aon-Bertolino, M.L.; Romero, J.I.; Galeano, P.; Holubiec, M.; Badorrey, M.S.; Saraceno, G.E.; Hanschmann, E.-M.; Lillig, C.H.; Capani, F. Thioredoxin and glutaredoxin system proteins—Immunolocalization in the rat central nervous system. Biochim. Biophys. Acta Gen. Subj. 2011, 1810, 93–110. [Google Scholar] [CrossRef]
- Blouet, C.; Schwartz, G.J. Nutrient-Sensing Hypothalamic TXNIP Links Nutrient Excess to Energy Imbalance in Mice. J. Neurosci. 2011, 31, 6019–6027. [Google Scholar] [CrossRef]
- Blouet, C.; Liu, S.-M.; Jo, Y.-H.; Chua, S.; Schwartz, G.J. TXNIP in Agrp Neurons Regulates Adiposity, Energy Expenditure, and Central Leptin Sensitivity. J. Neurosci. 2012, 32, 9870–9877. [Google Scholar] [CrossRef] [PubMed]
- Hand, L.E.; Saer, B.R.C.; Hui, S.T.; Jinnah, H.A.; Steinlechner, S.; Loudon, A.S.I.; Bechtold, D.A. Induction of the Metabolic Regulator Txnip in Fasting-Induced and Natural Torpor. Endocrinology 2013, 154, 2081–2091. [Google Scholar] [CrossRef] [PubMed]
- Ishrat, T.; Mohamed, I.N.; Pillai, B.; Soliman, S.; Fouda, A.Y.; Ergul, A.; El-Remessy, A.B.; Fagan, S.C. Thioredoxin-Interacting Protein: A Novel Target for Neuroprotection in Experimental Thromboembolic Stroke in Mice. Mol. Neurobiol. 2015, 51, 766–778. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, R.; Wang, X.; Fu, Q.; Ma, S. Umbelliferone ameliorates cerebral ischemia–reperfusion injury via upregulating the PPAR gamma expression and suppressing TXNIP/NLRP3 inflammasome. Neurosci. Lett. 2015, 600, 182–187. [Google Scholar] [CrossRef]
- Gamdzyk, M.; Doycheva, D.M.; Malaguit, J.; Enkhjargal, B.; Tang, J.; Zhang, J.H. Role of PPAR-β/δ/miR-17/TXNIP pathway in neuronal apoptosis after neonatal hypoxic-ischemic injury in rats. Neuropharmacology 2018, 140, 150–161. [Google Scholar] [CrossRef]
- Gamdzyk, M.; Doycheva, D.M.; Kang, R.; Tang, H.; Travis, Z.D.; Tang, J.; Zhang, J.H. GW0742 activates miR-17-5p and inhibits TXNIP/NLRP3-mediated inflammation after hypoxic-ischaemic injury in rats and in PC12 cells. J. Cell. Mol. Med. 2020, 24, 12318–12330. [Google Scholar] [CrossRef]
- Ding, R.; Ou, W.; Chen, C.; Liu, Y.; Li, H.; Zhang, X.; Chai, H.; Ding, X.; Wang, Q. Endoplasmic reticulum stress and oxidative stress contribute to neuronal pyroptosis caused by cerebral venous sinus thrombosis in rats: Involvement of TXNIP/peroxynitrite-NLRP3 inflammasome activation. Neurochem. Int. 2020, 141, 104856. [Google Scholar] [CrossRef]
- Su, C.-J.; Feng, Y.; Liu, T.-T.; Liu, X.; Bao, J.-J.; Shi, A.-M.; Hu, D.-M.; Liu, T.; Yu, Y.-L. Thioredoxin-interacting protein induced α-synuclein accumulation via inhibition of autophagic flux: Implications for Parkinson’s disease. CNS Neurosci. Ther. 2017, 23, 717–723. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Bharti, V.; Zhou, H.; Hoi, V.; Tan, H.; Wu, Z.; Nagakannan, P.; Eftekharpour, E.; Wang, J.-F. Upregulation of Thioredoxin-Interacting Protein in Brain of Amyloid-β Protein Precursor/Presenilin 1 Transgenic Mice and Amyloid-β Treated Neuronal Cells. J. Alzheimers Dis. 2019, 72, 139–150. [Google Scholar] [CrossRef]
- Zhou, H.; Tan, H.; Letourneau, L.; Wang, J.-F. Increased thioredoxin-interacting protein in brain of mice exposed to chronic stress. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 88, 320–326. [Google Scholar] [CrossRef]
- Pan, Q.; Guo, K.; Xue, M.; Tu, Q. Estrogen protects neuroblastoma cell from amyloid-β 42 (Aβ42)-induced apoptosis via TXNIP/TRX axis and AMPK signaling. Neurochem. Int. 2020, 135, 104685. [Google Scholar] [CrossRef] [PubMed]
- Fertan, E.; Rodrigues, G.J.; Wheeler, R.V.; Goguen, D.; Wong, A.A.; James, H.; Stadnyk, A.; Brown, R.E.; Weaver, I.C.G. Cognitive Decline, Cerebral-Spleen Tryptophan Metabolism, Oxidative Stress, Cytokine Production, and Regulation of the Txnip Gene in a Triple Transgenic Mouse Model of Alzheimer Disease. Am. J. Pathol. 2019, 189, 1435–1450. [Google Scholar] [CrossRef] [PubMed]
- Melone, M.A.B.; Dato, C.; Paladino, S.; Coppola, C.; Trebini, C.; Giordana, M.T.; Perrone, L. Verapamil Inhibits Ser202/Thr205 Phosphorylation of Tau by Blocking TXNIP/ROS/p38 MAPK Pathway. Pharm. Res. 2018, 35, 44. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Lučiūnaitė, A.; McManus, R.M.; Jankunec, M.; Rácz, I.; Dansokho, C.; Dalgėdienė, I.; Schwartz, S.; Brosseron, F.; Heneka, M.T. Soluble Aβ oligomers and protofibrils induce NLRP3 inflammasome activation in microglia. J. Neurochem. 2019, e14945. [Google Scholar] [CrossRef]
- Beyer, M.M.S.; Lonnemann, N.; Remus, A.; Latz, E.; Heneka, M.; Korte, M. Enduring Changes in Neuronal Function upon Systemic Inflammation Are NLRP3 Inflammasome Dependent. J. Neurosci. 2020, 40, 5480–5494. [Google Scholar] [CrossRef]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef]
- Mahmoudian, E.; Khalilnezhad, A.; Gharagozli, K.; Amani, D. Thioredoxin-1, redox factor-1 and thioredoxin-interacting protein, mRNAs are differentially expressed in Multiple Sclerosis patients exposed and non-exposed to interferon and immunosuppressive treatments. Gene 2017, 634, 29–36. [Google Scholar] [CrossRef]
- Patwari, P.; Chutkow, W.A.; Cummings, K.; Verstraeten, V.L.R.M.; Lammerding, J.; Schreiter, E.R.; Lee, R.T. Thioredoxin-independent Regulation of Metabolism by the α-Arrestin Proteins. J. Biol. Chem. 2009, 284, 24996–25003. [Google Scholar] [CrossRef]
- Wu, N.; Zheng, B.; Shaywitz, A.; Dagon, Y.; Tower, C.; Bellinger, G.; Shen, C.-H.; Wen, J.; Asara, J.; McGraw, T.E.; et al. AMPK-Dependent Degradation of TXNIP upon Energy Stress Leads to Enhanced Glucose Uptake via GLUT1. Mol. Cell 2013, 49, 1167–1175. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.F.; Kuhl, D.E.; Hawkins, R.A.; Phelps, M.E.; Cummings, J.L.; Tsai, S.Y. The Fluorodeoxyglucose 18F Scan in Alzheimer’s Disease and Multi-infarct Dementia. Arch. Neurol. 1983, 40, 711–714. [Google Scholar] [CrossRef] [PubMed]
- Koepsell, H. Glucose transporters in brain in health and disease. Pflügers Arch. Eur. J. Physiol. 2020, 472, 1299–1343. [Google Scholar] [CrossRef] [PubMed]
- De La Monte, S.M. The Full Spectrum of Alzheimer’s Disease Is Rooted in Metabolic Derangements That Drive Type 3 Diabetes. Adv. Exp. Med. Biol. 2019, 1128, 45–83. [Google Scholar] [CrossRef]
- Bendlin, B.B. Antidiabetic therapies and Alzheimer disease. Dialogues Clin. Neurosci. 2019, 21, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Malek-Ahmadi, M.; Beach, T.; Obradov, A.; Sue, L.; Belden, C.M.; Davis, K.; Walker, D.G.; Lue, L.-F.; Adem, A.; Sabbagh, M.N. Increased Alzheimer’s Disease Neuropathology is Associated with Type 2 Diabetes and ApoE ε4 Carrier Status. Curr. Alzheimer Res. 2013, 10, 654–659. [Google Scholar] [CrossRef]
- Cardoso, S.; Moreira, P.I. Antidiabetic drugs for Alzheimer’s and Parkinson’s diseases: Repurposing insulin, metformin, and thiazolidinediones. Int. Rev. Neurobiol. 2020, 155, 37–64. [Google Scholar] [CrossRef]
- Wu, C.-Y.; Ouk, M.; Wong, Y.Y.; Anita, N.Z.; Edwards, J.D.; Yang, P.; Shah, B.R.; Herrmann, N.; Lanctôt, K.L.; Kapral, M.K.; et al. Relationships between memory decline and the use of metformin or DPP4 inhibitors in people with type 2 diabetes with normal cognition or Alzheimer’s disease, and the role APOE carrier status. Alzheimers Dement. 2020. [Google Scholar] [CrossRef]
- Shi, Q.; Liu, S.; Fonseca, V.A.; Thethi, T.K.; Shi, L. Effect of metformin on neurodegenerative disease among elderly adult US veterans with type 2 diabetes mellitus. BMJ Open 2019, 9, e024954. [Google Scholar] [CrossRef]
- Tang, G.; Duan, F.; Li, W.; Wang, Y.; Zeng, C.; Hu, J.; Li, H.; Zhang, X.; Chen, Y.; Tan, H. Metformin inhibited Nod-like receptor protein 3 inflammasomes activation and suppressed diabetes-accelerated atherosclerosis in apoE(-/-) mice. Biomed. Pharmacother. 2019, 119, 109410. [Google Scholar] [CrossRef]
- Hou, X.; Song, J.; Li, X.-N.; Zhang, L.; Wang, X.; Chen, L.; Shen, Y.H. Metformin reduces intracellular reactive oxygen species levels by upregulating expression of the antioxidant thioredoxin via the AMPK-FOXO3 pathway. Biochem. Biophys. Res. Commun. 2010, 396, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Shaked, M.; Ketzinel-Gilad, M.; Cerasi, E.; Kaiser, N.; Leibowitz, G. AMP-Activated Protein Kinase (AMPK) Mediates Nutrient Regulation of Thioredoxin-Interacting Protein (TXNIP) in Pancreatic Beta-Cells. PLoS ONE 2011, 6, e28804. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Cui, R.; Wang, C.; Feng, Y.; Li, Z.; Tong, Y.; Qu, K.; Liu, C.; Zhang, J. Metformin protects against intestinal ischemia-reperfusion injury and cell pyroptosis via TXNIP-NLRP3-GSDMD pathway. Redox Biol. 2020, 32, 101534. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, J.S.; Chatterjee, A.; Castellani, L.W.; Ross, D.A.; Ohmen, J.; Cavalcoli, J.; Wu, C.; Dains, K.M.; Catanese, J.; Chu, M.; et al. Positional cloning of the combined hyperlipidemia gene Hyplip1. Nat. Genet. 2002, 30, 110–116. [Google Scholar] [CrossRef]
- Donnelly, K.L.; Margosian, M.R.; Sheth, S.S.; Lusis, A.J.; Parks, E.J. Increased lipogenesis and fatty acid reesterification contribute to hepatic triacylglycerol stores in hyperlipidemic Txnip-/- mice. J. Nutr. 2004, 134, 1475–1480. [Google Scholar] [CrossRef]
- Lee, K.N.; Kang, H.-S.; Jeon, J.-H.; Kim, E.-M.; Yoon, S.-R.; Song, H.; Lyu, C.-Y.; Piao, Z.-H.; Kim, S.-U.; Han, Y.-H.; et al. VDUP1 Is Required for the Development of Natural Killer Cells. Immunity 2005, 22, 195–208. [Google Scholar] [CrossRef]
- Okuyama, H.; Yoshida, T.; Son, A.; Oka, S.-I.; Wang, D.; Nakayama, R.; Masutani, H.; Nakamura, H.; Nabeshima, Y.-I.; Yodoi, J. Thioredoxin Binding Protein 2 Modulates Natural Killer T Cell-Dependent Innate Immunity in the Liver: Possible Link to Lipid Metabolism. Antioxid. Redox Signal. 2009, 11, 2585–2593. [Google Scholar] [CrossRef]
- Son, A.; Nakamura, H.; Okuyama, H.; Oka, S.-I.; Yoshihara, E.; Liu, W.; Matsuo, Y.; Kondo, N.; Masutani, H.; Ishii, Y.; et al. Dendritic cells derived from TBP-2-deficient mice are defective in inducing T cell responses. Eur. J. Immunol. 2008, 38, 1358–1367. [Google Scholar] [CrossRef]
- Oka, S.-I.; Yoshihara, E.; Bizen-Abe, A.; Liu, W.; Watanabe, M.; Yodoi, J.; Masutani, H. Thioredoxin Binding Protein-2/Thioredoxin-Interacting Protein Is a Critical Regulator of Insulin Secretion and Peroxisome Proliferator-Activated Receptor Function. Endocrinology 2008, 150, 1225–1234. [Google Scholar] [CrossRef]
- Chutkow, W.A.; Birkenfeld, A.L.; Brown, J.D.; Lee, H.-Y.; Frederick, D.W.; Yoshioka, J.; Patwari, P.; Kursawe, R.; Cushman, S.W.; Plutzky, J.; et al. Deletion of the α-Arrestin Protein Txnip in Mice Promotes Adiposity and Adipogenesis while Preserving Insulin Sensitivity. Diabetes 2010, 59, 1424–1434. [Google Scholar] [CrossRef]
- Shi, Y.; Ren, Y.; Zhao, L.; Du, C.; Wang, Y.; Zhang, Y.; Li, Y.; Zhao, S.; Duan, H. Knockdown of thioredoxin interacting protein attenuates high glucose-induced apoptosis and activation of ASK1 in mouse mesangial cells. FEBS Lett. 2011, 585, 1789–1795. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Shi, Y.; Wang, Y.; Li, Y.; Wu, S.; Li, H.; Zhang, Y.; Duan, H. p38 MAPK pathway is involved in high glucose-induced thioredoxin interacting protein induction in mouse mesangial cells. FEBS Lett. 2010, 584, 3480–3485. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Xia, L.; Masson, E.A.Y.; Gui, C.; Momen, A.; Shikatani, E.A.; Husain, M.; Quaggin, S.; John, R.; Fantus, I.G. Thioredoxin-Interacting Protein Deficiency Protects against Diabetic Nephropathy. J. Am. Soc. Nephrol. 2015, 26, 2963–2977. [Google Scholar] [CrossRef] [PubMed]
- Elshaer, S.L.; Mohamed, I.N.; Coucha, M.; Altantawi, S.; Eldahshan, W.; Bartasi, M.L.; Shanab, A.Y.; Lorys, R.; El-Remessy, A.B. Deletion of TXNIP Mitigates High-Fat Diet-Impaired Angiogenesis and Prevents Inflammation in a Mouse Model of Critical Limb Ischemia. Antioxidants 2017, 6, 47. [Google Scholar] [CrossRef] [PubMed]
- Katsu-Jiménez, Y.; Vázquez-Calvo, C.; Maffezzini, C.; Halldin, M.; Peng, X.; Freyer, C.; Wredenberg, A.; Giménez-Cassina, A.; Wedell, A.; Arnér, E.S.J. Absence of TXNIP in Humans Leads to Lactic Acidosis and Low Serum Methionine Linked to Deficient Respiration on Pyruvate. Diabetes 2019, 68, 709–723. [Google Scholar] [CrossRef]
- Kaya, B.; Erdi, F.; Kılınc, I.; Keskin, F.; Feyzıoglu, B.; Esen, H.; Karataş, Y.; Uyar, M.; Kalkan, E.; Kilinc, I.; et al. Alterations of the thioredoxin system during subarachnoid hemorrhage-induced cerebral vasospasm. Acta Neurochir. 2015, 157, 793–800. [Google Scholar] [CrossRef]
- Yamagishi, K.; Yamamoto, K.; Mochizuki, Y.; Nakano, T.; Yamada, S.; Tokiwa, H. Flexible ligand recognition of peroxisome proliferator-activated receptor-gamma (PPARgamma). Bioorg. Med. Chem. Lett. 2010, 20, 3344–3347. [Google Scholar] [CrossRef]
- Qi, W.; Chen, X.; Holian, J.; Tan, C.Y.R.; Kelly, D.J.; Pollock, C.A. Transcription Factors Krüppel-Like Factor 6 and Peroxisome Proliferator-Activated Receptor-γ Mediate High Glucose-Induced Thioredoxin-Interacting Protein. Am. J. Pathol. 2009, 175, 1858–1867. [Google Scholar] [CrossRef]
- Shaked, M.; Ketzinel-Gilad, M.; Ariav, Y.; Cerasi, E.; Kaiser, N.; Leibowitz, G. Insulin counteracts glucotoxic effects by suppressing thioredoxin-interacting protein production in INS-1E beta cells and in Psammomys obesus pancreatic islets. Diabetologia 2009, 52, 636–644. [Google Scholar] [CrossRef][Green Version]
- Nagaraj, K.; Lapkina-Gendler, L.; Sarfstein, R.; Gurwitz, D.; Pasmanik-Chor, M.; Laron, Z.; Yakar, S.; Werner, H. Identification of thioredoxin-interacting protein (TXNIP) as a downstream target for IGF1 action. Proc. Natl. Acad. Sci. USA 2018, 115, 1045–1050. [Google Scholar] [CrossRef]
- Chen, J.; Cha-Molstad, H.; Szabo, A.; Shalev, A. Diabetes induces and calcium channel blockers prevent cardiac expression of proapoptotic thioredoxin-interacting protein. Am. J. Physiol. Metab. 2009, 296, E1133–E1139. [Google Scholar] [CrossRef] [PubMed]
- Cha-Molstad, H.; Xu, G.; Chen, J.; Jing, G.; Young, M.E.; Chatham, J.C.; Shalev, A. Calcium channel blockers act through nuclear factor Y to control transcription of key cardiac genes. Mol. Pharmacol. 2012, 82, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Buyandelger, U.; Walker, D.G.; Taguchi, H.; Yanagisawa, D.; Tooyama, I. Novel fluorinated derivative of curcumin negatively regulates thioredoxin-interacting protein expression in retinal pigment epithelial and macrophage cells. Biochem. Biophys. Res. Commun. 2020, 532, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Yang, Z.; Sun, Z.; Zhang, W.; Chen, X.; Nie, S. Curcumin relieves paraquat-induced lung injury through inhibiting the thioredoxin interacting protein/NLR pyrin domain containing 3-mediated inflammatory pathway. Mol. Med. Rep. 2019, 20, 5032–5040. [Google Scholar] [CrossRef]
- Lu, X.; Wu, F.; Jiang, M.; Sun, X.; Tian, G. Curcumin ameliorates gestational diabetes in mice partly through activating AMPK. Pharm. Biol. 2019, 57, 250–254. [Google Scholar] [CrossRef]
- Hoshikawa, H.; Kamitori, K.; Indo, K.; Mori, T.; Kamata, M.; Takahashi, T.; Tokuda, M. Combined treatment with D-allose, docetaxel and radiation inhibits the tumor growth in an in vivo model of head and neck cancer. Oncol. Lett. 2018, 15, 3422–3428. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).