Recent Discoveries on the Involvement of Krüppel-Like Factor 4 in the Most Common Cancer Types

 , and
, and
Abstract
1. Introduction
2. Colorectal Cancer
3. Breast Cancer
4. Hepatocellular Carcinoma
5. Lung Cancer
6. Gastric Cancer
7. Prostate Cancer
8. Links between KLF4, Development, Tissue-Specific Conditional Deletion and Cancer
9. KLF4 Loss and Secondary Insult—A Paradigm for the Induction of Malignancy?
10. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Ghaleb, A.M.; Yang, V.W. Kruppel-like factor 4 (KLF4): What we currently know. Gene 2017, 611, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Xiu, D.H.; Chen, Y.; Liu, L.; Yang, H.S.; Liu, G.F. Tumor-suppressive role of Kruppel-like factor 4 (KLF-4) in colorectal cancer. Genet. Mol. Res. 2017, 16, gmr1601927. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wu, L.; Liu, X.; Xu, Y.; Shi, W.; Liang, Y.; Yao, L.; Zheng, J.; Zhang, J. KLF4 inhibits colorectal cancer cell proliferation dependent on NDRG2 signaling. Oncol. Rep. 2017, 38, 975–984. [Google Scholar] [CrossRef] [PubMed]
- Rong, Z.; Luo, Z.; Zhang, J.; Li, T.; Zhu, Z.; Yu, Z.; Fu, Z.; Qiu, Z.; Huang, C. GINS complex subunit 4, a prognostic biomarker and reversely mediated by Kruppel-like factor 4, promotes the growth of colorectal cancer. Cancer Sci. 2020, 111, 1203–1217. [Google Scholar] [CrossRef]
- Agbo, K.C.; Huang, J.Z.; Ghaleb, A.M.; Williams, J.L.; Shroyer, K.R.; Bialkowska, A.B.; Yang, V.W. Loss of the Kruppel-like factor 4 tumor suppressor is associated with epithelial-mesenchymal transition in colorectal cancer. J. Cancer Metastasis Treat. 2019, 5, 743443. [Google Scholar] [CrossRef]
- Yang, V.W.; Liu, Y.; Kim, J.; Shroyer, K.R.; Bialkowska, A.B. Increased Genetic Instability and Accelerated Progression of Colitis-Associated Colorectal Cancer through Intestinal Epithelium-specific Deletion of Klf4. Mol. Cancer Res. 2019, 17, 165–176. [Google Scholar] [CrossRef]
- Zhou, Q.; Fan, D.; Huang, K.; Chen, X.; Chen, Y.; Mai, Q. Activation of KLF4 expression by small activating RNA promotes migration and invasion in colorectal epithelial cells. Cell Biol. Int. 2018, 42, 495–503. [Google Scholar] [CrossRef]
- Halim, S.; Markert, E.K.; Vazquez, A. Analysis of cell proliferation and tissue remodelling uncovers a KLF4 activity score associated with poor prognosis in colorectal cancer. Br. J. Cancer 2018, 119, 855–863. [Google Scholar] [CrossRef]
- Brauer, P.R.; Kim, J.H.; Ochoa, H.J.; Stratton, E.R.; Black, K.M.; Rosencrans, W.; Stacey, E.; Hagos, E.G. Kruppel-like factor 4 mediates cellular migration and invasion by altering RhoA activity. Cell Commun. Adhes. 2018, 24, 1–10. [Google Scholar] [CrossRef]
- Tokarz, P.; Blasiak, J. The role of microRNA in metastatic colorectal cancer and its significance in cancer prognosis and treatment. Acta Biochim. Pol. 2012, 59, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Zhai, F.; Cao, C.; Zhang, L.; Zhang, J. miR-543 promotes colorectal cancer proliferation and metastasis by targeting KLF4. Oncotarget 2017, 8, 59246–59256. [Google Scholar] [CrossRef]
- Chen, H.Y.; Lin, Y.M.; Chung, H.C.; Lang, Y.D.; Lin, C.J.; Huang, J.; Wang, W.C.; Lin, F.M.; Chen, Z.; Huang, H.D.; et al. miR-103/107 promote metastasis of colorectal cancer by targeting the metastasis suppressors DAPK and KLF4. Cancer Res. 2012, 72, 3631–3641. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Li, Y.; Pan, Y.; Lan, X.; Song, F.; Sun, J.; Zhou, K.; Liu, X.; Ren, X.; Wang, F.; et al. Cancer-derived exosomal miR-25-3p promotes pre-metastatic niche formation by inducing vascular permeability and angiogenesis. Nat. Commun. 2018, 9, 5395. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Xie, Y.; Xu, Y. miR-7-5p regulates the proliferation and migration of colorectal cancer cells by negatively regulating the expression of Kruppel-like factor 4. Oncol. Lett. 2019, 17, 3241–3246. [Google Scholar] [PubMed]
- Xie, Y.; Zhao, J.; Liang, Y.; Chen, M.; Luo, Y.; Cui, X.; Jiang, B.; Peng, L.; Wang, X. MicroRNA-10b controls the metastasis and proliferation of colorectal cancer cells by regulating Kruppel-like factor 4. Artif. Cells Nanomed. Biotechnol. 2019, 47, 1722–1729. [Google Scholar] [CrossRef]
- Shao, H.; Dong, D.; Shao, F. Long non-coding RNA TUG1-mediated down-regulation of KLF4 contributes to metastasis and the epithelial-to-mesenchymal transition of colorectal cancer by miR-153-1. Cancer Manag. Res. 2019, 11, 8699–8710. [Google Scholar] [CrossRef]
- Leng, Z.; Li, Y.; Zhou, G.; Lv, X.; Ai, W.; Li, J.; Hou, L. Kruppel-like factor 4 regulates stemness and mesenchymal properties of colorectal cancer stem cells through the TGF-beta1/Smad/snail pathway. J. Cell. Mol. Med. 2020, 24, 1866–1877. [Google Scholar] [CrossRef]
- Yadav, S.S.; Kumar, M.; Varshney, A.; Yadava, P.K. KLF4 sensitizes the colon cancer cell HCT-15 to cisplatin by altering the expression of HMGB1 and hTERT. Life Sci. 2019, 220, 169–176. [Google Scholar] [CrossRef]
- Jie, Y.; He, W.; Yang, X.; Chen, W. Kruppel-like factor 4 acts as a potential therapeutic target of Sijunzi decoction for treatment of colorectal cancer. Cancer Gene 2017, 24, 361–366. [Google Scholar] [CrossRef]
- Goldie, S.J.; Cottle, D.L.; Tan, F.H.; Roslan, S.; Srivastava, S.; Brady, R.; Partridge, D.D.; Auden, A.; Smyth, I.M.; Jane, S.M.; et al. Loss of GRHL3 leads to TARC/CCL17-mediated keratinocyte proliferation in the epidermis. Cell Death Dis. 2018, 9, 1072. [Google Scholar] [CrossRef] [PubMed]
- Parenti, S.; Montorsi, L.; Fantini, S.; Mammoli, F.; Gemelli, C.; Atene, C.G.; Losi, L.; Frassineti, C.; Calabretta, B.; Tagliafico, E.; et al. KLF4 Mediates the Effect of 5-ASA on the beta-Catenin Pathway in Colon Cancer Cells. Cancer Prev. Res. 2018, 11, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Hu, X.; Ni, C.; Jin, W.; Xu, Y.; Chang, L.; Zhou, H.; Jiang, J.; Yang, L. KLF4 p.A472D Mutation Contributes to Acquired Resistance to Cetuximab in Colorectal Cancer. Mol. Cancer 2020, 19, 956–965. [Google Scholar] [CrossRef]
- Anuja, K.; Kar, M.; Chowdhury, A.R.; Shankar, G.; Padhi, S.; Roy, S.; Akhter, Y.; Rath, A.K.; Banerjee, B. Role of telomeric RAP1 in radiation sensitivity modulation and its interaction with CSC marker KLF4 in colorectal cancer. Int. J. Radiat. Biol. 2020, 96, 790–802. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.C.; Luo, C.W.; Huang, W.L.; Wu, C.C.; Chou, C.L.; Chen, C.I.; Chang, S.J.; Chai, C.Y.; Wang, H.C.; Chen, T.Y.; et al. BMI1-KLF4 axis deficiency improves responses to neoadjuvant concurrent chemoradiotherapy in patients with rectal cancer. Radiother. Oncol. 2020, 149, 249–258. [Google Scholar] [CrossRef]
- Roberts, M.S.; Anstine, L.J.; Finke, V.S.; Bryson, B.L.; Webb, B.M.; Weber-Bonk, K.L.; Seachrist, D.D.; Majmudar, P.R.; Keri, R.A. KLF4 defines the efficacy of the epidermal growth factor receptor inhibitor, erlotinib, in triple-negative breast cancer cells by repressing the EGFR gene. Breast Cancer Res. 2020, 22, 66. [Google Scholar] [CrossRef]
- Nagata, T.; Shimada, Y.; Sekine, S.; Moriyama, M.; Hashimoto, I.; Matsui, K.; Okumura, T.; Hori, T.; Imura, J.; Tsukada, K. KLF4 and NANOG are prognostic biomarkers for triple-negative breast cancer. Breast Cancer 2017, 24, 326–335. [Google Scholar] [CrossRef]
- Nagata, T.; Shimada, Y.; Sekine, S.; Hori, R.; Matsui, K.; Okumura, T.; Sawada, S.; Fukuoka, J.; Tsukada, K. Prognostic significance of NANOG and KLF4 for breast cancer. Breast Cancer 2014, 21, 96–101. [Google Scholar] [CrossRef]
- Lu, M.; Wu, Y.; Zeng, B.; Sun, J.; Li, Y.; Luo, J.; Wang, L.; Yi, Z.; Li, H.; Ren, G. CircEHMT1 inhibits metastatic potential of breast cancer cells by modulating miR-1233-3p/KLF4/MMP2 axis. Biochem. Biophys. Res. Commun. 2020, 526, 306–313. [Google Scholar] [CrossRef]
- Cannizzaro, E.; Bannister, A.J.; Han, N.; Alendar, A.; Kouzarides, T. DDX3X RNA helicase affects breast cancer cell cycle progression by regulating expression of KLF4. FEBS Lett. 2018, 592, 2308–2322. [Google Scholar] [CrossRef]
- Lee, H.K.; Lee, D.S.; Park, J.C. Nuclear factor I-C regulates E-cadherin via control of KLF4 in breast cancer. BMC Cancer 2015, 15, 113. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, N.; Meyer-Schaller, N.; Arnold, P.; Antoniadis, H.; Pachkov, M.; van Nimwegen, E.; Christofori, G. Klf4 Is a Transcriptional Regulator of Genes Critical for EMT, Including Jnk1 (Mapk8). PLoS ONE 2013, 8, e57329. [Google Scholar] [CrossRef]
- Fan, S.H.; Wang, Y.Y.; Wu, Z.Y.; Zhang, Z.F.; Lu, J.; Li, M.Q.; Shan, Q.; Wu, D.M.; Sun, C.H.; Hu, B.; et al. AGPAT9 suppresses cell growth, invasion and metastasis by counteracting acidic tumor microenvironment through KLF4/LASS2/V-ATPase signaling pathway in breast cancer. Oncotarget 2015, 6, 18406–18417. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.; Jin, J.; Deng, Y.; Ke, R.H.; Shen, Q.J.; Fan, S.H.; Qin, W.X. LASS2 interacts with V-ATPase and inhibits cell growth of hepatocellular carcinoma. Sheng Li Xue Bao [Acta Physiol. Sin.] 2010, 62, 196–202. [Google Scholar] [PubMed]
- Fan, S.; Niu, Y.; Tan, N.; Wu, Z.; Wang, Y.; You, H.; Ke, R.; Song, J.; Shen, Q.; Wang, W.; et al. LASS2 enhances chemosensitivity of breast cancer by counteracting acidic tumor microenvironment through inhibiting activity of V-ATPase proton pump. Oncogene 2013, 32, 1682–1690. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Chen, H.; Zhou, Z.; Wan, Y.; Liu, Z. ATXN3 promotes breast cancer metastasis by deubiquitinating KLF4. Cancer Lett. 2019, 467, 19–28. [Google Scholar] [CrossRef]
- Zhou, H.; Liu, Y.; Zhu, R.; Ding, F.; Wan, Y.; Li, Y.; Liu, Z. FBXO32 suppresses breast cancer tumorigenesis through targeting KLF4 to proteasomal degradation. Oncogene 2017, 36, 3312–3321. [Google Scholar] [CrossRef]
- He, H.; Li, S.; Chen, H.; Li, L.; Xu, C.; Ding, F.; Zhan, Y.; Ma, J.; Zhang, S.; Shi, Y.; et al. 12-O-tetradecanoylphorbol-13-acetate promotes breast cancer cell motility by increasing S100A14 level in a Kruppel-like transcription factor 4 (KLF4)-dependent manner. J. Biol. Chem. 2014, 289, 9089–9099. [Google Scholar] [CrossRef]
- Moon, J.S.; Kim, H.E.; Koh, E.; Park, S.H.; Jin, W.J.; Park, B.W.; Park, S.W.; Kim, K.S. Krüppel-like factor 4 (KLF4) activates the transcription of the gene for the platelet isoform of phosphofructokinase (PFKP) in breast cancer. J. Biol. Chem. 2011, 286, 23808–23816. [Google Scholar] [CrossRef]
- Okuda, H.; Xing, F.; Pandey, P.R.; Sharma, S.; Watabe, M.; Pai, S.K.; Mo, Y.Y.; Iiizumi-Gairani, M.; Hirota, S.; Liu, Y.; et al. miR-7 suppresses brain metastasis of breast cancer stem-like cells by modulating KLF4. Cancer Res. 2013, 73, 1434–1444. [Google Scholar] [CrossRef]
- Akalay, I.; Tan, T.Z.; Kumar, P.; Janji, B.; Mami-Chouaib, F.; Charpy, C.; Vielh, P.; Larsen, A.K.; Thiery, J.P.; Sabbah, M.; et al. Targeting WNT1-inducible signaling pathway protein 2 alters human breast cancer cell susceptibility to specific lysis through regulation of KLF-4 and miR-7 expression. Oncogene 2015, 34, 2261–2271. [Google Scholar] [CrossRef] [PubMed]
- Mimoto, R.; Imawari, Y.; Hirooka, S.; Takeyama, H.; Yoshida, K. Impairment of DYRK2 augments stem-like traits by promoting KLF4 expression in breast cancer. Oncogene 2017, 36, 1862–1872. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Zhang, C.; Zhou, L.; Xu, H.; Shi, Y.; Tong, Z. Micheliolide overcomes KLF4-mediated cisplatin resistance in breast cancer cells by downregulating glutathione. Oncotargets Ther. 2015, 8, 2319–2327. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.J.; Wang, L.B.; Jiang, Z.N.; Jin, M.; Hu, W.X.; Shen, J.G. The transcription factor KLF4 as an independent predictive marker for pathologic complete remission in breast cancer neoadjuvant chemotherapy: A case-control study. Oncotargets Ther. 2014, 7, 1963–1969. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xue, M.; Zhou, C.; Zheng, Y.; Zhang, Z.; Wang, S.; Fu, Y.; Atyah, M.; Xue, X.; Zhu, L.; Dong, Q.; et al. The association between KLF4 as a tumor suppressor and the prognosis of hepatocellular carcinoma after curative resection. Aging 2020, 12, 15566. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ding, X.; Wu, Q.; Qi, J.; Zhu, M.; Miao, C. Monomethyltransferase SET8 facilitates hepatocellular carcinoma growth by enhancing aerobic glycolysis. Cell Death Dis. 2019, 10, 312. [Google Scholar] [CrossRef]
- Sun, H.; Peng, Z.; Tang, H.; Xie, D.; Jia, Z.; Zhong, L.; Zhao, S.; Ma, Z.; Gao, Y.; Zeng, L.; et al. Loss of KLF4 and consequential downregulation of Smad7 exacerbate oncogenic TGF-beta signaling in and promote progression of hepatocellular carcinoma. Oncogene 2017, 36, 2957–2968. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, L.; Ma, S.; Lu, G.; Wang, D. The aberrant expressions of MACC1, ZEB1, and KLF4 in hepatocellular carcinoma and their clinical significance. Int. J. Clin. Exp. Pathol. 2019, 12, 3653–3661. [Google Scholar]
- Zhang, Y.; Liu, Z.; Li, J.S. Identifying Biomarkers of Hepatocellular Carcinoma Based on Gene Co-Expression Network from High-Throughput Data. Stud. Health Technol. Inf. 2017, 245, 667–671. [Google Scholar]
- Wang, L.; Shen, F.; Stroehlein, J.R.; Wei, D. Context-dependent functions of KLF4 in cancers: Could alternative splicing isoforms be the key? Cancer Lett. 2018, 438, 10–16. [Google Scholar] [CrossRef]
- He, H.; Wu, Z.; Li, S.; Chen, K.; Wang, D.; Zou, H.; Chen, H.; Li, Y.; Liu, Z.; Qu, C. TRAF7 enhances ubiquitin-degradation of KLF4 to promote hepatocellular carcinoma progression. Cancer Lett. 2020, 469, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Cai, X.; Liu, E.; Tian, X.; Tian, C. MicroRNA-18a promotes proliferation and metastasis in hepatocellular carcinoma via targeting KLF4. Oncotarget 2017, 8, 68263–68269. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Wang, F.; Xue, Y.; Lin, Z.; Song, W.; Yang, N.; Li, Q. MicroRNA95p downregulates Klf4 and influences the progression of hepatocellular carcinoma via the AKT signaling pathway. Int. J. Mol. Med. 2019, 43, 1417–1429. [Google Scholar] [PubMed]
- Wu, L.P.; Wu, J.; Shang, A.; Yang, M.; Li, L.L.; Yu, J.; Xu, L.R.; Wang, C.B.; Wang, W.W.; Zhu, J.J.; et al. miR-124 inhibits progression of hepatocarcinoma by targeting KLF4 and promises a novel diagnostic marker. Artif. Cells Nanomed. Biotechnol. 2018, 46 (Suppl. 1), 159–167. [Google Scholar] [CrossRef] [PubMed]
- Hujie, G.; Zhou, S.H.; Zhang, H.; Qu, J.; Xiong, X.W.; Hujie, O.; Liao, C.G.; Yang, S.E. MicroRNA-10b regulates epithelial-mesenchymal transition by modulating KLF4/KLF11/Smads in hepatocellular carcinoma. Cancer Cell Int. 2018, 18, 10. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Jia, X.; Li, C.; Zhang, K.; Li, L.; Wu, J.; Yuan, J.; Li, Q. DDX17 promotes hepatocellular carcinoma progression via inhibiting Klf4 transcriptional activity. Cell Death Dis. 2019, 10, 814. [Google Scholar] [CrossRef]
- Shen, Q.; Eun, J.W.; Lee, K.; Kim, H.S.; Yang, H.D.; Kim, S.Y.; Lee, E.K.; Kim, T.; Kang, K.; Kim, S.; et al. Barrier to autointegration factor 1, procollagen-lysine, 2-oxoglutarate 5-dioxygenase 3, and splicing factor 3b subunit 4 as early-stage cancer decision markers and drivers of hepatocellular carcinoma. Hepatology 2018, 67, 1360–1377. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, D.; Liu, S.; Li, X.; Hu, W.; Han, C. KLF4 suppresses the migration of hepatocellular carcinoma by transcriptionally upregulating monoglyceride lipase. Am. J. Cancer Res. 2018, 8, 1019–1029. [Google Scholar]
- Rajasekaran, D.; Jariwala, N.; Mendoza, R.G.; Robertson, C.L.; Akiel, M.A.; Dozmorov, M.; Fisher, P.B.; Sarkar, D. Staphylococcal Nuclease and Tudor Domain Containing 1 (SND1 Protein) Promotes Hepatocarcinogenesis by Inhibiting Monoglyceride Lipase (MGLL). J. Biol. Chem. 2016, 291, 10736–10746. [Google Scholar] [CrossRef]
- Zhao, Q.; Cai, W.; Zhang, X.; Tian, S.; Zhang, J.; Li, H.; Hou, C.; Ma, X.; Chen, H.; Huang, B.; et al. RYBP Expression Is Regulated by KLF4 and Sp1 and Is Related to Hepatocellular Carcinoma Prognosis. J. Biol. Chem. 2017, 292, 2143–2158. [Google Scholar] [CrossRef]
- Tian, C.; Yao, S.; Liu, L.; Ding, Y.; Ye, Q.; Dong, X.; Gao, Y.; Yang, N.; Li, Q. Klf4 inhibits tumor growth and metastasis by targeting microRNA-31 in human hepatocellular carcinoma. Int. J. Mol. Med. 2017, 39, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yu, S.; Li, L.; Chen, J.; Quan, M.; Li, Q.; Gao, Y. KLF4-mediated upregulation of CD9 and CD81 suppresses hepatocellular carcinoma development via JNK signaling. Cell Death Dis. 2020, 11, 299. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yu, S.; Wu, Q.; Dou, N.; Li, Y.; Gao, Y. KLF4-Mediated CDH3 Upregulation Suppresses Human Hepatoma Cell Growth and Migration via GSK-3beta Signaling. Int. J. Biol. Sci. 2019, 15, 953–961. [Google Scholar] [CrossRef] [PubMed]
- Pang, L.; Xu, L.; Yuan, C.; Li, X.; Zhang, X.; Wang, W.; Guo, X.; Ouyang, Y.; Qiao, L.; Wang, Z.; et al. Activation of EGFR-KLF4 positive feedback loop results in acquired resistance to sorafenib in hepatocellular carcinoma. Mol. Carcinog. 2019, 58, 2118–2126. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Karagonlar, Z.F.; Akbari, S.; Karabicici, M.; Sahin, E.; Avci, S.T.; Ersoy, N.; Ates, K.E.; Balli, T.; Karacicek, B.; Kaplan, K.N.; et al. A Novel Function for KLF4 in Modulating the De-differentiation of EpCAM(-)/CD133(-) nonStem Cells into EpCAM(+)/CD133(+) Liver Cancer Stem Cells in HCC Cell Line HuH7. Cells 2020, 9, 1198. [Google Scholar] [CrossRef]
- Yu, T.; Chen, X.; Zhang, W.; Liu, J.; Avdiushko, R.; Napier, D.L.; Liu, A.X.; Neltner, J.M.; Wang, C.; Cohen, D.; et al. KLF4 regulates adult lung tumor-initiating cells and represses K-Ras-mediated lung cancer. Cell Death Differ. 2016, 23, 207–215. [Google Scholar] [CrossRef]
- Wu, Y.; Lin, L.; Wang, X.; Li, Y.; Liu, Z.; Ye, W.; Huang, W.; Lin, G.; Liu, H.; Zhang, J.; et al. Overexpression of Kruppel-Like Factor 4 Suppresses Migration and Invasion of Non-Small Cell Lung Cancer Through c-Jun-NH2-Terminal Kinase/Epithelial-Mesenchymal Transition Signaling Pathway. Front. Pharm. 2019, 10, 1512. [Google Scholar] [CrossRef]
- Ding, X.; Zhong, T.; Jiang, L.; Huang, J.; Xia, Y.; Hu, R. miR-25 enhances cell migration and invasion in non-small-cell lung cancer cells via ERK signaling pathway by inhibiting KLF4. Mol. Med. Rep. 2018, 17, 7005–7016. [Google Scholar] [CrossRef]
- Li, S.; Huang, L.; Gu, J.; Wu, J.; Ou, W.; Feng, J.; Liu, B.; Xu, X.; Zhou, Y. Restoration of KLF4 Inhibits Invasion and Metastases of Lung Adenocarcinoma through Suppressing MMP2. J. Cancer 2017, 8, 3480–3489. [Google Scholar] [CrossRef]
- Wang, X.; Xia, S.; Li, H.; Wang, X.; Li, C.; Chao, Y.; Zhang, L.; Han, C. The deubiquitinase USP10 regulates KLF4 stability and suppresses lung tumorigenesis. Cell Death Differ. 2020, 27, 1747–1764. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, X.; Peng, K.Z.; Gao, T.; Cui, Y.; Ma, N.; Zhou, Y.; Hou, G. Subcellular localization of Klf4 in non-small cell lung cancer and its clinical significance. Biomed. Pharm. 2018, 99, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wen, Q. miR31205p acts as a diagnostic biomarker in nonsmall cell lung cancer and promotes cancer cell proliferation and invasion by targeting KLF4. Mol. Med. Rep. 2018, 18, 4621–4628. [Google Scholar] [PubMed]
- Cui, Y.; Li, G.; Zhang, X.; Dai, F.; Zhang, R. Increased MALAT1 expression contributes to cisplatin resistance in non-small cell lung cancer. Oncol. Lett. 2018, 16, 4821–4828. [Google Scholar] [CrossRef] [PubMed]
- Zhuan, B.; Lu, Y.; Chen, Q.; Zhao, X.; Li, P.; Yuan, Q.; Yang, Z. Overexpression of the long noncoding RNA TRHDE-AS1 inhibits the progression of lung cancer via the miRNA-103/KLF4 axis. J. Cell. Biochem. 2019, 120, 17616–17624. [Google Scholar] [CrossRef]
- Li, Z.; Huang, J.; Shen, S.; Ding, Z.; Luo, Q.; Chen, Z.; Lu, S. SIRT6 drives epithelial-to-mesenchymal transition and metastasis in non-small cell lung cancer via snail-dependent transrepression of KLF4. J. Exp. Clin. Cancer Res. 2018, 37, 323. [Google Scholar] [CrossRef]
- Jia, Y.; Ying, X.; Zhou, J.; Chen, Y.; Luo, X.; Xie, S.; Wang, Q.C.; Hu, W.; Wang, L. The novel KLF4/PLAC8 signaling pathway regulates lung cancer growth. Cell Death Dis. 2018, 9, 603. [Google Scholar] [CrossRef]
- Xu, K.; Sun, G.; Li, M.; Chen, H.; Zhang, Z.; Qian, X.; Li, P.; Xu, L.; Huang, W.; Wang, X. Glibenclamide Targets Sulfonylurea Receptor 1 to Inhibit p70S6K Activity and Upregulate KLF4 Expression to Suppress Non-Small Cell Lung Carcinoma. Mol. Cancer Ther. 2019, 18, 2085–2096. [Google Scholar] [CrossRef]
- Feng, W.; Xie, Q.; Liu, S.; Ji, Y.; Li, C.; Wang, C.; Jin, L. Kruppel-like factor 4 promotes c-Met amplification-mediated gefitinib resistance in non-small-cell lung cancer. Cancer Sci. 2018, 109, 1775–1786. [Google Scholar] [CrossRef]
- Hashimoto, I.; Nagata, T.; Sekine, S.; Moriyama, M.; Shibuya, K.; Hojo, S.; Matsui, K.; Yoshioka, I.; Okumura, T.; Hori, T.; et al. Prognostic significance of KLF4 expression in gastric cancer. Oncol. Lett. 2017, 13, 819–826. [Google Scholar] [CrossRef]
- Chen, Z.; Gao, Y.; Gao, S.; Song, D.; Feng, Y. MiR-135b-5p promotes viability, proliferation, migration and invasion of gastric cancer cells by targeting Kruppel-like factor 4 (KLF4). Arch. Med. Sci. 2020, 16, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Han, T.; Li, Y.; Sun, J.; Zhang, S.; Liu, Y.; Shan, B.; Zheng, D.; Shi, J. The lncRNA SNHG5/miR-32 axis regulates gastric cancer cell proliferation and migration by targeting KLF4. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2017, 31, 893–903. [Google Scholar]
- Zheng, J.; Liu, Y.; Qiao, Y.; Zhang, L.; Lu, S. miR-103 Promotes Proliferation and Metastasis by Targeting KLF4 in Gastric Cancer. Int. J. Mol. Sci. 2017, 18, 910. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Chen, Z.; Soutto, M.; Zhu, S.; Lu, H.; Romero-Gallo, J.; Peek, R.; Zhang, S.; El-Rifai, W. Helicobacter pylori-induced miR-135b-5p promotes cisplatin resistance in gastric cancer. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 264–274. [Google Scholar] [CrossRef]
- Ou, Y.; Ren, H.; Zhao, R.; Song, L.; Liu, Z.; Xu, W.; Liu, Y.; Wang, S. Helicobacter pylori CagA promotes the malignant transformation of gastric mucosal epithelial cells through the dysregulation of the miR-155/KLF4 signaling pathway. Mol. Carcinog. 2019, 58, 1427–1437. [Google Scholar] [CrossRef]
- Ba, M.C.; Long, H.; Cui, S.Z.; Gong, Y.F.; Yan, Z.F.; Wu, Y.B.; Tu, Y.N. Long noncoding RNA LINC00673 epigenetically suppresses KLF4 by interacting with EZH2 and DNMT1 in gastric cancer. Oncotarget 2017, 8, 95542–95553. [Google Scholar] [CrossRef]
- Kong, F.; Sun, T.; Kong, X.; Xie, D.; Li, Z.; Xie, K. Kruppel-like Factor 4 Suppresses Serine/Threonine Kinase 33 Activation and Metastasis of Gastric Cancer through Reversing Epithelial-Mesenchymal Transition. Clin. Cancer Res. 2018, 24, 2440–2451. [Google Scholar] [CrossRef]
- Wang, L.; Li, Y.; Li, L.; Wu, Z.; Wu, Y.; Ma, H.; Yu, H.; Yang, D.; Wang, D. Role of Kruppel-like factor 4 in regulating inhibitor of apoptosis-stimulating protein of p53 in the progression of gastric cancer. Oncol. Lett. 2018, 15, 6865–6872. [Google Scholar]
- Zhang, J.; Zhu, Z.; Wu, H.; Yu, Z.; Rong, Z.; Luo, Z.; Xu, Y.; Huang, K.; Qiu, Z.; Huang, C. PODXL, negatively regulated by KLF4, promotes the EMT and metastasis and serves as a novel prognostic indicator of gastric cancer. Gastric Cancer 2019, 22, 48–59. [Google Scholar] [CrossRef]
- Zhu, M.; Zhang, N.; Lu, X.; He, S. Negative Regulation of Kruppel-Like Factor 4 on microRNA-106a at Upstream Transcriptional Level and the Role in Gastric Cancer Metastasis. Dig. Dis. Sci. 2018, 63, 2604–2616. [Google Scholar] [CrossRef]
- Zhu, M.; Zhang, N.; He, S. Transcription factor KLF4 modulates microRNA-106a that targets Smad7 in gastric cancer. Pathol. Res. Pract. 2019, 215, 152467. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Liu, Z.; Xu, W.; Song, L.; Ren, H.; Ou, Y.; Liu, Y.; Wang, S. Helicobacter pylori infection leads to KLF4 inactivation in gastric cancer through a TET1-mediated DNA methylation mechanism. Cancer Med. 2020, 9, 2551–2563. [Google Scholar] [CrossRef] [PubMed]
- Koliadenko, V.; Wilanowski, T. Additional functions of selected proteins involved in DNA repair. Free Radic. Biol. Med. 2020, 146, 1–15. [Google Scholar] [CrossRef]
- Wei, L.Z.; Wang, Y.Q.; Chang, Y.L.; An, N.; Wang, X.; Zhou, P.J.; Zhu, H.H.; Fang, Y.X.; Gao, W.Q. Imbalance of a KLF4-miR-7 auto-regulatory feedback loop promotes prostate cancer cell growth by impairing microRNA processing. Am. J. Cancer Res. 2018, 8, 226–244. [Google Scholar] [PubMed]
- Feng, F.; Liu, H.; Chen, A.; Xia, Q.; Zhao, Y.; Jin, X.; Huang, J. miR-148-3p and miR-152-3p synergistically regulate prostate cancer progression via repressing KLF4. J. Cell. Biochem. 2019, 120, 17228–17239. [Google Scholar] [CrossRef]
- Jiang, Z.; Zhang, Y.; Chen, X.; Wu, P.; Chen, D. Long non-coding RNA LINC00673 silencing inhibits proliferation and drug resistance of prostate cancer cells via decreasing KLF4 promoter methylation. J. Cell. Mol. Med. 2020, 24, 1878–1892. [Google Scholar] [CrossRef]
- Siu, M.K.; Suau, F.; Chen, W.Y.; Tsai, Y.C.; Tsai, H.Y.; Yeh, H.L.; Liu, Y.N. KLF4 functions as an activator of the androgen receptor through reciprocal feedback. Oncogenesis 2016, 5, e282. [Google Scholar] [CrossRef]
- Zhang, N.; Su, P.; Li, X.; Xi, J.; Li, X.; Xu, L. Downregulated Kruppellike factor 4 expression is associated with the aggressiveness of prostate cancer. Oncol. Rep. 2019, 41, 1789–1796. [Google Scholar]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Xiong, X.; Schober, M.; Tassone, E.; Khodadadi-Jamayran, A.; Sastre-Perona, A.; Zhou, H.; Tsirigos, A.; Shen, S.; Chang, M.; Melamed, J.; et al. KLF4, A Gene Regulating Prostate Stem Cell Homeostasis, Is a Barrier to Malignant Progression and Predictor of Good Prognosis in Prostate Cancer. Cell Rep. 2018, 25, 3006–3020.e7. [Google Scholar] [CrossRef]
- Tassone, E.; Bradaschia-Correa, V.; Xiong, X.; Sastre-Perona, A.; Josephson, A.M.; Khodadadi-Jamayran, A.; Melamed, J.; Bu, L.; Kahler, D.J.; Ossowski, L.; et al. KLF4 as a rheostat of osteolysis and osteogenesis in prostate tumors in the bone. Oncogene 2019, 38, 5766–5777. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, X.; Chao, Y.; He, R.; Liu, J.; Yuan, Y.; Zhao, W.; Han, C.; Song, X. KLF4, a miR-32-5p targeted gene, promotes cisplatin-induced apoptosis by upregulating BIK expression in prostate cancer. Cell Commun. Signal. 2018, 16, 53. [Google Scholar] [CrossRef] [PubMed]
- Lv, S.; Ji, L.; Chen, B.; Liu, S.; Lei, C.; Liu, X.; Qi, X.; Wang, Y.; Lai-Han Leung, E.; Wang, H.; et al. Histone methyltransferase KMT2D sustains prostate carcinogenesis and metastasis via epigenetically activating LIFR and KLF4. Oncogene 2018, 37, 1354–1368. [Google Scholar] [CrossRef] [PubMed]
- Schagdarsurengin, U.; Lammert, A.; Schunk, N.; Sheridan, D.; Gattenloehner, S.; Steger, K.; Wagenlehner, F.; Dansranjavin, T. Impairment of IGF2 gene expression in prostate cancer is triggered by epigenetic dysregulation of IGF2-DMR0 and its interaction with KLF4. Cell Commun. Signal. 2017, 15, 40. [Google Scholar] [CrossRef]
- Barakat, D.J.; Suresh, R.; Barberi, T.; Pienta, K.J.; Simons, B.W.; Friedman, A.D. Absence of myeloid Klf4 reduces prostate cancer growth with pro-atherosclerotic activation of tumor myeloid cells and infiltration of CD8 T cells. PLoS ONE 2018, 13, e0191188. [Google Scholar] [CrossRef]
- Segre, J.A.; Bauer, C.; Fuchs, E. Klf4 is a transcription factor required for establishing the barrier function of the skin. Nat. Genet. 1999, 22, 356–360. [Google Scholar] [CrossRef]
- Ye, B.; Liu, B.; Hao, L.; Zhu, X.; Yang, L.; Wang, S.; Xia, P.; Du, Y.; Meng, S.; Huang, G.; et al. Klf4 glutamylation is required for cell reprogramming and early embryonic development in mice. Nat. Commun. 2018, 9, 1261. [Google Scholar] [CrossRef]
- Katz, J.P.; Perreault, N.; Goldstein, B.G.; Lee, C.S.; Labosky, P.A.; Yang, V.W.; Kaestner, K.H. The zinc-finger transcription factor Klf4 is required for terminal differentiation of goblet cells in the colon. Development 2002, 129, 2619–2628. [Google Scholar]
- Moon, B.S.; Bai, J.; Cai, M.; Liu, C.; Shi, J.; Lu, W. Kruppel-like factor 4-dependent Staufen1-mediated mRNA decay regulates cortical neurogenesis. Nat. Commun. 2018, 9, 401. [Google Scholar] [CrossRef]
- Jean, J.C.; George, E.; Kaestner, K.H.; Brown, L.A.; Spira, A.; Joyce-Brady, M. Transcription factor Klf4, induced in the lung by oxygen at birth, regulates perinatal fibroblast and myofibroblast differentiation. PLoS ONE 2013, 8, e54806. [Google Scholar] [CrossRef]
- Katz, J.P.; Perreault, N.; Goldstein, B.G.; Actman, L.; McNally, S.R.; Silberg, D.G.; Furth, E.E.; Kaestner, K.H. Loss of Klf4 in mice causes altered proliferation and differentiation and precancerous changes in the adult stomach. Gastroenterology 2005, 128, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Swamynathan, S.; Buela, K.A.; Kinchington, P.; Lathrop, K.L.; Misawa, H.; Hendricks, R.L.; Swamynathan, S.K. Klf4 regulates the expression of Slurp1, which functions as an immunomodulatory peptide in the mouse cornea. Investig. Ophthalmol Vis. Sci. 2012, 53, 8433–8446. [Google Scholar] [CrossRef] [PubMed]
- Swamynathan, S.; Kenchegowda, D.; Piatigorsky, J.; Swamynathan, S. Regulation of corneal epithelial barrier function by Kruppel-like transcription factor 4. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1762–1769. [Google Scholar] [CrossRef] [PubMed]
- Swamynathan, S.K. Kruppel-like factors: Three fingers in control. Hum. Genom. 2010, 4, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Swamynathan, S.K.; Katz, J.P.; Kaestner, K.H.; Ashery-Padan, R.; Crawford, M.A.; Piatigorsky, J. Conditional deletion of the mouse Klf4 gene results in corneal epithelial fragility, stromal edema, and loss of conjunctival goblet cells. Mol. Cell. Biol. 2007, 27, 182–194. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.; Loughner, C.L.; Swamynathan, S.; Swamynathan, S.K. KLF4 Plays an Essential Role in Corneal Epithelial Homeostasis by Promoting Epithelial Cell Fate and Suppressing Epithelial-Mesenchymal Transition. Investig. Ophthalmol. Vis. Sci. 2017, 58, 2785–2795. [Google Scholar] [CrossRef]
- Klaewsongkram, J.; Yang, Y.; Golech, S.; Katz, J.; Kaestner, K.H.; Weng, N.P. Kruppel-like factor 4 regulates B cell number and activation-induced B cell proliferation. J. Immunol. 2007, 179, 4679–4684. [Google Scholar] [CrossRef]
- Yoshida, T.; Kaestner, K.H.; Owens, G.K. Conditional deletion of Kruppel-like factor 4 delays downregulation of smooth muscle cell differentiation markers but accelerates neointimal formation following vascular injury. Circ. Res. 2008, 102, 1548–1557. [Google Scholar] [CrossRef]
- Godmann, M.; Katz, J.P.; Guillou, F.; Simoni, M.; Kaestner, K.H.; Behr, R. Kruppel-like factor 4 is involved in functional differentiation of testicular Sertoli cells. Dev. Biol. 2008, 315, 552–566. [Google Scholar] [CrossRef]
- Tetreault, M.P.; Yang, Y.; Travis, J.; Yu, Q.C.; Klein-Szanto, A.; Tobias, J.W.; Katz, J.P. Esophageal squamous cell dysplasia and delayed differentiation with deletion of kruppel-like factor 4 in murine esophagus. Gastroenterology 2010, 139, 171–181.e9. [Google Scholar] [CrossRef]
- Liao, X.; Sharma, N.; Kapadia, F.; Zhou, G.; Lu, Y.; Hong, H.; Paruchuri, K.; Mahabeleshwar, G.H.; Dalmas, E.; Venteclef, N.; et al. Kruppel-like factor 4 regulates macrophage polarization. J. Clin. Investig. 2011, 121, 2736–2749. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Golech, S.; Klaewsongkram, J.; Zhang, Y.; Subedi, K.; Huston, G.E.; Wood, W.H., 3rd; Wersto, R.P.; Becker, K.G.; Swain, S.L.; et al. Kruppel-like factor 4 (KLF4) directly regulates proliferation in thymocyte development and IL-17 expression during Th17 differentiation. FASEB J. 2011, 25, 3634–3645. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Chen, X.; Zhang, W.; Li, J.; Xu, R.; Wang, T.C.; Ai, W.; Liu, C. Kruppel-like factor 4 regulates intestinal epithelial cell morphology and polarity. PLoS ONE 2012, 7, e32492. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zheng, H.; Wang, J.; Yu, F.; Morris, R.J.; Wang, T.C.; Huang, S.; Ai, W. Expression of Kruppel-like factor KLF4 in mouse hair follicle stem cells contributes to cutaneous wound healing. PLoS ONE 2012, 7, e39663. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zheng, H.; Yu, F.; Yu, T.; Liu, C.; Huang, S.; Wang, T.C.; Ai, W. Deficiency of the Kruppel-like factor KLF4 correlates with increased cell proliferation and enhanced skin tumorigenesis. Carcinogenesis 2012, 33, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Ghaleb, A.M.; McConnell, B.B.; Kaestner, K.H.; Yang, V.W. Altered intestinal epithelial homeostasis in mice with intestine-specific deletion of the Kruppel-like factor 4 gene. Dev. Biol. 2011, 349, 310–320. [Google Scholar] [CrossRef]
- Ghaleb, A.M.; Laroui, H.; Merlin, D.; Yang, V.W. Genetic deletion of Klf4 in the mouse intestinal epithelium ameliorates dextran sodium sulfate-induced colitis by modulating the NF-kappaB pathway inflammatory response. Inflamm. Bowel. Dis. 2014, 20, 811–820. [Google Scholar] [CrossRef]
- Talmasov, D.; Xinjun, Z.; Yu, B.; Nandan, M.O.; Bialkowska, A.B.; Elkarim, E.; Kuruvilla, J.; Yang, V.W.; Ghaleb, A.M. Kruppel-like factor 4 is a radioprotective factor for the intestine following gamma-radiation-induced gut injury in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G121–G138. [Google Scholar] [CrossRef]
- Ghaleb, A.M.; Elkarim, E.A.; Bialkowska, A.B.; Yang, V.W. KLF4 Suppresses Tumor Formation in Genetic and Pharmacological Mouse Models of Colonic Tumorigenesis. Mol. Cancer Res. 2016, 14, 385–396. [Google Scholar] [CrossRef]
- Li, Q.; Jia, Z.; Wang, L.; Kong, X.; Li, Q.; Guo, K.; Tan, D.; Le, X.; Wei, D.; Huang, S.; et al. Disruption of Klf4 in villin-positive gastric progenitor cells promotes formation and progression of tumors of the antrum in mice. Gastroenterology 2012, 142, 531–542. [Google Scholar] [CrossRef]
- Park, C.S.; Lee, P.H.; Yamada, T.; Burns, A.; Shen, Y.; Puppi, M.; Lacorazza, H.D. Kruppel-like factor 4 (KLF4) promotes the survival of natural killer cells and maintains the number of conventional dendritic cells in the spleen. J. Leukoc. Biol. 2012, 91, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Park, C.S.; Suppipat, K.; Mistretta, T.A.; Puppi, M.; Horton, T.M.; Rabin, K.; Gray, N.S.; Meijerink, J.P.P.; Lacorazza, H.D. Inactivation of KLF4 promotes T-cell acute lymphoblastic leukemia and activates the MAP2K7 pathway. Leukemia 2017, 31, 1314–1324. [Google Scholar] [CrossRef] [PubMed]
- Park, C.S.; Lewis, A.H.; Chen, T.J.; Bridges, C.S.; Shen, Y.; Suppipat, K.; Puppi, M.; Tomolonis, J.A.; Pang, P.D.; Mistretta, T.A.; et al. A KLF4-DYRK2-mediated pathway regulating self-renewal in CML stem cells. Blood 2019, 134, 1960–1972. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, K.; Youn, B.U.; Lee, J.; Kim, I.; Shin, H.I.; Akiyama, H.; Choi, Y.; Kim, N. Kruppel-like factor 4 attenuates osteoblast formation, function, and cross talk with osteoclasts. J. Cell Biol. 2014, 204, 1063–1074. [Google Scholar] [CrossRef]
- Abrigo, M.; Alvarez, R.; Paparella, M.L.; Calb, D.E.; Bal de Kier Joffe, E.; Gutkind, J.S.; Raimondi, A.R. Impairing squamous differentiation by Klf4 deletion is sufficient to initiate tongue carcinoma development upon K-Ras activation in mice. Carcinogenesis 2014, 35, 662–669. [Google Scholar] [CrossRef]
- Shi, Y.; Ou, L.; Han, S.; Li, M.; Pena, M.M.; Pena, E.A.; Liu, C.; Nagarkatti, M.; Fan, D.; Ai, W. Deficiency of Kruppel-like factor KLF4 in myeloid-derived suppressor cells inhibits tumor pulmonary metastasis in mice accompanied by decreased fibrocytes. Oncogenesis 2014, 3, e129. [Google Scholar] [CrossRef]
- Nimpong, J.A.; Gebregziabher, W.; Singh, U.P.; Nagarkatti, P.; Nagarkatti, M.; Hodge, J.; Liu, C.; Fan, D.; Ai, W. Deficiency of KLF4 compromises the lung function in an acute mouse model of allergic asthma. Biochem. Biophys. Res. Commun. 2017, 493, 598–603. [Google Scholar] [CrossRef]
- Wei, D.; Wang, L.; Yan, Y.; Jia, Z.; Gagea, M.; Li, Z.; Zuo, X.; Kong, X.; Huang, S.; Xie, K. KLF4 Is Essential for Induction of Cellular Identity Change and Acinar-to-Ductal Reprogramming during Early Pancreatic Carcinogenesis. Cancer Cell 2016, 29, 324–338. [Google Scholar] [CrossRef]
- Yan, Y.; Li, Z.; Kong, X.; Jia, Z.; Zuo, X.; Gagea, M.; Huang, S.; Wei, D.; Xie, K. KLF4-Mediated Suppression of CD44 Signaling Negatively Impacts Pancreatic Cancer Stemness and Metastasis. Cancer Res. 2016, 76, 2419–2431. [Google Scholar] [CrossRef]
- Ou, L.; Shi, Y.; Dong, W.; Liu, C.; Schmidt, T.J.; Nagarkatti, P.; Nagarkatti, M.; Fan, D.; Ai, W. Kruppel-like factor KLF4 facilitates cutaneous wound healing by promoting fibrocyte generation from myeloid-derived suppressor cells. J. Investig. Dermatol. 2015, 135, 1425–1434. [Google Scholar] [CrossRef]
- Majesky, M.W.; Horita, H.; Ostriker, A.; Lu, S.; Regan, J.N.; Bagchi, A.; Dong, X.R.; Poczobutt, J.; Nemenoff, R.A.; Weiser-Evans, M.C. Differentiated Smooth Muscle Cells Generate a Subpopulation of Resident Vascular Progenitor Cells in the Adventitia Regulated by Klf4. Circ. Res. 2017, 120, 296–311. [Google Scholar] [CrossRef] [PubMed]
- Tetreault, M.P.; Weinblatt, D.; Shaverdashvili, K.; Yang, Y.; Katz, J.P. KLF4 transcriptionally activates non-canonical WNT5A to control epithelial stratification. Sci. Rep. 2016, 6, 26130. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.; Park, E.; Jung, E.; Seong, Y.J.; Hong, M.; Lee, S.; Burford, J.; Gyarmati, G.; Peti-Peterdi, J.; Srikanth, S.; et al. ORAI1 Activates Proliferation of Lymphatic Endothelial Cells in Response to Laminar Flow Through Kruppel-Like Factors 2 and 4. Circ. Res. 2017, 120, 1426–1439. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Horrell, J.; Snitow, M.; Cui, J.; Gochnauer, H.; Syrett, C.M.; Kallish, S.; Seykora, J.T.; Liu, F.; Gaillard, D.; et al. WNT10A mutation causes ectodermal dysplasia by impairing progenitor cell proliferation and KLF4-mediated differentiation. Nat. Commun. 2017, 8, 15397. [Google Scholar] [CrossRef]
- Shankman, L.S.; Gomez, D.; Cherepanova, O.A.; Salmon, M.; Alencar, G.F.; Haskins, R.M.; Swiatlowska, P.; Newman, A.A.; Greene, E.S.; Straub, A.C.; et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat. Med. 2015, 21, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Haskins, R.M.; Nguyen, A.T.; Alencar, G.F.; Billaud, M.; Kelly-Goss, M.R.; Good, M.E.; Bottermann, K.; Klibanov, A.L.; French, B.A.; Harris, T.E.; et al. Klf4 has an unexpected protective role in perivascular cells within the microvasculature. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H402–H414. [Google Scholar] [CrossRef] [PubMed]
- Murgai, M.; Ju, W.; Eason, M.; Kline, J.; Beury, D.W.; Kaczanowska, S.; Miettinen, M.M.; Kruhlak, M.; Lei, H.; Shern, J.F.; et al. KLF4-dependent perivascular cell plasticity mediates pre-metastatic niche formation and metastasis. Nat. Med. 2017, 23, 1176–1190. [Google Scholar] [CrossRef]
- Zhou, G.; Hamik, A.; Nayak, L.; Tian, H.; Shi, H.; Lu, Y.; Sharma, N.; Liao, X.; Hale, A.; Boerboom, L.; et al. Endothelial Kruppel-like factor 4 protects against atherothrombosis in mice. J. Clin. Investig. 2012, 122, 4727–4731. [Google Scholar] [CrossRef]
- Shatat, M.A.; Tian, H.; Zhang, R.; Tandon, G.; Hale, A.; Fritz, J.S.; Zhou, G.; Martinez-Gonzalez, J.; Rodriguez, C.; Champion, H.C.; et al. Endothelial Kruppel-like factor 4 modulates pulmonary arterial hypertension. Am. J. Respir Cell Mol. Biol. 2014, 50, 647–653. [Google Scholar] [CrossRef]
- He, M.; Chen, Z.; Martin, M.; Zhang, J.; Sangwung, P.; Woo, B.; Tremoulet, A.H.; Shimizu, C.; Jain, M.K.; Burns, J.C.; et al. miR-483 Targeting of CTGF Suppresses Endothelial-to-Mesenchymal Transition: Therapeutic Implications in Kawasaki Disease. Circ. Res. 2017, 120, 354–365. [Google Scholar] [CrossRef]
- Liao, X.; Zhang, R.; Lu, Y.; Prosdocimo, D.A.; Sangwung, P.; Zhang, L.; Zhou, G.; Anand, P.; Lai, L.; Leone, T.C.; et al. Kruppel-like factor 4 is critical for transcriptional control of cardiac mitochondrial homeostasis. J. Clin. Investig. 2015, 125, 3461–3476. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.K.; Liu, W.; Zi, M.; Li, Y.; Wang, S.; Tsui, H.; Prehar, S.; Castro, S.; Zhang, H.; Ji, Y.; et al. Stress-Activated Kinase Mitogen-Activated Kinase Kinase-7 Governs Epigenetics of Cardiac Repolarization for Arrhythmia Prevention. Circulation 2017, 135, 683–699. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Shen, Y.; Zhou, L.; Sangwung, P.; Fujioka, H.; Zhang, L.; Liao, X. Short-term administration of Nicotinamide Mononucleotide preserves cardiac mitochondrial homeostasis and prevents heart failure. J. Mol. Cell. Cardiol. 2017, 112, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Chen, X.; Lin, T.; Liu, J.; Li, M.; Zhang, W.; Xu, X.; Zhao, W.; Liu, M.; Napier, D.L.; et al. KLF4 deletion alters gastric cell lineage and induces MUC2 expression. Cell Death Dis. 2016, 7, e2255. [Google Scholar] [CrossRef]
- Moore, D.L.; Apara, A.; Goldberg, J.L. Kruppel-like transcription factors in the nervous system: Novel players in neurite outgrowth and axon regeneration. Mol. Cell. Neurosci. 2011, 47, 233–243. [Google Scholar] [CrossRef]
- Qin, S.; Zou, Y.; Zhang, C.L. Cross-talk between KLF4 and STAT3 regulates axon regeneration. Nat. Commun. 2013, 4, 2633. [Google Scholar] [CrossRef]
- Talla, V.; Koilkonda, R. Targeted Kruppel-Like Factor 4 Gene Knock-Out in Retinal Ganglion Cells Improves Visual Function in Multiple Sclerosis Mouse Model. eNeuro 2020, 7. [Google Scholar] [CrossRef]
- Godmann, M.; Gashaw, I.; Katz, J.P.; Nagy, A.; Kaestner, K.H.; Behr, R. Kruppel-like factor 4, a “pluripotency transcription factor” highly expressed in male postmeiotic germ cells, is dispensable for spermatogenesis in the mouse. Mech. Dev. 2009, 126, 650–664. [Google Scholar] [CrossRef]
- Yoshida, T.; Gan, Q.; Franke, A.S.; Ho, R.; Zhang, J.; Chen, Y.E.; Hayashi, M.; Majesky, M.W.; Somlyo, A.V.; Owens, G.K. Smooth and cardiac muscle-selective knock-out of Kruppel-like factor 4 causes postnatal death and growth retardation. J. Biol. Chem. 2010, 285, 21175–21184. [Google Scholar] [CrossRef]
- Hayashi, K.; Sasamura, H.; Nakamura, M.; Azegami, T.; Oguchi, H.; Sakamaki, Y.; Itoh, H. KLF4-dependent epigenetic remodeling modulates podocyte phenotypes and attenuates proteinuria. J. Clin. Investig. 2014, 124, 2523–2537. [Google Scholar] [CrossRef]
- Tussiwand, R.; Everts, B.; Grajales-Reyes, G.E.; Kretzer, N.M.; Iwata, A.; Bagaitkar, J.; Wu, X.; Wong, R.; Anderson, D.A.; Murphy, T.L.; et al. Klf4 expression in conventional dendritic cells is required for T helper 2 cell responses. Immunity 2015, 42, 916–928. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, A.Q.; Misra, A.; Rosas, I.O.; Adams, R.H.; Greif, D.M. Smooth muscle cell progenitors are primed to muscularize in pulmonary hypertension. Sci. Transl. Med. 2015, 7, 308ra159. [Google Scholar] [CrossRef] [PubMed]
- Cuttano, R.; Rudini, N.; Bravi, L.; Corada, M.; Giampietro, C.; Papa, E.; Morini, M.F.; Maddaluno, L.; Baeyens, N.; Adams, R.H.; et al. KLF4 is a key determinant in the development and progression of cerebral cavernous malformations. EMBO Mol. Med. 2016, 8, 6–24. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Shaw, P.X.; Wang, Y.; Goldberg, J.L. Kruppel-Like Factor 4 (KLF4) Is Not Required for Retinal Cell Differentiation. eNeuro 2016, 3. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Tang, A.T.; Wong, W.Y.; Bamezai, S.; Goddard, L.M.; Shenkar, R.; Zhou, S.; Yang, J.; Wright, A.C.; Foley, M.; et al. Cerebral cavernous malformations arise from endothelial gain of MEKK3-KLF2/4 signalling. Nature 2016, 532, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Rocha-Martins, M.; de Toledo, B.C.; Santos-Franca, P.L.; Oliveira-Valenca, V.M.; Vieira-Vieira, C.H.; Matos-Rodrigues, G.E.; Linden, R.; Norden, C.; Martins, R.A.P.; Silveira, M.S. De novo genesis of retinal ganglion cells by targeted expression of Klf4 in vivo. Development 2019, 146, dev176586. [Google Scholar] [CrossRef]
- Park, Y.K.; Wang, L.; Giampietro, A.; Lai, B.; Lee, J.E.; Ge, K. Distinct Roles of Transcription Factors KLF4, Krox20, and Peroxisome Proliferator-Activated Receptor gamma in Adipogenesis. Mol. Cell. Biol. 2017, 37, e00554-16. [Google Scholar] [CrossRef]
- Goddard, L.M.; Duchemin, A.L.; Ramalingan, H.; Wu, B.; Chen, M.; Bamezai, S.; Yang, J.; Li, L.; Morley, M.P.; Wang, T.; et al. Hemodynamic Forces Sculpt Developing Heart Valves through a KLF2-WNT9B Paracrine Signaling Axis. Dev. Cell 2017, 43, 274–289.e5. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef]


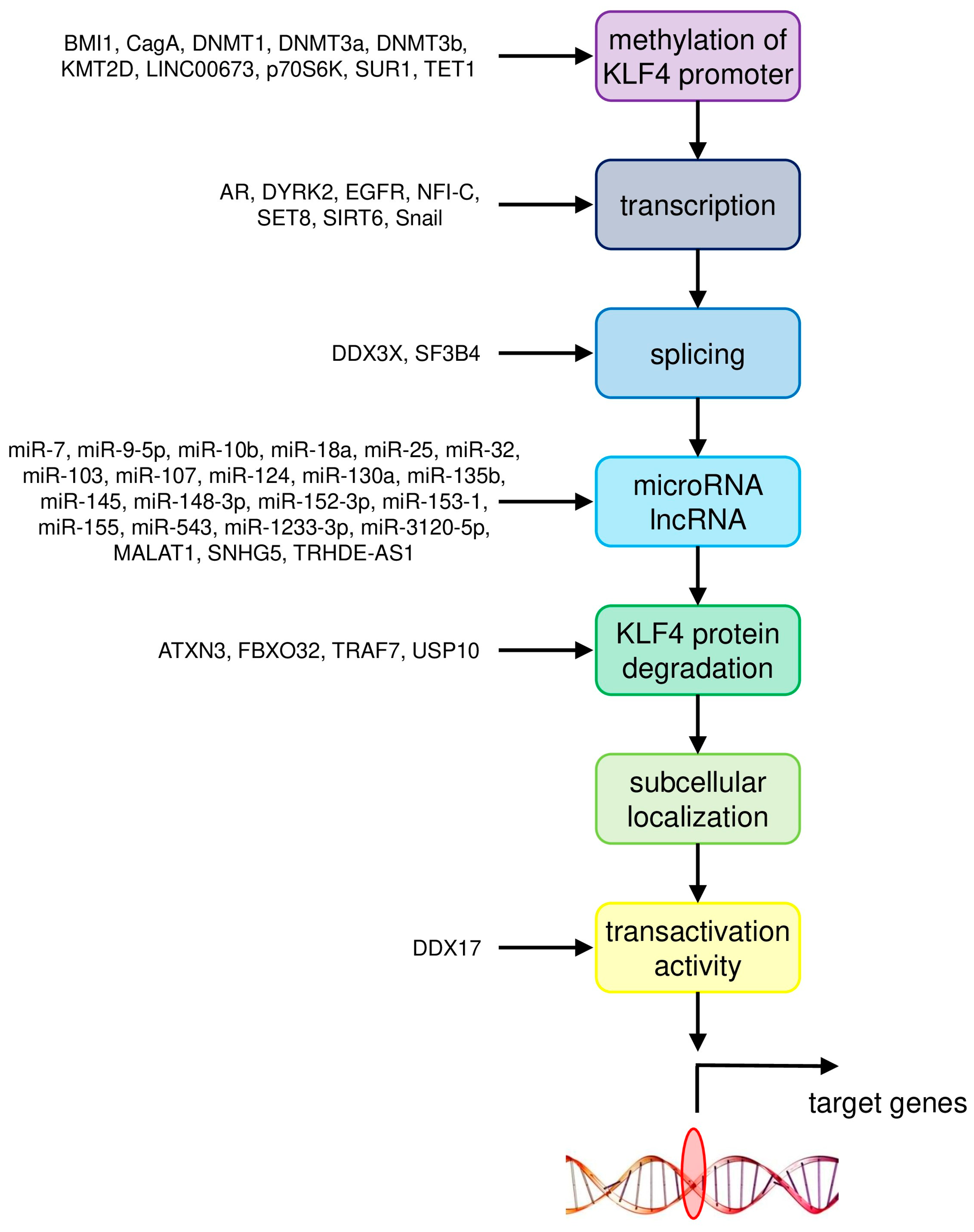{kind=link}
| Cancer Type | Upstream Regulator/Downstream Target | Gene/Pathway | Molecular Mechanism | Reference |
|---|---|---|---|---|
| Colorectal cancer | upstream regulators | BMI1 | methylation of KLF4 promoter | [25] |
| miR-7-5p, miR-10b, miR-25-3p, miR-103/107, miR-130a, miR-135b, miR-153-1, miR-543 | negative regulation by microRNA | [12,13,14,15,16,17,22] | ||
| downstream targets | GINS4 | negative | [5] | |
| NDRG2 | positive | [4] | ||
| tumor microenvironment | myeloid cell infiltration | [9] | ||
| Breast cancer | upstream regulators | AR, DYRK2 | transcription of KLF4 | [42] |
| NFI-C | transcription of KLF4 | [31] | ||
| DDX3X | splicing of KLF4 primary transcript | [30] | ||
| miR-7, miR-1233-3p | negative regulation by microRNA | [29,40,41] | ||
| ATXN3 | KLF4 protein degradation | [36] | ||
| FBXO32 | KLF4 protein degradation | [37] | ||
| downstream targets | E-cadherin | positive | [32] | |
| LASS2 | positive | [33] | ||
| PFKP | positive | [39] | ||
| S100A14 | positive | [38] | ||
| Hepatocellular carcinoma | upstream regulators | EGFR | transcription of KLF4 | [64] |
| SET8 | transcription of KLF4 | [46] | ||
| SF3B4 | splicing of KLF4 primary transcript | [57] | ||
| miR-9-5p, miR-10b, miR-18a, miR-124 | negative regulation by microRNA | [52,53,54,55] | ||
| TRAF7 | KLF4 protein degradation | [51] | ||
| DDX17 | transcriptional transactivation activity of KLF4 | [56] | ||
| downstream targets | CD9, CD81 | positive | [62] | |
| EGFR | positive | [64] | ||
| EpCAM, CD133/Prominin-1 | positive | [66] | ||
| KLF11 | negative | [55] | ||
| MGLL | positive | [58,59] | ||
| miR-31 | positive | [61] | ||
| P-cadherin | positive | [63] | ||
| RYBP | positive | [60] | ||
| SIRT4 | positive | [46] | ||
| Smad7 | positive | [47] | ||
| Lung cancer | upstream regulators | SUR1, p70S6K, DNMT1 | methylation of KLF4 promoter | [78] |
| SIRT6, Snail | transcription of KLF4 | [76] | ||
| miR-25, miR-103, miR-145, miR-3120-5p | negative regulation by microRNA | [69,73,74,75] | ||
| MALAT1, TRHDE-AS1 | positive regulation by lncRNA | [74,75] | ||
| USP10 | KLF4 protein degradation | [71] | ||
| downstream targets | β-catenin | negative | [79] | |
| β-catenin, c-Met | inhibition of binding between c-Met and β-catenin | [79] | ||
| MMP2, PLAC8 | negative | [70,77] | ||
| TIMP3 | positive | [71] | ||
| Gastric cancer | upstream regulators | CagA, TET1 | methylation of KLF4 promoter | [92] |
| LINC00673, EZH2, DNMT1 | methylation of KLF4 promoter | [86] | ||
| miR-32, miR-103, miR-135b-5p, miR-155 | negative regulation by microRNA | [81,82,83,84,85] | ||
| SNHG5 | positive regulation by lncRNA | [82] | ||
| downstream targets | iASPP | negative | [88] | |
| PODXL | negative | [89] | ||
| STK33 | negative | [87] | ||
| Prostate cancer | upstream regulators | KMT2D | methylation of KLF4 promoter | [103] |
| LINC00673, DNMT1, DNMT3a, DNMT3b | methylation of KLF4 promoter | [96] | ||
| AR | transcription of KLF4 | [97] | ||
| miR-7, miR-32-5p, miR-148-3p, miR-152-3p | negative regulation by microRNA | [94,95,102] | ||
| downstream targets | AR | positive | [97] | |
| BIK | positive | [102] | ||
| IGF2 | positive | [104] | ||
| miR-1 | positive | [97] | ||
| miR-7 | positive | [94] | ||
| tumor microenvironment | pro-inflammatory states | [105] |
| Allele Name | Cells Targeted | Genetic Modification | Phenotype | Reference |
|---|---|---|---|---|
| Klf4tm1.1Khk | The floxed region encompassing exons 2 and 3 was excised from Klf4tm1Khk via cre-mediated recombination in the germline. FULL KO MOUSE | Defective goblet cell differentiation in colonic epithelium | [108] | |
| Klf4tm1.1Khk | Neural Progenitor Cells (NPCs) | Nestin-Cre | Increased neurogenesis and reduced self-renewal in cortex. | [109] |
| Klf4tm1.1Khk | Fibroblasts | Klf4−/− cells from lung | p21 mRNA expression reduced prior to birth. Ongoing cell proliferation after birth. Impaired myofibroblast differentiation at tips of alveoli. | [110] |
| Klf4tm1.1Khk | Klf4+/−/Apc/Min+ | Increased incidence of intestinal adenomas. | ||
| Klf4tm1Khk | Gastric mucosa (glandular) | Foxa3 YAC used to direct expression of Cre recombinase | Increased proliferation of gastric epithelia. Defective epithelial differentiation and mucin production No increased inflammation, intestinal metaplasia, dysplasia, or cancer. | [111] |
| Klf4tm1Khk | Corneal Epithelia | Pax6-Cre (Le-Cre) Krt12rtTA/rtTA/Tet-O-Cre | Corneal epithelial fragility and increased proliferation. Disrupted corneal epithelial cell identity Promotion of mesenchymal over epithelial cell identity. Defects in lens formation. | [112,113,114,115,116] |
| Klf4tm1Khk | B-cells | CD19-Cre | Decrease in numbers of pre-B cells in bone marrow and mature B cells in spleen. | [117] |
| Klf4tm1Khk | Tamoxifen inducible—model of vascular injury | ERT-Cre | Enhanced neointimal formation in response to vascular injury caused by increased cellular proliferation. Transient delay in repression of SMC differentiation markers in response to vascular injury. | [118] |
| Klf4tm1Khk | Sertoli cells | Anti Müllerian hormone (AMH)-Cre | Disorganized germinal epithelium and delayed lumen formation. Impaired apical secretion. | [119] |
| Klf4tm1Khk | Squamous epithelia of the tongue, esophagus, and forestomach | ED-L2 promoter of Epstein-Barr virus to drive Cre (ED-L2-Cre) | Increased basal cell proliferation and a delay in cellular maturation. Epithelial hypertrophy and subsequent dysplasia by 6 months of age. | [120] |
| Klf4tm1Khk | Myeloid cells | LysM-Cre | Critical in regulating M1/M2 macrophage polarization. Promotes M1 (pro-inflammatory) macrophage differentiation. Loss of klf4 in myeloid cells slows growth of subcutaneously transplanted prostate cancer cell line. | [121] |
| Klf4tm1Khk | CD4+ Th1 thymocytes (T-cells) | CD4-Cre | Modest reduction of thymocytes due to the reduced proliferation of double-negative (DN) thymocytes. Significant reduction of IL-17-expressing CD4+ T cells. | [105,122] |
| Klf4tm1Khk | Differentiated (adult) intestinal epithelial cells | KLF4/CreER (endogenous locus) | Increase in cell proliferation Increased number of goblet cells in small intestine. Mispositioning of Paneth cells along the small intestinal crypts | [123] |
| Klf4tm1Khk | Hair-follicle stem cells | KLF4/CreER (endogenous locus) | Bulge stem cell-enriched population decreased. Delayed cutaneous wound healing. | [124] |
| Klf4tm1Khk | Skin | KLF4/CreER (BAC) | Increased migration and adhesion of primary keratinocytes. Increased cell proliferation and skin carcinogenesis in DMBA/TPA skin cancer model | [125] |
| Klf4tm1Khk | Villus and crypt epithelial cells of the small and large intestine | Villin-Cre | Increased epithelial cell proliferation and migration in small intestine. Mispositioning of Paneth cells in SI Impaired goblet cell differentiation in colon Protective against development and progression of colitis-associated colorectal cancer (CAC) by guarding against genetic instability. Significantly less sensitive to Dextran Sodium Sulfate (DSS)-induced colitis. Significantly increased mortality following irradiation. Increased tumour formation following genetic mutation (ApcMin/+) or pharmacological treatment (azoxymethane) | [7,126,127,128,129] |
| Klf4tm1Khk | Antral mucosa cells (Stomach) | Villin-Cre | Increased gastric tumor development, exclusively in the lesser curvature of the antrum. | [130] |
| Klf4tm1Khk | Hemopoietic cells | Mx1-Cre Vav-iCre RosaCreER transduced with NOTCH1 retrovirus | Significant reduction of NK cells (NK1.1+ TCR-β−) in the blood and spleen. Increased apoptosis of CD27+/− CD11b+ NK cells in the spleen. Accelerated development of NOTCH1-induced T-ALL by promoting expansion of leukemia-initiating cells. Impaired self-renewal and survival in CML stem/progenitor cells. Impaired maintenance of leukemia in a model of CML-like myeloproliferative neoplasia. De-repression of DYRK2. | [131,132,133] |
| Klf4tm1Khk | Osteoblasts/Osteoclasts | Col1α-Cre | Increased bone mass and enhanced bone formation. Significantly increased numbers of osteoclasts and osteoblasts. | [134] |
| Klf4tm1Khk | Oral cavity epithelia | K14-CreER | Dysplastic lesions, increased cell proliferation and abnormal differentiation in the tongue. Develop oral SCC following Ras activation | [135] |
| Klf4tm1Khk | Bone Marrow (esp. monocytes) | Rosa26-Cre ERFSP-1-Cre | Significantly reduced pulmonary metastasis. Compromised the generation of fibrocytes from MDSCs (myeloid-derived suppressor cells) Decreased expression of epithelial andTh2 cytokines. Impaired fibrocyte generation. Decreased airway hyperresponsiveness. | [136,137] |
| Klf4tm1Khk | Pancreas (esp. B-cells) | Pdx-Cre | Low incidence of hyperplasia in ductal epithelial cells. Reduced pancreatic intraepithelial neoplasia induced by mutant KrasG12D. | [138] |
| Klf4tm1Khk | Pancreatic cancer primary cell lines | AdCre viruses | Promoted acquisition of stem-like properties. | [139] |
| Klf4tm1Khk | Myeloid-derived CCR2+ suppressor cells | Fsp-1-Cre | Increased number of infiltrated lymphocytes in skin granule tissue. Significant hair and weight loss. | [140] |
| Klf4tm1Khk | Smooth Muscle Cells (inducible; adult) | SM22α-CreKI-YFP knockout (activated late in development) | Significant loss of multipotent adventitial Sca1+ cells. Premature death (by 4 weeks of age). | [141] |
| Klf4tm1Khk | Squamous epithelia of the tongue, esophagus, and forestomach | ED-L2-Cre | Hyperplastic esophageal epithelia with evidence of abnormal differentiation and stratification. | [142] |
| Klf4tm1Khk | Lung | Ad5CMVCre-eGFP (together with K-Ras activation) | Significantly increased lung tumorigenesis. Altered differentiation of lung tissue. Increased inflammation in lung. | [67] |
| Klf4tm1Khk | Developing lymphatic vessels | Prox1-CreERT2 | Defects in lymphatic branching morphogenesis. Decreased lymphatic density. | [143] |
| Klf4tm1Khk | Epithelial tissue | Krt5-rtTA tetO-Cre | Differentiation defects in palmoplantar and tongue epithelia. Defects in filiform papilla structure. | [144] |
| Klf4tm1.1Khk | Perivascular Smooth Muscle Cells (SMCs) within large arteries | Myh11-CreERT2 in WT or Apoe−/− mice | Reduced numbers of SMC-derived MSC- and macrophage-like cells. Decreased formation of a pre-metastatic niche and reduced metastasis. Reduction in atheroma size with concomitant increased stability. Significant cardiac dilatation. Impaired smooth muscle coverage of arteries. Arterial dilatation. | [145,146,147] |
| Klf4tm1Khk | Endothelial cells (with some leakiness in macrophages and lymphocytes) | VE-cadherin–Cre (on either wild-type or Apoe−/− backgrounds) | Promoted endothelial to mesenchymal transition (EndoMT) Significantly enhanced development of atherosclerosis after 20 weeks of high-fat diet. Significantly increased right ventricular and pulmonary artery pressures (after hypoxia). More severe pulmonary vascular muscularization and right ventricular hypertrophy (after hypoxia). | [148,149,150] |
| Klf4tm1Khk | Cardiac myocytes | Myh6-Cre αMHC-Cre | Impaired mitochondrial biogenesis and maturation. Reduced mitochondrial respiration. Hyperacetylation of mitochondrial proteins. Cardiac dysfunction with aging or in response to pressure overload. Postnatal premature mortality. Altered ion channel (esp. K+) expression following Transverse Aortic Constriction-induced stress. | [151,152,153] |
| Klf4tm1Khk | Gastric epithelia and antral stem cells | Rosa26-Cre Lgr5-Cre | Increased proliferating cells and decreased pit mucous cells. Induction of MUC2 (goblet cell marker) in antrum. | [154] |
| Klf4tm1Khk | Retinal Ganglion Cells (RGCs). | Thy1-Cre AAV–GFPCre (adenovirus) | Increased axon growth both in vitro and after optic nerve injury in vivo. No difference in survival, but increased neurite length. Increased axon regeneration of adult RGCs. Prevented visual loss and increased neuroprotection in the chronic experimental autoimmune encephalomyelitis (EAE) mouse model of multiple sclerosis | [155,156,157] |
| Klf4tm1Khk | primordial germ cells (PGC) at E9.5–10.5 | TNAP-Cre | No evident phenotype with regard to testicular histology, sperm maturation and fertility. | [158] |
| Klf4tm1Khk | Smooth Muscle | SM22α-Cre | Cardiac output significantly decreased. Marked growth retardation | [159] |
| Klf4tm1Khk | kidney glomerular podocytes | Podocin-Cre | Substantially exacerbated adriamycin-induced proteinuria (minimal phenotype otherwise). | [160] |
| Klf4tm1Khk | Conventional dendritic cells (cDCs) | Itgax-Cre | Impaired Th2 cell responses during challenge or infection. Selective loss of IRF4-expressing cDCs subsets | [161] |
| Klf4tm1Khk | Smooth Muscle | SMA-CreERT2 | Prevented Pulmonary Hypertension (PH) and right ventricle (RV) hypertrophy. Reduced both distal pulmonary arteriole muscularization and PH. | [162] |
| Klf4tm1Khk | Endothelium | Cdh5(PAC)-CreERT2; Ccm1fl/fl; (double Ccm/Klf4 conditional deletion) | Reduction in number, size, and extension of the Cerebral Cavernous Malformations (CCM) in cerebellum. 70% reduction in cavernomas. 75% reduced mortality in Ccm1-deficient pups | [163] |
| Klf4tm1Khk | Retinal progenitor cells | Chx10-Cre | Increased thickness of axon bundles in the nerve fiber layer. No significant difference in cell number among any retinal cell types. No significant difference in photoreceptor layer thickness. | [164] |
| Klf4tm1Khk | Vasculature (endothelial cells); Model of Cerebral cavernous malformations (CCMs) | iECre; Krit1fl/fl | Reduced lesion formation. Rescued lethality. | [165] |
| Klf4tm1Khk | progenitor cells of the peripheral retina | α-Cre | Not essential for generation or differentiation of RGCs during retinogenesis. | [166] |
| Klf4tm1Khk | Pre-adipocytes Brown adipose tissue Back muscles | Retroviral-Cre/Adenoviral-cre (in vitro) Myf5-Cre (in vivo) | Not required for induction of brown adipose tissue. Musculature of back unaffected. | [167] |
| Klf4tm1Khk | Endocardium | Nfatc1-Cre | Required for remodeling of cardiac cushions to mature heart valves. | [168] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taracha-Wisniewska, A.; Kotarba, G.; Dworkin, S.; Wilanowski, T. Recent Discoveries on the Involvement of Krüppel-Like Factor 4 in the Most Common Cancer Types. Int. J. Mol. Sci. 2020, 21, 8843. https://doi.org/10.3390/ijms21228843
Taracha-Wisniewska A, Kotarba G, Dworkin S, Wilanowski T. Recent Discoveries on the Involvement of Krüppel-Like Factor 4 in the Most Common Cancer Types. International Journal of Molecular Sciences. 2020; 21(22):8843. https://doi.org/10.3390/ijms21228843
Chicago/Turabian StyleTaracha-Wisniewska, Agnieszka, Grzegorz Kotarba, Sebastian Dworkin, and Tomasz Wilanowski. 2020. "Recent Discoveries on the Involvement of Krüppel-Like Factor 4 in the Most Common Cancer Types" International Journal of Molecular Sciences 21, no. 22: 8843. https://doi.org/10.3390/ijms21228843
APA StyleTaracha-Wisniewska, A., Kotarba, G., Dworkin, S., & Wilanowski, T. (2020). Recent Discoveries on the Involvement of Krüppel-Like Factor 4 in the Most Common Cancer Types. International Journal of Molecular Sciences, 21(22), 8843. https://doi.org/10.3390/ijms21228843