Age-Dependent Decline in Synaptic Mitochondrial Function Is Exacerbated in Vulnerable Brain Regions of Female 3xTg-AD Mice

 , ,
, ,
Abstract
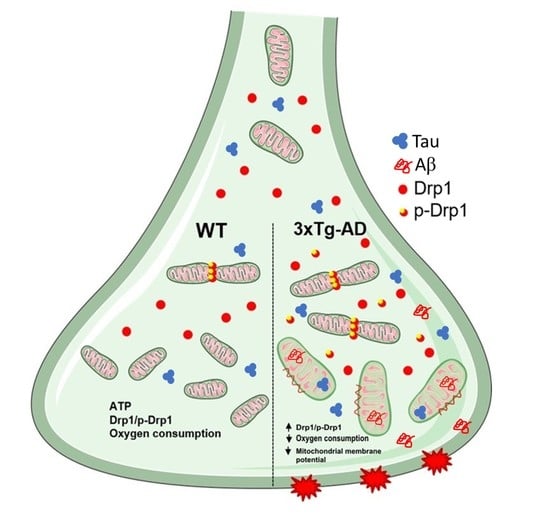
1. Introduction
2. Results
2.1. Evaluation of Mitochondrial Redox Capacity
2.2. Reduced Mitochondrial Bioenergetics Associated with Age
2.3. Mitochondrial Fission Proteins Are Activated by Aging in 3xTg-AD Mice
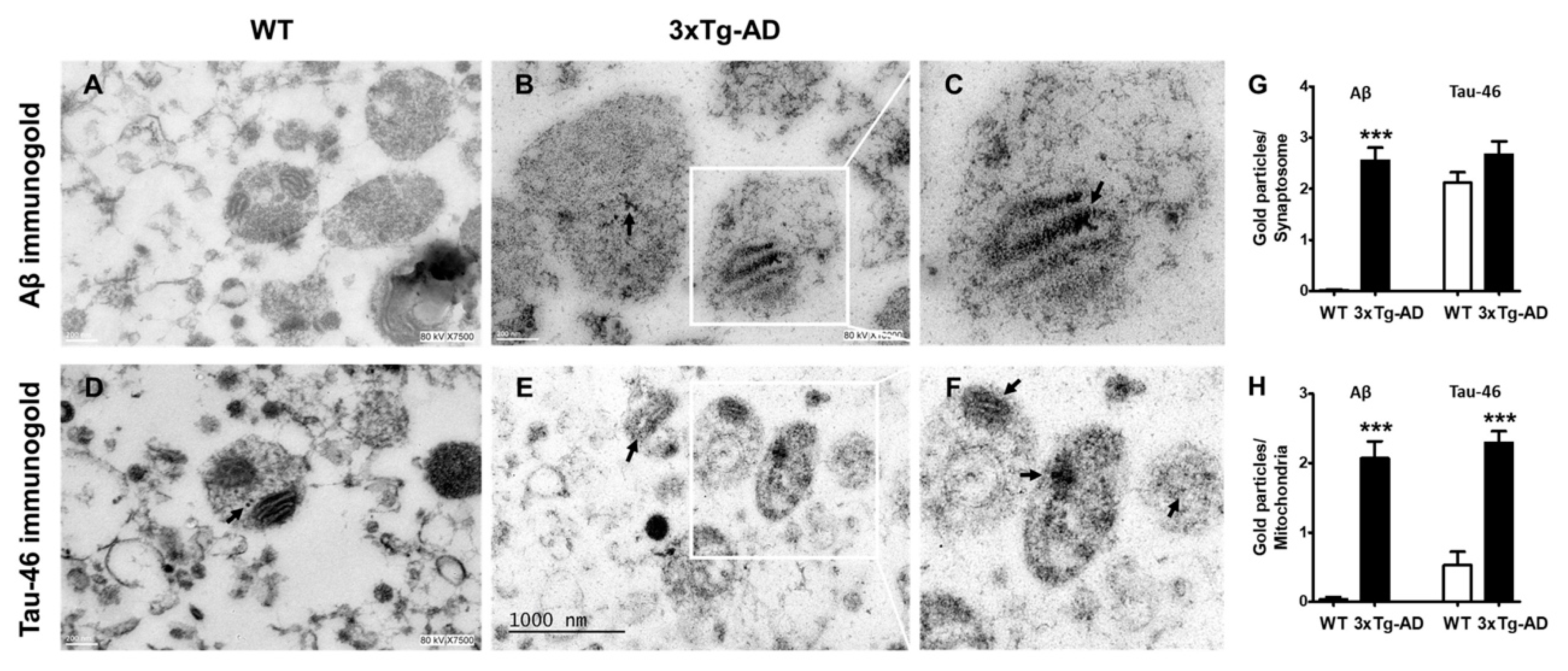
2.4. Aβ and Tau Accumulation in Synaptic Mitochondria
3. Discussion
4. Material and Methods
4.1. Animal Model
4.2. Synaptosomal Preparation
4.3. Evaluation of Mitochondrial Redox Activity in Synaptosomes
4.4. Synaptosomal Mitochondria Bioenergetics
4.5. Mitochondrial Membrane Potential
4.6. Electrophoresis and Immunoblot
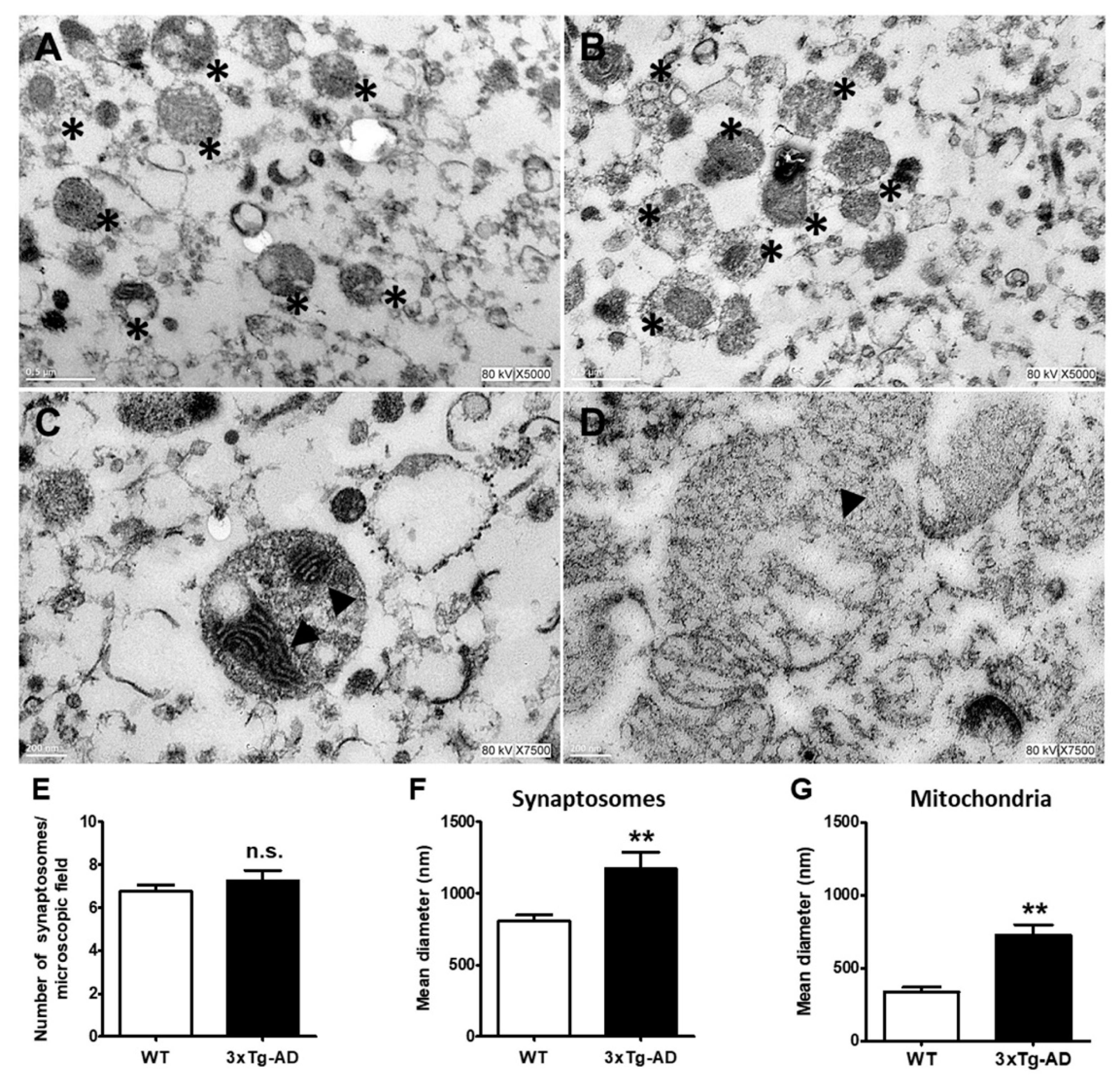
4.7. Electron Microscopy Analysis
4.8. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Morrison, J.H.; Baxter, M.G. The ageing cortical synapse: Hallmarks and implications for cognitive decline. Nat. Rev. Neurosci. 2012, 13, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Morrison, J.H.; Baxter, M.G. Synaptic health. JAMA Psychiatry 2014, 71, 835–837. [Google Scholar] [CrossRef] [PubMed]
- Petralia, R.S.; Mattson, M.P.; Yao, P.J. Communication breakdown: The impact of ageing on synapse structure. Ageing Res. Rev. 2014, 14, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.N.; Barnes, C.A. Neural plasticity in the ageing brain. Nat. Rev. Neurosci. 2006, 7, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Pannese, E. Morphological changes in nerve cells during normal aging. Brain Struct. Funct. 2011, 216, 85–89. [Google Scholar] [CrossRef] [PubMed]
- DeKosky, S.T.; Scheff, S.W. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Ann. Neurol. 1990, 27, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Castilla, P.; Rodriguez-Duran, L.F.; Guzman-Ramos, K.; Barcenas-Femat, A.; Escobar, M.L.; Bermudez-Rattoni, F. Dopaminergic neurotransmission dysfunction induced by amyloid-β transforms cortical long-term potentiation into long-term depression and produces memory impairment. Neurobiol. Aging 2016, 41, 187–199. [Google Scholar] [CrossRef]
- Mucke, L.; Selkoe, D.J. Neurotoxicity of amyloid β-protein: Synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2012, 2, a006338. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef]
- Li, Z.; Okamoto, K.I.; Hayashi, Y.; Sheng, M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 2004, 119, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Scheff, S.W.; Price, D.A. Synaptic pathology in Alzheimer’s disease: A review of ultrastructural studies. In Proceedings of the Neurobiology of Aging. Neurobiol. Aging 2003, 24, 1029–1046. [Google Scholar] [CrossRef] [PubMed]
- Gillingwater, T.H.; Wishart, T.M. Mechanisms underlying synaptic vulnerability and degeneration in neurodegenerative disease. Neuropathol. Appl. Neurobiol. 2013, 39, 320–334. [Google Scholar] [CrossRef] [PubMed]
- Devine, M.J.; Kittler, J.T. Mitochondria at the neuronal presynapse in health and disease. Nat. Rev. Neurosci. 2018, 19, 63–80. [Google Scholar] [CrossRef]
- Chen, H.; McCaffery, J.M.; Chan, D.C. Mitochondrial Fusion Protects against Neurodegeneration in the Cerebellum. Cell 2007, 130, 548–562. [Google Scholar] [CrossRef]
- Züchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J.; et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004, 36, 449–451. [Google Scholar] [CrossRef]
- Tanaka, A.; Cleland, M.M.; Xu, S.; Narendra, D.P.; Suen, D.F.; Karbowski, M.; Youle, R.J. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 2010, 191, 1367–1380. [Google Scholar] [CrossRef]
- Manczak, M.; Kandimalla, R.; Fry, D.; Sesaki, H.; Reddy, P.H. Protective effects of reduced dynamin-related protein 1 against amyloid beta-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum. Mol. Genet. 2016, 25, 5148–5166. [Google Scholar] [CrossRef]
- Baek, S.H.; Park, S.J.; Jeong, J.I.; Kim, S.H.; Han, J.J.-W.W.; Kyung, J.W.; Baik, S.-H.H.; Choi, Y.; Choi, B.Y.; Park, J.S.; et al. Inhibition of Drp1 Ameliorates Synaptic Depression, Aβ Deposition, and Cognitive Impairment in an Alzheimer’s Disease Model. J. Neurosci. 2017, 37, 5099–5110. [Google Scholar] [CrossRef]
- Hirai, K.; Aliev, G.; Nunomura, A.; Fujioka, H.; Russell, R.L.; Atwood, C.S.; Johnson, A.B.; Kress, Y.; Vinters, H.V.; Tabaton, M.; et al. Mitochondrial abnormalities in Alzheimer’s disease. J. Neurosci. 2001, 21, 3017–3023. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Lee, H.-G.H.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Tripathi, R.; Troung, Q.; Tirumala, K.; Reddy, T.P.; Anekonda, V.; Shirendeb, U.P.; Calkins, M.J.; Reddy, A.P.; Mao, P.; et al. Abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer’s disease: Implications to mitochondria-targeted antioxidant therapeutics. Biochim. Biophys. Acta 2012, 1822, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Su, B.; Siedlak, S.L.; Moreira, P.I.; Fujioka, H.; Wang, Y.; Casadesus, G.; Zhu, X. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 19318–19323. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Guo, L.; Yan, S.; Sosunov, A.A.; McKhann, G.M.; Yan, S.S. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc. Natl. Acad. Sci. USA 2010, 107, 18670–18675. [Google Scholar] [CrossRef]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675. [Google Scholar] [CrossRef]
- Adami, P.V.M.; Quijano, C.; Magnani, N.; Galeano, P.; Evelson, P.; Cassina, A.; Do Carmo, S.; Leal, M.C.; Castaño, E.M.; Cuello, A.C.; et al. Synaptosomal bioenergetic defects are associated with cognitive impairment in a transgenic rat model of early Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2017, 37, 69–84. [Google Scholar] [CrossRef]
- Wang, L.; Guo, L.; Lu, L.; Sun, H.; Shao, M.; Beck, S.J.; Li, L.; Ramachandran, J.; Du, Y.; Du, H. Synaptosomal mitochondrial dysfunction in 5xFAD mouse model of Alzheimer’s disease. PLoS ONE 2016, 11, e0150441. [Google Scholar] [CrossRef]
- Mungarro-Menchaca, X.; Morán, P.F.J.; Clorinda, A. β-amyloid peptide induces ultrastructural changes in synaptosomes and potentiates mitochondrial dysfunction in the presence of ryanodine. J. Neurosci. Res. 2002, 68, 89–96. [Google Scholar] [CrossRef]
- Quiroz-Baez, R.; Flores-Domínguez, D.; Arias, C. Synaptic aging is associated with mitochondrial dysfunction, reduced antioxidant contents and increased vulnerability to amyloid-β toxicity. Curr. Alzheimer Res. 2013, 10, 324–331. [Google Scholar] [CrossRef]
- Nicholls, D.G. Mitochondrial membrane potential and aging. Aging Cell 2004, 3, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Gower, A.J.; Lamberty, Y. The aged mouse as a model of cognitive decline with special emphasis on studies in NMRI mice. Behav. Brain Res. 1993, 57, 163–173. [Google Scholar] [CrossRef]
- Mavroudis, I.A.; Fotiou, D.F.; Adipepe, L.F.; Manani, M.G.; Njau, S.D.; Psaroulis, D.; Costa, V.G.; Baloyannis, S.J. Morphological changes of the human purkinje cells and deposition of neuritic plaques and neurofibrillary tangles on the cerebellar cortex of Alzheimer’s disease. Am. J. Alzheimers. Dis. Other Demen. 2010, 25, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda-Falla, D.; Barrera-Ocampo, A.; Hagel, C.; Korwitz, A.; Vinueza-Veloz, M.F.; Zhou, K.; Schonewille, M.; Zhou, H.; Velazquez-Perez, L.; Rodriguez-Labrada, R.; et al. Familial Alzheimer’s disease-associated presenilin-1 alters cerebellar activity and calcium homeostasis. J. Clin. Investig. 2014, 124, 1552–1567. [Google Scholar] [CrossRef] [PubMed]
- Andersen, B.B.; Gundersen, H.J.G.; Pakkenberg, B. Aging of the Human Cerebellum: A Stereological Study. J. Comp. Neurol. 2003, 466, 356–365. [Google Scholar] [CrossRef]
- Rogers, J.; Zornetzer, S.F.; Bloom, F.E.; Mervis, R.E. Senescent microstructural changes in rat cerebellum. Brain Res. 1984, 292, 23–32. [Google Scholar] [CrossRef]
- Stamer, K.; Vogel, R.; Thies, E.; Mandelkow, E.; Mandelkow, E.M. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol. 2002, 156, 1051–1063. [Google Scholar] [CrossRef]
- Morfini, G.; Pigino, G.; Beffert, U.; Busciglio, J.; Brady, S.T. Fast axonal transport misregulation and Alzheimer’s disease. NeuroMol. Med. 2002, 2, 89–99. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Fujioka, H.; Zhu, X. Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer’s disease patients. Am. J. Pathol. 2008, 173, 470–482. [Google Scholar] [CrossRef]
- Cho, D.-H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S. a S-Nitrosylation of Drp1 Mediates β-Amyloid: Related Mitochondrial Fission and Neuronal Injury. Science 2009, 324, 102–105. [Google Scholar] [CrossRef]
- Kandimalla, R.; Reddy, P.H. Multiple faces of dynamin-related protein 1 and its role in Alzheimer’s disease pathogenesis. Biochim. Biophys. Acta-Mol. Basis Dis. 2016, 1862, 814–828. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Wang, P.; Zhang, H.; Gong, G.; Gutierrez Cortes, N.; Zhu, W.; Yoon, Y.; Tian, R.; Wang, W. CaMKII induces permeability transition through Drp1 phosphorylation during chronic β-AR stimulation. In Proceedings of the Nature Communications; Nature Publishing Group: Berlin, Germany, 2016; Volume 7, pp. 1–13. [Google Scholar]
- Divakaruni, S.S.; Van Dyke, A.M.; Chandra, R.; LeGates, T.A.; Contreras, M.; Dharmasri, P.A.; Higgs, H.N.; Lobo, M.K.; Thompson, S.M.; Blanpied, T.A. Long-Term Potentiation Requires a Rapid Burst of Dendritic Mitochondrial Fission during Induction. Neuron 2018, 100, 860–875.e7. [Google Scholar] [CrossRef] [PubMed]
- Nikoletopoulou, V.; Tavernarakis, N. Calcium homeostasis in aging neurons. Front. Genet. 2012, 3, 200. [Google Scholar] [CrossRef]
- Manczak, M.; Reddy, P.H. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: Implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 2012, 21, 2538–2547. [Google Scholar] [CrossRef]
- Bender, T.; Lewrenz, I.; Franken, S.; Baitzel, C.; Voos, W. Mitochondrial enzymes are protected from stress-induced aggregation by mitochondrial chaperones and the Pim1/LON protease. Mol. Biol. Cell 2011, 22, 541–554. [Google Scholar] [CrossRef]
- Ruan, L.; Zhou, C.; Jin, E.; Kucharavy, A.; Zhang, Y.; Wen, Z.; Florens, L.; Li, R. Cytosolic proteostasis through importing of misfolded proteins into mitochondria. Nature 2017, 543, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s Disease with plaques and tangles: Intracellular Aβ and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Belfiore, R.; Rodin, A.; Ferreira, E.; Velazquez, R.; Branca, C.; Caccamo, A.; Oddo, S. Temporal and regional progression of Alzheimer’s disease-like pathology in 3xTg-AD mice. Aging Cell 2019, 18, e12873. [Google Scholar] [CrossRef]
- Löscher, W.; Böhme, G.; Müller, F.; Pagliusi, S.; Loscher, W.; Bohme, G.; Muller, F.; Pagliusi, S. Improved method for isolating synaptosomes from 11 regions of one rat brain: Electron microscopic and biochemical characterization and use in the study of drug effects on nerve terminal gamma-aminobutyric acid in vivo. J. Neurochem. 1985, 45, 879–889. [Google Scholar] [CrossRef]
- Quiroz-Baez, R.; Ferrera, P.; Rosendo-Gutiérrez, R.; Morán, J.; Bermúdez-Rattoni, F.; Arias, C. Caspase-12 activation is involved in amyloid-β protein-induced synaptic toxicity. J. Alzheimer’s Dis. 2011, 26, 467–476. [Google Scholar] [CrossRef]
- Israël, M.; Whittaker, V.P. The isolation of mossy fibre endings from the granular layer of the cerebellar cortex. Experientia 1965, 21, 325–326. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, F.; Gorini, A.; Villa, R.F. Energy metabolism of synaptosomes from different neuronal systems of rat cerebellum during aging: A functional proteomic characterization. Neurochem. Res. 2015, 40, 172–185. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Young | Old | |||
|---|---|---|---|---|
| WT | 3xTg-AD | WT | 3xTg-AD | |
| Cortex | ||||
| Glucose | 8.002 | 6.969 | 3.563 ** | 2.531 **& |
| Oligomycin | 5.393 | 3.869 | 2.102 ** | 1.626 ** |
| FCCP | 12.800 | 14.130 | 4.093 ** | 3.318 ** |
| Hippocampus | ||||
| Glucose | 9.801 | 8.022 | 4.458 ** | 3.036 **& |
| Oligomycin | 6.798 | 6.748 | 3.515 ** | 1.594 **& |
| FCCP | 13.742 | 12.907 | 7.120 ** | 5.119 **& |
| Cerebellum | ||||
| Glucose | 7.005 | 5.843 | 2.704 *** | 2.787 *** |
| Oligomycin | 3.258 | 4.277 | 2.601 *** | 1.726 *** |
| FCCP | 10.794 | 14.426 | 2.993 *** | 3.070 *** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Espino de la Fuente-Muñoz, C.; Rosas-Lemus, M.; Moreno-Castilla, P.; Bermúdez-Rattoni, F.; Uribe-Carvajal, S.; Arias, C. Age-Dependent Decline in Synaptic Mitochondrial Function Is Exacerbated in Vulnerable Brain Regions of Female 3xTg-AD Mice. Int. J. Mol. Sci. 2020, 21, 8727. https://doi.org/10.3390/ijms21228727
Espino de la Fuente-Muñoz C, Rosas-Lemus M, Moreno-Castilla P, Bermúdez-Rattoni F, Uribe-Carvajal S, Arias C. Age-Dependent Decline in Synaptic Mitochondrial Function Is Exacerbated in Vulnerable Brain Regions of Female 3xTg-AD Mice. International Journal of Molecular Sciences. 2020; 21(22):8727. https://doi.org/10.3390/ijms21228727
Chicago/Turabian StyleEspino de la Fuente-Muñoz, César, Mónica Rosas-Lemus, Perla Moreno-Castilla, Federico Bermúdez-Rattoni, Salvador Uribe-Carvajal, and Clorinda Arias. 2020. "Age-Dependent Decline in Synaptic Mitochondrial Function Is Exacerbated in Vulnerable Brain Regions of Female 3xTg-AD Mice" International Journal of Molecular Sciences 21, no. 22: 8727. https://doi.org/10.3390/ijms21228727
APA StyleEspino de la Fuente-Muñoz, C., Rosas-Lemus, M., Moreno-Castilla, P., Bermúdez-Rattoni, F., Uribe-Carvajal, S., & Arias, C. (2020). Age-Dependent Decline in Synaptic Mitochondrial Function Is Exacerbated in Vulnerable Brain Regions of Female 3xTg-AD Mice. International Journal of Molecular Sciences, 21(22), 8727. https://doi.org/10.3390/ijms21228727