Integrated Proteo-Transcriptomic Analyses Reveal Insights into Regulation of Pollen Development Stages and Dynamics of Cellular Response to Apple Fruit Crinkle Viroid (AFCVd)-Infection in Nicotiana tabacum


 , , , ,
, , , ,  ,
, 
Abstract
1. Introduction
2. Results
2.1. Transcriptome Sequencing and Assembly
2.2. Overview of Quantitative Proteomics Analysis
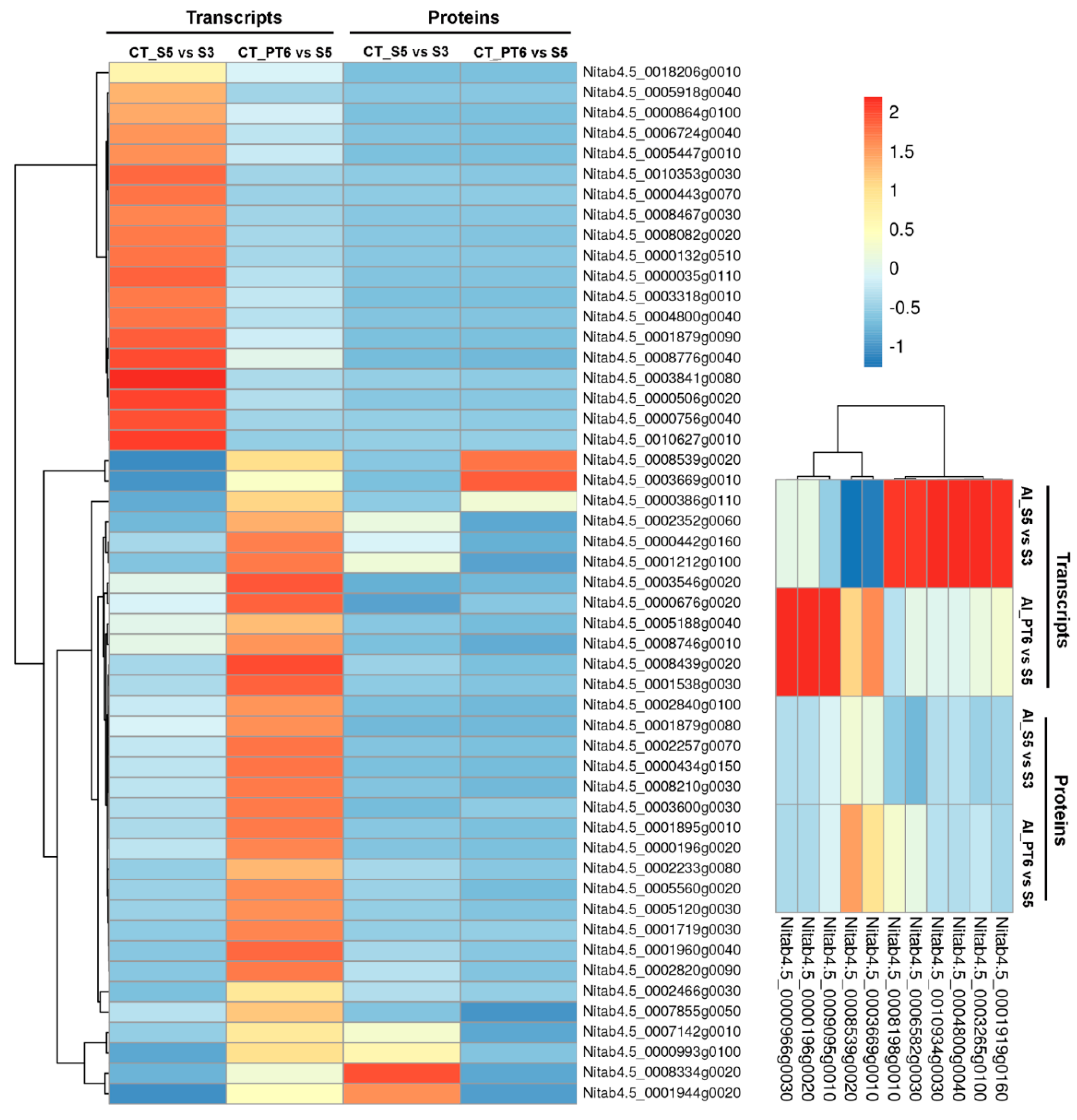
2.3. Correlation of Transcript and Protein Profiles
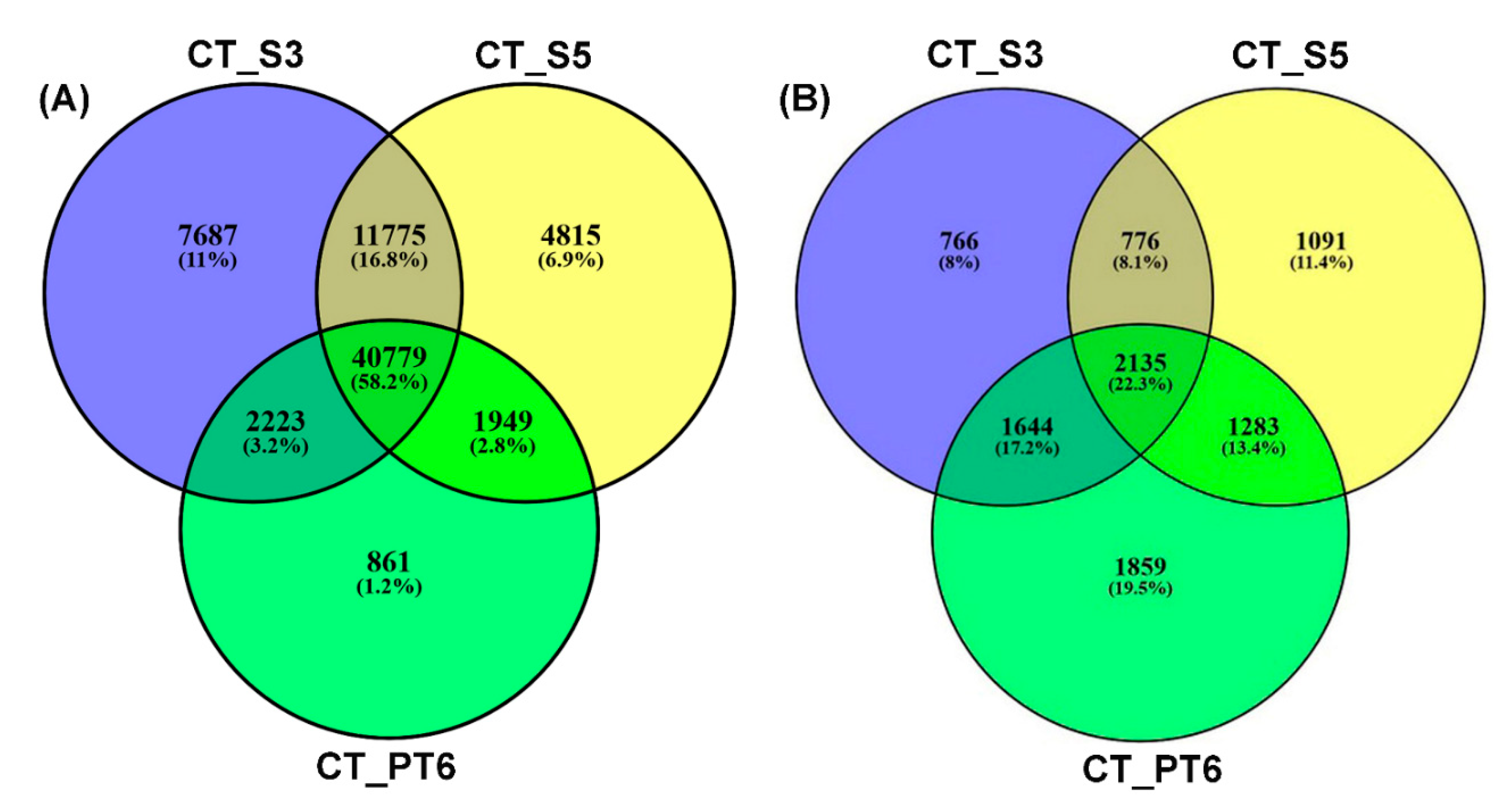
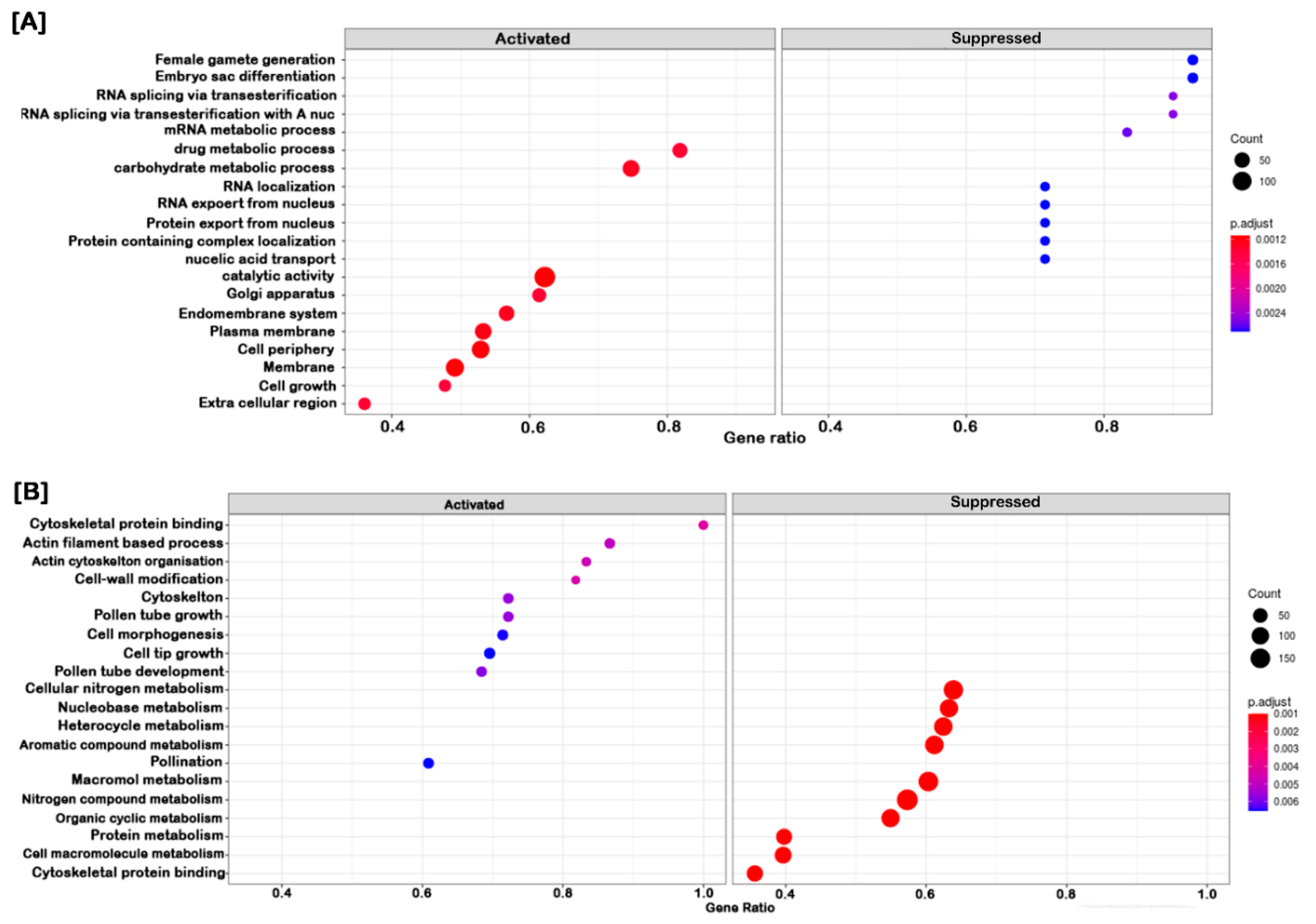
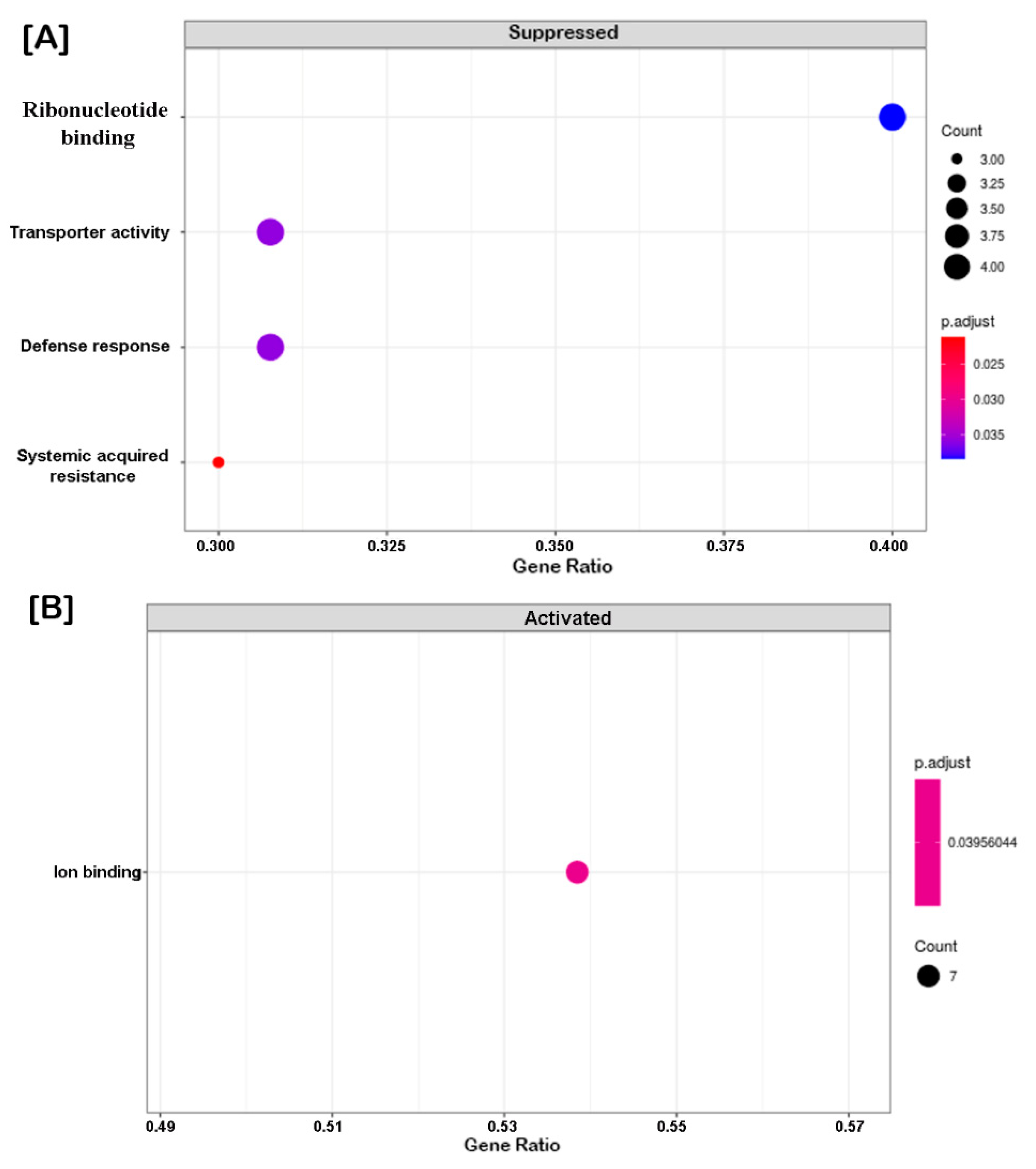
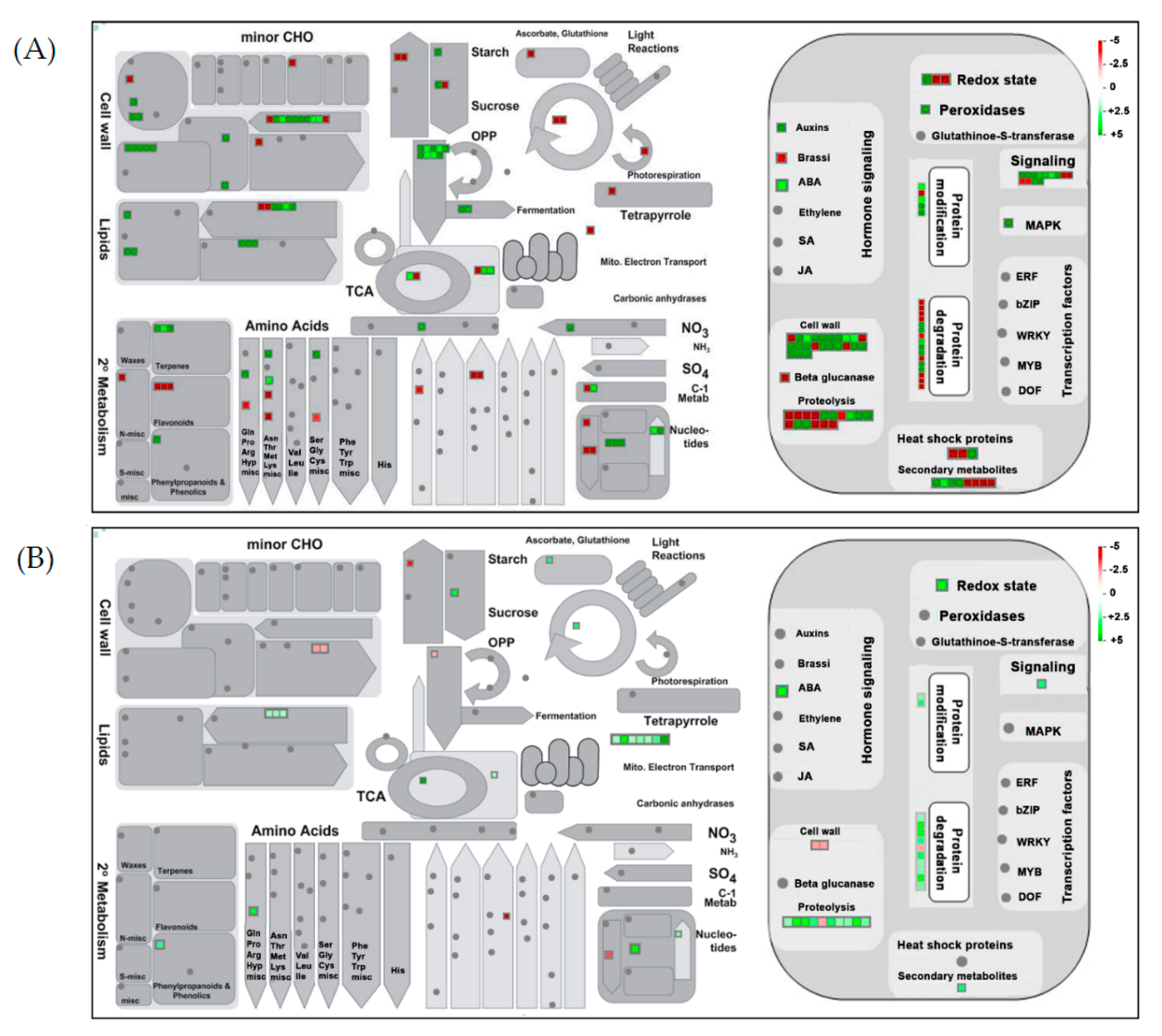
2.4. Clustering, Functional Annotation, and Enrichment Analysis of Correlated DEGS and DEPs
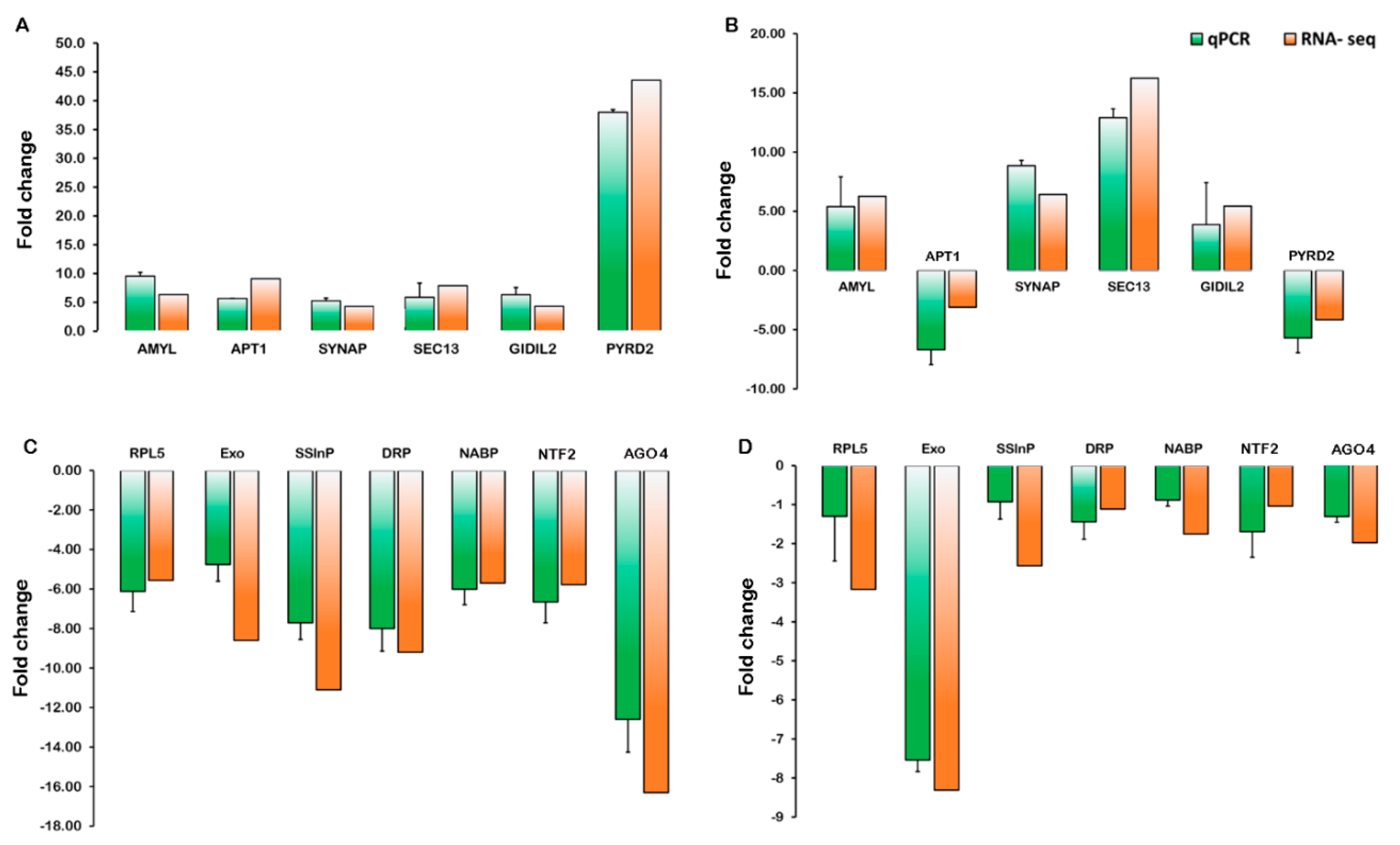
2.5. Validation of Gene Expression Levels
3. Discussion
3.1. Transcript-Protein Correlation Depicts Precise Expression Landscapes
3.2. Pollen Development Involves a Dynamic Plethora of Transcriptome and Proteome Alterations
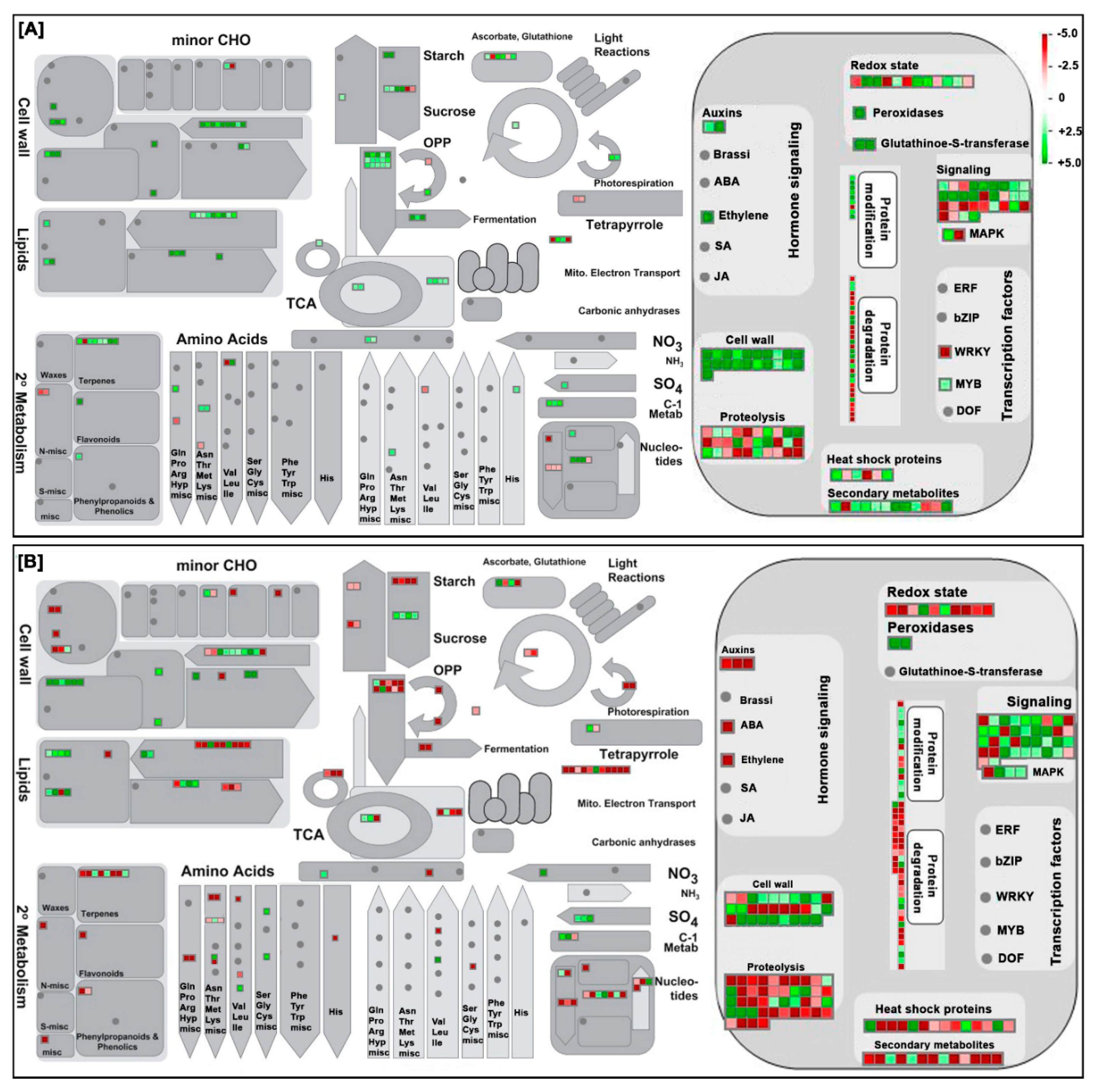
3.3. AFCVd Infected Pollen Exhibit Several Changes in Expression of Genes and Proteins in Stagewise Comparisons
3.4. RNA Stabilization/Destabilization Proteins Participate in Viroid Eradication in Developing Pollen
4. Materials and Methods
4.1. Plant Materials, Viroid Infection, and Detection in Pollen
4.2. RNA Extraction, Illumina Sequencing, and Data Processing
4.3. Protein Isolation and LC-MS/MS Analysis
4.4. Correlation Analyses of Transcriptome and Proteome Profiles and Functional Analysis
4.5. Validation of cor-DEGs/DEPs by Quantitative Real-Time PCR (RT-qPCR) Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Škorić, D. Viroid biology. In Viroids and Satellites; Elsevier: Amsterdam, The Netherlands, 2017; pp. 53–61. [Google Scholar]
- Flores, R.; Hernández, C.; De Alba, A.E.M.; Daròs, J.-A.; Serio, F.D. Viroids and viroid-host interactions. Annu. Rev. Phytopathol. 2005, 43, 117–139. [Google Scholar] [CrossRef]
- Qi, Y.; Ding, B. Differential subnuclear localization of RNA strands of opposite polarity derived from an autonomously replicating viroid. Plant Cell 2003, 15, 2566–2577. [Google Scholar] [CrossRef]
- Tsagris, E.M.; de Alba, Á.E.M.; Gozmanova, M.; Kalantidis, K. Viroids. Cell. Microbiol. 2008, 10, 2168–2179. [Google Scholar] [CrossRef]
- Rodio, M.-E.; Delgado, S.; De Stradis, A.; Gómez, M.-D.; Flores, R.; Di Serio, F. A viroid RNA with a specific structural motif inhibits chloroplast development. Plant Cell 2007, 19, 3610–3626. [Google Scholar] [CrossRef]
- Gas, M.-E.; Hernández, C.; Flores, R.; Daròs, J.-A. Processing of nuclear viroids in vivo: An interplay between RNA conformations. PLoS Pathog. 2007, 3, e182. [Google Scholar] [CrossRef] [PubMed]
- Eiras, M.; Nohales, M.A.; Kitajima, E.W.; Flores, R.; Daròs, J.A. Ribosomal protein L5 and transcription factor IIIA from Arabidopsis thaliana bind in vitro specifically Potato spindle tuber viroid RNA. Arch. Virol. 2011, 156, 529–533. [Google Scholar] [CrossRef]
- De Alba, A.E.M.; Sägesser, R.; Tabler, M.; Tsagris, M. A bromodomain-containing protein from tomato specifically binds potato spindle tuber viroid RNA in vitro and in vivo. J. Virol. 2003, 77, 9685–9694. [Google Scholar] [CrossRef] [PubMed]
- Gómez, G.; Pallás, V. Identification of an in vitro ribonucleoprotein complex between a viroid RNA and a phloem protein from cucumber plants. Mol. Plant Microbe Interact. 2001, 14, 910–913. [Google Scholar] [CrossRef] [PubMed]
- Voinnet, O. Use, tolerance and avoidance of amplified RNA silencing by plants. Trends Plant Sci. 2008, 13, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Minoia, S.; Navarro, B.; Covelli, L.; Barone, M.; García-Becedas, M.; Ragozzino, A.; Alioto, D.; Flores, R.; Di Serio, F. Viroid-like RNAs from cherry trees affected by leaf scorch disease: Further data supporting their association with mycoviral double-stranded RNAs. Arch. Virol. 2014, 159, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, Y.; Yanagisawa, H. Distribution of Tomato planta macho viroid in germinating pollen and transmitting tract. Virus Genes 2018, 54, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Flores, R.; Grubb, D.; Elleuch, A.; Nohales, M.-Á.; Delgado, S.; Gago, S. Rolling-circle replication of viroids, viroid-like satellite RNAs and hepatitis delta virus: Variations on a theme. RNA Biol. 2011, 8, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Hadidi, A.; Giunchedi, L.; Osaki, H.; Shamloul, A.; Crescenzi, A.; Gentit, P.; Nemchinov, L.; Piazzolla, P.; Kyriakopoulou, P. Peach latent mosaic viroid in temperate fruit hosts. Viroids 2003, 8, 161–164. [Google Scholar]
- Matoušek, J.; Orctova, L.; Škopek, J.; Pešina, K.; Steger, G. Elimination of hop latent viroid upon developmental activation of pollen nucleases. Biol. Chem. 2008, 389, 905–918. [Google Scholar] [CrossRef] [PubMed]
- Hadidi, A.; Hansen, A.; Parish, C.; Yang, X. Scar skin and dapple apple viroids are seed-borne and persistent in infected apple trees. Res. Virol. 1991, 142, 289–296. [Google Scholar] [CrossRef]
- Matoušek, J.; Steinbachová, L.; Drábková, L.Z.; Kocábek, T.; Potěšil, D.; Mishra, A.K.; Honys, D.; Steger, G. Elimination of Viroids from Tobacco Pollen Involves a Decrease in Propagation Rate and an Increase of the Degradation Processes. Int. J. Mol. Sci. 2020, 21, 3029. [Google Scholar] [CrossRef] [PubMed]
- McCormick, S. Male gametophyte development. Plant Cell 1993, 5, 1265. [Google Scholar] [CrossRef]
- Rotsch, A.H.; Kopka, J.; Feussner, I.; Ischebeck, T. Central metabolite and sterol profiling divides tobacco male gametophyte development and pollen tube growth into eight metabolic phases. Plant J. 2017, 92, 129–146. [Google Scholar] [CrossRef]
- Gómez, J.F.; Talle, B.; Wilson, Z.A. Anther and pollen development: A conserved developmental pathway. J. Integr. Plant Biol. 2015, 57, 876–891. [Google Scholar] [CrossRef]
- Wilson, Z.A.; Zhang, D.-B. From Arabidopsis to rice: Pathways in pollen development. J. Exp. Bot. 2009, 60, 1479–1492. [Google Scholar] [CrossRef]
- Bokvaj, P.; Hafidh, S.; Honys, D. Transcriptome profiling of male gametophyte development in Nicotiana tabacum. Genom. Data 2015, 3, 106–111. [Google Scholar] [CrossRef]
- Conze, L.L.; Berlin, S.; Le Bail, A.; Kost, B. Transcriptome profiling of tobacco (Nicotiana tabacum) pollen and pollen tubes. BMC Genom. 2017, 18, 1–11. [Google Scholar] [CrossRef]
- Grant-Downton, R.; Le Trionnaire, G.; Schmid, R.; Rodriguez-Enriquez, J.; Hafidh, S.; Mehdi, S.; Twell, D.; Dickinson, H. MicroRNA and tasiRNA diversity in mature pollen of Arabidopsis thaliana. BMC Genom. 2009, 10, 643. [Google Scholar] [CrossRef] [PubMed]
- Mergner, J.; Frejno, M.; List, M.; Papacek, M.; Chen, X.; Chaudhary, A.; Samaras, P.; Richter, S.; Shikata, H.; Messerer, M. Mass-spectrometry-based draft of the Arabidopsis proteome. Nature 2020, 579, 409–414. [Google Scholar] [CrossRef]
- Fíla, J.; Drábková, L.Z.; Gibalová, A.; Honys, D. When simple meets complex: Pollen and the-omics. In Pollen Tip Growth; Springer: Berlin/Heidelberg, Germany, 2017; pp. 247–292. [Google Scholar]
- Galland, M.; Huguet, R.; Arc, E.; Cueff, G.; Job, D.; Rajjou, L. Dynamic proteomics emphasizes the importance of selective mRNA translation and protein turnover during Arabidopsis seed germination. Mol. Cell. Proteom. 2014, 13, 252–268. [Google Scholar] [CrossRef] [PubMed]
- Osorio, S.; Alba, R.; Nikoloski, Z.; Kochevenko, A.; Fernie, A.R.; Giovannoni, J.J. Integrative comparative analyses of transcript and metabolite profiles from pepper and tomato ripening and development stages uncovers species-specific patterns of network regulatory behavior. Plant Physiol. 2012, 159, 1713–1729. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.; Simm, S.; Consortium, S.-I. The coupling of transcriptome and proteome adaptation during development and heat stress response of tomato pollen. BMC Genom. 2018, 19, 447. [Google Scholar] [CrossRef]
- Peng, X.; Qin, Z.; Zhang, G.; Guo, Y.; Huang, J. Integration of the proteome and transcriptome reveals multiple levels of gene regulation in the rice dl2 mutant. Front. Plant Sci. 2015, 6, 351. [Google Scholar] [CrossRef]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef]
- Honys, D.; Twell, D. Transcriptome analysis of haploid male gametophyte development in Arabidopsis. Genome Biol. 2004, 5, R85. [Google Scholar] [CrossRef]
- Hafidh, S.; Potěšil, D.; Müller, K.; Fíla, J.; Michailidis, C.; Herrmannová, A.; Feciková, J.; Ischebeck, T.; Valášek, L.S.; Zdráhal, Z. Dynamics of the pollen sequestrome defined by subcellular coupled omics. Plant Physiol. 2018, 178, 258–282. [Google Scholar] [CrossRef] [PubMed]
- Hirose, T.; Zhang, Z.; Miyao, A.; Hirochika, H.; Ohsugi, R.; Terao, T. Disruption of a gene for rice sucrose transporter, OsSUT1, impairs pollen function but pollen maturation is unaffected. J. Exp. Bot. 2010, 61, 3639–3646. [Google Scholar] [CrossRef] [PubMed]
- García, C.C.; Nepi, M.; Pacini, E. It is a matter of timing: Asynchrony during pollen development and its consequences on pollen performance in angiosperms—A review. Protoplasma 2017, 254, 57–73. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Hu, C.; Zhu, Y.; Cheng, K.; Li, X.; Wei, Z.; Xue, L.; Lin, F.; Shi, H.; Yi, J. CIK receptor kinases determine cell fate specification during early anther development in Arabidopsis. Plant Cell 2018, 30, 2383–2401. [Google Scholar] [CrossRef] [PubMed]
- Hord, C.L.; Sun, Y.-J.; Pillitteri, L.J.; Torii, K.U.; Wang, H.; Zhang, S.; Ma, H. Regulation of Arabidopsis early anther development by the mitogen-activated protein kinases, MPK3 and MPK6, and the ERECTA and related receptor-like kinases. Mol. Plant 2008, 1, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Yang, X.; Hu, G.; Liu, Q.; Li, W.; Zhang, L.; Song, X. Genome-Wide Investigation of Heat Shock Transcription Factor Family in Wheat (Triticum aestivum L.) and Possible Roles in Anther Development. Int. J. Mol. Sci. 2020, 21, 608. [Google Scholar] [CrossRef]
- Attallah, C.V.; Welchen, E.; Gonzalez, D.H. The promoters of Arabidopsis thaliana genes AtCOX17-1 and-2, encoding a copper chaperone involved in cytochrome c oxidase biogenesis, are preferentially active in roots and anthers and induced by biotic and abiotic stress. Physiol. Plant. 2007, 129, 123–134. [Google Scholar] [CrossRef]
- Sancenón, V.; Puig, S.; Mateu-Andrés, I.; Dorcey, E.; Thiele, D.J.; Peñarrubia, L. The Arabidopsis copper transporter COPT1 functions in root elongation and pollen development. J. Biol. Chem. 2004, 279, 15348–15355. [Google Scholar] [CrossRef]
- Sánchez-Bel, P.; Egea, I.; Sánchez-Ballesta, M.T.; Martinez-Madrid, C.; Fernandez-Garcia, N.; Romojaro, F.; Olmos, E.; Estrella, E.; Bolarín, M.C.; Flores, F.B. Understanding the mechanisms of chilling injury in bell pepper fruits using the proteomic approach. J. Proteom. 2012, 75, 5463–5478. [Google Scholar] [CrossRef]
- Tena, G.; Boudsocq, M.; Sheen, J. Protein kinase signaling networks in plant innate immunity. Curr. Opin. Plant Biol. 2011, 14, 519–529. [Google Scholar] [CrossRef]
- Denancé, N.; Sánchez-Vallet, A.; Goffner, D.; Molina, A. Disease resistance or growth: The role of plant hormones in balancing immune responses and fitness costs. Front. Plant Sci. 2013, 4, 155. [Google Scholar] [CrossRef]
- Pérez-Llorca, M.; Muñoz, P.; Müller, M.; Munné-Bosch, S. Biosynthesis, metabolism and function of auxin, salicylic acid and melatonin in climacteric and non-climacteric fruits. Front. Plant Sci. 2019, 10, 136. [Google Scholar] [CrossRef] [PubMed]
- He, S.-L.; Hsieh, H.-L.; Jauh, G.-Y. SMALL AUXIN UP RNA62/75 are required for the translation of transcripts essential for pollen tube growth. Plant Physiol. 2018, 178, 626–640. [Google Scholar] [CrossRef] [PubMed]
- Smakowska, E.; Kong, J.; Busch, W.; Belkhadir, Y. Organ-specific regulation of growth-defense tradeoffs by plants. Curr. Opin. Plant Biol. 2016, 29, 129–137. [Google Scholar] [CrossRef]
- Di Serio, F.; Flores, R.; Verhoeven, J.T.J.; Li, S.-F.; Pallás, V.; Randles, J.; Sano, T.; Vidalakis, G.; Owens, R. Current status of viroid taxonomy. Arch. Virol. 2014, 159, 3467–3478. [Google Scholar] [CrossRef] [PubMed]
- Frank, G.; Pressman, E.; Ophir, R.; Althan, L.; Shaked, R.; Freedman, M.; Shen, S.; Firon, N. Transcriptional profiling of maturing tomato (Solanum lycopersicum L.) microspores reveals the involvement of heat shock proteins, ROS scavengers, hormones, and sugars in the heat stress response. J. Exp. Bot. 2009, 60, 3891–3908. [Google Scholar] [CrossRef] [PubMed]
- Cottilli, P.; Belda-Palazón, B.; Adkar-Purushothama, C.R.; Perreault, J.-P.; Schleiff, E.; Rodrigo, I.; Ferrando, A.; Lisón, P. Citrus exocortis viroid causes ribosomal stress in tomato plants. Nucleic Acids Res. 2019, 47, 8649–8661. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-Y.; Brown, R.; Damann, K.; Cleveland, T. Identification of unique or elevated levels of kernel proteins in aflatoxin-resistant maize genotypes through proteome analysis. Phytopathology 2002, 92, 1084–1094. [Google Scholar] [CrossRef] [PubMed]
- Hundertmark, M.; Hincha, D.K. LEA (late embryogenesis abundant) proteins and their encoding genes in Arabidopsis thaliana. BMC Genom. 2008, 9, 118. [Google Scholar] [CrossRef]
- Sun, S.; Yu, J.-P.; Chen, F.; Zhao, T.-J.; Fang, X.-H.; Li, Y.-Q.; Sui, S.-F. TINY, a dehydration-responsive element (DRE)-binding protein-like transcription factor connecting the DRE-and ethylene-responsive element-mediated signaling pathways in Arabidopsis. J. Biol. Chem. 2008, 283, 6261–6271. [Google Scholar] [CrossRef]
- Fan, C.; Wang, G.; Wu, L.; Liu, P.; Huang, J.; Jin, X.; Zhang, G.; He, Y.; Peng, L.; Luo, K. Distinct cellulose and callose accumulation for enhanced bioethanol production and biotic stress resistance in OsSUS3 transgenic rice. Carbohydr. Polym. 2020, 232, 115448. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, A.S.; Moffat, C.S.; Lopez-Ruiz, F.J.; Gibberd, M.R.; Hamblin, J.; Zerihun, A. Host–multi-pathogen warfare: Pathogen interactions in co-infected plants. Front. Plant Sci. 2017, 8, 1806. [Google Scholar] [CrossRef]
- Jiang, J.; Smith, H.N.; Ren, D.; Mudiyanselage, S.D.D.; Dawe, A.L.; Wang, L.; Wang, Y. Potato spindle tuber viroid modulates its replication through a direct interaction with a splicing regulator. J. Virol. 2018, 92, e01004-18. [Google Scholar] [CrossRef] [PubMed]
- Dissanayaka Mudiyanselage, S.D.; Qu, J.; Tian, N.; Jiang, J.; Wang, Y. Potato spindle tuber viroid RNA-templated transcription: Factors and regulation. Viruses 2018, 10, 503. [Google Scholar] [CrossRef]
- Minoia, S.; Carbonell, A.; Di Serio, F.; Gisel, A.; Carrington, J.C.; Navarro, B.; Flores, R. Specific argonautes selectively bind small RNAs derived from potato spindle tuber viroid and attenuate viroid accumulation in vivo. J. Virol. 2014, 88, 11933–11945. [Google Scholar] [CrossRef]
- Dubé, A.; Bisaillon, M.; Perreault, J.-P. Identification of proteins from Prunus persica that interact with peach latent mosaic viroid. J. Virol. 2009, 83, 12057–12067. [Google Scholar] [CrossRef]
- Liu, L.; Chen, X. RNA quality control as a key to suppressing RNA silencing of endogenous genes in plants. Mol. Plant 2016, 9, 826–836. [Google Scholar] [CrossRef] [PubMed]
- Matoušek, J.; Siglová, K.; Jakše, J.; Radišek, S.; Brass, J.R.; Tsushima, T.; Guček, T.; Duraisamy, G.; Sano, T.; Steger, G. Propagation and some physiological effects of Citrus bark cracking viroid and Apple fruit crinkle viroid in multiple infected hop (Humulus lupulus L.). J. Plant Physiol. 2017, 213, 166–177. [Google Scholar]
- Tupý, J.; Süss, J.; Hrabětová, E.; Říhova, L. Developmental changes in gene expression during pollen differentiation and maturation in Nicotiana tabacum L. Biol. Plant. 1983, 25, 231. [Google Scholar] [CrossRef]
- Tupý, J.; Hrabětová, E.; Balatková, V. A simple rapid method of determining pollen tube growth in mass culture. Plant Sci. Lett. 1977, 9, 285–290. [Google Scholar] [CrossRef]
- Matoušek, J.; Schröder, A.R.; Trněná, L.; Reimers, M.; Baumstark, T.; Dědič, P.; Vlasák, J.; Becker, I.; Kreuzaler, F.; Fladung, M. Inhibition of viroid infection by antisense RNA expression in transgenic plants. Biol. Chem. Hoppe Seyler 1994, 375, 765–778. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. In Babraham Bioinformatics; Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Wisniewski, J.R.; Ostasiewicz, P.; Mann, M. High recovery FASP applied to the proteomic analysis of microdissected formalin fixed paraffin embedded cancer tissues retrieves known colon cancer markers. J. Proteome Res. 2011, 10, 3040–3049. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Yeung, Y.G.; Stanley, E.R. Rapid detergent removal from peptide samples with ethyl acetate for mass spectrometry analysis. Curr. Protoc. Protein Sci. 2010, 59, 16.12.1–16.12.5. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef]
- Thimm, O.; Bläsing, O.; Gibon, Y.; Nagel, A.; Meyer, S.; Krüger, P.; Selbig, J.; Müller, L.A.; Rhee, S.Y.; Stitt, M. MAPMAN: A user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004, 37, 914–939. [Google Scholar] [CrossRef] [PubMed]
- Reich, M.; Ohm, K.; Angelo, M.; Tamayo, P.; Mesirov, J.P. GeneCluster 2.0: An advanced toolset for bioarray analysis. Bioinformatics 2004, 20, 1797–1798. [Google Scholar] [CrossRef] [PubMed]
- Marshall, O.J. PerlPrimer: Cross-platform, graphical primer design for standard, bisulphite and real-time PCR. Bioinformatics 2004, 20, 2471–2472. [Google Scholar] [CrossRef] [PubMed]
- Nath, V.S.; Shrestha, A.; Awasthi, P.; Mishra, A.K.; Kocábek, T.; Matoušek, J.; Sečnik, A.; Jakše, J.; Radišek, S.; Hallan, V. Mapping the Gene Expression Spectrum of Mediator Subunits in Response to Viroid Infection in Plants. Int. J. Mol. Sci. 2020, 21, 2498. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Number of Raw Reads (Million) | Number of Clean Reads (Million) | Total Bases of Raw Reads (GB) | Total Bases of Clean Reads (GB) | GC Content of Raw Reads (%) | GC Content of Clean Reads (%) | Mean Length of Raw Reads (bp) | Mean Length of Raw Reads (bp) | Number of Mapped Reads (Million) |
|---|---|---|---|---|---|---|---|---|---|
| CT_S3 | 100.90 | 96.65 | 15.23 | 10.87 | 43.05 | 41.75 | 151 | 112 | 86.69 (89.96%) |
| CT_S5 | 102.13 | 97.00 | 15.41 | 11.08 | 42.67 | 41.05 | 151 | 113 | 88.36 (91.09%) |
| CT_PT6 | 193.22 | 154.70 | 14.41 | 9.98 | 45.97 | 41.88 | 74 | 64 | 151.20 (97.30%) |
| AI_S3 | 65.70 | 60.31 | 9.91 | 6.63 | 40.70 | 38.78 | 151 | 110 | 50.58 (83.86%) |
| AI_S5 | 119.96 | 115.24 | 18.11 | 13.56 | 44.14 | 42.62 | 151 | 118 | 107.37 (93.17%) |
| AI_PT6 | 229.70 | 203.75 | 17.13 | 14.44 | 44.25 | 42.29 | 74 | 71 | 185.27 (90.93%) |
| Single Stage Comparison | Transcripts (T) | Proteins (P) | Corresponding Number of TP Pairs | |||
|---|---|---|---|---|---|---|
| CT_S3 | 64,033 | 5321 | 2660 (4.0%) | |||
| CT_S5 | 60,597 | 5286 | 2639 (4.2%) | |||
| CT_PT6 | 46,640 | 6923 | 3428 (6.8%) | |||
| AI_S3 | 63,335 | 5371 | 2672 (4.4%) | |||
| AI_S5 | 60,278 | 5267 | 2618 (4.2%) | |||
| AI_PT6 | 46,461 | 6923 | 3438 (6.9%) | |||
| Pairwise stage comparisons | DE_Transcripts | DE_Proteins | DE_Correlated | |||
| UR | DR | UR | DR | UR | DR | |
| CT_S5 vs. CT_S3 | 5003 | 4304 | 869 | 1365 | 412 | 395 |
| CT_PT6 vs. CT_S5 | 6309 | 7756 | 1094 | 1014 | 301 | 545 |
| AI_S5 vs. AI_S3 | 3014 | 1752 | 829 | 1395 | 237 | 248 |
| AI_PT6 vs. AI_S5 | 2533 | 3307 | 709 | 600 | 68 | 24 |
| KEGG Categories | CT S5 vs. S3 | CT PT6 vs. S5 | AI S5 vs. S3 | AI PT6 vs. S5 | ||||
|---|---|---|---|---|---|---|---|---|
| Metabolism | UR | DR | UR | DR | UR | DR | UR | DR |
| Carbohydrate Metabolism | 92 | 8 | 41 | 71 | 47 | 18 | 12 | 6 |
| Energy metabolism | 19 | 3 | 12 | 26 | 9 | 5 | 7 | 3 |
| Lipid metabolism | 29 | 1 | 16 | 17 | 14 | 6 | 2 | 0 |
| Nucleotide metabolism | 8 | 4 | 8 | 8 | 9 | 3 | 3 | 1 |
| Amino acid metabolism | 42 | 10 | 21 | 35 | 25 | 16 | 2 | 2 |
| Metabolism of other amino acids | 8 | 4 | 6 | 6 | 4 | 2 | 2 | 1 |
| Glycan biosynthesis and metabolism | 0 | 0 | 2 | 7 | 3 | 2 | 0 | 0 |
| Metabolism of cofactors and vitamins | 8 | 6 | 6 | 11 | 3 | 4 | 4 | 0 |
| Metabolism of terpenoids and polyketides | 8 | 2 | 2 | 3 | 3 | 1 | 0 | 0 |
| Biosynthesis of other secondary metabolites | 13 | 6 | 7 | 9 | 5 | 6 | 3 | 1 |
| Xenobiotics biodegradation and metabolism | 6 | 1 | 2 | 2 | 3 | 1 | 2 | 0 |
| Enzyme families | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Genetic information processing | ||||||||
| Transcription | 0 | 21 | 1 | 13 | 0 | 10 | 2 | 1 |
| Translation | 5 | 45 | 8 | 67 | 2 | 33 | 8 | 3 |
| Folding, sorting and degradation | 14 | 14 | 12 | 37 | 5 | 8 | 4 | 0 |
| Replication and repair | 0 | 9 | 4 | 9 | 0 | 6 | 5 | 0 |
| RNA family | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Cellular Process | ||||||||
| Transport and catabolism | 25 | 7 | 27 | 11 | 7 | 3 | 2 | 0 |
| Cell growth and death | 9 | 11 | 9 | 23 | 2 | 8 | 5 | 0 |
| Cellular community - eukaryotes | 3 | 1 | 8 | 5 | 2 | 1 | 0 | 0 |
| Cellular community - prokaryotes | 2 | 0 | 2 | 0 | 1 | 1 | 0 | 1 |
| Cell motility | 3 | 1 | 3 | 1 | 3 | 1 | 0 | 0 |
| Environmental information processing | ||||||||
| Membrane transport | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Signal transduction | 44 | 13 | 56 | 56 | 17 | 11 | 5 | 0 |
| Signaling molecules and interaction | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Others | 140 | 96 | 160 | 341 | 52 | 74 | 80 | 4 |
| Annotated | 282 | 255 | 203 | 380 | 131 | 171 | 50 | 16 |
| Query dataset | 412 | 395 | 301 | 542 | 237 | 248 | 67 | 24 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shrestha, A.; Mishra, A.K.; Matoušek, J.; Steinbachová, L.; Potěšil, D.; Nath, V.S.; Awasthi, P.; Kocábek, T.; Jakse, J.; Drábková, L.Z.; et al. Integrated Proteo-Transcriptomic Analyses Reveal Insights into Regulation of Pollen Development Stages and Dynamics of Cellular Response to Apple Fruit Crinkle Viroid (AFCVd)-Infection in Nicotiana tabacum. Int. J. Mol. Sci. 2020, 21, 8700. https://doi.org/10.3390/ijms21228700
Shrestha A, Mishra AK, Matoušek J, Steinbachová L, Potěšil D, Nath VS, Awasthi P, Kocábek T, Jakse J, Drábková LZ, et al. Integrated Proteo-Transcriptomic Analyses Reveal Insights into Regulation of Pollen Development Stages and Dynamics of Cellular Response to Apple Fruit Crinkle Viroid (AFCVd)-Infection in Nicotiana tabacum. International Journal of Molecular Sciences. 2020; 21(22):8700. https://doi.org/10.3390/ijms21228700
Chicago/Turabian StyleShrestha, Ankita, Ajay Kumar Mishra, Jaroslav Matoušek, Lenka Steinbachová, David Potěšil, Vishnu Sukumari Nath, Praveen Awasthi, Tomáš Kocábek, Jernej Jakse, Lenka Záveská Drábková, and et al. 2020. "Integrated Proteo-Transcriptomic Analyses Reveal Insights into Regulation of Pollen Development Stages and Dynamics of Cellular Response to Apple Fruit Crinkle Viroid (AFCVd)-Infection in Nicotiana tabacum" International Journal of Molecular Sciences 21, no. 22: 8700. https://doi.org/10.3390/ijms21228700
APA StyleShrestha, A., Mishra, A. K., Matoušek, J., Steinbachová, L., Potěšil, D., Nath, V. S., Awasthi, P., Kocábek, T., Jakse, J., Drábková, L. Z., Zdráhal, Z., Honys, D., & Steger, G. (2020). Integrated Proteo-Transcriptomic Analyses Reveal Insights into Regulation of Pollen Development Stages and Dynamics of Cellular Response to Apple Fruit Crinkle Viroid (AFCVd)-Infection in Nicotiana tabacum. International Journal of Molecular Sciences, 21(22), 8700. https://doi.org/10.3390/ijms21228700