Expanding Phenotype of Schimke Immuno-Osseous Dysplasia: Congenital Anomalies of the Kidneys and of the Urinary Tract and Alteration of NK Cells


 , , , , ,
, , , , ,  , , and
, , and 
Abstract
1. Introduction
2. Results
2.1. Clinical Report
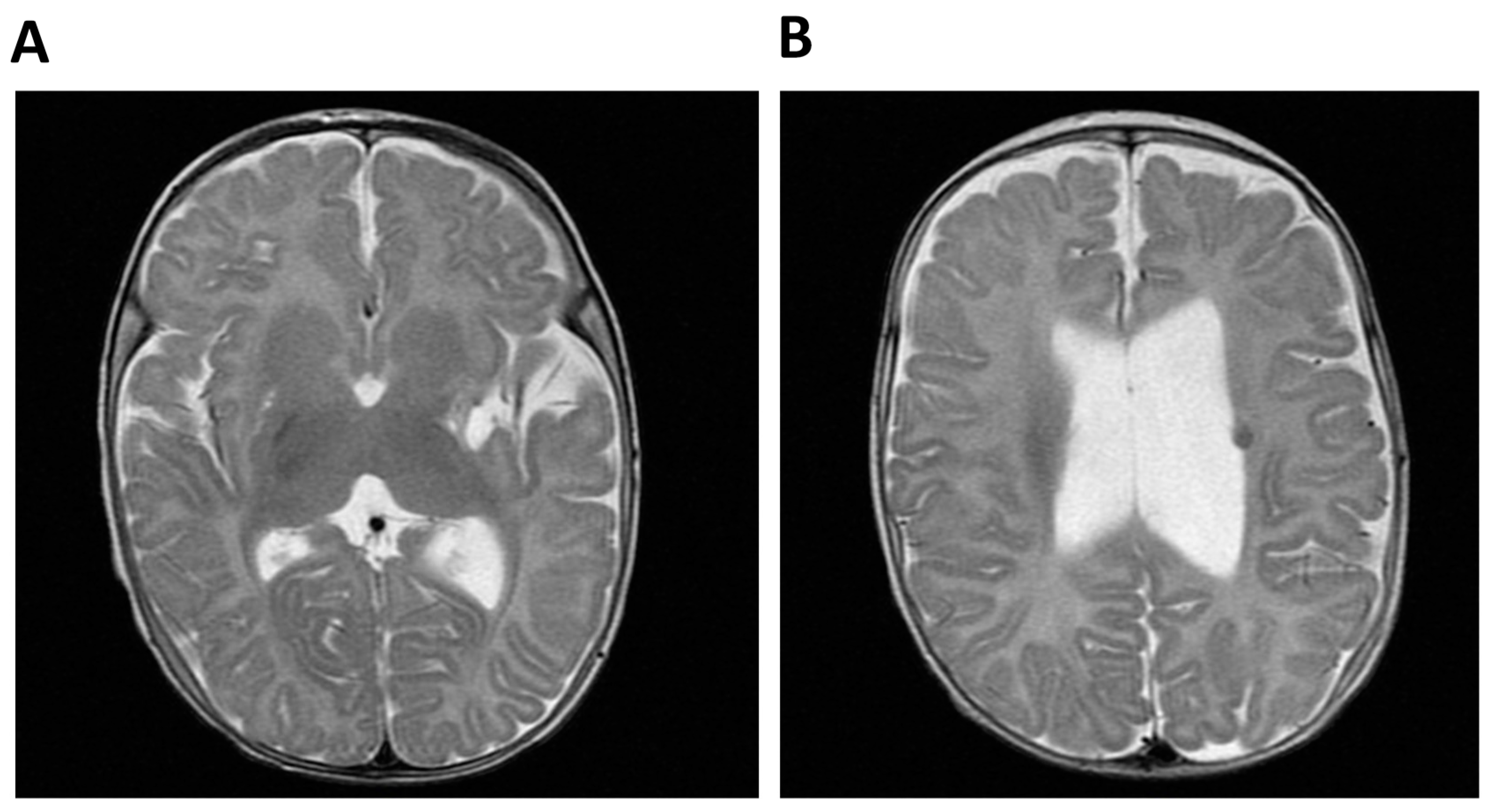
2.1.1. Patient 1 (P1)
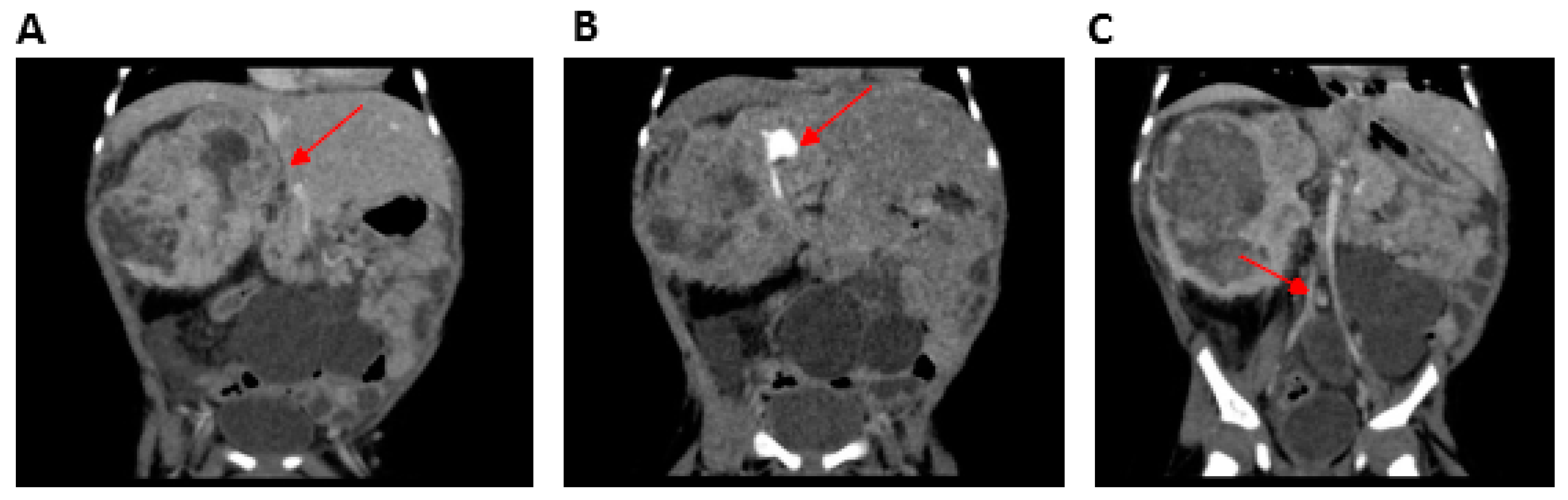
2.1.2. Patient 2 (P2)
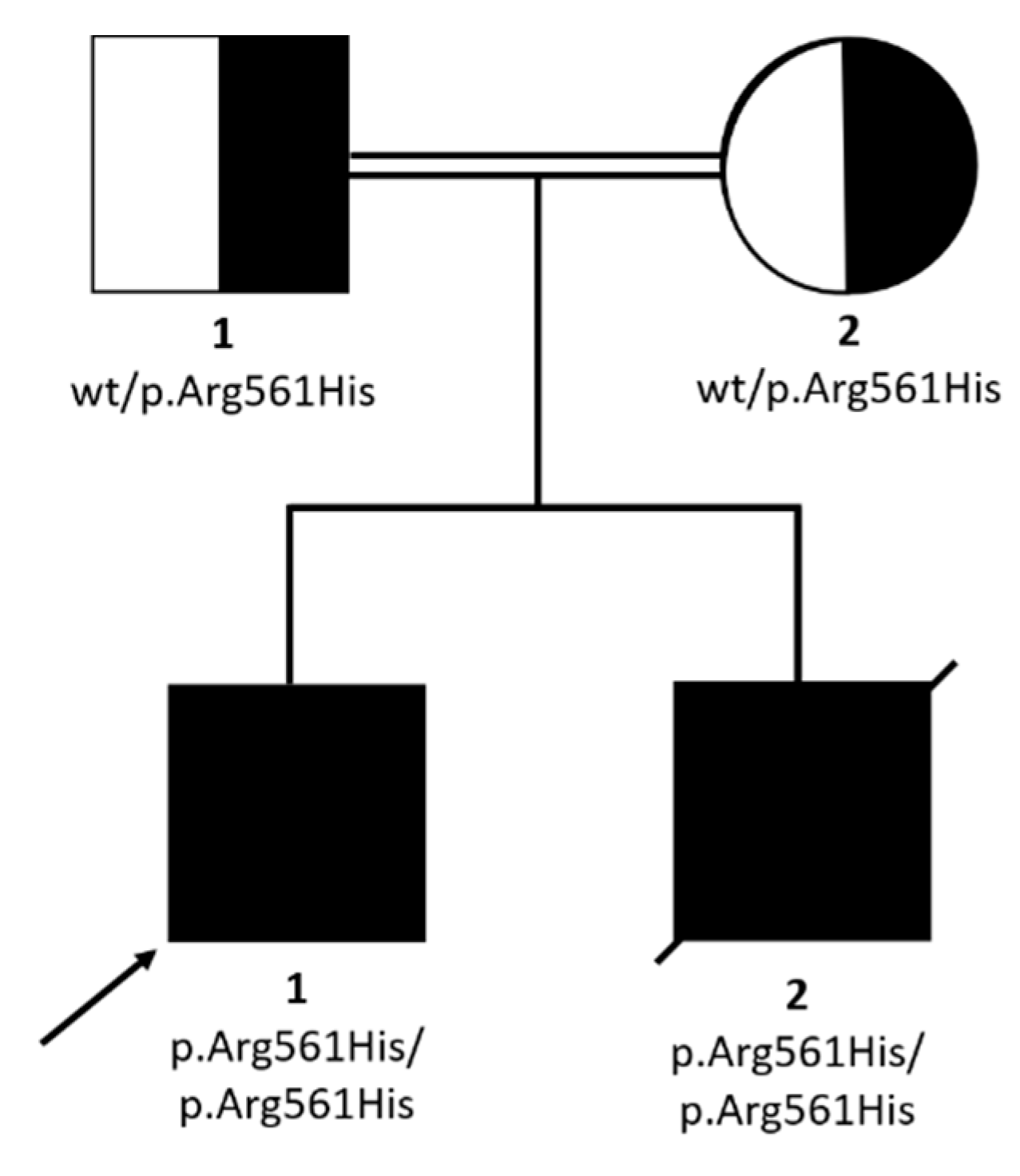
2.2. WES Findings
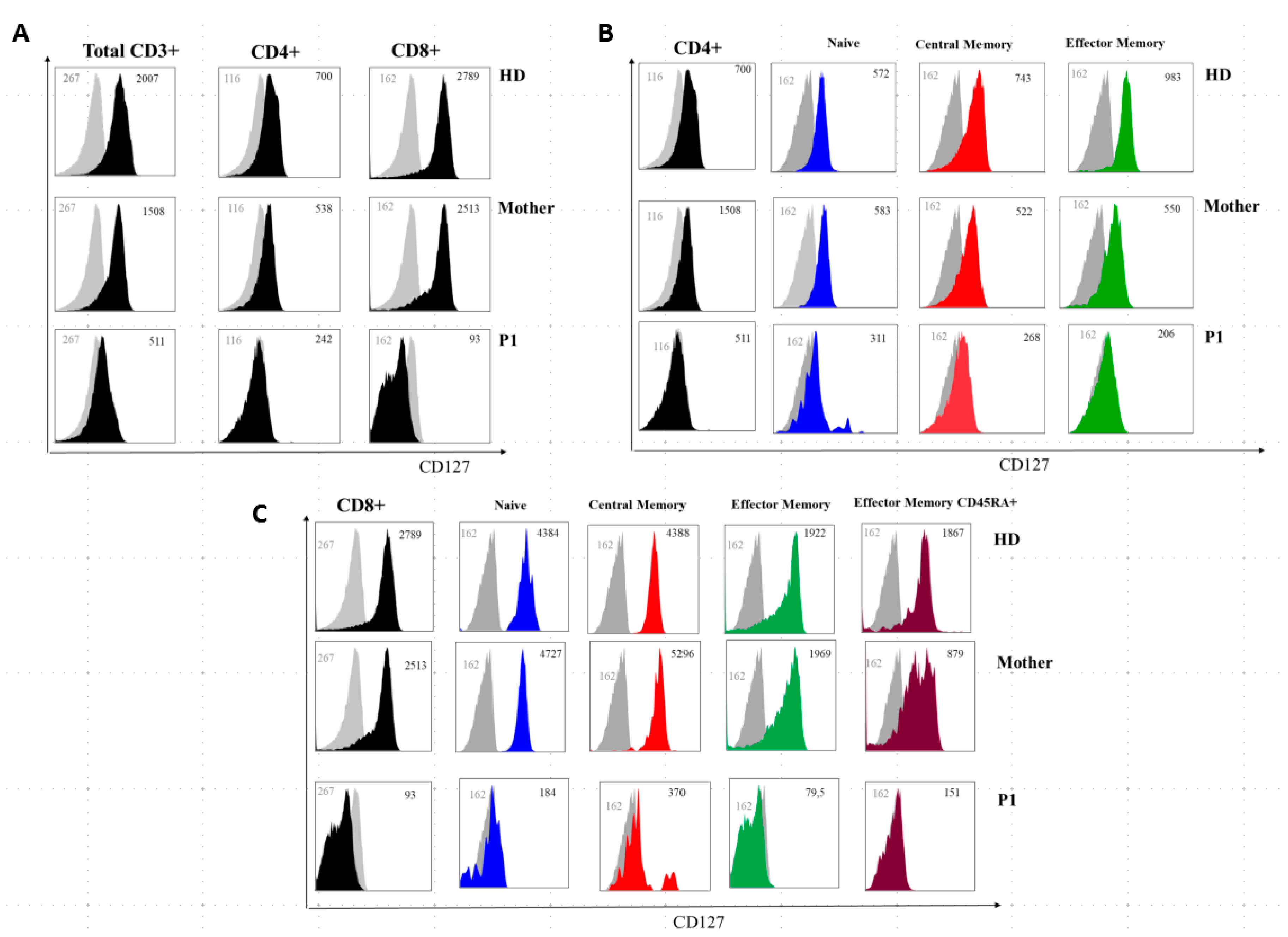
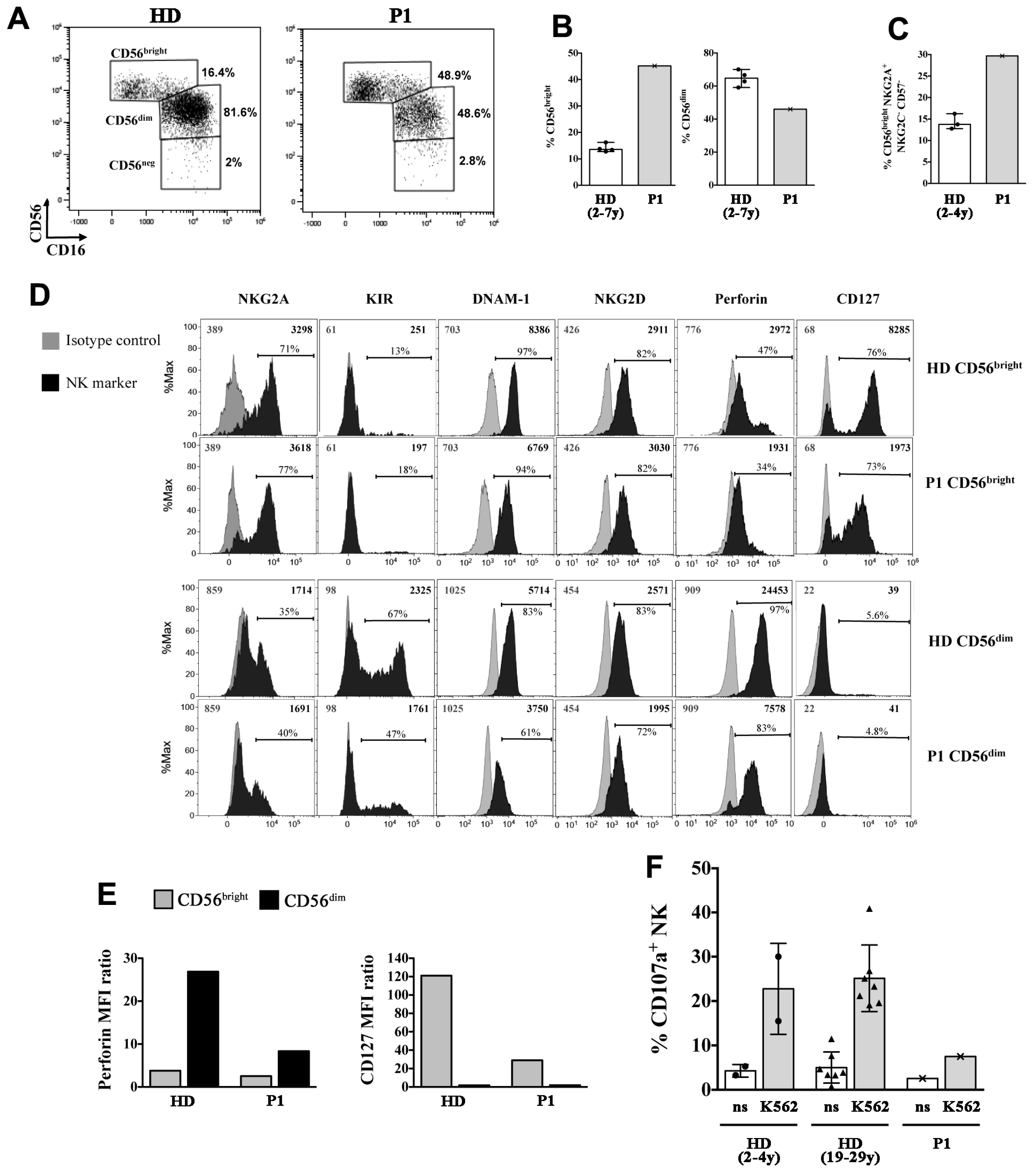
2.3. P1 and P2 Showed a Combined Immunodeficiency Phenotype
3. Discussion
4. Materials and Methods
4.1. Subjects and Families/Collection of Samples and Informed Consent
4.2. Whole Exome Sequencing/Sanger Sequencing
4.3. Flow Cytometry Analysis
4.3.1. Immunophenotype and IL7Ra Membrane Expression
4.3.2. NK-cell Degranulation Assay
4.4. Cell Preparation and B Cell Proliferation Assay
4.4.1. Flow Cytometric Analysis
4.4.2. ELISA Immunoassay for IgM, IgA, and IgG
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schimke, R.N.; Horton, W.A.; King, C.R.; Martin, N.L. Chondroitin-6-sulfate mucopoly-saccharidosis in conjunction with lymphopenia, defective cellular immunity and the nephrotic syndrome. Birth Defects Orig. Artic. Ser. 1974, 10, 258–266. [Google Scholar]
- Boerkoel, C.F.; O’Neill, S.; André, J.L.; Benke, P.J.; Bogdanovíć, R.; Bulla, M.; Burguet, A.; Cockfield, S.; Cordeiro, I.; Ehrich, J.H.; et al. Manifestations and treatment of Schimke immuno-osseous dysplasia: 14 new cases and a review of the literature. Eur. J. Pediatr. 2000, 159, 1–7. [Google Scholar] [CrossRef]
- Spranger, J.; Hinkel, G.K.; Stöss, H.; Thoenes, W.; Wargowski, D.; Zepp, F. Schimke immuno-osseous dysplasia: A newly recognized multisystem disease. J. Pediatr. 1991, 119 Pt 1, 64–72. [Google Scholar] [CrossRef]
- Morimoto, M.; Lewis, D.B.; Lücke, T.; Boerkoel, C.F.; Adam, M.P.; Ardinger, H.H.; Pagon, R.A.; Wallace, S.E.; Bean, L.J.H.; Stephens, K. Schimke Immunoosseous Dysplasia. In GeneReviews®; University of Washington: Seattle, WA, USA, 1993–2020. [Google Scholar]
- Carroll, C.; Badu-Nkansah, A.; Hunley, T.; Baradaran-Heravi, A.; Cortez, D.; Frangoul, H. Schimke Immunoosseous Dysplasia associated with undifferentiated carcinoma and a novel SMARCAL1 variant in a child. Pediatr. Blood Cancer 2013, 60, E88–E90. [Google Scholar] [CrossRef] [PubMed]
- Lev, A.; Amariglio, N.; Levy, Y.; Spirer, Z.; Anikster, Y.; Rechavi, G.; Dekel, B.; Somech, R. Molecular assessment of thymic capacities in patients with Schimke immuno-osseous dysplasia. Clin. Immunol. 2009, 133, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Lücke, T.; Clewing, J.M.; Boerkoel, C.F.; Hartmann, H.; Das, A.M.; Knauth, M.; Becker, H.; Donnerstag, F. Cerebellar atrophy in Schimke-immuno-osseous dysplasia. Am. J. Med. Genet. A 2007, 143, 2040–2045. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, M.; Kérourédan, O.; Gendronneau, M.; Shuen, C.; Baradaran-Heravi, A.; Asakura, Y.; Basiratnia, M.; Bogdanovic, R.; Bonneau, D.; Buck, A.; et al. Dental abnormalities in Schimke immuno-osseous dysplasia. J. Dent. Res. 2012, 91 (Suppl. 7), S29–S37. [Google Scholar] [CrossRef]
- Zieg, J.; Krepelova, A.; Baradaran-Heravi, A.; Levtchenko, E.; Guillén-Navarro, E.; Balascakova, M.; Sukova, M.; Seeman, T.; Dusek, J.; Simankova, N.; et al. Rituximab resistant evans syndrome and autoimmunity in Schimke immuno-osseous dysplasia. Pediatr. Rheumatol. Online J. 2011, 9, 27. [Google Scholar] [CrossRef]
- Bansbach, C.E.; Bétous, R.; Lovejoy, C.A.; Glick, G.G.; Cortez, D. The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Genes Dev. 2009, 23, 2405–2414. [Google Scholar] [CrossRef]
- Coleman, M.A.; Eisen, J.A.; Mohrenweiser, H.W. Cloning and characterization of HARP/SMARCAL1: A prokaryotic HepA-related SNF2 helicase protein from human and mouse. Genomics 2000, 65, 274–282. [Google Scholar] [CrossRef]
- Elizondo, L.I.; Huang, C.; Northrop, J.L.; Deguchi, K.; Clewing, J.M.; Armstrong, D.L.; Boerkoel, C.F. Schimke immuno-osseous dysplasia: A cell autonomous disorder? Am. J. Med. Genet. A 2006, 140, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Elizondo, L.I.; Cho, K.S.; Zhang, W.; Yan, J.; Huang, C.; Huang, Y.; Choi, K.; Sloan E, A.; Deguchi, K.; Lou, S.; et al. Schimke immuno-osseous dysplasia: SMARCAL1 loss-of-function and phenotypic correlation. J. Med. Genet. 2009, 46, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Baradaran-Heravi, A.; Raams, A.; Lubieniecka, J.; Cho, K.S.; DeHaai, K.A.; Basiratnia, M.; Mari, P.O.; Xue, Y.; Rauth, M.; Olney, A.H.; et al. SMARCAL1 deficiency predisposes to non-Hodgkin lymphoma and hypersensitivity to genotoxic agents in vivo. Am. J. Med. Genet. A 2012, 158, 2204–2213. [Google Scholar] [CrossRef] [PubMed]
- Boerkoel, C.F.; Takashima, H.; John, J.; Yan, J.; Stankiewicz, P.; Rosenbarker, L.; André, J.L.; Bogdanovic, R.; Burguet, A.; Cockfield, S.; et al. Mutant chromatin remodeling protein SMARCAL1 causes Schimke immuno-osseous dysplasia. Nat. Genet. 2002, 30, 215–220. [Google Scholar] [CrossRef]
- Dekel, B.; Metsuyanim, S.; Goldstein, N.; Pode-Shakked, N.; Kovalski, Y.; Cohen, Y.; Davidovits, M.; Anikster, Y. Schimke immuno-osseous dysplasia: Expression of SMARCAL1 in blood and kidney provides novel insight into disease phenotype. Pediatr. Res. 2008, 63, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, M.; Morimoto, M.; Baradaran-Heravi, A.; Choi, K.; Kambham, N.; Jensen, K.; Dutt, S.; Dionis-Petersen, K.Y.; Liu, L.X.; Felix, K.; et al. Lack of IL7Rα expression in T cells is a hallmark of T-cell immunodeficiency in Schimke immuno-osseous dysplasia (SIOD). Clin. Immunol. Orlando Fla. 2015, 161, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Yue, Z.; Xiong, S.; Sun, L.; Huang, W.; Mo, Y.; Huang, L.; Jiang, X.; Chen, S.; Hu, B.; Wang, Y. Novel compound variants of SMARCAL1 associated with severe Schimke immuno-osseous dysplasia in a Chinese patient. Nephrol. Dial. Transplant. 2010, 25, 1697–1702. [Google Scholar] [CrossRef]
- Lipska-Ziętkiewicz, B.S.; Gellermann, J.; Boyer, O.; Gribouval, O.; Ziętkiewicz, S.; Kari, J.A.; Shalaby, M.A.; Ozaltin, F.; Dusek, J.; Melk, A.; et al. PodoNet Consortium. Low renal but high extrarenal phenotype variability in Schimke immuno-osseous dysplasia. PLoS ONE 2017, 12, e0180926. [Google Scholar]
- Basiratnia, M.; Baradaran-Heravi, A.; Yavarian, M.; Geramizadeh, B.; Karimi, M. Non-hodgkin lymphoma in a child with schimke immuno-osseous dysplasia. Iran. J. Med. Sci. 2011, 36, 222–225. [Google Scholar]
- Aksu, G.; Genel, F.; Koturoğlu, G.; Kurugöl, Z.; Kütükçüler, N. Serum immunoglobulin (IgG, IgM, IgA) and IgG subclass concentrations in healthy children: A study using nephelometric technique. Turk. J. Pediatr. 2006, 48, 19–24. [Google Scholar] [PubMed]
- Schatorjé, E.J.H.; Gemen, E.F.A.; Driessen, G.J.A.; Leuvenink, J.; van Hout, R.W.N.M.; van der Burg, M.; de Vries, E. Age-matched reference values for B-lymphocyte subpopulations and CVID classifications in children. Scand. J. Immunol. 2011, 74, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Piątosa, B.; Wolska-Kuśnierz, B.; Pac, M.; Siewiera, K.; Gałkowska, E.; Bernatowska, E. B cell subsets in healthy children: Reference values for evaluation of B cell maturation process in peripheral blood. Cytom. B Clin. Cytom. 2010, 78, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Dobbs, K.; Tabellini, G.; Calzoni, E.; Patrizi, O.; Martinez, P.; Giliani, S.C.; Moratto, D.; Al-Herz, W.; Cancrini, C.; Cowan, M.; et al. Natural Killer Cells from Patients with Recombinase-Activating Gene and Non-Homologous End Joining Gene Defects Comprise a Higher Frequency of CD56bright NKG2A+++ Cells, and Yet Display Increased Degranulation and Higher Perforin Content. Front. Immunol. 2017, 8, 798. [Google Scholar] [CrossRef] [PubMed]
- Bökenkamp, A.; deJong, M.; van Wijk, J.A.; Block, D.; van Hagen, J.M.; Ludwig, M. R561C missense variant in the SMARCAL1 gene associated with mild Schimke immuno-osseous dysplasia. Pediatr. Nephrol. 2005, 20, 1724–1728. [Google Scholar] [CrossRef] [PubMed]
- Lücke, T.; Billing, H.; Sloan, E.A.; Boerkoel, C.F.; Franke, D.; Zimmering, M.; Ehrich, J.H.H.; Das, A.M. Schimke-immuno-osseous dysplasia: New variant with weak genotype-phenotype correlation in siblings. Am. J. Med. Genet. A 2005, 135, 202–205. [Google Scholar] [CrossRef]
- Gooskens, S.L.; Houwing, M.E.; Vujanic, G.M.; Dome J, S.; Diertens, T.; Coulomb-l’Herminé, A.; Godzinski, J.; Pritchard-Jones, K.; Graf, N.; van den Heuvel-Eibrink, M.M. Congenital mesoblastic nephroma 50 years after its recognition: A narrative review. Pediatr. Blood Cancer 2017, 64, e26437. [Google Scholar] [CrossRef]
- Vokuhl, C.; Nourkami-Tutdibi, N.; Furtwängler, R.; Gessler, M.; Graf, N.; Leuschner, I. ETV6-NTRK3 in congenital mesoblastic nephroma: A report of the SIOP/GPOH nephroblastoma study. Pediatr. Blood Cancer 2018, 65, e26925. [Google Scholar] [CrossRef]
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef]
- Aranburu, A.; Piano Mortari, E.; Baban, A.; Giorda, E.; Cascioli, S.; Marcellini, V.; Scarsella, M.; Ceccarelli, S.; Corbelli, S.; Cantarutti, N.; et al. Human B-cell memory is shaped by age- and tissue-specific T-independent and GC-dependent events. Eur. J. Immunol. 2017, 47, 327–344. [Google Scholar] [CrossRef]
- Scharer, C.D.; Barwick, B.G.; Guo, M.; Bally, A.P.R.; Boss, J.M. Plasma cell differentiation is controlled by multiple cell division-coupled epigenetic programs. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Poli, A.; Michel, T.; Thérésine, M.; Andrès, E.; Hentges, F.; Zimmer, J. CD56bright natural killer (NK) cells: An important NK cell subset. Immunology 2009, 126, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Judge, C.J.; Kostadinova, L.; Sherman, K.E.; Butt, A.A.; Falck-Ytter, Y.; Funderburg, N.T.; Landay, A.L.; Lederman, M.M.; Sieg, S.F.; Sandberg, J.K.; et al. CD56bright NK IL-7Rα expression negatively associates with HCV level, and IL-7-induced NK function is impaired during HCV and HIV infections. J. Leukoc. Biol. 2017, 102, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, E.; Vassena, L.; Di Cesare, S.; Malagnino, V.; Desimio, M.G.; Andreoni, M.; Barnaba, V.; Doria, M. NK cells of HIV-1-infected patients with poor CD4+ T-cell reconstitution despite suppressive HAART show reduced IFN-γ production and high frequency of autoreactive CD56bright cells. Immunol. Lett. 2017, 190, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Petty, E.M.; Yanik, G.A.; Hutchinson, R.J.; Alter B, P.; Schmalstieg F, C.; Levine J, E.; Ginsburg, D.; Robillard, J.E.; Castle, V.P. Successful bone marrow transplantation in a patient with Schimke immuno-osseous dysplasia. J. Pediatr. 2000, 137, 882–886. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.E.; Hutchinson, R.J.; DebRoy, M.; Magee, J.C. Successful renal transplantation following prior bone marrow transplantation in pediatric patients. Pediatr. Transplant. 2004, 8, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Magi, A.; Tattini, L.; Palombo, F.; Benelli, M.; Gialluisi, A.; Giusti, B.; Abbate, R.; Seri, M.; Gensini, G.F.; Romeo, G.; et al. H3M2: Detection of runs of homozygosity from whole-exome sequencing data. Bioinformatics 2014, 30, 2852–2859. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical and Genetic Features | P1 (6 Years) | P2 (18 Months) | Basiratnia 2011 (8 Years) | Yue 2010 (8 Years) | Bökenkamp 2005 (12 Years) | |
|---|---|---|---|---|---|---|
| SMARCAL1 genotype | p.Arg561His/ p.Arg561His | p.Arg561His/ p.Arg561His | p.Arg561His/ p.Arg561His | p.Arg561His/ p.Met1Ile | p.Arg561Cys/ p.Arg561Cys | |
| Clinical features | ||||||
| Growth | IUGR | + | + | NR | + | − |
| Short stature | + | + | + | + | + | |
| Skeletal features | Short neck | + | + | + | + | + |
| Short trunk | + | + | NR | + | + | |
| Thoracic scoliosis | + | NR | NR | + (kyphosis) | ||
| Lumbar lordosis | − | − | NR | NR | + | |
| Dysmorphic vertebrae | + | − | + | + | + | |
| Hypoplastic pelvis | − | − | NR | NR | NR | |
| Abnormal femoral heads | + | − | + | NR | + | |
| Microcephaly | + | + | NR | NR | NR | |
| Osteopenia | − | − | + | + | NR | |
| Renal disease and malformations | Proteinuria or nephropathy | + | + | + | + | + |
| FSGS | − | − | NR | + | + | |
| Ectopic kidney | + | − | NR | NR | NR | |
| Multicystic kidney | − | + | NR | NR | NR | |
| Heart defects | Atrial septal defects | + | + | NR | NR | NR |
| Pulmonary valve stenosis | − | + | NR | NR | NR | |
| Cerebral anomalies | Ventriculomegaly | + | − | NR | NR | NR |
| Hypoplasia corpus callosum | + | − | NR | NR | NR | |
| Physical features | Broad nasal tip | − | − | NR | + | NR |
| Wide and depressed nasal bridge | − | − | NR | + | + | |
| Protruding abdomen | − | + | + | + | + | |
| Pigmented macules | − | + | NR | + | NR | |
| Unusual hair | − | − | NR | NR | NR | |
| Microdontia | − | − | NR | NR | NR | |
| Corneal opacities | + | − | NR | NR | NR | |
| Development | Neuro-developmental delay | + | + | NR | − | − |
| Language development delay | + | − | NR | − | − | |
| Vasculature | Headaches | + | − | NR | NR | − |
| TIAs | − | − | NR | NR | − | |
| Strokes | + | − | NR | NR | − | |
| Other | Hypothyroidism | − | + | + | − | − |
| Non−Hodgkin lymphoma | − | − | + | NR | NR | |
| Recurrent infections | + | + | − | + | − | |
| Mesoblastic nephroma | − | + | − | NR | NR | |
| Extremity edema | − | − | + | + | + (minimal) | |
| Hypertension | − | − | + | NR | NR | |
| Genomic Position (hg19) | cDNA/ Protein Position | Gene | ROH Size (Mb) | GnomAD Frequency | PhyloP100way Vertebrate | CADD | Disease |
|---|---|---|---|---|---|---|---|
| chr2: | NM_001127207.2:c.1682G>A/ p.Arg561His | SMARCAL1 | 5.081 | 0.00001593 | 7.994 | 32 | SIOD |
| 217303180 | |||||||
| chr11: | NM_001198810.2:c.1471G>A/ p.Ala491Thr | SLC43A1 | 14.523 | 0.00003184 | 7.537 | 23.2 | / |
| 57254630 |
| P1 | P2 | |
|---|---|---|
| Sex | Male | Male |
| Age | 7 years | Died (15 months of age) |
| Age at diagnosis | 5 years 6 month | 1 year |
| White blood cells, 103/μL | 3.04 | 4.1 |
| Hemoglobin, g/L | 10.5 | 7.4 |
| Platelets, 103/μL | 305 | 49 |
| Neutrophils, 103/μL | 2.06 | 2.92 |
| Lymphocytes, 103/μL | 0.57 | 0.55 |
| (1.2–4.7) | (3.2–12.3) | |
| CD3+ (PAN T), % (cells/µL) | 64.2% (0.36) | 30% (0.16) |
| (0.77–4.0) | (2.4–8.3) | |
| CD3+/α+β | 86.60% | 75.40% |
| CD3+/γ+σ+ | 12.70% | 24.6% |
| CD3+CD4-CD8-, % | 1.70% | 27% |
| CD4, % (cells/µL) | 24.6% (0.14) | 14% (0.16) |
| (0.4–2.5) | (1.3–7.1) | |
| CD4+CD45 RA+ (naïve), % | 2.40% | 17% |
| (46–99) | (77–96) | |
| CD4+CD45 RA-CCR7+ (central memory), % | 55.60% | ND |
| (0.35–100) | ||
| CD4+CD45 RA-CCR7- (effector memory), % | 39.40% | ND |
| (0.27–18) | ||
| CD4+CD45 RA+CCR7- (terminal effector memory), % | 2.37% | ND |
| (0.0031–1.8) | ||
| CD3+CD4+CD31+CD45 RA+ (recent thymic emigrant) % | 2% | ND |
| (41–81) | ||
| CD8, % (cells/µL) | 26.6% (0.15) | 10% (0.05) |
| (0.2–1.7) | (0.4–4.1) | |
| CD8+CD45 RA+ (naïve), % | 2% | ND |
| (16–100) | ||
| CD8+CD45 RA-CCR7+ (central memory), % | 1.57% | ND |
| (1–6) | ||
| CD8+CD45 RA-CCR7- (effector memory), % | 80% | ND |
| (5–100) | ||
| CD8+CD45 RA+CCR7- (terminal effector memory), % | 16.50% | ND |
| (15–41) | ||
| CD56+16+CD3- (NK), % (cells/µL) | 15% (0.08) | 26% (0.14) |
| (0.012–0.34) | (0.0075–0.33) | |
| CD19 (PAN B), % (cells/µL) | 18.4% (0.1) | 42% (0.23) |
| (0.10–0.80) | (0.11–7.7) | |
| CD19+IgD+CD27- (B naïve) | 85% | 94% |
| (47.3–77.0) | (76.5–94.7) | |
| CD19+IgD+CD27+ (B memory) | 8.80% | 1% |
| (5.2–20.4) | (3.0–10.7) | |
| CD19+IgD-CD27+ (switched B memory) | 6.16% | 1.8% |
| (4.7–21.2) | (1.4–11.9) | |
| CD19+CD21+CD38- (B CD21+low) | 0.80% | ND |
| (5.9–25.8) | ||
| CD19+IgM++CD38++ (B transitional) | 18.40% | 30% |
| (4.6–8.3) | (3.6–12.7) | |
| CD19+IgM-+CD38++ (B plasmablast) | 0.10% | 1% |
| (0.6–5.3) | (0.4–5.5) | |
| IgM | 0.10 g/L | 0.56 g/L |
| (0.03-0.20) | (0.02–0.18) | |
| IgA | 0.10 g/L | 0.10 g/L |
| (0.02–0.20) | (0.02–0.15) | |
| IgG | 0.64 g/L * | 1.16 g/L * |
| (0.52–1.49) | (0.42-1-1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bertulli, C.; Marzollo, A.; Doria, M.; Di Cesare, S.; La Scola, C.; Mencarelli, F.; Pasini, A.; Affinita, M.C.; Vidal, E.; Magini, P.; et al. Expanding Phenotype of Schimke Immuno-Osseous Dysplasia: Congenital Anomalies of the Kidneys and of the Urinary Tract and Alteration of NK Cells. Int. J. Mol. Sci. 2020, 21, 8604. https://doi.org/10.3390/ijms21228604
Bertulli C, Marzollo A, Doria M, Di Cesare S, La Scola C, Mencarelli F, Pasini A, Affinita MC, Vidal E, Magini P, et al. Expanding Phenotype of Schimke Immuno-Osseous Dysplasia: Congenital Anomalies of the Kidneys and of the Urinary Tract and Alteration of NK Cells. International Journal of Molecular Sciences. 2020; 21(22):8604. https://doi.org/10.3390/ijms21228604
Chicago/Turabian StyleBertulli, Cristina, Antonio Marzollo, Margherita Doria, Silvia Di Cesare, Claudio La Scola, Francesca Mencarelli, Andrea Pasini, Maria Carmen Affinita, Enrico Vidal, Pamela Magini, and et al. 2020. "Expanding Phenotype of Schimke Immuno-Osseous Dysplasia: Congenital Anomalies of the Kidneys and of the Urinary Tract and Alteration of NK Cells" International Journal of Molecular Sciences 21, no. 22: 8604. https://doi.org/10.3390/ijms21228604
APA StyleBertulli, C., Marzollo, A., Doria, M., Di Cesare, S., La Scola, C., Mencarelli, F., Pasini, A., Affinita, M. C., Vidal, E., Magini, P., Dimartino, P., Masetti, R., Greco, L., Palomba, P., Conti, F., & Pession, A. (2020). Expanding Phenotype of Schimke Immuno-Osseous Dysplasia: Congenital Anomalies of the Kidneys and of the Urinary Tract and Alteration of NK Cells. International Journal of Molecular Sciences, 21(22), 8604. https://doi.org/10.3390/ijms21228604